SpCas9 vs SaCas9: A Comprehensive Performance Comparison for Precision Genome Editing
This article provides a detailed comparative analysis of the two most prominent CRISPR-Cas9 systems, Streptococcus pyogenes Cas9 (SpCas9) and Staphylococcus aureus Cas9 (SaCas9), tailored for researchers and drug development professionals.
SpCas9 vs SaCas9: A Comprehensive Performance Comparison for Precision Genome Editing
Abstract
This article provides a detailed comparative analysis of the two most prominent CRISPR-Cas9 systems, Streptococcus pyogenes Cas9 (SpCas9) and Staphylococcus aureus Cas9 (SaCas9), tailored for researchers and drug development professionals. We explore their foundational characteristics, including PAM requirements and molecular structure, then delve into practical applications across various cell types and model organisms. The content addresses common efficiency challenges and presents cutting-edge optimization strategies, such as gRNA scaffold engineering. Finally, we synthesize validation data on editing fidelity, specificity, and distinct mutational outcomes to guide nuclease selection for both basic research and therapeutic development, incorporating the most recent findings from 2025.
Understanding the Core Machinery: PAM Specificity, Size, and Origin of SpCas9 and SaCas9
The CRISPR-Cas9 system has revolutionized genetic engineering, offering unprecedented precision in manipulating genomes across diverse organisms. Among the numerous Cas9 proteins discovered, two bacterial nucleases stand out for their widespread adoption and unique characteristics: Streptococcus pyogenes Cas9 (SpCas9) and Staphylococcus aureus Cas9 (SaCas9). These two enzymes, while serving similar functional roles in bacterial immunity, have evolved distinct structural features that translate into significantly different performance profiles in research and therapeutic applications. Understanding their molecular origins and structural differences is crucial for researchers, scientists, and drug development professionals seeking to harness their capabilities for precision genome editing.
This guide provides a comprehensive objective comparison of SpCas9 and SaCas9, drawing upon recent structural studies and experimental data to elucidate how their evolutionary origins shape their contemporary applications. We examine their comparative performance across multiple parameters including editing efficiency, specificity, and practical utility in therapeutic contexts, providing a scientific foundation for informed nuclease selection in research and clinical applications.
Molecular Origins and Structural Evolution
SpCas9 and SaCas9 originate from evolutionarily distinct bacterial species, resulting in significant structural divergence despite their shared fundamental function as RNA-guided DNA endonucleases.
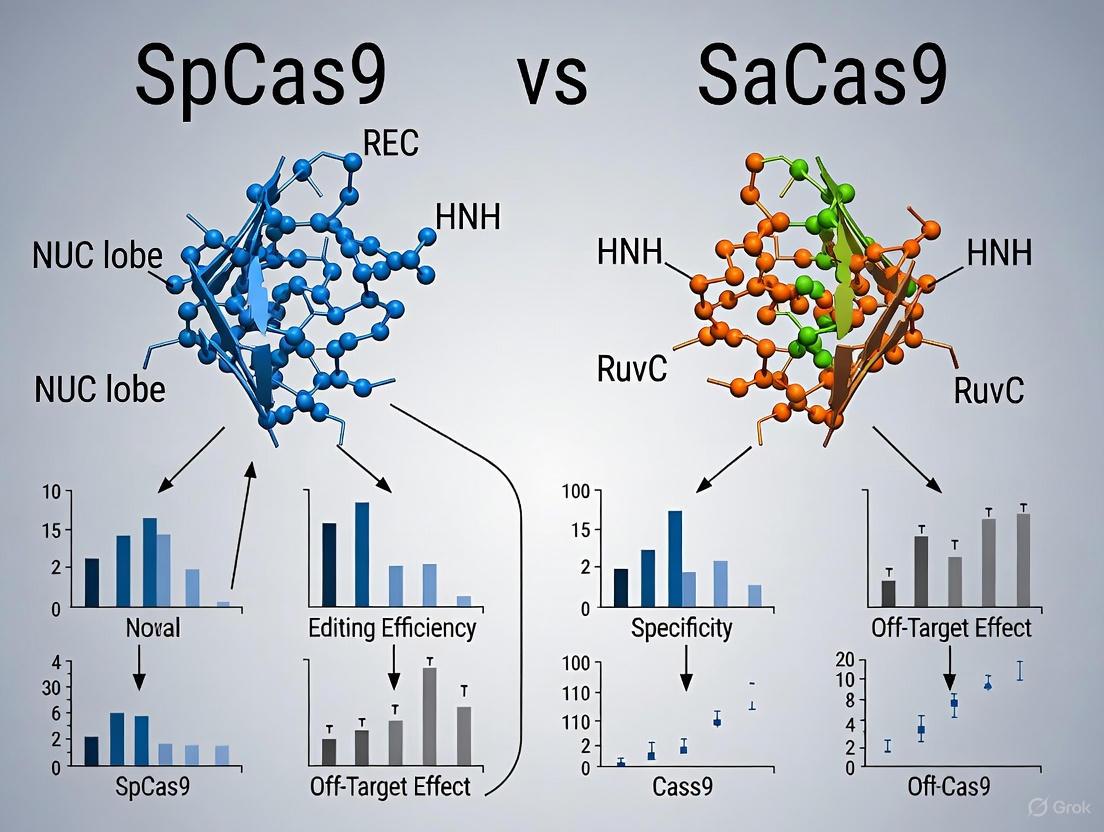
SpCas9, derived from Streptococcus pyogenes, is the pioneering CRISPR-associated nuclease that launched the genome editing revolution. With 1368 amino acid residues, it represents the larger of the two nucleases and has served as the foundational platform for numerous engineering efforts. Its structure reveals a bilobed architecture consisting of a recognition (REC) lobe and a nuclease (NUC) lobe, with a central channel accommodating the RNA-DNA heteroduplex [1]. The REC lobe, composed of REC1, REC2, and REC3 domains, facilitates guide RNA and target DNA recognition, while the NUC lobe contains the HNH and RuvC nuclease domains responsible for DNA cleavage [1].
SaCas9, isolated from Staphylococcus aureus, shares only approximately 17% sequence identity with SpCas9 yet maintains the core bilobed architecture [2]. At 1053 amino acids, it is significantly more compact—a critical advantage for viral delivery applications. Structural analyses reveal that SaCas9 similarly comprises REC and NUC lobes connected by an arginine-rich bridge helix and linker loop [2]. The NUC lobe contains RuvC, HNH, WED, and PI domains, with the WED domain representing a distinctive structural feature not initially characterized in SpCas9 [2].
The following table summarizes the key structural differences between these two nucleases:
Table 1: Fundamental Structural Characteristics of SpCas9 and SaCas9
| Structural Feature | SpCas9 | SaCas9 |
|---|---|---|
| Amino Acid Length | 1368 aa | 1053 aa |
| Sequence Identity | Reference | ~17% to SpCas9 |
| REC Lobe Composition | REC1, REC2, REC3 domains | REC domain (residues 41-425) |
| NUC Lobe Composition | HNH, RuvC domains, PI domain | RuvC, HNH, WED, PI domains |
| Notable Structural Elements | REC2/REC3 conformational flexibility in high-fidelity variants [1] | Unique WED domain, compact arrangement [2] |
| PAM-Interacting Domain | Recognizes 5'-NGG-3' PAM [1] | Recognizes 5'-NNGRRT-3' PAM [2] |
The structural divergence between these nucleases is particularly evident in their REC lobes. While SpCas9 features three distinct REC domains (REC1-3) that undergo conformational changes upon target binding, SaCas9 exhibits a more compact REC lobe organization [1] [2]. These differences in domain architecture and conformational flexibility have profound implications for their DNA recognition mechanisms and editing outcomes.
PAM Specificity and DNA Recognition Mechanisms
The Protospacer Adjacent Motif (PAM) requirement represents a fundamental constraint in CRISPR-Cas9 applications, and SpCas9 and SaCas9 recognize distinct PAM sequences through different structural mechanisms.
SpCas9 requires a 5'-NGG-3' PAM sequence immediately downstream of the target site. Structural studies reveal that this recognition is mediated primarily by residues R1333 and R1335 in the PI domain, which form specific hydrogen bonds with the guanine bases in the PAM sequence [1]. The stringent recognition mechanism involves salt bridge-stabilized conformations of these arginine residues, with E1219 playing a key role in maintaining R1335 in a conformation optimal for GG recognition [1].
SaCas9 recognizes a more complex 5'-NNGRRT-3' PAM (where R is A or G), providing both constraints and opportunities for targeting different genomic regions. Structural analyses of SaCas9 in complex with target DNA have revealed mechanisms for relaxed PAM recognition compared to SpCas9, though the specific residues involved differ [2]. The structural basis for this expanded PAM recognition involves distinct conformational arrangements in the PI domain.
Table 2: PAM Specificity and DNA Recognition Mechanisms
| PAM Characteristic | SpCas9 | SaCas9 |
|---|---|---|
| Primary PAM | 5'-NGG-3' | 5'-NNGRRT-3' |
| PAM Recognition Domain | PI domain with R1333/R1335 | PI domain with distinct residue interactions |
| Key Recognition Residues | R1333, R1335, E1219 [1] | Structure-specific interactions [2] |
| Recognition Mechanism | Salt bridge-stabilized R1335 for GG recognition [1] | Flexible accommodation of purine-rich PAM sequences [2] |
| Engineered PAM Variants | VQR (NGA), EQR (NGAG), VRER (NGCG) [3] [4] | Naturally broader PAM recognition |
The following diagram illustrates the key structural differences in the DNA recognition mechanisms between SpCas9 and SaCas9:
Comparative Performance and Editing Outcomes
Direct comparative studies reveal significant differences in the editing efficiencies, fidelity, and mutational profiles generated by SpCas9 versus SaCas9.
Editing Efficiency and Fidelity
Recent comprehensive comparisons across 11 genomic sites in human induced pluripotent stem cells (iPSCs) and K562 cells demonstrated that SaCas9 achieved higher editing efficiencies than SpCas9 at most target sites [5] [6]. Perhaps more significantly, SaCas9 exhibited superior fidelity with significantly reduced off-target effects compared to SpCas9, as validated by GUIDE-seq analysis [5] [6]. This enhanced specificity makes SaCas9 particularly valuable for therapeutic applications where minimizing off-target mutations is crucial.
The optimal spacer lengths also differ between the two systems: 20 nucleotides for SpCas9 compared to 21 nucleotides for SaCas9, though optimal length for individual guides can vary (18-21 nt for SpCas9 versus 21-22 nt for SaCas9) [5] [6].
Editing Patterns and Repair Outcomes
The two nucleases generate distinct mutational profiles that influence their suitability for different applications:
- SpCas9 exhibits a strong bias for +1 insertions at the fourth nucleotide upstream of the PAM, characteristic of a staggered cut pattern [5] [6].
- SaCas9 produces more varied indel patterns and demonstrates higher efficiency in knock-in applications, including both non-homologous end joining (NHEJ)-mediated double-stranded oligodeoxynucleotide insertion and homology-directed repair (HDR)-mediated donor integration [5] [6].
Table 3: Experimental Performance Comparison of SpCas9 vs. SaCas9
| Performance Metric | SpCas9 | SaCas9 |
|---|---|---|
| Editing Efficiency | High, but lower than SaCas9 at most sites [6] | Superior efficiency at most of 11 tested sites [6] |
| Off-Target Effects | Significant off-target effects [6] | Significantly reduced off-target effects [6] |
| Optimal Spacer Length | 20 nt (range: 18-21 nt) [6] | 21 nt (range: 21-22 nt) [6] |
| Indel Pattern Bias | Strong +1 insertion bias at 4th nt upstream of PAM [6] | More varied indel patterns [6] |
| Knock-in Efficiency | Moderate | Higher efficiency for dsODN insertion and HDR [6] |
| Therapeutic Applications | Limited by size and specificity | Preferred for AAV delivery and therapeutic knock-in [6] |
Guide RNA Optimization Strategies
A critical factor influencing the performance of both nucleases is guide RNA (gRNA) expression levels. Recent research has identified optimization strategies that significantly enhance editing efficiency, particularly under the constrained conditions typical of therapeutic applications.
The standard gRNA scaffold contains a sequence of four thymine nucleotides (4T) that can inhibit transcription from Pol III promoters such as the U6 promoter [7] [8]. While this inhibition doesn't significantly affect editing efficiency under standard transfection protocols with abundant vector availability, it becomes limiting when vector quantities are constrained [7].
Reducing the poly-T tract from 4T to 3TC (by replacing the fourth thymine with cytosine in the tetraloop) significantly increases gRNA transcript levels [7] [8]. This optimization:
- Boosted gRNA levels by 8.1–13.5 doublings (271-11,349-fold increases) in experimental systems [7]
- Enhanced editing efficiency for previously suboptimal gRNAs to over 95% [7]
- Proved particularly beneficial under conditions of limited vector availability [7]
- Was compatible with both SpCas9 and SaCas9, as well as high-fidelity variants and base editors [7]
The following diagram illustrates the experimental workflow for comparing Cas9 performance and optimizing gRNA expression:
Research and Therapeutic Applications
The structural and functional differences between SpCas9 and SaCas9 translate into distinct advantages for specific research and therapeutic applications.
Therapeutic Implementation
The compact size of SaCas9 (1053 aa) enables efficient packaging into adeno-associated virus (AAV) vectors for in vivo gene therapy applications, a significant advantage over the larger SpCas9 [2]. This property has made SaCas9 the nuclease of choice for therapeutic approaches like EDIT-101, a clinical candidate for treating CEP290-related retinal degeneration, where the 3TC scaffold modification demonstrated marked improvements in performance [7] [8].
The superior fidelity of SaCas9, combined with its efficient knock-in capabilities, makes it particularly valuable for therapeutic applications requiring precise gene integration while minimizing off-target effects [5] [6].
Experimental Design Considerations
For researchers designing CRISPR experiments, several practical considerations emerge from these comparative analyses:
- Target Selection: The distinct PAM requirements of SpCas9 (NGG) versus SaCas9 (NNGRRT) significantly influence targetable sites within the genome
- Delivery Method: SaCas9 is strongly preferred for AAV delivery due to its smaller size
- Specificity Requirements: For applications requiring high specificity, SaCas9's superior fidelity makes it the preferred choice
- Desired Editing Outcome: The distinct indel patterns and knock-in efficiencies may guide nuclease selection based on the specific genetic modification desired
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents and Experimental Tools for Cas9 Studies
| Reagent/Tool | Function/Application | Examples/References |
|---|---|---|
| CATS Bioinformatic Tool | Automated detection of overlapping PAM sequences for comparing Cas9 nucleases | Identifies shared target sites for fair nuclease comparison [9] |
| PX459.v2 Plasmid | All-in-one CRISPR plasmid (CBh-SpCas9-T2A-Puro + U6-sgRNA) | Standard SpCas9 expression system [7] |
| 3TC-Modified Scaffold | gRNA scaffold with reduced poly-T tract for enhanced transcription | Increases gRNA levels and editing efficiency under limited vector availability [7] |
| High-Fidelity Variants | Engineered Cas9 versions with reduced off-target effects | SpCas9-HF1, eSpCas9(1.1) [7] [1] |
| GUIDE-seq | Comprehensive method for profiling genome-wide off-target effects | Validated SaCas9's superior fidelity [6] |
| AAV Vectors | Viral delivery system for in vivo genome editing | SaCas9's compact size enables efficient packaging [2] |
SpCas9 and SaCas9 represent two powerful but distinct genome editing platforms with complementary strengths and limitations. SpCas9, the pioneering enzyme, offers well-characterized behavior and extensive engineering variants but is limited by its larger size and higher off-target effects. SaCas9, with its compact architecture, superior fidelity, and efficient knock-in capabilities, has emerged as particularly valuable for therapeutic applications where delivery and specificity are paramount.
The molecular origins of these nucleases in different bacterial species have engendered structural differences that translate directly to their performance characteristics. Understanding these differences enables researchers to make informed decisions about nuclease selection based on their specific experimental or therapeutic requirements. As CRISPR technology continues to evolve, both nucleases will likely play important roles in advancing research and developing genetic therapies.
The CRISPR-Cas9 system has revolutionized biomedical research and therapeutic development by providing unprecedented capability for targeted genome manipulation. At the core of this technology lies a critical recognition element: the protospacer adjacent motif (PAM). This short DNA sequence adjacent to the target site serves as a binding signal for Cas enzymes, enabling them to distinguish between self and non-self DNA in bacterial adaptive immunity [10]. In engineered CRISPR systems, the PAM requirement represents both a targeting mechanism and a fundamental constraint—dictating which genomic loci can be accessed for editing [11] [12].
For researchers and drug development professionals, understanding PAM specifications is crucial for experimental design and therapeutic application. The PAM sequences for the two most widely used Cas9 orthologs—Streptococcus pyogenes Cas9 (SpCas9) and Staphylococcus aureus Cas9 (SaCas9)—create dramatically different targeting landscapes across the genome [7] [10]. This comparison guide provides a comprehensive analysis of their distinct PAM requirements, targeting capabilities, and experimental considerations to inform strategic nuclease selection for research and clinical applications.
PAM Fundamentals and Molecular Recognition Mechanisms
The Biological Function of PAM Sequences
In native bacterial CRISPR systems, PAM sequences solve a critical self/non-self discrimination problem. When Cas enzymes store fragments of viral DNA in the CRISPR array for future immunity, they exclude the PAM sequence. This ensures that the bacterial genome itself, which contains the matching spacer sequences but lacks the flanking PAM, is not recognized as a target [10]. In engineered CRISPR systems, this biological mechanism translates to a simple design rule: any target site must be adjacent to the appropriate PAM sequence for the Cas nuclease being used.
Structural Basis of PAM Recognition
The molecular mechanism of PAM recognition differs significantly between Cas9 variants. For SpCas9, structural studies have revealed that two arginine residues (R1333 and R1335) in the PAM-interacting domain form specific contacts with the nucleobases of the NGG PAM sequence [13]. This interaction enforces strict specificity through rigid structural constraints that favor guanine bases. The recent evolution of xCas9, an SpCas9 variant with broadened PAM compatibility, demonstrates how introducing flexibility into this recognition interface (particularly at R1335) can expand targeting capability while maintaining specificity [11] [13].
Table 1: Key Structural Features Governing PAM Recognition
| Feature | SpCas9 | SaCas9 | xCas9 (SpCas9 variant) |
|---|---|---|---|
| Key PAM-Interacting Residues | R1333, R1335 | Not specified in results | E1219V, R1333, R1335 |
| Recognition Mechanism | Rigid arginine dyad enforcing guanine specificity | Not specified | Flexible R1335 enabling broader recognition |
| Recognition Domain | C-terminal PAM-interacting domain | Not specified | C-terminal PAM-interacting domain with distributed mutations |
Comparative Analysis of SpCas9 and SaCas9 PAM Requirements
Defining the PAM Sequences and Targeting Densities
The PAM requirements for SpCas9 and SaCas9 create fundamentally different genomic targeting landscapes:
SpCas9 recognizes a simple 5'-NGG-3' PAM sequence immediately following the target protospacer [10]. This 3-nucleotide motif occurs approximately once every 16 base pairs in random DNA sequence, creating a relatively dense targeting landscape across the genome [11].
SaCas9 requires a more complex 5'-NNGRRT-3' (where R is A or G) PAM sequence [7] [10]. This longer, more specific 6-nucleotide motif occurs less frequently, substantially reducing the density of potential target sites but potentially increasing specificity.
Table 2: Comprehensive PAM and Targeting Comparison
| Parameter | SpCas9 | SaCas9 | Experimental Evidence |
|---|---|---|---|
| PAM Sequence | 5'-NGG-3' | 5'-NNGRRT-3' or 5'-NNGRRN-3' | GenomePAM validation in human cells [14] [10] |
| PAM Length | 3 nucleotides | 5-6 nucleotides | Established characterization [7] [10] |
| PAM Position | 3' of protospacer | 3' of protospacer | Consistent for type II Cas nucleases [14] |
| Theoretical Targeting Density | ~1 in 16 bp | ~1 in 1024 bp (for NNGRRT) | Calculated from sequence probability |
| Canonical PAM Recognition | Rigid specificity for GG dinucleotide | Specificity for G-rich sequences | Structural studies [13] |
| Engineered PAM Flexibility | xCas9: NG, GAA, GAT [11] | Limited data in results | PACE evolution [11] [12] |
Targeting Scope Implications for Genome Editing Applications
The different PAM requirements directly impact which genomic regions can be targeted, with particular significance for precision editing applications:
Base Editing: Cytosine and adenine base editors require precise positioning of the Cas9 relative to the target nucleotide (typically within a ~13-17 nucleotide window from the PAM) [12]. The more frequent SpCas9 PAM sites offer greater flexibility for positioning base editors optimally.
Therapeutic Targeting: The compact size of SaCas9 (~1 kilobase smaller than SpCas9) enables packaging into adeno-associated virus (AAV) vectors for in vivo gene therapy, making its PAM requirements a critical consideration for therapeutic target selection [7].
Allele-Specific Editing: Single nucleotide polymorphisms (SNPs) that create or disrupt PAM sequences enable allele-specific targeting. The longer SaCas9 PAM may offer advantages for discriminating between disease-associated and wild-type alleles in dominant disorders [15].
Experimental Characterization of PAM Requirements
Methodologies for PAM Determination
Recent advances in PAM characterization methods have enabled more accurate profiling of nuclease specificity in mammalian cells:
GenomePAM: A Novel Method for Direct PAM Characterization in Mammalian Cells
The recently developed GenomePAM method addresses key limitations of previous approaches by leveraging naturally occurring repetitive sequences in the mammalian genome [14]. This innovative methodology:
- Utilizes genomic repeats (e.g., a 20-nt Alu-derived sequence occurring ~16,942 times per diploid human cell) as naturally occurring target libraries
- Eliminates the need for synthetic oligo libraries or protein purification
- Captures cleavage events via GUIDE-seq integration followed by sequencing
- Identifies functional PAMs through statistical enrichment of sequences flanking cleaved sites
Experimental workflow:
- Target Identification: Bioinformatic identification of highly repetitive genomic sequences with diverse flanking regions
- gRNA Design: Construction of guide RNAs targeting the selected repetitive element
- Editing & Capture: Transfection of Cas-gRNA complex followed by GUIDE-seq to capture cleavage sites
- PAM Analysis: Sequencing and motif analysis to determine enriched flanking sequences (PAMs)
This method has been validated for SpCas9, SaCas9, and Cas12a nucleases, accurately reproducing their known PAM requirements and enabling characterization in physiologically relevant contexts [14].
Quantitative PAM Preference Profiling
Advanced characterization methods have revealed that PAM recognition is not binary but exists on a spectrum of binding affinity and cleavage efficiency. For example, while SpCas9 strongly prefers NGG PAMs, engineered variants like xCas9 show measurable activity on NG, GAA, and GAT PAMs [11]. Similarly, SaCas9 demonstrates a preference hierarchy within the NNGRRT motif, with some sequences supporting more efficient editing than others [14].
Practical Implications for Experimental Design
Nuclease Selection Framework
The choice between SpCas9 and SaCas9 involves balancing multiple factors beyond PAM requirements:
gRNA Design Optimization
Recent studies have revealed that gRNA expression levels significantly impact editing efficiency, particularly for SaCas9 and high-fidelity SpCas9 variants [7]. Optimization strategies include:
- Scaffold Modification: Shortening the poly-T tract in the gRNA scaffold from 4T to 3TC to enhance transcription from U6 promoters
- Expression Tuning: Implementing dual promoter systems or modulating delivery dosage to achieve optimal gRNA levels
- Sequence Optimization: Avoiding T-rich gRNA sequences that exacerbate transcription termination issues
These optimizations are particularly valuable in therapeutic contexts where vector payload limits constrain delivery [7].
Research Reagent Solutions and Computational Tools
Table 3: Essential Research Tools for PAM Analysis and Nuclease Comparison
| Tool/Reagent | Function | Application Context |
|---|---|---|
| GenomePAM Method [14] | Direct PAM characterization using genomic repeats | Determining PAM specificity in mammalian cells |
| CATS Bioinformatic Tool [15] | Automated detection of overlapping PAM sequences for different nucleases | Comparing targeting capabilities across Cas variants |
| 3TC gRNA Scaffold [7] | Enhanced gRNA expression by reducing transcription termination | Improving editing efficiency for SaCas9 and high-fidelity variants |
| Phage-Assisted Continuous Evolution (PACE) [11] [12] | Directed evolution of novel PAM specificities | Engineering Cas variants with expanded targeting scope |
| xCas9 Variants [11] [13] | SpCas9 derivatives with broad PAM recognition (NG, GAA, GAT) | Targeting sites inaccessible to wild-type SpCas9 |
The PAM requirements for SpCas9 (NGG) and SaCas9 (NNGRRT) create distinct targeting landscapes with significant implications for experimental design and therapeutic development. While SpCas9 offers greater targeting density due to its simpler PAM, SaCas9 provides advantages in viral delivery and potentially higher specificity. Recent methodological advances, including GenomePAM for characterization and CATS for bioinformatic comparison, are empowering researchers to make more informed nuclease selections based on empirical data rather than theoretical considerations.
The ongoing evolution of Cas9 variants with expanded PAM compatibility, such as xCas9, continues to blur the historical tradeoffs between targeting scope, specificity, and efficiency. For researchers and drug developers, understanding these fundamental recognition mechanisms and their practical implications remains essential for harnessing the full potential of CRISPR-based genome editing.
The development of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-based therapeutics faces a significant delivery challenge, particularly for in vivo applications. The preferred delivery vehicle, Adeno-Associated Virus (AAV), has a strict packaging limit of approximately 4.7 kilobases (kb), which includes the essential regulatory elements, the Cas9 coding sequence, and the guide RNA expression cassette [16] [17]. This physical constraint directly pits the size of the CRISPR machinery against delivery efficiency. Within this context, Staphylococcus aureus Cas9 (SaCas9) emerges as a critical solution due to its compact structure, while the more commonly used Streptococcus pyogenes Cas9 (SpCas9) exceeds AAV packaging capacity. This comparison guide objectively analyzes the structural and functional advantages of SaCas9, providing researchers with experimental data and methodologies to inform therapeutic development decisions.
Structural and Functional Comparison of SaCas9 and SpCas9
The fundamental difference between SaCas9 and SpCas9 lies in their molecular dimensions, which dictates their compatibility with viral delivery systems.
Table 1: Fundamental Characteristics of SaCas9 and SpCas9
| Feature | SaCas9 | SpCas9 |
|---|---|---|
| Amino Acids | 1,053 aa [18] [19] [20] | 1,368 aa [19] [20] |
| Coding Sequence Size | ~3.2 kb [16] | ~4.2 kb [16] |
| AAV Co-packaging with sgRNA | Feasible [16] | Not feasible (exceeds capacity) [16] [17] |
| PAM Sequence | NNGRRT (where R is A or G) [19] [20] | NGG [19] |
| PAM Occurrence in Genome | ~1 in 32 base pairs [19] | ~1 in 8 base pairs [19] |
| Target Sequence Length | 21-22 nucleotides [19] | 20 nucleotides [20] |
The 1053-amino-acid length of SaCas9 is its most defining advantage, making it over 300 amino acids shorter than SpCas9 [19]. This size difference translates directly into a coding sequence that is approximately 1 kb smaller, allowing it to be packaged into a single AAV vector alongside its single-guide RNA (sgRNA) and necessary regulatory elements [16]. In contrast, the SpCas9 coding sequence alone consumes 4.2 kb of the 4.7 kb AAV capacity, leaving insufficient space for the sgRNA expression cassette [17]. This fundamental limitation necessitates complex and less efficient workarounds for SpCas9 delivery, such as using dual AAV vectors, which can reduce therapeutic editing potential [17].
The following diagram illustrates how SaCas9's compact size enables efficient AAV packaging, a key advantage for in vivo therapeutic applications.
Diagram 1: AAV Packaging Advantage of SaCas9. SaCas9's compact size allows co-packaging with its sgRNA in a single AAV vector, while the larger SpCas9 system exceeds viral capacity.
Beyond size, the Protospacer Adjacent Motif (PAM) requirements of the two nucleases differ significantly and influence their targeting scope. SaCas9 recognizes the NNGRRT PAM (where R is A or G), which appears approximately once every 32 base pairs in the genome [19]. While this offers a lower theoretical target density than the SpCas9 NGG PAM (appearing every 8 base pairs), it provides a distinct sequence recognition landscape that can be advantageous for targeting specific genomic regions and may contribute to higher specificity [19]. Furthermore, SaCas9 utilizes a slightly longer guide sequence (21-22 nt versus 20 nt for SpCas9), which potentially enhances its target discrimination capability [19] [20].
Experimental Performance and Editing Outcomes
Editing Efficiency and Fidelity
Multiple independent studies have systematically compared the editing performance of SaCas9 and SpCas9. A rigorous 2022 study in Genomics, Proteomics & Bioinformatics compared both nucleases at 11 target sites in human induced pluripotent stem cells (iPSCs) and K562 cells [19]. The research employed a standardized experimental protocol: nuclease plasmids were electroporated into cells, target loci were amplified with barcoded primers 48-72 hours post-transfection, and editing efficiency was quantified via high-throughput sequencing followed by CRISPResso2 analysis [19]. The study found that SaCas9 not only achieved editing efficiencies comparable to SpCas9 but often exceeded them, with the optimal spacer length being 21-22 nucleotides for SaCas9 and 20 nucleotides for SpCas9 [19].
Perhaps more importantly, the study revealed fundamental differences in editing outcomes. SpCas9 exhibited a more substantial bias for nonhomologous end-joining (NHEJ) +1 insertion at the fourth nucleotide upstream of the PAM, characteristic of a staggered cut [19]. In contrast, SaCas9 produced different indel patterns, which resulted in higher efficiencies for NHEJ-mediated double-stranded oligodeoxynucleotide (dsODN) insertion and homology-directed repair (HDR) using AAV6 donors [19]. This makes SaCas9 particularly valuable for knock-in applications where precise gene integration is required.
Table 2: Experimental Performance Comparison of SaCas9 and SpCas9
| Performance Metric | SaCas9 | SpCas9 | Experimental Context |
|---|---|---|---|
| Average Editing Efficiency | Superior at multiple sites [19] | Variable [19] | K562 cells and iPSCs, 11 target sites [19] |
| Optimal Spacer Length | 21-22 nt [19] | 20 nt [19] | Systematic testing of spacer lengths [19] |
| Indel Pattern | Lower +1 insertion bias [19] | High +1 insertion bias [19] | CRISPResso2 analysis of NHEJ outcomes [19] |
| HDR/Knock-in Efficiency | Higher [19] | Lower [19] | AAV6 donor delivery in human cells [19] |
| Off-Target Effects | Significantly reduced [19] | Higher [19] | GUIDE-seq analysis [19] |
| Plant Editing Efficiency | 75.6% (NtPDS), 65.1% (NtFT4) [20] | Comparable to SaCas9 [20] | Tobacco transformation [20] |
The fidelity of SaCas9 represents another significant advantage. GUIDE-seq analysis revealed that SaCas9 exhibited significantly reduced off-target effects compared to SpCas9 [19]. This enhanced specificity, combined with its more restricted PAM requirement, makes SaCas9 a preferable choice for therapeutic applications where minimizing unintended genomic alterations is paramount.
Performance in Challenging Conditions: The Impact of gRNA Optimization
Recent research has revealed that SaCas9 performance can be substantially enhanced through guide RNA (gRNA) scaffold optimization. A 2025 study demonstrated that the standard gRNA scaffold contains a sequence of four thymine nucleotides (4T) that can inhibit transcription from U6 promoters [7]. The researchers tested a simple modification—shortening the 4T tract to 3TC by replacing the fourth T in the tetraloop with a C and its complementary A with a G [7].
The experimental protocol involved:
- Constructing modified gRNA scaffolds containing the 3TC mutation in the PX459.v2 plasmid backbone [7].
- Transfecting cells with both original (4T) and modified (3TC) scaffolds under varying vector availability conditions [7].
- Quantifying gRNA transcript levels using qPCR and measuring editing efficiency through sequencing analysis [7].
This modification dramatically increased gRNA transcript levels by 8.1–13.5 doublings (271-11,349 fold changes) and subsequently enhanced editing efficiency, particularly for initially low-performing gRNAs [7]. The benefit was most pronounced under conditions of limited vector availability, a common scenario in therapeutic applications where viral vector doses are constrained [7]. This optimization strategy also proved compatible with SaCas9 and was successfully applied to the EDIT-101 therapeutic strategy for treating a form of inherited blindness, demonstrating its clinical relevance [7].
Diagram 2: gRNA Scaffold Optimization Enhances SaCas9 Efficiency. Modifying the gRNA scaffold to reduce poly-T tracts boosts transcription and improves editing, especially when vector doses are low.
Therapeutic Applications and Clinical Translation
Addressing Immunogenicity Challenges
A significant hurdle for all CRISPR-based therapeutics, including those utilizing SaCas9, is pre-existing immunity in human populations. As bacterial-derived proteins, Cas nucleases can trigger immune responses that may compromise therapy safety and efficacy. Recent research has quantified this challenge, revealing that approximately 78% of healthy individuals have class-switched immunoglobulin G (IgG) antibodies against SaCas9 [21] [22].
A 2025 study in Nature Communications addressed this problem by rationally engineering reduced immunogenicity (Redi) variants of SaCas9 [21] [22]. The research methodology involved:
- Identifying immunogenic epitopes through MHC-associated peptide proteomics (MAPPs) analysis on HLA-A*02:01-expressing cells transfected with SaCas9 [21] [22].
- Computationally designing mutants using Rosetta protein design package to reduce MHC-binding propensity while preserving nuclease activity [21] [22].
- Validating variants through ELISpot assays to measure T-cell recognition and transfection-based editing efficiency tests [21] [22].
This process identified three immunodominant epitopes in SaCas9 and successfully generated triple mutants (e.g., SaCas9.Redi.1 - L9A/I934T/L1035A) that maintained wild-type levels of nuclease activity across multiple target sites while significantly reducing CD8+ T cell reactivity [21] [22]. In vivo editing of PCSK9 with SaCas9.Redi.1 demonstrated efficiency comparable to wild-type SaCas9 but with substantially reduced immune responses [21] [22]. This engineering breakthrough provides a promising path forward for clinical applications by mitigating immunogenicity concerns.
Expanding the Toolkit: Split-SaCas9 Systems
Further enhancing its therapeutic potential, researchers have developed split-SaCas9 systems where the protein is divided into two inactive fragments that reassemble inside cells [18]. This approach offers multiple advantages:
- Further reduces individual component sizes for flexible vector packaging [18].
- Enables temporal and spatial control of editing activity [18].
- Permits delivery via multiple vectors, including plant virus vectors for integration-free genome editing [18].
Experimental work in Nicotiana benthamiana leaves demonstrated that two split-SaCas9 systems (430N/431C and 739N/740C) expressed via Agrobacterium infiltration successfully induced targeted mutagenesis, with the 739N/740C system exhibiting activity almost identical to full-length SaCas9 [18]. This split-protein strategy represents an innovative solution for overcoming delivery constraints while maintaining editing efficiency.
Essential Research Reagent Solutions
The following table catalogs key reagents and tools essential for conducting SaCas9-based research, particularly for therapeutic development applications.
Table 3: Research Reagent Solutions for SaCas9 Applications
| Reagent/Tool | Function/Application | Key Features |
|---|---|---|
| Optimized SaCas9 gRNA (Sa-v2) | Enhanced editing efficiency [19] | Contains T4>C mutation and UGUCG extension; increases transcription [19] |
| SaCas9.Redi Variants | Reduced immunogenicity for therapeutic use [21] [22] | Triple-point mutations (e.g., L9A/I934T/L1035A); retain activity while evading immune detection [21] [22] |
| Split-SaCas9 Systems | Flexible delivery and spatial/temporal control [18] | 739N/740C fragment system maintains near full-length activity [18] |
| Dual AAV-SaCas9 System | In vivo delivery of large cargo [17] | Co-packaging of SaCas9 and gRNA in separate AAVs; requires high viral dose [17] |
| CATS Bioinformatic Tool | Comparing Cas9 nucleases with different PAM requirements [9] | Automates detection of overlapping PAM sequences; integrates ClinVar data for therapeutic target identification [9] |
The compact structure of SaCas9 provides a definitive advantage for viral delivery applications, particularly within the stringent packaging constraints of AAV vectors. While SpCas9 remains a valuable research tool for basic science applications, SaCas9 demonstrates superior characteristics for therapeutic development, including robust editing efficiency, enhanced specificity with reduced off-target effects, and compatibility with single-AAV delivery. Recent innovations in gRNA scaffold optimization, split-protein systems, and engineered low-immunogenicity variants have further strengthened SaCas9's position as a premier platform for in vivo genome editing. For researchers and drug development professionals designing CRISPR-based therapeutics, SaCas9 represents a versatile and clinically viable nuclease that effectively balances size constraints with editing performance.
The CRISPR-Cas9 system has revolutionized genome engineering, with SpCas9 from Streptococcus pyogenes and SaCas9 from Staphylococcus aureus emerging as two of the most prominent editors. While both function as RNA-guided endonucleases, they differ significantly in molecular size, protospacer adjacent motif (PAM) requirements, and their optimal guide RNA configurations. Among these parameters, spacer length—the sequence in the guide RNA that determines target specificity—plays a crucial role in balancing editing efficiency with specificity. Systematic comparative studies have revealed that SpCas9 and SaCas9 perform best with distinct spacer lengths: primarily 20 nucleotides (nt) for SpCas9 and 21 nt for SaCas9 [5] [6]. This guide objectively compares the experimental evidence supporting these optimal spacer lengths, providing researchers with validated protocols and data-driven recommendations for implementing both systems effectively.
Comparative Performance Data: Spacer Length Directly Influences Editing Outcomes
Direct, side-by-side comparisons of SpCas9 and SaCas9 editing across multiple genomic loci in human cells have quantified the distinct spacer length requirements for each nuclease. The table below synthesizes key performance metrics from these comparative studies.
Table 1: Performance Comparison of SpCas9 and SaCas9 at Their Optimal Spacer Lengths
| Parameter | SpCas9 (20-nt spacer) | SaCas9 (21-nt spacer) | Experimental Context |
|---|---|---|---|
| Optimal Spacer Length | 20 nt [5] [6] | 21 nt [5] [6] [23] | 11 target sites in human iPSCs and K562 cells [5] [6] |
| Functional Spacer Range | 18–21 nt [5] [6] | 21–22 nt (high efficiency); 19–23 nt (functional) [5] [6] [23] | Systematic screening of 88,692 guide-target pairs in HEK293FT cells [23] |
| Cleavage Efficiency | High efficiency with 17-23 nt spacers, best with 17-20 nt [24] | High efficiency with 21-23 nt spacers [24] | 90 target sites in genes with varying expression levels in HEK293T cells [24] |
| Mismatch Tolerance | N/A | 20-nt spacers are markedly less tolerant of mismatches than 21-nt or 22-nt spacers [23] | Pairwise library screen with single and double mismatches [23] |
| Knock-in Efficiency | Lower efficiency for HDR-mediated AAV6 donor knock-in [5] | Higher efficiency for HDR-mediated AAV6 donor knock-in [5] | Characterization of editing outcomes in human iPSCs and K562 cells [5] |
| Off-target Effects | Significantly higher off-target effects [5] | Significantly reduced off-target effects [5] | GUIDE-seq analysis [5] |
The Impact of Spacer Length on Specificity
Beyond pure efficiency, spacer length is a critical determinant of specificity. For SaCas9, high-efficiency 20-nt spacers show markedly reduced tolerance to mismatched target sequences compared to 21-nt or 22-nt spacers [23]. This makes 20-nt SaCas9 guides inherently more specific, though this comes with the trade-off of requiring a perfectly matched target site for activity. This relationship is less pronounced for SpCas9, though the general principle that longer spacers can accommodate more mismatches holds true across systems. The PAM-proximal "seed" region remains critical for both nucleases, but the longer optimal spacer for SaCas9 extends its sequence-specific recognition capacity, contributing to its observed superior fidelity with reduced off-target effects in GUIDE-seq analyses [5] [6].
Experimental Protocols for Determining Optimal Spacer Length
Large-Scale Pairwise Library Screening for SaCas9
A high-throughput method was developed to systematically interrogate SaCas9 specificity and spacer length preferences in human cells [23].
- Library Design: A library of 88,692 guide-target pairs was synthesized. The design included sgRNAs with spacer lengths ranging from 19 to 24 nt, paired with target sites containing all possible single mismatches, double mismatches, and bulges.
- Delivery and Expression: The library was packaged into lentivirus and transduced into HEK293FT cells at a low multiplicity of infection to ensure single-copy integrations. SaCas9 was delivered via a separate transduction step.
- Editing Measurement: Genomic DNA was harvested at days 3 and 14 post-transduction. The integrated target site was amplified and deep-sequenced. A unique Hamming barcode embedded in the cassette allowed accurate tracking of original guide-target pairs even after editing-induced indels.
- Data Analysis: Indel frequencies were calculated for each guide-target pair. On-target efficiency was compared across spacer lengths, and off-target ratios were computed for mismatched targets.
Figure 1: Workflow for high-throughput pairwise library screen to determine SaCas9 spacer length preferences and specificity.
Multi-Locus Comparison in Human Cell Lines
A more targeted approach directly compared both nucleases across a panel of endogenous sites [5] [6] [24].
- Cell Lines and Transfection: Studies were conducted in human induced pluripotent stem cells (iPSCs), K562 cells, and HEK293T cells [5] [24]. All-in-one CRISPR plasmids expressing the Cas9 nuclease and sgRNA were transfected using standard protocols.
- Spacer Length Testing: For a set of 11 target sites, sgRNAs with varying spacer lengths (18–21 nt for SpCas9 and 19–23 nt for SaCas9) were designed and tested [5] [6].
- Efficiency Quantification: Editing efficiency was quantified by next-generation sequencing of PCR-amplified target loci. Indel patterns and frequencies were analyzed bioinformatically.
- Specificity Assessment: Off-target effects were profiled using genome-wide methods like GUIDE-seq [5].
Figure 2: Experimental workflow for direct multi-locus comparison of SpCas9 and SaCas9 spacer lengths in human cells.
Successful spacer optimization requires a suite of reliable reagents and tools. The table below lists key solutions used in the cited studies.
Table 2: Key Research Reagent Solutions for Spacer Length Optimization Studies
| Reagent / Resource | Function in Experiment | Example Use Case |
|---|---|---|
| All-in-one CRISPR Plasmid (e.g., PX459.v2) | Co-expresses Cas9 (CBh promoter) and sgRNA (U6 promoter) from a single vector [7]. | Standardized testing of sgRNA activity across different cell lines [5] [7]. |
| Modified gRNA Scaffold (3TC) | Replaces the 4T sequence in the standard scaffold to enhance gRNA transcription by RNA Pol III, boosting editing efficiency, especially for T-rich guides [7]. | Improving editing efficiency of low-performing gRNAs and under conditions of limited vector availability [7]. |
| Lentiviral Pairwise Library | Enables high-throughput screening of thousands of guide-target pairs in a controlled genomic context [23]. | Systematic profiling of SaCas9 mismatch tolerance and optimal spacer length [23]. |
| Error-Correcting Barcodes | Short DNA sequences embedded in library constructs that allow accurate identification of guide-target pairs after editing-induced mutations [23]. | Ensuring high-fidelity recovery of original guide-target relationships in deep sequencing data [23]. |
| U6 Promoter-driven sgRNA Cassette | Provides high-level, Pol III-mediated expression of sgRNAs with precise start and end sites, maintaining designed spacer length [23]. | Faithful expression of sgRNAs with spacer lengths from 18 to 24 nt in human cells [20] [23]. |
The empirical data clearly dictates that 20-nt and 21-nt spacers are optimal for SpCas9 and SaCas9, respectively. This one-nucleotide difference reflects deeper mechanistic variations in how these orthologs engage with their target DNA. For researchers, adhering to these guidelines is a critical first step in designing effective editing experiments. The choice between SpCas9 and SaCas9 should be guided by the target sequence availability (dictated by their distinct PAMs), delivery constraints (where SaCas9's smaller size is advantageous for viral packaging), and the requirement for high fidelity, where SaCas9 holds a demonstrated advantage [5] [25]. Future efforts will continue to refine these systems through engineered high-fidelity variants and novel scaffold designs [7] [25], but the foundational principle of nuclease-specific spacer length optimization will remain a cornerstone of efficient and precise genome editing.
From Bench to Bedside: Practical Applications in Cell Lines, Animal Models, and Therapeutics
The selection of a CRISPR-Cas9 nuclease is a critical determinant of success in mammalian genome editing. While the most widely used nuclease, Streptococcus pyogenes Cas9 (SpCas9), has become a laboratory staple, its smaller ortholog, Staphylococcus aureus Cas9 (SaCas9), presents distinct advantages for specific applications. Framed within a broader thesis comparing SpCas9 and SaCas9 performance, this guide provides an objective, data-driven comparison of their editing efficiencies. We focus on experimentally quantified outcomes in HEK293T cells, human induced pluripotent stem cells (iPSCs), K562 cells, and touch upon considerations for mouse embryos, offering researchers a clear framework for nuclease selection in drug development and basic research.
Head-to-Head Performance Comparison in Mammalian Cell Lines
Direct comparative studies reveal that SaCas9 can achieve not only high but often superior editing efficiencies compared to SpCas9 across multiple human cell types.
Quantitative Efficiency and Off-Target Profile
A rigorous 2022 study systematically compared SpCas9 and SaCas9 at 11 target sites within 8 genes (including AAVS1, CCR5, and TRAC) with clinical application prospects in human iPSCs and K562 cells [19] [5]. The research found that SaCas9 edited the genome with greater efficiencies than SpCas9 in these systems [19]. Furthermore, GUIDE-seq analysis, a sensitive method for detecting off-target sites, revealed a significant finding: SaCas9 exhibited significantly reduced off-target effects compared with SpCas9 [19] [5].
Table 1: Comparative Performance of SpCas9 and SaCas9 in Human Cell Lines
| Performance Metric | SpCas9 | SaCas9 | Experimental Context |
|---|---|---|---|
| On-Target Editing Efficiency | Baseline | Greater | 11 target sites in iPSCs & K562 cells [19] |
| Off-Target Effect (GUIDE-seq) | Higher | Significantly Reduced | Analysis in human iPSCs and K562 cells [19] [5] |
| Knock-in Efficiency (HDR) | Lower | Higher | AAV6 donor knock-in in iPSCs and K562 cells [19] |
| Optimal sgRNA Spacer Length | 20 nt | 21 nt | Determined across 11 target sites [19] |
| Indel Pattern | More +1 insertions | More balanced | Characteristic of a staggered cut for SpCas9 [19] |
Editing Outcomes and Knock-in Superiority
Beyond raw indel rates, the nature of the editing outcomes is crucial. The same study reported that SpCas9 exhibited a more substantial bias for nonhomologous end-joining (NHEJ)-mediated +1 insertion at a specific position upstream of the protospacer adjacent motif (PAM), indicating a characteristic of a staggered cut [19]. In contrast, editing with SaCas9 led to higher efficiencies of both NHEJ-mediated oligodeoxynucleotide insertion and HDR-mediated adeno-associated virus (AAV) donor knock-in [19]. This makes SaCas9 particularly attractive for therapeutic gene editing that requires precise transgene integration.
Detailed Experimental Protocols for Key Comparisons
The following section outlines the core methodologies used to generate the comparative data cited in this guide, providing a blueprint for researchers seeking to reproduce or validate these findings.
Protocol for Comparing SpCas9 vs. SaCas9 in iPSCs and K562 Cells
This protocol is adapted from the 2022 study that provided a direct comparison of nuclease performance [19].
- 1. Vector Construction: The study used optimized Cas9 constructs. Specifically, both HMGA2-SpCas9-BPNLS and HMGA2-SaCas9-BPNLS fusion proteins were utilized to enhance nuclear localization and activity. A modified sgRNA scaffold (Sa-v2) with a T4>C mutation was employed to prevent premature transcriptional termination and increase efficiency [19] [7].
- 2. Cell Culture and Transfection:
- Human iPSCs and K562 cells were maintained in their respective standard culture conditions.
- Cells were electroporated with the Cas9-sgRNA plasmid constructs using appropriate electroporation systems (e.g., Neon for iPSCs).
- 3. Editing Efficiency Analysis:
- Time Course: Target loci were amplified 48h and 72h post-electroporation. Maximum editing was observed at 48h with no significant change thereafter [19].
- Sequencing: The target loci were amplified with barcoded primers and pooled for high-throughput sequencing.
- Data Analysis: Indel frequencies were determined using CRISPResso2 analysis. Good reproducibility was confirmed with Pearson correlation (R² = 0.9546 for iPSCs and 0.8923 for K562) [19].
- 4. Off-Target Assessment:
- GUIDE-seq was performed according to standard protocols to identify and quantify potential off-target sites genome-wide [19].
Protocol for Optimizing gRNA Transcription for Enhanced Editing
A 2025 study demonstrated that enhancing gRNA transcript levels is critical for optimal efficiency, especially for challenging gRNAs or under limited vector availability [7]. The following workflow details the key optimization step.
- Key Modification: The conventional gRNA scaffold contains a sequence of four thymine nucleotides (4T) that can inhibit transcription from the U6 promoter. The optimization involves shortening this to a 3TC sequence by replacing the fourth 'T' in the tetraloop with a 'C' and its complementary 'A' with a 'G' [7].
- Application: This 3TC scaffold modification is compatible with both SpCas9 and SaCas9 systems and has been shown to boost the editing efficiency of high-fidelity SpCas9 variants and base editors (e.g., ABEmax), particularly when vector availability is limited, as in therapeutic applications like the EDIT-101 strategy [7].
The Scientist's Toolkit: Essential Research Reagents
The following table catalogues key reagents and their functions that are fundamental to conducting robust comparisons of CRISPR nucleases in mammalian systems.
Table 2: Key Research Reagent Solutions for CRISPR-Cas9 Performance Comparison
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| HMGA2-Cas9-BPNLS Fusion | Fusion protein combining a chromatin-modulating peptide (HMGA2) and a bipartite nuclear localization signal (BPNLS) to enhance nuclear import and editing activity. | Used to boost SaCas9 and SpCas9 activity in K562 and iPSCs [19]. |
| 3TC gRNA Scaffold | A modified gRNA scaffold where the native 4T tract is shortened to 3TC to prevent premature transcription termination and increase gRNA yield. | Optimizing editing efficiency for SaCas9 and SpCas9, especially with low-activity or T-rich gRNAs [7]. |
| CRISPResso2 | A software tool for the analysis of high-throughput sequencing data from CRISPR genome editing experiments. Quantifies indel frequencies and characterizes repair outcomes. | Used to calculate and compare editing efficiencies and indel patterns between SpCas9 and SaCas9 [19]. |
| GUIDE-seq | (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) A molecular assay to detect off-target sites of CRISPR nucleases genome-wide. | Demonstrated the superior fidelity and reduced off-target profile of SaCas9 compared to SpCas9 [19] [5]. |
| AAV6 Donor Template | Adeno-associated virus serotype 6 used as a donor vector for homology-directed repair (HDR). Highly efficient for gene knock-in in hematopoietic cells and iPSCs. | Showcased SaCas9's higher HDR-mediated knock-in efficiency compared to SpCas9 [19]. |
The direct, empirical comparison of SpCas9 and SaCas9 reveals a nuanced landscape for nuclease selection. While SpCas9 remains a powerful and versatile tool, SaCas9 demonstrates compelling advantages in specific contexts relevant to therapeutic development. The data indicate that SaCas9 offers superior on-target editing efficiency, reduced off-target effects, and higher knock-in rates in human iPSC and K562 models [19].
The choice between these nucleases should be guided by the specific experimental goal: SaCas9 is an excellent candidate for therapeutic gene editing where fidelity and efficient transgene integration are paramount, and its smaller size is beneficial for AAV delivery [19] [16]. SpCas9, with its NGG PAM, might offer greater target site flexibility for basic research applications. Ultimately, this comparison underscores that optimal genome editing requires not only choosing a nuclease but also implementing optimized experimental parameters, such as sgRNA spacer length and scaffold design, to achieve desired outcomes reliably [19] [7].
The generation of gene-modified mice is a cornerstone of biomedical research, enabling the study of gene function and the modeling of human diseases. The CRISPR-Cas9 system has revolutionized this process, with Streptococcus pyogenes Cas9 (SpCas9) serving as the initial gold standard. However, its application is constrained by its large size and specific NGG PAM requirement, which can limit targeting options [26]. Staphylococcus aureus Cas9 (SaCas9) presents a powerful alternative with distinct advantages, primarily due to its significantly smaller size and unique NNGRRT PAM recognition [27]. This guide provides an objective, data-driven comparison of SaCas9 and SpCas9 performance in mouse model generation, offering detailed experimental protocols and resource information to assist researchers in selecting the optimal nuclease for their specific applications.
Performance Comparison: SaCas9 vs. SpCas9
Direct, quantitative comparison of SaCas9 and SpCas9 in mouse zygotes reveals key differences in efficiency, specificity, and practical application. The data below summarize findings from a foundational study that tested both nucleases under identical conditions [26].
Table 1: Direct Comparison of SaCas9 and SpCas9 Editing in Mouse Zygotes
| Parameter | SaCas9 | SpCas9 | Experimental Context |
|---|---|---|---|
| Editing Efficiency (Slx2 locus) | 88.8% (24/27 embryos) | 53.8% (14/26 embryos) | T7EI assay & sequencing of cultured embryos [26] |
| Editing Efficiency (Zp1 locus) | 92.0% (23/25 embryos) | 96.3% (26/27 embryos) | T7EI assay & sequencing of cultured embryos [26] |
| Founder Mutation Rate (Slx2) | 94.1% (16/17 pups) | 47.1% (8/17 pups) | Sequencing of live-born pups [26] |
| Founder Mutation Rate (Zp1) | 77.7% (14/18 pups) | 83.3% (10/12 pups) | Sequencing of live-born pups [26] |
| PAM Sequence | 5'-NNGRRT-3' | 5'-NGG-3' | Dictates available target sites [26] [27] |
| Protein Size | ~1053 amino acids [27] | ~1368 amino acids [27] | Critical for AAV packaging [27] |
| Observed Mosaicism | Present (in Tyr gene targeting) | Present (in Tyr gene targeting) | Coat color mosaicism in C57BL/6J mice [26] |
The data demonstrates that SaCas9 can achieve editing efficiencies comparable to, and sometimes exceeding, SpCas9. However, efficiency is highly dependent on the specific target locus [26]. A significant advantage of SaCas9 is its smaller size, which facilitates efficient packaging into Adeno-Associated Virus (AAV) vectors for in vivo delivery, a common challenge for the larger SpCas9 [27].
Experimental Protocol: SaCas9-Mediated Gene Editing in Mouse Zygotes
The following detailed methodology is adapted from the proven protocol used to generate the comparative data in Section 2 [26].
The diagram below illustrates the key steps for generating gene-modified mice using SaCas9.
Detailed Methodological Steps
Step 1: gRNA Design and Preparation
Step 2: Microinjection Mix Preparation
- Prepare the injection mixture containing SaCas9 mRNA (typically 50-100 ng/μL) and the target-specific Sa-gRNA (10-50 ng/μL) in nuclease-free microinjection buffer [26].
- For knock-in experiments, include a single-stranded oligodeoxynucleotide (ssODN) or double-stranded DNA donor template (50-200 ng/μL) for Homology-Directed Repair (HDR).
Step 3: Zygote Collection and Microinjection
- Collect zygotes from super-ovulated female mice.
- Using a standard microinjection setup, inject the mixture from Step 2 into the cytoplasm or pronucleus of the zygotes.
Step 4: Embryo Transfer
- Shortly after microinjection (typically within 2 hours), transfer the viable zygotes into the oviducts of pseudopregnant foster female mice [26].
- Alternatively, culture the injected zygotes in vitro for 48 hours to assess editing efficiency at the blastocyst stage via the T7 Endonuclease I (T7EI) assay or PCR followed by sequencing [26].
Step 5: Genotyping of Founders
- After birth, genotype the founder mice (F0) to assess mutation rates. This can be done via:
- T7EI Assay: A quick method to detect indels, though it may miss low-efficiency events [26].
- Sanger Sequencing: Provides a definitive confirmation of editing but can underestimate mosaicism [26].
- Deep Sequencing: The most sensitive method, capable of quantifying editing efficiency and detecting mosaic events with high accuracy [26].
- After birth, genotype the founder mice (F0) to assess mutation rates. This can be done via:
Optimization Strategies for Enhanced SaCas9 Performance
Recent advances have identified specific modifications to further improve SaCas9 efficiency. A primary focus has been on optimizing the gRNA scaffold to increase transcript levels.
gRNA Scaffold Engineering
The standard gRNA scaffold contains a sequence of four thymine nucleotides (4T), which can act as a termination signal for RNA Polymerase III (e.g., U6 promoter), thereby inhibiting transcription. Research has shown that shortening this poly-T tract is a highly effective optimization strategy [7].
Table 2: Impact of gRNA Scaffold Modification on Editing Efficiency
| gRNA Scaffold Type | Transcript Level | Editing Efficiency | Application Context |
|---|---|---|---|
| Standard Scaffold (4T) | Baseline | High when vector is abundant | Standard plasmid transfection with selection [7] |
| Modified Scaffold (3TC) | 271- to 11,349-fold increase | Enhanced under limited vector availability | Low-dose delivery; therapeutic contexts (e.g., AAV) [7] |
Replacing the fourth thymine (T) in the tetraloop with a cytosine (C)—creating a "3TC" scaffold—significantly boosts gRNA transcript levels by reducing premature transcription termination [7]. This modification is particularly beneficial in scenarios where delivery efficiency is a limiting factor, such as with AAV vectors or low-dose plasmid transfection, and it is compatible with SaCas9, SpCas9 high-fidelity variants, and base editors [7].
The following diagram illustrates the logical decision process for choosing and optimizing SaCas9.
The Scientist's Toolkit: Essential Research Reagents
Successful execution of SaCas9-mediated mouse model generation requires a set of core reagents. The following table lists these essential materials and their functions.
Table 3: Essential Research Reagents for SaCas9 Mouse Model Generation
| Reagent / Resource | Function | Example/Note |
|---|---|---|
| SaCas9 mRNA | The nuclease component; introduces double-strand breaks at the target DNA site. | Can be purchased as synthetic, codon-optimized mRNA ready for microinjection. |
| SaCas9 gRNA | Guides the nuclease to the specific genomic locus via base-pairing. | Must be designed with a 5'-NNGRRT-3' PAM. Use the 3TC-modified scaffold for enhanced expression [7]. |
| Donor DNA Template | Provides the homology sequence for precise HDR-mediated knock-in. | Single-stranded oligodeoxynucleotide (ssODN) or double-stranded DNA vector. |
| Microinjection Buffer | A stable, nuclease-free solution for diluting and delivering CRISPR components. | Standardized buffers ensure zygote viability during microinjection. |
| Mouse Zygotes | The starting biological material for generating founders. | Typically obtained from super-ovulated C57BL/6 or other desired strains. |
| Bioinformatics Tool (CATS) | Compares PAM sites for different Cas9 nucleases to identify optimal targets and alleles [9]. | Automates finding overlapping PAMs and identifies pathogenic mutations for allele-specific targeting from ClinVar [9]. |
SaCas9 has firmly established itself as a powerful and efficient tool for the generation of gene-modified mice. Its compact size makes it the nuclease of choice for in vivo applications requiring AAV delivery, while its unique NNGRRT PAM expands the range of targetable genomic sites. While editing efficiency is locus-dependent and mosaicism remains a consideration—as it does with SpCas9—the empirical data confirms that SaCas9 delivers performance on par with the older standard. The ongoing optimization of its components, such as the 3TC gRNA scaffold, continues to push the boundaries of its efficiency, particularly in challenging delivery contexts. For researchers designing new mouse models, SaCas9 is an indispensable and highly effective alternative within the expanding CRISPR arsenal.
The advent of CRISPR-Cas9 technology has revolutionized genetic research and therapeutic development, creating an expanding toolkit of engineered editors for precise genome modification. Among these, adenine base editors (ABEs) like ABEmax and high-fidelity Cas9 variants represent significant advancements toward safer, more accurate genetic interventions. ABEmax enables precise A•T to G•C base conversions without inducing double-strand breaks (DSBs), addressing a key safety concern associated with earlier CRISPR systems [28]. Concurrently, high-fidelity Cas9 variants such as SpCas9-HF1 and eSpCas9(1.1) have been engineered to minimize off-target effects while maintaining on-target efficiency [7] [29].
This guide objectively compares the compatibility and performance of these advanced therapeutic toolkits, focusing specifically on their integration with the broader SpCas9 versus SaCas9 performance comparison research. For therapeutic applications, particularly those involving viral vector delivery, understanding these compatibility relationships is crucial for developing effective treatment strategies with optimal safety profiles. The following sections provide detailed experimental data and methodologies to guide researchers in selecting appropriate toolkits for specific therapeutic contexts.
Toolkit Components and Technological Foundations
Core Editor Systems
- ABEmax: An optimized adenine base editor that combines an evolved tRNA-specific adenosine deaminase (TadA*) with a Cas9 nickase (nCas9) to catalyze precise A•T to G•C conversions without double-strand breaks [28]. The system operates by deaminating adenine to inosine in DNA, which is subsequently read as guanine during DNA replication [28].
- High-Fidelity SpCas9 Variants: Engineered versions of Streptococcus pyogenes Cas9 with reduced off-target activity. Key examples include SpCas9-HF1 and eSpCas9(1.1), which incorporate mutations that reduce non-specific interactions with the DNA backbone while maintaining on-target cleavage efficiency [7] [29].
- SaCas9: The Staphylococcus aureus Cas9 ortholog, which recognizes the NNGRRT PAM and has a smaller size than SpCas9, allowing for easier packaging into adeno-associated virus (AAV) vectors for in vivo delivery [7].
Essential Research Reagent Solutions
Table 1: Key Research Reagents for Base Editor and High-Fidelity Nuclease Studies
| Reagent/Solution | Function | Application Notes |
|---|---|---|
| All-in-one CRISPR Plasmid (e.g., PX459.v2) | Expresses both Cas9 and guide RNA from different promoters (CBh and U6) [7] | Contains original gRNA scaffold with 4T sequence; suitable for initial efficiency testing |
| Modified gRNA Scaffold (3TC) | Enhanced gRNA scaffold with reduced poly-T tract (3 thymines followed by cytosine) [7] | Improves transcription from U6 promoter; critical for T-rich gRNAs and limited vector scenarios |
| Dual gRNA Expression Plasmid (e.g., pDG459) | Contains two U6 expression cassettes for increased gRNA expression [7] | Alternative approach to boost gRNA transcript levels for difficult-to-edit targets |
| Chemical Modifications (2'-O-Me, PS) | Synthetic gRNA modifications that reduce off-target effects and improve editing efficiency [30] | Particularly valuable for in vivo therapeutic applications where precision is critical |
| Bioinformatic Tools (e.g., CATS, CRISPOR) | Computational tools for gRNA design, off-target prediction, and Cas9 nuclease comparison [9] [30] | Essential for pre-experimental planning and identifying optimal targeting strategies |
Comparative Performance Analysis
Editing Efficiency Across Systems
Table 2: Quantitative Comparison of Editing Efficiency Across CRISPR Systems
| Editor System | Therapeutic Context | Editing Efficiency | Key Factors Influencing Efficiency |
|---|---|---|---|
| ABEmax with Standard gRNA | Plasmid transfection with selection | 83-99% across 55 gRNAs tested [7] | gRNA sequence composition; T-rich gRNAs show reduced efficiency (~83-89%) |
| ABEmax with 3TC gRNA | Plasmid transfection with selection | >95% for previously suboptimal gRNAs [7] | Enhanced gRNA transcript levels address limitations of T-rich sequences |
| High-Fidelity SpCas9 Variants | Standard plasmid transfection | High efficiency with reduced off-targets [7] | Benefits significantly from 3TC scaffold modification |
| SaCas9 with Standard gRNA | AAV delivery contexts | Moderate to high efficiency [7] | NNGRRT PAM requirement limits targeting scope compared to NGG PAM |
| SaCas9 with 3TC gRNA | AAV delivery contexts | Marked improvement in EDIT-101 strategy [7] | Enhanced gRNA transcription particularly beneficial in vector-limited scenarios |
Compatibility and Performance in Constrained Environments
Table 3: Performance Under Therapeutic-Relevant Constraints
| Constraint Condition | ABEmax Performance | High-Fidelity SpCas9 Performance | SaCas9 Performance |
|---|---|---|---|
| Limited Vector Availability | 3TC scaffold significantly improves editing efficiency [7] | 3TC scaffold provides substantial benefit [7] | 3TC modification particularly valuable for in vivo applications [7] |
| Therapeutic AAV Packaging | Requires smaller editors (ABE8e) or dual AAV | Too large for single AAV with regulatory elements | Compatible with single AAV packaging [7] |
| Off-Target Considerations | Reduced RNA off-targeting in optimized ABEmax [7] | Engineered for minimal off-target effects [7] [29] | Naturally different off-target profile due to distinct PAM requirement [7] |
| T-rich Target Sequences | Initially problematic, resolved with 3TC scaffold [7] | Initially problematic, resolved with 3TC scaffold [7] | Initially problematic, resolved with 3TC scaffold [7] |
Experimental Protocols for Compatibility Assessment
gRNA Scaffold Optimization Protocol
Objective: To evaluate and enhance editing efficiency of ABEmax and high-fidelity variants through gRNA scaffold modifications.
Materials:
- All-in-one CRISPR plasmid (e.g., PX459.v2 with CBh-driven SpCas9-T2A-Puro and U6-driven gRNA)
- 3TC scaffold-modified plasmid (replacing fourth T in tetraloop with C nucleotide)
- Target cell lines (HEK293T, C2C12, or mES cells)
- Transfection reagent
- Puromycin for selection
- PCR reagents and sequencing primers for editing assessment
Methodology:
- Vector Preparation: Clone target gRNAs into both standard (4T) and modified (3TC) scaffold vectors [7].
- Cell Transfection: Transfect cells using standard protocols with varying vector quantities (high: 1-2μg, low: 100-200ng per well in 24-well plate) [7].
- Selection and Analysis: Apply puromycin selection 24-48 hours post-transfection (optional for limited vector experiments) [7].
- Efficiency Assessment: Harvest cells 3-5 days post-transfection, isolate genomic DNA, amplify target regions via PCR, and quantify editing efficiency through next-generation sequencing or T7E1 assay [7].
- gRNA Expression Analysis: For low-efficiency gRNAs, perform qPCR to measure gRNA transcript levels comparing 4T and 3TC scaffolds [7].
Expected Outcomes: The 3TC scaffold typically increases gRNA transcript levels by 8.1-13.5 doubling cycles (271-11,349 fold changes), with the most significant improvements observed for T-rich gRNAs and under limited vector availability [7].
Off-Target Assessment Protocol
Objective: To comprehensively evaluate off-target effects of ABEmax and high-fidelity variants in therapeutic contexts.
Materials:
- Designed gRNAs with minimal predicted off-targets
- Editor proteins (ABEmax, SpCas9-HF1, eSpCas9[1.1])
- Target cells with known genomic sequence
- Whole genome sequencing services or targeted sequencing reagents
Methodology:
- In Silico Prediction: Use bioinformatic tools (CRISPOR, Cas-OFFinder) to identify potential off-target sites with up to 5 nucleotide mismatches [29] [30].
- Editor Delivery: Transfect cells with editor components using optimal conditions determined in efficiency experiments.
- Off-Target Detection: Employ one or more of the following methods:
- Candidate Site Sequencing: Amplify and sequence predicted off-target sites [30].
- GUIDE-seq: Detect in vivo nuclease cutting sites through integration of double-stranded oligodeoxynucleotides [30].
- Whole Genome Sequencing: Most comprehensive approach to identify all potential off-target effects, including chromosomal rearrangements [30].
- Risk Assessment: Compare off-target profiles between standard editors and high-fidelity variants, noting particularly any edits in coding regions or oncogenic loci.
Therapeutic Considerations: For clinical applications, the FDA recommends thorough off-target characterization, with special attention to individuals carrying rare genetic variants who may be at higher risk [30].
Integration with SpCas9 vs SaCas9 Performance Research
The comparison between SpCas9 and SaCas9 represents a fundamental framework in therapeutic genome editing, primarily centered on the trade-offs between targeting scope and deliverability. The PAM requirements of each system directly influence their compatibility with base editors and high-fidelity variants. SpCas9 (NGG PAM) offers broader targeting range, while SaCas9 (NNGRRT PAM) provides superior deliverability due to its smaller size [7].
Bioinformatic tools like CATS (Comparing Cas9 Activities by Target Superimposition) enable researchers to identify overlapping PAM sequences for different Cas9 nucleases, facilitating direct comparison in identical genomic contexts [9]. This approach is particularly valuable for therapeutic development, as it allows for the selection of optimal editors based on both efficiency and deliverability constraints.
For therapeutic applications, the editor selection strategy should follow a decision pathway that considers both target sequence constraints and delivery requirements:
The compatibility between base editors like ABEmax and high-fidelity Cas9 variants represents a critical consideration in therapeutic development. The experimental data demonstrates that gRNA scaffold optimization through 3TC modification significantly enhances editing efficiency across all systems, particularly under the vector-limited conditions frequently encountered in therapeutic applications [7].
For researchers developing therapeutic strategies, the optimal toolkit configuration involves:
- Editor Selection: Choosing between SpCas9 and SaCas9 systems based on PAM availability and delivery constraints
- Fidelity Enhancement: Incorporating high-fidelity variants for applications requiring minimal off-target effects
- Scaffold Optimization: Implementing 3TC-modified gRNAs to maximize editing efficiency, particularly for challenging targets
This systematic approach to toolkit compatibility ensures that therapeutic developers can maximize editing efficiency while maintaining the safety profile required for clinical applications, ultimately accelerating the development of effective genetic therapies.
The ability to simultaneously modify multiple genetic loci, known as multiplexed genome editing, represents a paradigm shift in genetic engineering. This approach has become indispensable for addressing complex biological questions where single-gene manipulations are insufficient, including synthetic lethality studies, polygenic disease modeling, and metabolic pathway engineering [31] [32]. The CRISPR-Cas system, with its programmable RNA-guided mechanism, has emerged as the most versatile platform for multiplexed editing due to its simplicity and scalability compared to previous technologies like ZFNs and TALENs [31] [33]. While the original Streptococcus pyogenes Cas9 (SpCas9) remains the most characterized nuclease, its smaller ortholog from Staphylococcus aureus (SaCas9) offers distinct advantages for therapeutic applications. This guide provides an objective comparison of these two key systems within the context of multiplexed genome editing, detailing their performance characteristics, experimental implementation, and practical applications for researchers and drug development professionals.
Comparative Performance: SpCas9 vs. SaCas9 in Multiplexed Editing
Table 1: Fundamental Characteristics of SpCas9 and SaCas9
| Parameter | SpCas9 | SaCas9 |
|---|---|---|
| Origin | Streptococcus pyogenes | Staphylococcus aureus |
| Size (aa) | ~1,368 | ~1,053 [16] |
| PAM Requirement | NGG [34] | NNGRRT (where R = A or G) [35] [16] |
| Primary Advantage | High efficiency, extensive characterization | Smaller size enables AAV delivery [16] |
| Key Limitation | Large size challenges delivery, off-target concerns [16] | More restrictive PAM limits targeting sites [16] |
| Therapeutic Delivery | Challenging in AAV vectors [16] | Compatible with AAV delivery [35] [16] |
Table 2: Quantitative Performance Comparison in Multiplexed Editing
| Performance Metric | SpCas9 | SaCas9 | Experimental Context |
|---|---|---|---|
| Typical Editing Efficiency | Up to 95%+ at multiple loci [35] | High efficiency with optimized scaffolds [35] | HEK293T cells with optimized gRNA expression |
| Off-target Profile | Can be significant with standard variant [34] | Improved specificity with engineered variants [16] | High-fidelity variants show reduced off-targets |
| Multiplexing Capacity (Demonstrated) | Up to 10-plex in HEK293T [31] [32] | Effective in multiplexed configurations [35] | Dependent on expression system |
| Base Editing Efficiency | Variable at difficult loci [34] | Improved with scaffold optimization (3TC) [35] | EDIT-101 therapeutic strategy demonstrated enhancement |
| gRNA Transcript Requirement | High levels needed for optimal editing [35] | Benefits from enhanced transcript expression [35] | Particularly critical under limited vector availability |
Implementing Multiplexed Editing Systems: Methodologies and Workflows
gRNA Scaffold Engineering for Enhanced Efficiency
A critical factor in achieving efficient multiplexed editing with both SpCas9 and SaCas9 is ensuring sufficient gRNA transcript levels. Research has demonstrated that the conventional gRNA scaffold containing four thymine nucleotides (4T) can inhibit transcription from Pol III promoters. Modifying this sequence to a 3TC scaffold (replacing the fourth T with C) significantly increases gRNA expression levels, particularly beneficial for SaCas9 and under conditions of limited vector availability [35]. In one study, this modification increased gRNA levels by 8.1–13.5 doublings (271-11,349 fold changes), boosting editing efficiency of suboptimal gRNAs from approximately 83-89% to over 95% [35]. This optimization is particularly valuable for therapeutic applications like the EDIT-101 strategy, where vector quantity is often constrained [35].
Strategies for Multiplexed gRNA Expression
Successful multiplexed editing requires coordinated expression of multiple gRNAs. Three primary architectural strategies have been developed for this purpose:
Figure 1: Multiplexed gRNA expression architectures for simultaneous multi-gene targeting.
- Individual Promoter Approach: Each gRNA is driven by a separate RNA Polymerase III promoter (e.g., U6). While reliable, this method becomes impractical beyond approximately 5 gRNAs due to promoter availability and construct size [36].
- Endogenous Processing Systems: Utilizing natural CRISPR mechanisms, such as Cas12a's inherent ability to process crRNA arrays from a single transcript [36]. This approach enabled simultaneous cleavage of five target genes and transcriptional regulation of ten additional targets in human cells [36].
- Artificial Processing Systems: Incorporating synthetic processing elements including:
- tRNA-gRNA arrays: Exploits ubiquitous RNase P and Z enzymes to process multiple gRNAs from a single transcript [33] [36].
- Ribozyme-flanked gRNAs: Hammerhead and hepatitis delta virus ribozymes flank each gRNA for self-cleavage [36].
- Csy4-processing: The Cas protein Csy4 cleaves at specific 28-nt recognition sites [36].
Experimental Workflow for Multiplexed Editing
Figure 2: Core workflow for implementing multiplexed CRISPR genome editing.
The implementation of multiplexed editing follows a systematic workflow:
- gRNA Design: Select target sequences with minimal off-target potential and optimal efficiency. For SaCas9, designs must accommodate the NNGRRT PAM requirement [35] [16].
- Vector Assembly: Constructs are typically assembled using methods like Golden Gate assembly, which facilitates the modular integration of multiple gRNA units [31] [32]. For example, this approach has been used to construct functional cassettes with up to seven gRNAs [31] [32].
- Delivery System: Choice depends on application:
- Editing Validation: Next-generation sequencing (NGS) is required to detect complex editing outcomes, including large deletions, inversions, and translocations that may occur with multiplexed targeting [37].
- Outcome Analysis: Assess both intended edits and potential unintended effects, such as chromosomal rearrangements, which become more likely as the number of simultaneous edits increases [38].
Advanced Applications and Therapeutic Implementation
Research and Therapeutic Applications
Multiplexed editing has enabled sophisticated applications across biological research and therapeutic development:
- Functional Genomics Screening: CRISPR-based double-knockout (CDKO) libraries allow systematic interrogation of genetic interactions and synthetic lethality [31] [32]. One such screen identified synthetic lethal targets in K562 cells from 490,000 gRNA pairs [31] [32].
- Noncoding Element Characterization: Paired gRNA libraries enable functional screening of long noncoding RNAs and regulatory elements by creating large deletions [31] [32]. One study identified 51 lncRNAs regulating liver cancer proliferation from 700 targeted lncRNAs [31] [32].
- Cancer-Specific Cell Targeting: Multiplexed DSBs can be designed to accumulate selective stress in cancer cells while sparing normal cells, offering a potential therapeutic strategy [31] [32].
- Agricultural Trait Stacking: Simultaneous editing of multiple genes facilitates rapid improvement of complex traits such as disease resistance and nutritional quality in crops [37] [38].
Research Reagent Solutions Toolkit
Table 3: Essential Reagents for Multiplexed Genome Editing Experiments
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Cas9 Expression Plasmids | PX459.v2 (SpCas9) [35] | All-in-one vectors with Cas9 and gRNA expression |
| gRNA Scaffold Variants | 4T scaffold, 3TC scaffold [35] | Optimized gRNA backbones for enhanced transcription |
| Multiplex Assembly Systems | Golden Gate assembly [31] [32] | Modular construction of multi-gRNA arrays |
| Specialized Cas9 Variants | eSpOT-ON, hfCas12Max [16] | High-fidelity nucleases with reduced off-target effects |
| Delivery Vehicles | AAV-SaCas9 constructs [16] | Therapeutic delivery of editing components |
| Editing Detection Kits | NGS library preparation kits | Validation of editing outcomes and off-target assessment |
Future Directions and Experimental Considerations
The field of multiplexed genome editing continues to evolve rapidly. Emerging challenges include managing potential unintended effects such as chromosomal rearrangements and large deletions, which current research suggests may become significant when editing numerous loci simultaneously [38]. Future innovations will likely focus on:
- Novel Cas Variants: Orthologs like SeqCas9 with simplified NNG PAM requirements are expanding targeting range [34].
- Enhanced Specificity Systems: High-fidelity variants and engineered nucleases with reduced off-target effects [16] [34].
- Advanced Delivery Platforms: Lipid nanoparticles, virus-like particles, and metal-organic frameworks that overcome conventional delivery barriers [33].
- Computational Design Tools: AI-optimized gRNA designs and outcome prediction algorithms [33] [37].
When designing multiplexed editing experiments, researchers should consider the trade-offs between editing scale and potential unintended effects. Current evidence suggests that simultaneously editing approximately ten genes may be achievable with minimal unintended consequences, while editing twenty or more loci significantly increases risks of chromosomal alterations [38]. The choice between SpCas9 and SaCas9 should be guided by specific application requirements: SpCas9 for maximal targeting flexibility and efficiency, SaCas9 for therapeutic applications requiring viral delivery.
The advent of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and associated protein 9 (Cas9) systems has revolutionized biomedical research and therapeutic development. Among the diverse Cas9 orthologs available, Streptococcus pyogenes Cas9 (SpCas9) and Staphylococcus aureus Cas9 (SaCas9) have emerged as leading platforms for clinical applications. The fundamental distinction driving their therapeutic selection is packaging size: SpCas9 (1368 amino acids) exceeds the adeno-associated virus (AAV) packaging capacity of ~4.7 kb when combined with its necessary expression elements, whereas the more compact SaCas9 (1053 amino acids) readily fits within AAV constraints [39] [40]. This critical difference positioned SaCas9 as the foundation for EDIT-101, the first in vivo CRISPR therapy to enter clinical trials for an inherited retinal disease [41] [42].
EDIT-101 represents a landmark case study in translating CRISPR technology to human therapeutics. This strategy utilizes SaCas9 to address Leber Congenital Amaurosis type 10 (LCA10) caused by mutations in the CEP290 gene. The CEP290 coding sequence exceeds AAV packaging capacity, making conventional gene augmentation therapy impossible. Instead, EDIT-101 employs a single guide RNA (sgRNA) and SaCas9 to remove a mutation-bearing intronic region, thereby restoring normal CEP290 protein expression [41]. This review examines the experimental data, design optimization, and clinical outcomes of the EDIT-101 strategy, providing a comparative framework for SaCas9 versus SpCas9 performance in therapeutic contexts.
The EDIT-101 Therapeutic Strategy: From Concept to Clinic
Molecular Pathogenesis and Therapeutic Mechanism
Leber Congenital Amaurosis type 10 results from autosomal recessive mutations in the CEP290 gene, which encodes a centrosomal protein critical for photoreceptor function. A prevalent pathogenic mutation is an IVS26 intronic point mutation that creates a cryptic splice site, leading to the insertion of a pseudoexon containing a premature stop codon. The resultant CEP290 protein is truncated and non-functional, causing progressive retinal degeneration and severe visual impairment beginning in infancy [41].
EDIT-101 employs a single AAV5 vector to deliver two key components: (1) the SaCas9 endonuclease and (2) a guide RNA (gRNA) targeting the pathological intronic region. Unlike SpCas9, which recognizes an NGG protospacer adjacent motif (PAM), SaCas9 recognizes an NNGRRT PAM sequence [35] [40], which was strategically available flanking the target site. The therapeutic mechanism involves dual incision of the DNA strands upstream and downstream of the mutation, resulting in excision of the aberrant splice site. With the pathogenic element removed, the cell's DNA repair machinery rejoins the DNA ends, restoring normal CEP290 splicing and protein expression in photoreceptor cells [41].
gRNA Scaffold Optimization for Enhanced Efficacy
A critical advancement in the EDIT-101 development was the optimization of gRNA transcription efficiency. The standard gRNA scaffold contains a sequence of four thymine nucleotides (4T), which functions as a termination signal for RNA Polymerase III, thereby potentially limiting gRNA yield [35].
Research demonstrated that modifying this scaffold to shorten the T-string significantly enhanced editing efficiency, particularly under conditions of limited vector availability – a common scenario in AAV-mediated delivery. Replacing the fourth thymine with cytosine (3TC scaffold) increased gRNA transcript levels by 8.1–13.5 doublings (271- to 11,349-fold increases) compared to the original scaffold [35]. This modification proved compatible with SaCas9 and was incorporated into the EDIT-101 design, boosting gRNA transcription and subsequent editing activity – a crucial optimization for achieving therapeutic efficacy with the limited AAV payload [35].
Table 1: Key Components of the EDIT-101 Therapeutic Strategy
| Component | Description | Role in Therapy |
|---|---|---|
| Cas Nuclease | Staphylococcus aureus Cas9 (SaCas9) | DNA endonuclease for targeted gene editing |
| Delivery Vector | Adeno-associated virus serotype 5 (AAV5) | Vehicle for delivering SaCas9 and gRNA to retinal cells |
| Target Gene | CEP290 (IVS26 mutation) | Disease-causing gene in Leber Congenital Amaurosis type 10 |
| Therapeutic Action | Excision of mutant intronic region | Restoration of normal CEP290 splicing and protein function |
| gRNA Scaffold | Modified 3TC scaffold | Enhanced gRNA transcription efficiency for improved editing |
Experimental Protocols and Performance Data
In Vitro and Preclinical Assessment
The development of EDIT-101 followed a rigorous preclinical pathway to establish proof-of-concept, efficacy, and safety. Initial in vitro assays in human cell lines confirmed the molecular mechanism – specifically, the ability of the selected gRNA to guide SaCas9 to the target sequence and precisely excise the mutation, with editing efficiency quantified via next-generation sequencing [41].
Subsequent studies progressed to animal models, including mice and non-human primates, to evaluate both therapeutic effect and safety profile. These investigations employed:
- Subretinal injections to deliver the AAV5 vector directly to the retinal space.
- Molecular analyses (PCR, sequencing) to confirm target engagement and editing accuracy in photoreceptors.
- Immunohistochemistry and Western blotting to verify restored CEP290 protein expression.
- Electroretinography (ERG) and visual behavior tests to assess functional visual improvement.
- GUIDE-seq and whole-genome sequencing to comprehensively profile potential off-target effects at the genome-wide level [41] [43].
The preclinical data demonstrated efficient target editing, restoration of CEP290 protein, functional visual improvement in animal models, and a favorable safety profile with no significant off-target effects, supporting the advancement to human clinical trials [41].
Clinical Trial Outcomes: BRILLIANCE Trial
The Phase I/II BRILLIANCE clinical trial (NCT03872479) represented the first-in-human test of in vivo CRISPR genome editing. The trial enrolled 14 participants with LCA10 caused by the IVS26 mutation in CEP290 [41].
The recently reported outcomes indicated that EDIT-101 demonstrated a good safety and tolerability profile, with no serious adverse events related to the treatment. However, the clinical efficacy was modest and variable. Only 3 out of 14 enrolled patients showed clinically meaningful improvements in visual function, with the most significant benefits observed in homozygous participants [41]. Consequently, the trial was recently paused, highlighting the challenges in achieving consistent therapeutic efficacy across a genetically diverse patient population [41].
Comparative Performance: SaCas9 vs. SpCas9
Direct Comparison of Editing Capabilities
While direct head-to-head comparisons of SaCas9 and SpCas9 in the EDIT-101 context are not available, broader studies illuminate their relative performance characteristics. A comprehensive evaluation of programmable nucleases targeting human papillomavirus (HPV) genes revealed that SpCas9 demonstrated superior efficiency and specificity compared to other nuclease platforms, including TALENs and ZFNs [43]. In this study, SpCas9 achieved comparable or better on-target editing while generating fewer off-target events across multiple genomic targets [43].
Table 2: Functional Comparison of SaCas9 and SpCas9 Properties
| Property | SaCas9 | SpCas9 |
|---|---|---|
| Size (amino acids) | 1053 [40] | 1368 [40] |
| PAM Sequence | NNGRRT [35] [40] | NGG [44] [40] |
| AAV Packaging | Compatible (fits with gRNA) [35] [39] | Incompatible (too large) [39] [40] |
| Target Space | Smaller (due to more restrictive PAM) [40] | Larger (relaxed NGG PAM) [40] |
| gRNA Scaffold | Benefits from 3TC optimization [35] | Benefits from 3TC optimization [35] |
| Editing Efficiency | Efficient in EDIT-101 context [41] | Generally high; tool of choice where deliverable [43] |
| Specificity | Favorable off-target profile in preclinical studies [41] | High; HF variants further reduce off-targets [43] [40] |
The smaller size of SaCas9 comes with a trade-off: its more complex NNGRRT PAM sequence reduces the number of potential target sites in the genome compared to SpCas9's simpler NGG PAM [40]. This constraint can limit targeting flexibility during therapeutic design. However, for applications where the target locus contains an appropriate PAM, and AAV delivery is essential – as with EDIT-101 – SaCas9 provides the necessary combination of compact size and efficient editing.
Application Scope and Therapeutic Versatility
The EDIT-101 case demonstrates that SaCas9 is the nuclease of necessity for AAV-delivered in vivo editing. Its compact size enables complete packaging within a single AAV vector alongside its gRNA expression cassette, making it indispensable for targeting tissues like the retina, where AAV is the delivery vehicle of choice [39] [40].
In contrast, SpCas9 offers greater versatility for ex vivo applications and in vivo approaches not constrained by AAV packaging. SpCas9's simpler PAM requirement provides more potential target sites for any given gene, and its extensive characterization has led to numerous high-fidelity variants (e.g., SpCas9-HF1, eSpCas9(1.1)) that further minimize off-target activity [35] [40]. Furthermore, the development of SpCas9-derived base editors (ABEmax) and prime editors expands the therapeutic scope beyond double-strand breaks to include precise single-base changes [35] [45].
Diagram Title: EDIT-101 Molecular Mechanism of Action
Research Reagent Solutions for Therapeutic CRISPR Development
The development and optimization of CRISPR-based therapeutics like EDIT-101 rely on specialized reagents and methodologies. The table below outlines key tools derived from the EDIT-101 case study and related research for scientists developing similar gene editing strategies.
Table 3: Essential Research Reagents and Methods for Therapeutic CRISPR Development
| Reagent/Method | Function | Application in EDIT-101 |
|---|---|---|
| AAV Serotype 5 | Viral delivery vector with retinal tropism | Delivers SaCas9 and gRNA to photoreceptor cells [41] |
| 3TC gRNA Scaffold | Enhanced Pol III transcription termination | Increases gRNA transcript levels and editing efficiency [35] |
| GUIDE-seq | Genome-wide unbiased off-target detection | Profiles nuclease specificity and identifies potential off-target sites [43] |
| High-Fidelity Cas9 Variants | Engineered nucleases with reduced off-target effects | eSpCas9(1.1), SpCas9-HF1; not used in EDIT-101 but relevant for safety [35] [40] |
| Base Editors (ABEmax) | Catalytically impaired Cas9 fused to deaminase | Enables precise single-base conversions without double-strand breaks [35] [45] |
| Dual gRNA Vector (pDG459) | Plasmid with two U6 expression cassettes | Increases gRNA dosage; tested as alternative to 3TC optimization [35] |
The EDIT-101 clinical program represents a pioneering effort in in vivo CRISPR genome editing, establishing SaCas9 as a viable nuclease for human therapeutics where AAV delivery is essential. The case study demonstrates that strategic optimization, such as the 3TC gRNA scaffold modification, can significantly enhance molecular efficacy. However, the modest clinical outcomes in the BRILLIANCE trial underscore the considerable challenges in translating efficient molecular editing to consistent patient benefit.
Future directions will likely focus on optimizing delivery efficiency to photoreceptors, refining patient selection criteria (potentially focusing on homozygotes who showed better response), and developing next-generation editing tools like base and prime editors that could offer enhanced precision for different mutation types [45]. As CRISPR technology evolves, the EDIT-101 case will stand as a critical milestone that provided both the promise and the practical roadmap for the coming generation of genomic medicines.
Maximizing Editing Success: Overcoming Efficiency Barriers and gRNA Design Challenges
The CRISPR-Cas9 system has emerged as a revolutionary tool for genome engineering, with SpCas9 and SaCas9 being two of the most widely used nucleases in research and therapeutic development. A critical but often overlooked component of this system is the single-guide RNA (sgRNA) scaffold, which contains structural elements essential for Cas9 binding and function. The canonical sgRNA scaffold includes a sequence of four thymine nucleotides (4T), which is recognized as a transcription termination signal for RNA polymerase III (Pol III) promoters such as the U6 promoter commonly used to express gRNAs [7]. This 4T sequence creates a fundamental transcription limitation, potentially reducing gRNA yield and consequently impairing genome editing efficiency.
Recent research has demonstrated that shortening the poly-T tract from 4T to 3TC (where the fourth T is replaced with a C) represents a straightforward yet powerful optimization strategy to enhance gRNA transcript levels [7] [46]. This review provides a comprehensive comparison of how this scaffold modification impacts the performance of both SpCas9 and SaCas9 systems, presenting quantitative experimental data to guide researchers in implementing this optimization for improved editing outcomes across various applications.
Molecular Mechanism: How Poly-T Reduction Enhances Transcription
The Transcription Termination Problem
RNA polymerase III initiates transcription at the U6 promoter and terminates upon encountering a run of thymine nucleotides in the DNA template, producing a gRNA with a 3' poly-U tail [7] [47]. The standard gRNA scaffold contains an inherent sequence of four thymine nucleotides (4T) within its structural elements. When transcribed into the gRNA, this region can be misinterpreted during transcription as a termination signal, leading to premature transcription cessation and reduced yield of full-length, functional gRNA molecules [7].
The 3TC Scaffold Solution
The 3TC modification addresses this limitation through a minimalistic alteration: replacing the fourth thymine (T) in the tetraloop of the gRNA scaffold with a cytosine (C) nucleotide, with a corresponding change from adenine (A) to guanine (G) in the complementary position to preserve base pairing and maintain scaffold structural integrity [7]. This single-nucleotide substitution reduces the consecutive T-stretch, thereby minimizing Pol III recognition as a termination signal while preserving the gRNA's secondary structure and function.
The diagram below illustrates the structural context and nucleotide change involved in the 4T to 3TC modification.
Comparative Performance Analysis: 3TC Scaffold Impact on SpCas9 and SaCas9
Quantitative Assessment of Editing Efficiency Enhancement
Table 1: Comparative Editing Efficiencies of 4T vs. 3TC Scaffolds for SpCas9 and SaCas9
| Cas9 Variant | gRNA Scaffold | Baseline Editing Efficiency (%) | Enhanced Editing Efficiency (%) | Fold Increase in gRNA Transcripts | Experimental Context |
|---|---|---|---|---|---|
| SpCas9 | 4T | 83.0-89.0 | 90.0-95.0+ | 271-11,349 | Low-efficiency gRNAs in C2C12 cells [7] |
| SpCas9 | 3TC | 92.0 | 97.4 | Not reported | hDMD-B gRNA in HEK293T cells [7] |
| SpCas9-HF1 | 4T | Moderate | Significantly improved | Not reported | High-fidelity editing [7] |
| SpCas9-HF1 | 3TC | Not reported | Enhanced | Not reported | High-fidelity editing [7] |
| eSpCas9(1.1) | 4T | Moderate | Significantly improved | Not reported | High-fidelity editing [7] |
| eSpCas9(1.1) | 3TC | Not reported | Enhanced | Not reported | High-fidelity editing [7] |
| SaCas9 | 4T | Variable | Significantly improved | Not reported | Multiple cell types [7] |
| SaCas9 | 3TC | Not reported | Enhanced | Not reported | EDIT-101 therapeutic strategy [7] |
| ABEmax | 4T | Moderate | Significantly improved | Not reported | Base editing application [7] |
| ABEmax | 3TC | Not reported | Enhanced | Not reported | Base editing application [7] |
Context-Dependent Performance Benefits
The performance advantage of the 3TC scaffold varies significantly depending on experimental conditions:
- Under standard transfection conditions with abundant vector availability, the 4T scaffold often achieves near-perfect editing efficiency (>95%) for most gRNAs, making the 3TC advantage minimal in these scenarios [7].
- With limited vector availability, the 3TC scaffold demonstrates markedly superior performance, yielding significantly higher editing efficiencies compared to the 4T scaffold [7].
- For T-rich gRNA sequences, the 4T scaffold shows substantially reduced activity due to compounded transcription inhibition, while the 3TC modification effectively rescues editing efficiency by boosting transcript levels [7].
- In therapeutic contexts where delivery efficiency is constrained (e.g., AAV vector capacity limitations), the 3TC scaffold provides critical enhancements, as demonstrated in the EDIT-101 therapeutic strategy [7].
Table 2: Performance of 3TC Scaffold Across Different CRISPR Applications
| Application | 4T Scaffold Limitations | 3TC Scaffold Benefits | Key Findings |
|---|---|---|---|
| Standard Gene Knockout | Moderate efficiency for low-activity gRNAs | Rescues efficiency of low-activity gRNAs | 8.1-13.5 additional doublings in gRNA levels for problematic gRNAs [7] |
| High-Fidelity Editing | Reduced efficiency with SpCas9-HF1 and eSpCas9(1.1) | Improves editing efficiency of high-fidelity variants | Maintains target specificity while enhancing on-target activity [7] |
| Base Editing | Suboptimal efficiency with ABEmax | Enhances base editing performance | Increases base conversion rates without compromising product purity [7] |
| SaCas9 Applications | Variable performance across targets | Improves editing activity and consistency | Particularly beneficial for AAV-delivered therapeutic applications [7] |
| Low Vector Availability | Significant reduction in editing efficiency | Maintains high efficiency with limited vector doses | Critical advantage for therapeutic delivery scenarios [7] |
Experimental Protocols and Methodologies
Key Experimental Workflow for 3TC Scaffold Validation
The following diagram outlines the comprehensive experimental approach used to validate the 3TC scaffold enhancement:
Detailed Methodological Approaches
Plasmid Construction and gRNA Scaffold Modification
The 3TC scaffold was implemented in the PX459.v2 backbone (Addgene #62988) through site-directed mutagenesis to replace the fourth T in the tetraloop with C and the complementary A with G [7]. This preserved the secondary structure while eliminating one T in the problematic T-stretch. Modified plasmids containing the 3TC scaffold are available through Addgene, including PX459v3 (plasmid #178799) and SaCas9v3-Puro (plasmid #178813) [48].
Cell Culture and Transfection
- Cell lines: HEK293T (human), C2C12 (mouse myoblast), and mouse embryonic stem cells (mES) were maintained under standard conditions [7].
- Transfection: Lipofectamine-based transfection with varying plasmid quantities (10-500 ng) was performed to assess efficiency under different vector availability conditions [7].
- Selection: Puromycin selection (2-5 μg/mL for 24-48 hours) was applied post-transfection to enrich for transfected cells, except in limited vector experiments where selection was omitted [7].
gRNA Transcript Quantification
Total RNA was extracted 48 hours post-transfection using TRIzol reagent. Reverse transcription followed by quantitative PCR (qPCR) with sgRNA-specific primers was performed to measure gRNA transcript levels, normalized to U6 snRNA as an internal control [7].
Editing Efficiency Analysis
Genomic DNA was extracted 72-96 hours post-transfection. Target loci were amplified by PCR and subjected to next-generation sequencing (Illumina platform) to quantify indel frequencies. Editing efficiency was calculated as the percentage of modified reads containing indels at the target site [7].
Research Reagent Solutions
Table 3: Essential Research Reagents for Implementing 3TC Scaffold Modifications
| Reagent/Resource | Function/Application | Availability |
|---|---|---|
| PX459v3 (Plasmid #178799) | SpCas9 with 2A-Puro and modified sgRNA scaffold with 3TC | Addgene [48] |
| SaCas9v3-Puro (Plasmid #178813) | SaCas9 with 2A-Puro and modified sgRNA scaffold | Addgene [48] |
| ABEmax-Puro V3 (Plasmid #226956) | ABEmax base editor with 3TC scaffold for enhanced base editing | Addgene [48] |
| PDG459 V3 (Plasmid #226958) | Dual gRNA vector with 3TC scaffolds for multiplexed editing | Addgene [48] |
| U6 Promoter | RNA Pol III promoter for high-level gRNA expression | Standard molecular biology suppliers |
| BbsI Restriction Enzyme | Golden Gate cloning for gRNA insertion into expression vectors | Common enzyme suppliers |
Discussion and Research Implications
Strategic Implementation Guidelines
The 3TC scaffold modification represents a simple yet effective optimization that researchers should consider incorporating into their CRISPR workflows, particularly for:
- Therapeutic applications where delivery efficiency is limited and maximal editing from minimal vector is crucial [7].
- Difficult-to-edit targets with T-rich sequences or historically low efficiency [7].
- High-fidelity applications using engineered Cas9 variants that typically suffer from reduced activity [7].
- Multiplexed editing where sufficient expression of multiple gRNAs is challenging [7] [48].
Compatibility with Advanced CRISPR Systems
The 3TC scaffold modification demonstrates broad compatibility across diverse CRISPR platforms:
- Base editing systems: ABEmax efficiency was enhanced with the 3TC scaffold, improving base conversion rates without increasing off-target effects [7].
- High-fidelity SpCas9 variants: Both SpCas9-HF1 and eSpCas9(1.1) showed improved editing efficiency with the 3TC scaffold while maintaining their enhanced specificity [7].
- SaCas9 platforms: The modification proved particularly valuable for SaCas9, which is favored for therapeutic applications due to its smaller size and AAV compatibility [7] [5].
Research Applications and Future Directions
The significant enhancement in gRNA transcription provided by the 3TC scaffold (ranging from 271 to over 11,000-fold increases in transcript levels for problematic gRNAs) enables more reliable experimentation across diverse research contexts [7]. This optimization facilitates the generation of animal models with higher efficiency, enhances functional genomics screens by improving knockout consistency, and supports therapeutic development by increasing the potency of gene editing formulations, particularly in challenging delivery contexts where vector quantity is constrained.
Future research directions include exploring additional scaffold modifications that could further enhance gRNA stability and functionality, optimizing this approach for emerging CRISPR systems beyond Cas9, and conducting comprehensive in vivo studies to validate the therapeutic benefits of this optimization in clinical-relevant models.
Achieving high editing efficiency is pivotal for successful CRISPR-Cas9 applications in both basic research and therapeutic development [7]. The single guide RNA (gRNA) serves as the targeting component of the CRISPR system, and its sufficient expression is a critical determinant of editing success. The standard gRNA scaffold contains a sequence of four thymine nucleotides (4T), which is known to inhibit transcription from Pol III promoters such as the U6 promoter commonly used to drive gRNA expression [7] [46]. This transcriptional suppression presents a particular challenge for gRNAs that are already T-rich in their targeting sequence, as these guides suffer from compounded transcriptional deficiencies that substantially reduce their editing efficiency [7].
The performance implications of suboptimal gRNA expression extend across the CRISPR toolkit, affecting standard SpCas9 systems, high-fidelity variants, base editors, and the smaller SaCas9 ortholog preferred for therapeutic viral delivery [7]. This review comprehensively compares optimization strategies for T-rich gRNAs, with particular focus on scaffold engineering solutions that enhance transcript levels and subsequently improve editing outcomes across both SpCas9 and SaCas9 platforms. Through systematic analysis of experimental data and methodologies, we provide researchers with evidence-based protocols for overcoming the limitations of T-rich gRNAs in their editing workflows.
The Poly-T Problem: Mechanistic Insights and Performance Impact
Transcription Inhibition by Scaffold Poly-T Tracts
The molecular basis of the poly-T problem lies in the termination signal function of T-tracts for RNA polymerase III (Pol III). In native contexts, consecutive thymine nucleotides serve as transcription termination signals for Pol III promoters, including the U6 promoter widely used in CRISPR systems [7]. When the gRNA scaffold contains a 4T sequence, this creates a premature termination signal that reduces the yield of full-length gRNA transcripts. The issue is particularly pronounced for gRNAs with T-rich target sequences because the combined effect of genomic T-content and scaffold T-content creates an additive inhibitory effect on transcription.
Experimental evidence demonstrates that gRNAs with lower activity were frequently T-rich and exhibited reduced gRNA transcript levels as measured by quantitative PCR [7]. This transcriptional deficiency directly correlated with reduced editing efficiencies in human and mouse cell lines. For instance, while many gRNAs achieved near-perfect editing (>95%) with standard scaffolds, T-rich gRNAs such as mDmd-E, -H, -I, and -K showed only moderate editing efficiency of 83-89% in C2C12 cells [7], highlighting the significant performance penalty associated with T-rich sequences.
Quantitative Assessment of Editing Deficiency
Table 1: Performance Comparison of Standard vs. T-Rich gRNAs
| gRNA ID | Cell Line | Target Gene | Editing Efficiency (4T Scaffold) | Editing Efficiency (3TC Scaffold) | Fold Improvement |
|---|---|---|---|---|---|
| hDMD-B | HEK293T | DMD | ~92.0% | ~97.4% | ~1.06x |
| mDmd-E | C2C12 | Dmd | ~85% | >95% | >1.12x |
| mDmd-H | C2C12 | Dmd | ~84% | >95% | >1.13x |
| mDmd-I | C2C12 | Dmd | ~83% | >95% | >1.14x |
| mDmd-K | C2C12 | Dmd | ~89% | >95% | >1.07x |
| mPrl-D | mES | Prl | ~93.6% | >95% | ~1.02x |
The performance data reveal a consistent pattern where T-rich gRNAs underperform compared to their non-T-rich counterparts when using standard scaffold configurations [7]. The deficiency is most pronounced in challenging delivery contexts, including hard-to-transfect cell types and in vivo applications where vector quantity is limited. This underscores the importance of scaffold optimization particularly for therapeutic applications where delivery efficiency is often a bottleneck.
Scaffold Engineering Solutions: The 3TC Modification
Molecular Design and Mechanism
The most direct solution to the poly-T transcription problem involves engineering the gRNA scaffold to reduce consecutive thymine residues while maintaining structural integrity and Cas9 binding capability. The 3TC scaffold modification represents a particularly effective approach, wherein the fourth T nucleotide in the tetraloop is replaced with a C nucleotide, effectively shortening the poly-T tract from 4T to 3TC [7]. To maintain base pairing in the tetraloop structure, the corresponding complementary A nucleotide in the scaffold is replaced with a G nucleotide [7].
This minimal modification preserves the functional architecture of the gRNA while eliminating the Pol III termination signal that impedes transcription efficiency. The 3TC scaffold can be incorporated into standard CRISPR vectors through site-directed mutagenesis of the scaffold region, making it readily implementable in existing laboratory systems without requiring specialized reagents or protocols.
Quantitative Performance Enhancement
Table 2: 3TC Scaffold Efficacy Across Editing Platforms
| Editing System | Enhancement with 3TC Scaffold | Key Applications | Notable Findings |
|---|---|---|---|
| SpCas9 (standard) | 1.02-1.14x editing improvement | Basic gene knockout | Rescues inefficient T-rich gRNAs to >95% efficiency [7] |
| SpCas9-HF1 | Significant enhancement reported | High-specificity editing | Improves efficiency without compromising specificity [7] |
| eSpCas9(1.1) | Significant enhancement reported | High-specificity editing | Improves efficiency without compromising specificity [7] |
| ABEmax | Compatible with efficiency gain | Base editing | Enhances A•T to G•C conversion efficiency [7] |
| SaCas9 | Increased gRNA transcription | Therapeutic applications | Improves performance of EDIT-101 strategy [7] |
The 3SC modification demonstrates remarkable versatility across CRISPR platforms. For standard SpCas9 systems, the scaffold enhancement proved particularly beneficial under conditions of limited vector availability, where the 3TC scaffold yielded significantly higher editing efficiency compared to the 4T scaffold [7]. This advantage positions the 3TC modification as particularly valuable for therapeutic applications where delivery efficiency is often constrained.
Experimental Comparison: SpCas9 vs. SaCas9 Performance with T-Rich gRNAs
Methodology for Editing Efficiency Assessment
Researchers employ multiple methods to quantify genome editing efficiency, each with distinct advantages and limitations [49]. The T7 Endonuclease I (T7EI) assay detects alleles with small insertions or deletions (indels) by cleaving heteroduplex DNA fragments at mismatch sites, though it provides only semi-quantitative results [49]. Tracking of Indels by Decomposition (TIDE) and Inference of CRISPR Edits (ICE) methods analyze Sanger sequencing chromatograms through sequence trace decomposition algorithms to yield more quantitative estimations of editing frequencies [49]. Droplet digital PCR (ddPCR) offers highly precise and quantitative measurements using differentially labeled fluorescent probes, while fluorescent reporter systems enable live-cell tracing of editing events [49].
For the comparative analysis of SpCas9 and SaCas9 performance with T-rich gRNAs, the cited studies primarily utilized T7EI and sequencing-based approaches (TIDE/ICE) in conjunction with qPCR measurement of gRNA transcript levels [7]. The experimental workflow typically involved:
- Design of T-rich gRNAs targeting model genes (EMX1, VEGFA, RUNX1, DMD)
- Cloning into CRISPR vectors with 4T and 3TC scaffold variants
- Transfection into relevant cell lines (HEK293T, C2C12, mES)
- Selection with puromycin (unless testing limited vector conditions)
- Genomic DNA extraction and PCR amplification of target loci
- Efficiency quantification via T7EI or sequencing methods
- gRNA transcript measurement by qPCR
Comparative Performance Data
The performance differential between standard and optimized scaffolds reveals context-dependent advantages. For SpCas9 systems, the 3TC modification provided modest improvements (2-5%) for already efficient gRNAs under standard transfection conditions with selection [7]. However, the enhancement became substantially more significant (exceeding 10% absolute improvement) for problematic T-rich gRNAs and under limited vector conditions where transcription efficiency becomes the rate-limiting factor [7].
SaCas9 systems similarly benefited from the 3TC scaffold modification, showing increased gRNA transcription and improved editing activity [7]. This enhancement is particularly valuable for SaCas9 applications, which often involve viral delivery where payload size constraints and limited transduction efficiency create challenging environments for editing efficacy. The 3TC modification was successfully applied to the EDIT-101 therapeutic strategy, demonstrating marked improvements in performance [7].
Diagram 1: Strategic workflow for addressing T-rich gRNA limitations through scaffold engineering, demonstrating the causal relationship between the poly-T problem, 3TC solution, and resulting performance enhancements across CRISPR platforms.
Table 3: Research Reagent Solutions for gRNA Optimization
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| PX459.v2 (4T scaffold) | All-in-one CRISPR plasmid with original scaffold | Baseline for comparison; contains CBh-driven SpCas9-T2A-Puro and U6-driven gRNA [7] |
| 3TC scaffold vectors | Modified gRNA expression vectors | Created by site-directed mutagenesis of fourth T to C in tetraloop with complementary A to G change [7] |
| pDG459 (dual U6) | Dual gRNA expression vector | Alternative approach to boost gRNA levels through promoter doubling [7] |
| SpCas9-HF1/eSpCas9(1.1) | High-fidelity Cas9 variants | Benefit from 3TC modification for improved efficiency [7] |
| ABEmax | Adenine base editor | Compatible with 3TC scaffold for enhanced base editing [7] |
| SaCas9 constructs | Compact Cas9 for therapeutic delivery | Shows improved performance with 3TC modification [7] |
| T7 Endonuclease I | Editing efficiency detection | Semi-quantitative assessment of indels [49] |
| TIDE/ICE analysis | Sequencing-based efficiency quantification | Quantitative editing assessment from Sanger sequencing [49] |
| ddPCR systems | High-precision editing quantification | Absolute quantification of editing frequencies [49] |
Discussion and Strategic Implementation Guidelines
The comparative analysis of scaffold optimization strategies reveals that 3TC modification provides a robust solution to the T-rich gRNA problem across both SpCas9 and SaCas9 platforms. The enhancement is most valuable in contexts where transcription efficiency limits editing outcomes, including:
- Therapeutic applications with limited vector availability [7]
- Hard-to-transfect cell types where delivery efficiency is low
- High-fidelity editing systems where reduced off-targeting comes at an efficiency cost [7]
- Viral delivery contexts where payload size constraints favor smaller constructs [16]
For researchers designing CRISPR experiments, particularly those involving T-rich target sites, the 3TC scaffold modification represents a simple yet effective optimization that can rescue otherwise problematic gRNAs. The modification is straightforward to implement through standard molecular biology techniques and maintains compatibility with existing CRISPR workflows without requiring specialized equipment or reagents.
Future directions in gRNA optimization will likely involve more sophisticated scaffold engineering combined with chemical modifications to enhance stability and performance. However, the 3TC solution currently offers an immediately accessible and highly effective strategy for overcoming the limitations of T-rich gRNAs in both basic research and therapeutic development contexts.
In the therapeutic application of CRISPR-Cas9 systems, efficient delivery often represents the most significant bottleneck. The packaging capacity of viral vectors, particularly adeno-associated viruses (AAVs), imposes strict limitations on the quantity of genetic material that can be delivered to target cells [7] [16]. This constraint is especially pertinent when comparing the two most widely used Cas9 orthologs: the canonical Streptococcus pyogenes Cas9 (SpCas9) and the smaller Staphylococcus aureus Cas9 (SaCas9). While SaCas9's compact size facilitates easier packaging into AAVs, both systems face the fundamental challenge of achieving sufficient editing efficiency when vector copies are limited [7] [16].
Emerging research demonstrates that a critical factor in overcoming this challenge lies in optimizing gRNA expression levels. The conventional gRNA scaffold contains a sequence of four thymine nucleotides (4T) that can prematurely terminate transcription by RNA polymerase III (Pol III) promoters, such as the U6 promoter [7]. This review objectively compares the performance of SpCas9 and SaCas9 systems under vector-limited conditions, with a specific focus on how enhanced gRNA expression through scaffold engineering transforms editing outcomes, supported by direct experimental evidence.
gRNA Scaffold Engineering: A Solution to Transcriptional Limitation
The 4T Transcriptional Termination Problem
The standard gRNA scaffold contains a sequence of four thymine nucleotides (4T), which functions as a termination signal for Pol III transcription [7]. Under standard laboratory conditions using plasmid transfection with subsequent selection, this limitation may be masked by high vector copy numbers. However, in therapeutic contexts where vector availability is constrained, this transcriptional impairment becomes a critical determinant of editing success [7].
The 3TC Scaffold Modification
A simple yet powerful modification to the gRNA scaffold involves shortening the 4T sequence by replacing the fourth thymine in the tetraloop with a cytosine, creating a 3TC scaffold, with a corresponding complementary change from adenine to guanine [7]. This minimal alteration reduces the premature transcription termination while maintaining the scaffold's structural integrity.
Table 1: Impact of 3TC Scaffold Modification on gRNA Expression and Editing Efficiency
| Parameter | 4T Scaffold (Standard) | 3TC Scaffold (Modified) | Experimental Context |
|---|---|---|---|
| gRNA Transcript Levels | Baseline | 271-11,349-fold increase (8.1-13.5 doublings) [7] | qPCR measurement of suboptimal gRNAs (mDmd-E, -H, -I) |
| Editing Efficiency (Low-Performing gRNAs) | 83-89% | >95% [7] | Problematic gRNAs in C2C12 cells |
| Editing Efficiency (Moderate gRNAs) | ~92.0% | ~97.4% [7] | hDMD-B gRNA in HEK293T cells |
| Performance Under Vector Limitation | Significantly reduced editing | Maintained high editing efficiency [7] | Low plasmid transfection without selection |
Comparative Performance of SpCas9 and SaCas9 with Enhanced gRNA Expression
SpCas9 Systems Benefit from Transcript Enhancement
Research demonstrates that SpCas9 editing efficiency under vector-limited conditions improves markedly with the 3TC scaffold modification [7]. In experiments where plasmid quantity was restricted by omitting selection steps, the 3TC scaffold yielded significantly higher editing efficiencies compared to the standard 4T scaffold [7]. This enhancement proves particularly valuable for challenging gRNAs that are T-rich in sequence and would otherwise produce suboptimal editing outcomes.
The 3TC scaffold modification also benefits high-fidelity SpCas9 variants (e.g., SpCas9-HF1, eSpCas9[1].1) and base editing systems such as ABEmax, which typically exhibit reduced activity compared to the wild-type nuclease [7]. By increasing gRNA availability, the modification helps overcome the inherent efficiency trade-offs of these more precise editing systems.
SaCas9 Displays Enhanced Therapeutic Compatibility
SaCas9 recognizes an NNGRRT PAM and possesses a naturally compact size that allows complete packaging in a single AAV vector, making it particularly advantageous for therapeutic applications [7] [16]. The 3TC scaffold modification significantly increases gRNA transcription for SaCas9 systems, subsequently improving editing activity [7]. This combination of smaller nuclease size and enhanced gRNA expression creates a powerful platform for in vivo genome editing.
Notably, the 3TC modification was applied to the EDIT-101 therapeutic strategy, where it demonstrated marked improvements in performance [7]. This successful translation from basic optimization to therapeutic application underscores the clinical relevance of gRNA scaffold engineering.
Table 2: SpCas9 vs. SaCas9 Characteristics with gRNA Optimization
| Characteristic | SpCas9 | SaCas9 | Impact of Enhanced gRNA |
|---|---|---|---|
| Size | 4.2 kb (theoretically challenging for AAV packaging) [16] | ~1 kb smaller than SpCas9 (compatible with AAV) [16] | More critical for SpCas9 due to packaging constraints |
| PAM Requirement | NGG [7] | NNGRRT [7] | gRNA enhancement benefits both PAM specificities |
| Therapeutic Application | Limited by delivery options | Well-suited for AAV delivery [7] [16] | SaCas9-3TC combination ideal for vector-limited contexts |
| Editing Efficiency with Limited Vectors | Moderate with 4T, significantly enhanced with 3TC [7] | Moderate with 4T, significantly enhanced with 3TC [7] | Both benefit, but SaCas9 has inherent delivery advantages |
Experimental Protocols for Validating Enhanced gRNA Expression
gRNA Scaffold Modification Protocol
The modification from 4T to 3TC scaffold involves precise molecular cloning techniques:
- Plasmid Selection: Use standard CRISPR plasmids (e.g., PX459.v2 for SpCas9) containing the U6-driven gRNA expression cassette [7].
- Scaffold Modification: Employ site-directed mutagenesis to replace the fourth thymine (T) in the tetraloop of the gRNA scaffold with cytosine (C) [7].
- Complementary Change: Make a corresponding change from adenine (A) to guanine (G) in the complementary sequence to maintain scaffold stability [7].
- Validation: Sequence the modified scaffold to confirm the 3TC alteration before proceeding to experimental applications.
Quantifying gRNA Transcript Levels
Accurate measurement of gRNA expression is essential for validating the impact of scaffold modifications:
- RNA Extraction: Harvest transfected cells 48-72 hours post-transfection using appropriate RNA isolation methods [7].
- cDNA Synthesis: Perform reverse transcription with gRNA-specific primers.
- qPCR Analysis: Implement quantitative PCR with probes targeting the constant region of the gRNA scaffold [7].
- Normalization: Use appropriate endogenous controls for normalization and calculate fold-changes using the ΔΔCt method [7].
Assessing Editing Efficiency Under Vector Limitation
To objectively compare editing performance in vector-restricted conditions:
- Vector Titration: Transfect cells with serially diluted plasmid quantities (e.g., from 500 ng to 10 ng per well in 24-well plates) without implementing antibiotic selection [7].
- Genomic DNA Extraction: Harvest cells 72-96 hours post-transfection and isolate genomic DNA.
- Target Amplification: PCR-amplify the target region using gene-specific primers.
- Editing Quantification: Analyze INDEL frequencies using next-generation sequencing or T7 endonuclease I assays [7] [50].
- Data Analysis: Compare editing efficiencies across vector quantities and between scaffold types to determine the enhancement factor provided by the 3TC modification.
Visualization of gRNA Enhancement Strategy
The Scientist's Toolkit: Essential Reagents for gRNA Optimization
Table 3: Research Reagent Solutions for gRNA Enhancement Studies
| Reagent / Tool | Function | Application Notes |
|---|---|---|
| PX459.v2 Plasmid | All-in-one vector with CBh-driven SpCas9-T2A-Puro and U6-driven gRNA [7] | Base plasmid for 3TC scaffold modification |
| Dual gRNA Plasmid (pDG459) | Contains dual U6 expression cassettes for increased gRNA dosage [7] | Alternative approach to enhance gRNA expression |
| 3TC-Modified Scaffold | gRNA scaffold with reduced poly-T tract [7] | Core modification to reduce Pol III termination |
| SaCas9 Expression System | Compact Cas9 ortholog for AAV delivery [7] [16] | Ideal for therapeutic applications with size constraints |
| High-Fidelity Cas9 Variants | Engineered nucleases with reduced off-target effects (e.g., SpCas9-HF1, eSpCas9[1].1) [7] | Benefit from enhanced gRNA expression to compensate for reduced activity |
| ABEmax Base Editor | Adenine base editor fusion protein [7] | Improved efficiency with 3TC scaffold |
| U6 Promoter Plasmids | RNA Pol III promoters for gRNA expression [7] | Standard system sensitive to poly-T termination signals |
The objective comparison of SpCas9 and SaCas9 systems under vector-limited conditions reveals that enhanced gRNA expression through scaffold optimization provides a critical advantage for therapeutic applications. While SaCas9 offers inherent benefits for AAV packaging due to its smaller size, both nucleases significantly benefit from the 3TC modification that boosts gRNA transcript levels [7].
The experimental data consistently demonstrates that reducing the poly-T tract in the gRNA scaffold increases transcription efficiency, leading to markedly improved editing outcomes when vector copies are limited. This optimization approach proves compatible with diverse editing platforms, including high-fidelity nucleases and base editors, and has already shown success in therapeutic strategies like EDIT-101 [7].
For researchers and drug development professionals, these findings highlight the importance of gRNA scaffold engineering as a fundamental optimization strategy in CRISPR therapeutic development. By implementing the 3TC modification, scientists can maximize editing efficiency within the stringent constraints of viral delivery systems, accelerating the translation of CRISPR technologies from laboratory tools to clinical solutions.
The CRISPR-Cas9 system has revolutionized genetic engineering, offering unprecedented precision in genome editing. Among the growing portfolio of Cas9 nucleases, two variants stand out for their distinct characteristics and widespread application: the well-established Streptococcus pyogenes Cas9 (SpCas9) and the compact Staphylococcus aureus Cas9 (SaCas9). While SpCas9 serves as the gold standard with its NGG PAM requirement, SaCas9 recognizes an NNGRRT PAM and possesses a significantly smaller size, making it ideal for viral vector packaging such as adeno-associated viruses (AAVs) [25]. This fundamental difference in PAM requirements, however, presents a significant methodological challenge for researchers seeking to objectively compare their performance within the same genomic context.
Direct comparisons between Cas9 nucleases with different PAM specifications are inherently biased when performed across different genomic sites, as editing efficiency is strongly influenced by both the guide RNA sequence and the local genomic environment [15] [9]. Traditional bioinformatic tools like CHOPCHOP and CRISPOR are designed to identify optimal gRNAs for a single nuclease within a gene of interest but lack the functionality for fair cross-nuclease comparison [15]. This technological gap necessitates a specialized tool that can identify overlapping targetable sites, enabling unbiased experimental comparisons. The Comparing Cas9 Activities by Target Superimposition (CATS) bioinformatic tool was developed specifically to address this challenge, automating the detection of overlapping PAM sequences and facilitating direct performance comparisons between SpCas9, SaCas9, and other Cas9 variants [15] [9].
CATS is a specialized bioinformatic tool designed to automate the detection of genomic regions where PAM sequences for different Cas9 nucleases overlap or are in close proximity. This functionality addresses the critical need for standardized comparison by minimizing sequence composition bias that would otherwise confound nuclease performance evaluations [15]. The tool operates through a structured workflow that integrates multiple data sources and user-defined parameters to identify comparable target sites.
The core analysis is driven by user-specified PAM sequences input using standard IUPAC nucleotide notation. CATS is not limited to predefined CRISPR-Cas9 systems and can accept any PAM sequence, though it is optimized for Cas9-like systems where the PAM is located immediately downstream (3′) of the spacer [15] [9]. Users can define the search window size, which determines the maximum distance between PAM sequences to be considered overlapping, and specify the amount of sequence context to report around each identified PAM. The tool comes with built-in references for human and mouse genomes based on GENCODE transcript sequences and can incorporate custom genomes via FASTA files [9].
A particularly powerful feature of CATS is its integration with the ClinVar database, which enables the identification of pathogenic mutations within the analyzed genomic regions [15] [9]. This functionality supports the development of allele-specific targeting strategies by highlighting mutations that either generate a de novo PAM or occur within the seed sequence (typically the first 10 nucleotides upstream of the PAM), both of which can be leveraged to discriminate between healthy and disease-causing alleles in autosomal dominant disorders [9]. The following diagram illustrates the complete CATS analytical workflow:
Figure 1: CATS Analytical Workflow for Cas9 Nuclease Comparison
Comparative Performance Analysis: SpCas9 vs. SaCas9
Key Characteristics and Experimental Data
When comparing SpCas9 and SaCas9 performance using standardized methodologies enabled by CATS, several critical differences emerge beyond their obvious PAM requirement variations. The table below summarizes the fundamental characteristics and comparative performance data for these two widely used nucleases:
Table 1: Direct Comparison of SpCas9 and SaCas9 Characteristics and Performance
| Parameter | SpCas9 | SaCas9 | Experimental Context |
|---|---|---|---|
| PAM Requirement | NGG [25] | NNGRRT [25] | Fundamental targeting specificity |
| Size (aa) | 1368 [25] | 1053 [25] | AAV packaging feasibility |
| Editing Efficiency | 83-100% with 4T scaffold [7] | Improved with 3TC scaffold [7] | Multiple genomic loci in HEK293T, C2C12, mES cells |
| gRNA Optimization | 3TC scaffold boosts efficiency under limited vector conditions [7] | 3TC scaffold increases editing activity [7] | EDIT-101 therapeutic strategy |
| Therapeutic Application | Wide research applications | Advantage: Fits single AAV vector with promoter and gRNA [25] | In vivo neuronal and liver disease models |
The editing efficiency data reveals that while both nucleases can achieve high performance, SpCas9 with the original gRNA scaffold containing four thymine nucleotides (4T) demonstrated near-perfect editing (83-100% modified alleles) across 55 different gRNAs targeting multiple genes in various cell lines [7]. However, gRNAs with T-rich sequences showed reduced efficiency, which was directly correlated with lower gRNA transcript levels. Modification of the gRNA scaffold to reduce the poly-T tract (3TC scaffold) significantly enhanced gRNA expression and improved editing efficiency for both SpCas9 and SaCas9, particularly under conditions of limited vector availability [7] [46].
gRNA Design Considerations and Optimization
gRNA design represents a critical factor in the performance of both SpCas9 and SaCas9. Recent research has demonstrated that the standard gRNA scaffold contains a sequence of four thymine nucleotides (4T) that can inhibit transcription from Pol III promoters such as the U6 promoter [7]. While this inhibitory effect didn't significantly impact editing efficiency for most gRNAs tested under standard transfection protocols with abundant vector quantities, it became a limiting factor for T-rich gRNAs and under conditions of limited vector availability.
The solution emerged through scaffold engineering: shortening the 4T sequence to 3TC (replacing the fourth T in the tetraloop with C and its complementary A with G) [7]. This modification markedly increased gRNA transcript levels—by 8.1–13.5 doublings (271-11,349 fold changes)—and subsequently enhanced editing efficiency for previously suboptimal gRNAs to over 95% [7]. This optimization proved beneficial for both SpCas9 high-fidelity variants and SaCas9, highlighting its broad applicability across nuclease platforms. The following experimental workflow diagram illustrates the process for validating Cas9 performance:
Figure 2: Experimental Workflow for Cas9 Nuclease Comparison
Experimental Design and Methodologies
Computational Protocol Using CATS
To implement CATS for systematic comparison of SpCas9 and SaCas9, researchers should follow this detailed computational protocol:
Input PAM Sequences: Specify the PAM sequences for both nucleases using IUPAC notation: "NGG" for SpCas9 and "NNGRRT" for SaCas9 [15] [9].
Define Genomic Regions: Input the target genomic sequence(s) either by selecting from built-in human or mouse genome references or by uploading custom FASTA files. For coding regions, restrict the analysis to specific genes of interest to enhance relevance and computational efficiency [9].
Set Analysis Parameters: Configure the maximum distance between PAM sequences (window size) to define what constitutes an overlapping site. The default of 25 nucleotides upstream and downstream of the PAM can be adjusted based on experimental needs [15].
Incorporate Pathogenic Variants (Optional): Enable the 'pathogenic' option to integrate ClinVar annotations. This identifies disease-causing mutations that create de novo PAM sequences or alter seed regions, enabling allele-specific targeting strategies for autosomal dominant disorders [9].
Execute Analysis and Interpret Results: Run the CATS algorithm to identify overlapping target sites. The output will list genomic coordinates where both nucleases can bind in proximity, facilitating direct experimental comparison.
Experimental Validation Protocol
Following the computational identification of overlapping target sites, this experimental protocol validates and compares nuclease performance:
Vector Preparation: Clone identified gRNA sequences into appropriate expression plasmids. For SpCas9, use PX459.v2 or similar vectors; for SaCas9, use KKHSaCas9 or similar backbones. Create both standard (4T) and modified (3TC) scaffold versions for each gRNA [7].
Cell Culture and Transfection: Culture relevant cell lines (HEK293T, C2C12, or others appropriate to the research context). Transfect with varying vector quantities (high: 2-4μg with selection; low: 0.1-0.5μg without selection) to assess efficiency under different resource conditions [7].
gRNA Expression Analysis: Isolate total RNA 48 hours post-transfection. Perform quantitative PCR (qPCR) with specific probes for gRNA transcripts to correlate expression levels with editing outcomes [7].
Editing Efficiency Assessment: Harvest cells 72-96 hours post-transfection. Extract genomic DNA and amplify target regions by PCR. Sequence amplicons using next-generation sequencing to precisely quantify indel frequencies and calculate editing efficiencies for both nucleases at comparable sites [7].
Data Analysis: Compare editing efficiencies between SpCas9 and SaCas9 at overlapping target sites using statistical tests (e.g., t-tests for paired comparisons). Correlate gRNA expression levels with editing efficiencies to validate the impact of scaffold modifications.
Research Reagent Solutions
Successful implementation of CATS-guided comparisons requires specific reagent systems optimized for both SpCas9 and SaCas9 nucleases:
Table 2: Essential Research Reagents for Cas9 Comparison Studies
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Expression Plasmids | PX459.v2 (SpCas9), pX601 (SaCas9) | Nuclease and gRNA delivery in mammalian systems [7]. |
| Modified gRNA Scaffolds | 3TC scaffold variants | Enhanced gRNA transcription, especially for T-rich guides and low vector conditions [7]. |
| Bioinformatic Tools | CATS, CRISPOR, CHOPCHOP | Target site identification, gRNA design, and nuclease comparison [15] [51]. |
| Delivery Vectors | AAV vectors (for in vivo) | Therapeutic delivery; SaCas9's smaller size allows packaging with promoter and gRNA in single AAV [25]. |
| Validation Assays | NGS amplicon sequencing, T7E1 assay | Quantitative assessment of editing efficiency and specificity [7]. |
The objective comparison of CRISPR-Cas9 nucleases with different PAM requirements represents a significant methodological challenge in genome engineering research. CATS addresses this fundamental need by enabling automated identification of overlapping target sites, thereby eliminating sequence composition bias and standardizing performance evaluations. Through systematic application of this tool combined with optimized experimental protocols, researchers can now generate reliable, directly comparable data on nuclease performance.
The comparison between SpCas9 and SaCas9 reveals a complex tradeoff landscape where factors beyond simple editing efficiency—including molecular size, PAM availability, and gRNA expression requirements—must be considered for specific applications. The development of scaffold modifications like the 3TC variant further enhances the performance of both nucleases, particularly under the constrained conditions typical of therapeutic applications. For research requiring maximal target flexibility and established protocols, SpCas9 remains the workhorse nuclease, while SaCas9 offers distinct advantages for viral delivery and specialized targeting contexts. As the CRISPR toolkit continues to expand, bioinformatic approaches like CATS will become increasingly vital for the rational selection and optimization of genome editing technologies across both basic research and clinical applications.
Head-to-Head Performance: Analyzing Editing Outcomes, Fidelity, and Specificity
The selection of a CRISPR-Cas system is a foundational decision in therapeutic development, with on-target efficiency being a critical performance metric. While the Cas9 nuclease from Streptococcus pyogenes (SpCas9) has been the workhorse of genome editing, its smaller ortholog from Staphylococcus aureus (SaCas9) offers distinct advantages for viral delivery due to its compact size. This guide provides a direct, data-driven comparison of their editing rates across standardized experimental setups, offering vital intelligence for researchers navigating the transition from benchtop to clinical application. The core distinction lies in their molecular architecture: SpCas9 (∼4.2 kb) recognizes an NGG PAM (Protospacer Adjacent Motif), while the smaller SaCas9 (∼3.2 kb) binds a more complex NNGRRT PAM, influencing their respective targeting scopes within the genome [7] [16]. Understanding their performance differential is essential for designing effective gene therapies, especially with the growing emphasis on in vivo applications where delivery constraints are paramount.
Quantitative Comparison of Editing Efficiency
Editing Performance at a Glance
The following table synthesizes quantitative editing efficiency data from comparable experimental conditions, primarily in human HEK293T cells, to enable a direct performance comparison.
Table 1: Direct Comparison of SpCas9 and SaCas9 On-Target Editing Efficiency
| Cas Nuclease | Target Locus/Gene | Cell Line | Editing Efficiency | Key Experimental Condition | Citation |
|---|---|---|---|---|---|
| SpCas9 | 55 different gRNAs (e.g., EMX1, VEGFA, RUNX1, DMD) | HEK293T | Most gRNAs: >95% modified reads [7] | Standard plasmid transfection with puromycin selection [7] | [7] |
| SpCas9 | mDmd-E, -H, -I, -K | Mouse C2C12 | Moderate efficiency: 83–89% modified alleles [7] | Standard plasmid transfection with puromycin selection [7] | [7] |
| SaCas9 | Multiple endogenous loci | HEK293T | Improved activity with 3TC scaffold modification [7] | Use of optimized 3TC gRNA scaffold [7] | [7] |
| SeqCas9 (SpCas9 ortholog, NNG PAM) | Multiple endogenous loci | HEK293T | Superior base editing efficiency vs. SpCas9-NG/SpCas9-NRRH [52] | GFP-activation assay & base editing assessment [52] | [52] |
Analysis of Comparative Performance
The data indicates that SpCas9, under standard laboratory conditions with high vector availability, achieves exceptionally high editing rates (often exceeding 95%) across a wide array of genomic loci [7]. However, its performance can be gRNA-dependent, with T-rich gRNAs sometimes showing reduced efficiency. SaCas9 demonstrates robust editing capability that is significantly enhanced by optimizing gRNA transcription. Specifically, reducing the poly-T tract in the gRNA scaffold from 4T to 3TC markedly increases gRNA transcript levels and subsequently improves SaCas9's editing activity, making it a critical optimization for therapeutic applications where vector doses are limited [7]. Furthermore, emerging SpCas9 orthologs like SeqCas9, which recognizes a simple NNG PAM, show promise by delivering high activity and enhanced specificity comparable to high-fidelity variants [52].
Detailed Experimental Protocols for Performance Comparison
Core Workflow for Efficiency Assessment
The following diagram outlines the general experimental workflow used to generate the comparative efficiency data cited in this guide.
Diagram 1: Workflow for comparing SpCas9 and SaCas9 efficiency. This general protocol, involving transfection, selection, and NGS analysis, underpins the data presented in this guide [7] [53].
Protocol Details: gRNA Optimization for Enhanced Transcription
A key methodological insight from recent studies is the protocol for boosting gRNA transcript levels, which is particularly beneficial for SaCas9 and suboptimal SpCas9 gRNAs [7].
- gRNA Scaffold Modification: The standard gRNA scaffold contains a sequence of four thymine nucleotides (4T), which can act as a termination signal for Pol III promoters like U6. The optimization involves shortening this to a 3TC sequence by:
- Replacing the fourth T nucleotide in the tetraloop with a C.
- Replacing its corresponding complementary A nucleotide with a G in the scaffold sequence [7].
- Vector Transfection: The modified CRISPR plasmid (e.g., PX459.v2 with 3TC scaffold) is transfected into cells using standard protocols.
- Efficiency Assessment under Limited Vector: To clearly demonstrate the benefit of the 3TC scaffold, editing efficiency is compared to the 4T scaffold under conditions of limited vector availability (e.g., low plasmid dose, no selection step), where the enhanced transcription provides a significant advantage [7].
The Scientist's Toolkit: Essential Research Reagents
Successful comparison and application of SpCas9 and SaCas9 require a suite of specialized reagents. The table below details key solutions used in the cited research.
Table 2: Essential Research Reagents for CRISPR-Cas9 Efficiency Studies
| Research Reagent | Function & Description | Application in Performance Comparison |
|---|---|---|
| All-in-One CRISPR Plasmid (e.g., PX459.v2) | Combines a CBh-driven Cas9 (Sp/Sa) and a U6-driven single gRNA expression cassette in a single vector [7]. | Serves as the standard delivery vehicle for both nucleases, enabling consistent transfection and selection. |
| Optimized gRNA Scaffold (3TC variant) | A modified gRNA scaffold where the 4T sequence is shortened to 3TC to enhance Pol III transcription and increase gRNA yield [7]. | Critical for maximizing SaCas9 and marginal SpCas9 gRNA activity; key differentiator in efficiency. |
| Puromycin Selection Marker | A selectable antibiotic marker (often co-expressed with Cas9 via a T2A peptide) to enrich for successfully transfected cells [7]. | Ensures high vector availability in edited cell populations, standardizing efficiency measurements. |
| High-Fidelity Cas9 Variants (e.g., SpCas9-HF1, eSpCas9(1.1)) | Engineered SpCas9 proteins with reduced off-target effects, though sometimes with a trade-off in on-target activity [7] [54]. | Used as a benchmark for comparing the specificity of new orthologs like SeqCas9 [52]. |
| AAV Delivery Vectors | Adenovirus vectors with limited cargo capacity (~4.5 kb), making them suitable for SaCas9 but not the larger SpCas9 [16]. | The primary delivery system for in vivo therapeutic applications, defining a key use-case for SaCas9. |
The choice between SpCas9 and SaCas9 is not a simple matter of declaring one superior to the other, but rather of matching nuclease properties to application requirements. SpCas9 remains a powerful and reliable choice for in vitro research where its high efficiency and well-characterized behavior are advantageous. However, for the burgeoning field of in vivo gene therapy, SaCas9's compact size is a decisive benefit, enabling efficient packaging into AAV vectors. The critical finding for therapeutic developers is that SaCas9's inherent efficiency can be optimized to rival that of SpCas9 through straightforward gRNA scaffold engineering (3TC modification) [7]. Furthermore, the exploration of novel orthologs like SeqCas9, which combines an NNG PAM with high activity and specificity, points to a future where the distinction between "standard" and "alternative" nucleases will blur, offering a expanded toolkit for precision genetic medicine [52].
The CRISPR-Cas9 system has revolutionized genome engineering, with Cas9 orthologs from Streptococcus pyogenes (SpCas9) and Staphylococcus aureus (SaCas9) emerging as two of the most widely used nucleases in both basic research and therapeutic development [25] [6]. While both enzymes catalyze double-strand breaks (DSBs) in DNA, their molecular mechanisms and resulting editing outcomes differ significantly. Understanding these differences is crucial for selecting the appropriate nuclease for specific research or therapeutic applications. This guide provides a comprehensive comparison of the indel patterns generated by these two nucleases, focusing on SpCas9's characteristic staggered cut bias and SaCas9's distinct editing profiles, to inform experimental design and nuclease selection in genome editing workflows.
Molecular Mechanisms and Cleavage Profiles
SpCas9's Staggered Cut Configuration
SpCas9 was initially characterized as generating blunt-ended double-strand breaks (DSBs). However, recent high-resolution analyses reveal that SpCas9 exhibits flexible cleavage profiles, with a significant proportion of cuts resulting in staggered ends [55]. BreakTag methodology, which enables genome-wide profiling of Cas9-induced DSBs, has demonstrated that approximately 35% of SpCas9 cuts create staggered ends rather than blunt breaks [55]. This staggered cleavage configuration often results in 5' overhangs of a single nucleotide, which profoundly influences the repair outcome.
The specific type of incision made by SpCas9 is influenced by multiple factors, including DNA:gRNA complementarity and the use of engineered Cas9 variants [55]. This flexible cut profile contradicts the earlier simplistic model of uniform blunt-end formation and provides a mechanistic explanation for the non-random repair outcomes observed with SpCas9 editing.
SaCas9's Cleavage Characteristics
In contrast to SpCas9, SaCas9 demonstrates different cleavage dynamics that contribute to its distinct editing outcomes. While detailed structural studies of SaCas9's cleavage configuration are less extensive than for SpCas9, comparative functional analyses reveal that SaCas9 exhibits a more balanced distribution of indel types without the strong +1 insertion bias characteristic of SpCas9 [6]. This fundamental difference in cleavage behavior and repair outcome patterning suggests that the molecular architecture of SaCas9 engages with DNA repair machinery differently than SpCas9.
The smaller size of SaCas9 (1053 amino acids vs. 1368 for SpCas9) contributes to its popularity for viral delivery applications, particularly in adeno-associated virus (AAV)-based therapeutic strategies [25] [56]. This size difference, while practically advantageous for delivery, also reflects evolutionary divergence in protein structure that likely underlies their distinct cleavage behaviors.
Comparative Analysis of Editing Outcomes
Indel Pattern Differences
Direct comparative studies of editing outcomes at multiple genomic loci reveal fundamental differences in the indel patterns generated by SpCas9 and SaCas9. The table below summarizes the key distinctions based on experimental evidence from human iPSCs and K562 cells [6]:
| Editing Characteristic | SpCas9 | SaCas9 |
|---|---|---|
| +1 Insertion Bias | Strong preference at 4th nucleotide upstream of PAM | More balanced indel distribution |
| Deletion Patterns | Diverse small deletions | Context-dependent variations |
| Overall Editing Efficiency | Variable across sites | Generally high efficiencies |
| Knock-in Efficiency | Moderate | Higher for both NHEJ-mediated and HDR-mediated integration |
SpCas9 exhibits a pronounced +1 insertion bias at the fourth nucleotide upstream of the protospacer adjacent motif (PAM), a characteristic consistent with its tendency to generate staggered cuts [6]. This non-random insertion pattern is mechanistically linked to its flexible scission profile, where staggered breaks can promote single-nucleotide insertions during non-homologous end joining (NHEJ) repair [55].
SaCas9 demonstrates distinct editing outcomes without the strong +1 insertion bias observed with SpCas9 [6]. This difference in indel patterning reflects fundamental variations in how these nucleases interact with DNA and how their cleavage products are processed by cellular repair machinery.
Figure 1: Mechanistic workflow of SpCas9 and SaCas9 editing outcomes. The diagram illustrates how identical CRISPR-Cas9 ribonucleoprotein (RNP) complexes lead to different editing outcomes through nuclease-specific processing pathways, highlighting the key difference in insertion bias.
Efficiency and Specificity Comparison
Beyond indel patterns, SpCas9 and SaCas9 differ significantly in their editing efficiencies and genome-wide specificities:
| Performance Metric | SpCas9 | SaCas9 |
|---|---|---|
| On-target Efficiency | Context-dependent | Generally high across multiple sites |
| Off-target Effects | Moderate to high | Significantly reduced |
| Spacer Length Optimization | 18-21 nt (optimal: 20 nt) | 19-23 nt (optimal: 21 nt) |
| Therapeutic Knock-in | Moderate efficiency | Higher efficiency |
Experimental evidence indicates that SaCas9 often achieves higher editing efficiencies than SpCas9 across multiple genomic loci in human cells [6]. Furthermore, GUIDE-seq analysis reveals that SaCas9 exhibits significantly reduced off-target effects compared to SpCas9, making it a preferable choice for applications requiring high genome-wide precision [6] [56].
The optimal spacer length for single-guide RNAs (sgRNAs) differs between the two systems: 20 nucleotides for SpCas9 versus 21 nucleotides for SaCas9 [6]. This distinction highlights the importance of nuclease-specific guide RNA optimization for achieving maximal editing efficiency.
Experimental Methodologies for Indel Analysis
CLEAR-time dPCR for DNA Repair Quantification
The CLEAR-time dPCR (Cleavage and Lesion Evaluation via Absolute Real-Time Digital PCR) method provides a modular approach to quantifying genome integrity after gene editing [57]. This technique enables absolute quantification of various editing outcomes through multiplexed dPCR assays:
- Edge Assay: Primers flanking the target site with two probes (cleavage and distal) quantify wild-type sequences, indels, and non-indel aberrations.
- Flanking and Linkage Assay: Two amplicons flanking the cleavage site with nested probes quantify DSBs, large deletions, and structural variations.
- Aneuploidy Assay: Primers and probes in sub-telomeric regions detect chromosomal gains or losses.
This method is particularly valuable for identifying unresolved DSBs and large deletions that conventional PCR-based methods often miss due to amplification biases [57].
BreakTag for DSB Profiling
BreakTag represents a advanced methodology for profiling Cas9-induced DSBs genome-wide at nucleotide resolution [55]. The protocol involves:
- End repair/A-tailing of free DSB ends in genomic DNA
- Adapter ligation with unique molecular identifiers (UMIs) for DSB counting
- Tagmentation with Tn5 transposase
- PCR amplification of ligated fragments for sequencing
BreakTag enables comprehensive mapping of cleavage sites and can distinguish between blunt and staggered breaks based on read signatures, making it ideal for characterizing the flexible cut profiles of different Cas9 nucleases [55].
GUIDE-seq for Off-target Assessment
GUIDE-seq (Genome-wide Unbiased Identification of DSBs Enabled by Sequencing) provides a sensitive method for detecting off-target activities in living cells [56]. The approach involves:
- Transfection of cells with Cas9-gRNA complex and a double-stranded oligodeoxynucleotide (dsODN) tag
- Integration of the dsODN tag into DSB sites during repair
- Amplification and sequencing of tag-integrated genomic regions
- Bioinformatic analysis to identify off-target sites
Studies utilizing GUIDE-seq have demonstrated superior genome-wide specificity for SaCas9 compared to SpCas9 [6] [56].
The Scientist's Toolkit: Essential Research Reagents
| Research Reagent | Function in Indel Analysis |
|---|---|
| CLEAR-time dPCR Assays | Absolute quantification of editing outcomes including DSBs, indels, and large deletions [57] |
| BreakTag Library Prep Kit | Genome-wide profiling of nuclease-induced DSBs and their end structures [55] |
| GUIDE-seq dsODN Tag | Detection of off-target cleavage activities in cellulo [56] |
| High-Fidelity SaCas9 Variants | Engineered SaCas9 (e.g., SaCas9-HF) with enhanced specificity [56] |
| Modified gRNA Scaffolds | gRNA with 3TC scaffold to enhance transcript levels and editing efficiency [7] |
| GenomePAM System | Characterization of PAM requirements using genomic repeats in mammalian cells [14] |
Figure 2: Experimental workflow for comparative indel analysis. The diagram outlines method selection based on research goals, with all pathways converging on SpCas9-SaCas9 comparison data generation.
Implications for Therapeutic Applications
The distinct indel patterns of SpCas9 and SaCas9 have significant implications for therapeutic genome editing:
Precision Editing Applications
SaCas9's reduced +1 insertion bias and higher knock-in efficiency make it particularly suitable for therapeutic applications requiring precise gene correction [6]. The more predictable editing outcomes and reduced off-target effects address critical safety concerns in clinical applications.
Engineered high-fidelity variants of SaCas9 (e.g., SaCas9-HF) further enhance specificity while maintaining robust on-target activity [56]. These variants demonstrate dramatically reduced off-target effects without compromising editing efficiency, making them valuable tools for therapeutic development.
Viral Delivery Considerations
The compact size of SaCas9 enables efficient packaging into adeno-associated virus (AAV) vectors, facilitating in vivo delivery for therapeutic applications [25] [56]. This practical advantage, combined with its favorable editing outcomes and specificity profile, has established SaCas9 as a preferred nuclease for many gene therapy approaches.
The comprehensive analysis of indel patterns reveals fundamental differences between SpCas9 and SaCas9 that significantly impact their research and therapeutic applications. SpCas9's characteristic staggered cut bias results in a strong +1 insertion preference, while SaCas9 produces more balanced editing outcomes with higher precision and reduced off-target effects. These distinctions, coupled with SaCas9's superior packaging compatibility with AAV delivery systems, make it increasingly attractive for therapeutic applications requiring high precision. Researchers should consider these nuclease-specific characteristics when designing gene editing experiments, particularly for applications where predictable outcomes and minimal off-target effects are paramount. The continued development of novel analytical methods and engineered nuclease variants will further enhance our ability to precisely control genome editing outcomes for both basic research and clinical applications.
The therapeutic application of CRISPR-Cas9 genome editing necessitates a comprehensive understanding of nuclease specificity to minimize potentially deleterious off-target effects. Among the various Cas9 orthologs developed for gene editing, Staphylococcus aureus Cas9 (SaCas9) and Streptococcus pyogenes Cas9 (SpCas9) represent two of the most widely used systems. This guide objectively compares their performance through the lens of Genome-wide Unbiased Identification of DSBs Enabled by Sequencing (GUIDE-seq), a sensitive, cell-based method for profiling nuclease activity genome-wide. The experimental data consistently reveal SaCas9's superior fidelity profile, providing critical insights for researchers and drug development professionals selecting nuclease systems for therapeutic applications.
Experimental Comparison of SaCas9 and SpCas9 Fidelity
Quantitative Assessment of Editing Profiles
Direct comparison of SaCas9 and SpCas9 editing outcomes across 11 sites in human induced pluripotent stem cells (iPSCs) and K562 cells demonstrated significant differences in their functional characteristics [5].
Table 1: Performance Comparison of SaCas9 and SpCas9
| Parameter | SaCas9 | SpCas9 | Experimental Context |
|---|---|---|---|
| Editing Efficiency | Greater efficiencies | Lower efficiencies | 11 sites in human iPSCs and K562 cells [5] |
| Optimal Spacer Length | 21 nt | 20 nt | sgRNA spacer optimization [5] |
| NHEJ +1 Insertion Bias | Reduced | Substantial | At fourth nucleotide upstream of PAM [5] |
| Knock-in Efficiency | Higher | Lower | NHEJ-mediated dsODN insertion & HDR-mediated AAV6 donor knock-in [5] |
| Off-target Effects | Significantly reduced | More prevalent | GUIDE-seq analysis [5] |
Spacer Length Optimization
The optimal spacer length for single-guide RNAs (sgRNAs) differs between these nucleases. While the optimal spacer length for a particular sgRNA was 18–21 nt for SpCas9, it was 21–22 nt for SaCas9, with the overall optimal lengths being 20 nt and 21 nt for SpCas9 and SaCas9, respectively [5].
GUIDE-Seq Methodology for Off-Target Profiling
Principles and Workflow
GUIDE-seq is a sensitive, unbiased method that enables genome-wide detection of nuclease-induced double-strand breaks (DSBs) in living cells. The technique relies on the integration of a double-stranded oligodeoxynucleotide (dsODN) tag into DSBs via the non-homologous end-joining (NHEJ) repair pathway [58] [59].
The core innovation of GUIDE-seq involves using phosphorothioate-modified dsODNs, which resist exonuclease degradation and integrate into nuclease-induced breaks with high efficiency. These integrated tags then serve as anchors for amplification and sequencing of the flanking genomic regions, allowing precise mapping of DSB locations throughout the genome [58].
GUIDE-seq Workflow: From Tag Integration to Off-target Identification
Critical Experimental Steps
- dsODN Design: The 34 bp dsODN contains phosphorothioate linkages at the 5' and 3' ends of both strands to enhance stability against exonuclease degradation [58]. For improved efficiency, 3'-only end-protected dsODN tags are recommended [59].
- Cell Transfection: Delivery of Cas9:gRNA ribonucleoprotein (RNP) complex along with dsODN tag into cells. Optimal dsODN integration frequency (>5% of nuclease-induced mutations) is critical for successful GUIDE-seq [59].
- Library Preparation: Genomic DNA is fragmented, followed by end-repair, A-tailing, and ligation of a single-tailed sequencing adapter containing an 8-bp unique molecular index (UMI) for PCR bias correction [59].
- Amplification & Sequencing: Two rounds of PCR with primers complementary to the sequencing adapter and dsODN tag enable specific amplification of tag-integrated regions [58].
- Bioinformatic Analysis: Consolidated sequencing reads are mapped to a reference genome, and off-target sites are identified by characteristic bi-directional mapping read signatures [60].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for GUIDE-seq Experiments
| Reagent/Resource | Function | Specifications |
|---|---|---|
| dsODN Tag | Integrates into DSBs for detection | 34 bp, blunt-ended, phosphorothioate-modified [58] [59] |
| Cas9 Nuclease | Creates targeted double-strand breaks | SaCas9 or SpCas9, as protein, plasmid, or mRNA [59] |
| Guide RNA | Directs Cas9 to specific genomic loci | Optimal length: 21 nt for SaCas9, 20 nt for SpCas9 [5] |
| U6-gRNA Expression Vector | Enables gRNA transcription | Modified scaffolds (e.g., 3TC) enhance transcript levels [7] |
| GUIDEseq Bioconductor Package | Analyzes GUIDE-seq datasets | Flexible platform with >60 parameters for different nuclease systems [60] |
| High-Fidelity Cas9 Variants | Reduces off-target activity | SaCas9-HF, eSpCas9(1.1), SpCas9-HF1 [61] [7] |
Mechanistic Insights into SaCas9's Enhanced Specificity
The superior fidelity of SaCas9 observed in GUIDE-seq profiling can be attributed to several molecular and mechanistic factors. SaCas9 recognizes a longer PAM sequence (NNGRRT) compared to SpCas9 (NGG), which statistically occurs less frequently in the genome, thereby reducing the potential off-target landscape [62] [25]. Furthermore, SpCas9 exhibits a more substantial bias for nonhomologous end-joining (NHEJ) +1 insertion at the fourth nucleotide upstream of the protospacer adjacent motif (PAM), indicating a characteristic of a staggered cut, whereas SaCas9 demonstrates reduced this bias [5].
The enhanced specificity of SaCas9 also makes it particularly valuable for therapeutic applications where minimizing off-target effects is critical. Its compact size (1053 amino acids) allows for packaging into adeno-associated virus (AAV) vectors, facilitating in vivo delivery for clinical applications [25]. When combined with its naturally higher fidelity, this delivery advantage positions SaCas9 as a preferred nuclease for therapeutic genome editing.
GUIDE-seq analysis provides compelling evidence for SaCas9's superior fidelity compared to SpCas9. The experimental data demonstrate that SaCas9 not only edits with greater efficiency but also produces significantly reduced off-target effects across multiple genomic loci. These findings, coupled with SaCas9's practical advantages for therapeutic delivery, establish it as a highly valuable nuclease for precision genome editing applications where specificity is paramount. Researchers can leverage these insights to make informed decisions when selecting CRISPR systems for both basic research and clinical development.
In the rapidly advancing field of therapeutic genome editing, achieving efficient and precise integration of exogenous DNA sequences—a process known as knock-in—represents a critical hurdle for both basic research and clinical applications. The challenge stems from a fundamental biological reality: mammalian cells predominantly repair double-strand breaks (DSBs) via the non-homologous end joining (NHEJ) pathway, which is highly efficient but error-prone, rather than the precise homology-directed repair (HDR) pathway [63]. This preference creates a significant bottleneck for researchers and drug development professionals seeking to develop precise genetic therapies. While HDR has traditionally been the preferred mechanism for achieving precise edits, recent investigations have revealed that NHEJ-mediated pathways, particularly those utilizing double-stranded oligodeoxynucleotides (dsODNs), can offer surprising advantages in certain contexts. Understanding the relative capabilities, efficiencies, and limitations of these competing pathways provides crucial insights for optimizing knock-in strategies in therapeutic development, particularly within the broader framework of comparing SpCas9 and SaCas9 systems.
DNA Repair Pathway Fundamentals: HDR and NHEJ Mechanisms
When CRISPR-Cas9 induces a DSB, the cell activates several competing DNA repair mechanisms. The two most relevant for knock-in are HDR and NHEJ, which operate via fundamentally distinct molecular processes.
Homology-Directed Repair (HDR) is a precise repair mechanism that uses a homologous DNA template—either a sister chromatid or an exogenously supplied donor template—to accurately repair the break [64]. This pathway is restricted to the S and G2 phases of the cell cycle when homologous templates are naturally available [63]. The process involves resection of the DNA ends to create single-stranded overhangs, followed by strand invasion and synthesis using the homologous template. For therapeutic applications, researchers supply designed donor templates containing the desired modification flanked by homology arms that match the sequences surrounding the cut site.
Non-Homologous End Joining (NHEJ) represents a faster, more efficient repair mechanism that simply rejoins broken DNA ends without requiring a homologous template [64]. This pathway operates throughout the cell cycle and often results in small insertions or deletions (indels) at the repair site. While traditionally viewed as unsuitable for precise editing, researchers have harnessed NHEJ for knock-in by co-delivering dsODN donors along with CRISPR components. In this approach, the dsODN is integrated at the break site through the cell's end-joining machinery.
The following diagram illustrates the key decision points in DNA repair pathway choice following a CRISPR-induced double-strand break:
Quantitative Comparison of Knock-in Efficiency and Fidelity
Direct comparative studies reveal significant differences in the performance characteristics of HDR versus NHEJ-mediated dsODN insertion strategies. The table below summarizes key efficiency and fidelity metrics based on experimental data from multiple studies:
Table 1: Performance comparison between HDR and NHEJ-mediated dsODN insertion
| Performance Metric | HDR-Mediated Knock-in | NHEJ-Mediated dsODN Insertion | Experimental Context |
|---|---|---|---|
| Knock-in Efficiency | Up to 30% in iPSCs [65] | Up to 20% in somatic cells [66] | GAPDH locus targeting in human cells |
| Template Type | ssODN, dsDNA, AAV vectors [67] | Double-stranded ODNs [68] | Varies by experimental design |
| Homology Requirement | 300-600 bp arms optimal [65] | No homology required [66] | Double cut HDR donor design |
| Large Deletion Reduction | ~80% with AAV donors [68] | ~60% with dsODN insertion [68] | Nanopore sequencing analysis |
| Cell Type Dependence | Highly variable (preferable in cycling cells) [63] | More consistent across cell types [66] | Comparison across primary cells |
| Pathway Interference | NHEJ/MMEJ inhibition boosts HDR to >90% purity [69] | Endogenous dominance without inhibition [64] | HDRobust method with repair inhibition |
The data reveals a complex efficiency landscape where NHEJ-mediated dsODN insertion can outperform HDR in certain contexts, particularly in non-cycling cells where HDR is naturally suppressed. However, HDR achieves superior precision when appropriate template design and cell cycle synchronization strategies are employed.
Experimental Protocols for Knock-in Efficiency Assessment
HDR Efficiency Quantification Protocol
The following methodology enables direct quantification and comparison of HDR-mediated DNA integration across different human cell types [66]:
Reporter System Construction: Develop a promoterless fluorescent reporter system (e.g., mCherry, eGFP) targeted to a safe harbor locus such as GAPDH or AAVS1.
Donor Template Design:
Cell Transfection/Electroporation:
Efficiency Quantification: Analyze knock-in efficiency 4-5 days post-transfection using flow cytometry for fluorescent reporters or NGS-based amplicon sequencing for specific sequence modifications.
NHEJ-Mediated dsODN Insertion Protocol
This protocol outlines the assessment of NHEJ-mediated integration of dsODN donors [66] [68]:
dsODN Donor Design: Design double-stranded ODNs containing the payload sequence without homology arms. The optimal length depends on the insertion size but typically ranges from 50-200 bp.
RNP Complex Formation: Pre-complex Cas9 protein with sgRNA at a 1:2 molar ratio in opti-MEM buffer and incubate for 10-20 minutes at room temperature to form ribonucleoprotein (RNP) complexes.
Primary Cell Electroporation:
- For human T cells and HSPCs: Use Cas9-RNP complexes with chemically modified sgRNAs combined with dsODN donors [68]
- Electroporation parameters: Cell-specific optimized protocols (e.g., Neon system for iPSCs)
Outcome Analysis:
- Assess insertion efficiency 72 hours post-delivery by PCR amplification of target loci followed by Illumina sequencing [68]
- Quantify large deletions using long-range PCR (4-6 kb amplicons) and nanopore sequencing [68]
- Calculate deletion index as: (deletion in editing group % − deletion in WT group %) [68]
Pathway Modulation Strategies to Enhance Knock-in Outcomes
HDR Enhancement Through Repair Pathway Interference
Significant improvements in HDR efficiency can be achieved through strategic interference with competing repair pathways. The HDRobust method demonstrates that combined inhibition of NHEJ and MMEJ dramatically increases HDR purity [69]:
NHEJ Inhibition: Introduce K3753R mutation in DNA-PKcs to inactivate kinase function while keeping other protein domains intact
MMEJ Inhibition: Introduce V896* stop codon in POLQ to eliminate the DNA polymerase domain and RAD51 binding
Small Molecule Implementation: Transient inhibition using substance mixes targeting both pathways simultaneously
This combined approach results in DSB repair predominantly through HDR (>90% purity) while dramatically reducing indels, large deletions, and off-target editing events [69].
Template Engineering Strategies
Optimizing donor template design significantly impacts both HDR and NHEJ-mediated knock-in efficiency:
Table 2: Template optimization strategies for enhanced knock-in
| Template Type | Optimization Strategy | Efficiency Impact | Key Considerations |
|---|---|---|---|
| Double-cut HDR Donor | Flank cassette with sgRNA-PAM sequences for in vivo linearization [65] | 2-5x increase vs. circular donors [65] | Requires careful sgRNA selection to avoid self-targeting |
| ssODN Design | 50 bp homology arms with blocking mutations to prevent re-cleavage [69] | 38-46% HDR efficiency in T cells [68] | Optimal for point mutations and short insertions |
| AAV Donors | Long homology arms (≥500 bp) for enhanced recombination [68] | ~80% reduction in large deletions [68] | Manufacturing complexity and cargo capacity limitations |
| dsODN for NHEJ | Short, double-stranded fragments without homology arms [68] | ~60% reduction in large deletions [68] | Limited cargo capacity compared to viral vectors |
The Scientist's Toolkit: Essential Reagents for Knock-in Experiments
Table 3: Key research reagents for HDR and NHEJ-mediated knock-in studies
| Reagent Category | Specific Examples | Function & Application | Key Features |
|---|---|---|---|
| CRISPR Nucleases | SpCas9, SaCas9, hfCas12Max, eSpOT-ON [16] | DSB induction at target genomic loci | SpCas9: NGG PAM; SaCas9: NNGRRN PAM, smaller size [16] |
| Donor Templates | ssODNs, dsODNs, AAV vectors, plasmid donors [67] | Providing repair template for precise edits | ssODNs: Short edits; AAV: Large inserts with long homology [68] |
| Repair Modulators | Nocodazole, CCND1, DNA-PKcs inhibitors, Polθ inhibitors [69] [65] | Shift repair balance toward HDR | Nocodazole: G2/M synchronizer; CCND1: G1/S cyclin [65] |
| Delivery Tools | Lipofectamine 2000, FuGENE HD, Amaxa Nucleofector [66] | Introducing editing components into cells | Chemical transfection: Cell lines; Electroporation: Primary cells [66] |
| Analysis Reagents | Long-range PCR kits, nanopore sequencing, NGS libraries | Assessing editing outcomes and detecting large deletions | Nanopore sequencing: Detects large structural variants [68] |
The comparative analysis of HDR and NHEJ-mediated dsODN insertion reveals a nuanced landscape where each approach offers distinct advantages depending on the experimental or therapeutic context. HDR remains the gold standard for precision, particularly when combined with pathway interference strategies that achieve remarkable purity (>90%) [69]. However, NHEJ-mediated dsODN insertion demonstrates compelling advantages in certain scenarios, including reduced large deletions, better performance in non-cycling cells, and simplified donor design [66] [68].
For therapeutic applications, the choice between these pathways should be guided by multiple factors: the dividing status of target cells, the size of the intended insertion, precision requirements, and safety considerations regarding potential large deletions. Emerging evidence suggests that NHEJ-mediated dsODN insertion presents a valuable alternative to HDR, particularly for applications where high efficiency in primary cells is more critical than absolute precision. As the field advances, the development of hybrid approaches that leverage the advantages of both pathways while mitigating their limitations will likely yield the most robust solutions for therapeutic genome editing.
The CRISPR-Cas9 system has revolutionized genome editing, yet the off-target activity of the wild-type Streptococcus pyogenes Cas9 (WT-SpCas9) poses significant challenges for therapeutic applications. To address this, protein engineering efforts have produced high-fidelity variants, with SpCas9-HF1 and eSpCas9(1.1) representing two of the most prominent first-generation solutions. This guide provides an objective comparison of their performance, drawing on direct benchmarking studies and experimental data to inform researchers and drug development professionals. Understanding the trade-offs between on-target efficiency and specificity in these variants is crucial for selecting the appropriate nuclease for specific applications, particularly within the broader context of optimizing CRISPR systems like SpCas9 and the smaller SaCas9 for therapeutic use.
Engineering Strategies and Molecular Mechanisms
The design of SpCas9-HF1 and eSpCas9(1.1) was guided by rational engineering approaches aimed at reducing non-specific interactions between the Cas9-sgRNA complex and the DNA substrate.
- eSpCas9(1.1): This variant was engineered based on the hypothesis that enhancing the stability of the Cas9-sgRNA complex with its target DNA could minimize off-target binding. It incorporates three mutations (K848A, K1003A, R1060A) strategically located in the Rec3 domain of Cas9. These mutations are proposed to weaken the protein's interaction with the non-target DNA strand, thereby reducing the tolerance for guide-target mismatches and decreasing off-target cleavage [61].
- SpCas9-HF1: Designed through structure-guided engineering, SpCas9-HF1 features four mutations (N497A, R661A, Q695A, Q926A) that target key residues involved in forming hydrogen bonds with the DNA phosphate backbone. By alanine substitution, these direct DNA-contact residues are neutralized, tightening the binding requirements and theoretically ensuring that only perfect matches between the guide RNA and target DNA result in cleavage [61].
Performance Benchmarking: On-Target Efficiency and Specificity
Direct comparative studies reveal distinct performance characteristics for each variant, highlighting a critical balance between activity and precision.
On-Target Activity
A comprehensive genome-scale screen measuring the indel rates of over 50,000 sgRNAs for each nuclease provided robust data on their on-target efficacy [70]. When assessed using a precision-recall curve framework, WT-SpCas9 demonstrated the highest on-target activity (90% recall at 95% precision). Under the same stringent conditions, the high-fidelity variants showed reduced activity, with eSpCas9(1.1) achieving 40% recall and SpCas9-HF1 achieving 25% recall [71].
A critical factor influencing this performance is the requirement for a perfectly matched guanine at the 5' end of the guide sequence. Both high-fidelity variants are highly sensitive to a mismatched 5'-G, a constraint that substantially reduces the number of potential target sites. When analysis is restricted to guides with a correctly paired 5'-G (G19 guides), the performance gap narrows significantly. In this scenario, eSpCas9(1.1) reaches 90% recall, SpCas9-HF1 reaches 76%, and WT-SpCas9 achieves 94% [71].
Table 1: Summary of On-Target Performance Metrics
| Metric | WT-SpCas9 | eSpCas9(1.1) | SpCas9-HF1 |
|---|---|---|---|
| Recall at 95% Precision (All guides) [71] | 90% | 40% | 25% |
| Recall at 95% Precision (G19 guides only) [71] | 94% | 90% | 76% |
| Sensitivity to 5'-G Mismatch | Low | High | High |
| PAM Flexibility | NGG | NGG | NGG |
Off-Target Susceptibility
The primary advantage of high-fidelity variants is their reduced off-target editing. In a systematic evaluation using a library of sgRNAs containing all possible single and double mismatches, both eSpCas9(1.1) and SpCas9-HF1 showed a significantly improved ability to distinguish between perfectly matched and mismatched guides compared to WT-SpCas9 [71]. The area under the receiver operating characteristic curve (ROC-AUC) for this separation was highest for the high-fidelity variants, with HiFi Cas9 (a later evolved variant) performing best, followed by eSpCas9(1.1) and then WT-Cas9 [71]. This demonstrates a superior specificity profile for the engineered variants.
Experimental Protocols for Performance Assessment
The data presented in this guide are derived from robust, high-throughput experimental methods that can serve as templates for future benchmarking.
Genome-Wide gRNA Activity Profiling
The foundational data for comparing on-target activity were generated through a large-scale, pooled screen [70] [72]. The protocol is summarized below:
Key Steps:
- gRNA Library Design: Four top-ranked sgRNAs per gene are designed, ensuring initiation with either A or G to accommodate the mouse U6 (mU6) promoter, which expands targeting scope compared to the traditional human U6 promoter [70].
- Library Delivery: The oligonucleotide library is cloned into lentiviral vectors and transduced at a low multiplicity of infection (MOI=0.3) into cells expressing the Cas9 variant of interest. This ensures most cells receive only a single guide, enabling clean activity measurement.
- Outcome Measurement: After 5 days, genomic DNA is harvested, and the integrated target sites are amplified and sequenced. The indel rate for each guide is calculated, providing a direct measure of its activity [70] [72].
Off-Target Mismatch Tolerance Assay
To quantitatively profile specificity, a specialized library is employed [71]:
- Library Design: A set of highly active sgRNAs is selected, and a library is constructed that includes the perfect-match guides along with all possible single-nucleotide mismatches and double mismatches.
- Screening: This library is screened in cells expressing WT-SpCas9, eSpCas9(1.1), or other high-fidelity variants.
- Data Analysis: The log-fold depletion of each guide is calculated. The separation between perfect-match guides (true positives) and mismatch guides (true negatives) is quantified using ROC-AUC analysis. Furthermore, the data is used to generate a Cutting Frequency Determination (CFD) matrix specific to each variant, which predicts the impact of mismatches at each position in the guide sequence [71].
Practical Application and Compatibility
For researchers implementing these tools, practical considerations around guide RNA design and compatibility with other systems are paramount.
- gRNA Scaffold Optimization: The standard gRNA scaffold contains a sequence of four thymines (4T) that can prematurely terminate transcription by RNA polymerase III. Modifying this scaffold to a "3TC" configuration (replacing the fourth T with a C) has been shown to significantly boost gRNA transcript levels. This enhancement is particularly beneficial for high-fidelity variants like SpCas9-HF1 and eSpCas9(1.1), especially when vector delivery is limited, as it can improve their sometimes lower on-target efficiency without compromising specificity [7].
- Therapeutic Specificity: High-fidelity variants are critical for discriminating between mutant and wild-type alleles in therapeutic contexts. For instance, HiFi Cas9 (a variant with R691A mutation) has been successfully used to selectively target oncogenic KRASG12C and KRASG12D mutations in non-small cell lung cancer models without editing the wild-type KRAS allele, a level of specificity unattainable with WT-SpCas9 [73].
- Specialized Editing Applications: Beyond standard knockout generation, SpCas9-HF1 has demonstrated utility in sophisticated editing protocols. When integrated into a cell cycle-dependent genome editing system that uses an anti-CRISPR-AcrIIA4-Cdt1 fusion protein to control Cas9 activity, SpCas9-HF1 achieved increased homology-directed repair (HDR) efficiency while maintaining low off-target effects [74].
Table 2: Comparison of Key Characteristics and Applications
| Feature | eSpCas9(1.1) | SpCas9-HF1 | Therapeutic Implication |
|---|---|---|---|
| Primary Engineering Strategy | Weaken non-target strand binding [61] | Neutralize DNA phosphate backbone contacts [61] | Different strategies achieve high specificity |
| Relative On-Target Efficiency | Higher | Lower | eSpCas9(1.1) may be preferable when activity is critical |
| gRNA Scaffold Preference | 3TC for low vector doses [7] | 3TC for low vector doses [7] | Optimized scaffolds can enhance efficacy |
| Compatibility with HDR | Effective in cell cycle editing [74] | High HDR efficiency in cell cycle editing [74] | Both are suitable for precise gene correction |
| Allele-Specific Discrimination | High | High | Enables targeting of dominant disease alleles |
The Scientist's Toolkit: Essential Reagents
Table 3: Key Research Reagents and Resources
| Reagent / Resource | Function and Importance in Benchmarking |
|---|---|
| Lentiviral gRNA Libraries | Enables high-throughput, parallel assessment of thousands of guide RNAs in a single experiment [70]. |
| Stable Cell Lines Expressing Cas9 Variants | Ensures consistent and comparable nuclease expression levels across different experimental conditions, critical for a fair comparison [71]. |
| Next-Generation Sequencing (NGS) | Provides deep, quantitative data on indel rates and the spectrum of editing outcomes at on-target and off-target sites [71] [73]. |
| CFD (Cutting Frequency Determination) Matrices | Algorithmic models derived from experimental data that predict the potential for off-target activity for a given sgRNA, specific to each Cas9 variant [71]. |
| U6 Promoter Plasmids (e.g., with 3TC scaffold) | Optimized vectors for gRNA expression that maximize transcript levels, helping to overcome the lower on-target activity of high-fidelity variants [7]. |
| Ribonucleoprotein (RNP) Complexes | Complexing purified Cas9 protein with sgRNA for direct delivery; reduces off-target effects by limiting exposure time and is a preferred method for therapeutic development [73]. |
The benchmarking of SpCas9-HF1 and eSpCas9(1.1) establishes a fundamental trade-off in CRISPR-Cas9 engineering: a gain in specificity is often accompanied by a loss in on-target potency. eSpCas9(1.1) generally maintains higher activity across a diverse set of guides, while SpCas9-HF1, though potentially less active, represents a stringent option for applications where utmost specificity is required. The choice between them should be guided by the specific experimental or therapeutic context, considering the tolerance for lower efficiency versus the risk of off-target effects. Furthermore, practical strategies such as optimized gRNA scaffold design and RNP delivery can help mitigate the efficiency loss. As the field progresses, these first-generation high-fidelity variants continue to provide a critical benchmark against which newer AI-designed and evolved editors, such as OpenCRISPR-1 and other next-generation tools, are measured [54] [75].
Conclusion
The choice between SpCas9 and SaCas9 is not a matter of superiority but of strategic application. SpCas9, with its well-characterized NGG PAM and extensive variant toolkit, remains a powerful workhorse for many laboratory applications. However, SaCas9 demonstrates compelling advantages for therapeutic development, including superior fidelity with significantly reduced off-target effects, more favorable insertion patterns for knock-in applications, and a compact size ideal for AAV delivery. Recent advancements in gRNA scaffold optimization (3TC) benefit both systems, particularly under the vector-limited conditions common in clinical settings. Future directions should focus on further engineering PAM flexibility without compromising fidelity, refining delivery systems for in vivo applications, and accumulating clinical data from ongoing trials like EDIT-101. This will solidify CRISPR-based therapies, leveraging the unique strengths of each nuclease to address diverse genetic challenges.
References
- 1. Structural insights into a high fidelity variant of SpCas9 [https://www.nature.com/articles/s41422-018-0131-6]
- 2. Crystal structure of Staphylococcus aureus Cas9 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4670267/]
- 3. Structural Basis for the Altered PAM Specificities of ... [https://www.sciencedirect.com/science/article/pii/S1097276516001295]
- 4. Structural Basis for the Altered PAM Specificities of ... [https://pubmed.ncbi.nlm.nih.gov/26990991/]
- 5. Superior Fidelity and Distinct Editing Outcomes of SaCas9 ... [https://www.sciencedirect.com/science/article/pii/S1672022922001681]
- 6. Superior Fidelity and Distinct Editing Outcomes of SaCas9 ... [https://pubmed.ncbi.nlm.nih.gov/36549468/]
- 7. Optimal SpCas9- and SaCas9-mediated gene editing by ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11317-2]
- 8. and SaCas9-mediated gene editing by enhancing gRNA ... [https://research.sahmri.org.au/en/publications/optimal-spcas9-and-sacas9-mediated-gene-editing-by-enhancing-grna/]
- 9. CATS: a bioinformatic tool for automated Cas9 nucleases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12328375/]
- 10. Importance of the PAM Sequence in CRISPR Experiments [https://www.synthego.com/guide/how-to-use-crispr/pam-sequence/]
- 11. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5951633/]
- 12. Continuous evolution of SpCas9 variants compatible with non-G PAMs - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7145744/]
- 13. Flexibility in PAM Recognition Expands DNA Targeting in xCas9 [https://elifesciences.org/reviewed-preprints/102538]
- 14. GenomePAM directs PAM characterization and ... [https://www.nature.com/articles/s41551-025-01464-y]
- 15. CATS: a bioinformatic tool for automated Cas9 nucleases ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1571023/full]
- 16. Alternatives to CRISPR-Cas9: Nucleases for Next-Gen ... [https://www.synthego.com/blog/alternatives-to-crispr-cas9-nucleases-for-next-gen-therapy/]
- 17. Viral Vectors for the in Vivo Delivery of CRISPR Components [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2022.895713/full]
- 18. A Split Staphylococcus aureus Cas9 as a Compact ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5444561/]
- 19. Superior Fidelity and Distinct Editing Outcomes of SaCas9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11082263/]
- 20. Highly specific targeted mutagenesis in plants using ... [https://www.nature.com/articles/srep26871]
- 21. Rational engineering of minimally immunogenic nucleases for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11696374/]
- 22. Rational engineering of minimally immunogenic nucleases ... [https://www.nature.com/articles/s41467-024-55522-1]
- 23. Pairwise library screen systematically interrogates ... [https://www.nature.com/articles/s41467-018-05391-2]
- 24. Systematic evaluation of CRISPR-Cas systems reveals design ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1445-x]
- 25. How to Choose the Right Cas Variant for Every CRISPR ... [https://www.synthego.com/guide/how-to-use-crispr/cas-nuclease-variants/]
- 26. Production of Efficient - Gene using Staphylococcus... Modified Mice [https://www.nature.com/articles/srep32565?error=cookies_not_supported&code=d01f9a8b-db9b-49de-a777-51550c377168]
- 27. Broad Institute-MIT team identifies highly efficient new... | Broad Institute [https://www.broadinstitute.org/news/broad-institute-mit-team-identifies-highly-efficient-new-cas9-vivo-genome-editing]
- 28. Pioneering Precision with ABEmax Technology [https://www.croyezbio.com/support/detail/id/128.html]
- 29. CRISPR/Cas Systems in Genome Editing: Methodologies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7080517/]
- 30. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 31. Applications of multiplexed CRISPR–Cas for genome ... [https://www.nature.com/articles/s12276-025-01500-6]
- 32. Applications of multiplexed CRISPR–Cas for genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322214/]
- 33. Programmable Multiplex Genome Editing: Innovations in ... [https://www.sciepublish.com/article/pii/680]
- 34. Enhanced genome editing with a Streptococcus equinus ... [https://www.nature.com/articles/s42003-025-07593-z]
- 35. Optimal SpCas9- and SaCas9-mediated gene editing by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11823040/]
- 36. Multiplexed CRISPR technologies for gene editing and ... [https://www.nature.com/articles/s41467-020-15053-x]
- 37. harnessing multiplex editing for polygenic trait engineering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12527382/]
- 38. USDA Grant Funds Study of Multiplex Genome Editing in ... [https://today.uconn.edu/2025/11/usda-grant-funds-study-of-multiplex-genome-editing-in-tomatoes/]
- 39. CRISPR/Cas9 therapeutics: progress and prospects [https://www.nature.com/articles/s41392-023-01309-7]
- 40. CRISPR Nucleases: The Ultimate Guide To Selection [https://bitesizebio.com/46146/crispr-nucleases-the-ultimate-guide/]
- 41. Shining light on CRISPR/Cas9 therapeutics for inherited ... [https://www.sciencedirect.com/science/article/abs/pii/S1350946225000497]
- 42. CRISPR-Cas technologies: Emerging tools from research ... [https://www.jmicrobiol.or.kr/journal/view.php?number=3016]
- 43. The comparison of ZFNs, TALENs, and SpCas9 by GUIDE ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8655392/]
- 44. CRISPR–Cas9 gRNA efficiency prediction: an overview of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9023298/]
- 45. Base and Prime Editing for Inherited Retinal Diseases [https://www.mdpi.com/1999-4923/17/11/1405]
- 46. and SaCas9-mediated gene editing by enhancing gRNA ... [https://pubmed.ncbi.nlm.nih.gov/39939860/]
- 47. A Guide to Guides: An Overview of SpCas9 sgRNA ... [https://www.mdpi.com/2674-0583/3/4/19]
- 48. Optimal SpCas9- and SaCas9-mediated gene editing by ... [https://www.addgene.org/browse/article/28248644/]
- 49. Comparative Analysis of Methods for Assessing On-Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11932317/]
- 50. Optimization of gene knockout approaches and sgRNA ... [https://www.nature.com/articles/s41598-025-24184-4]
- 51. A benchmark of computational CRISPR-Cas9 guide design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6738662/]
- 52. Enhanced genome editing with a Streptococcus equinus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11806022/]
- 53. State of CRISPR Gene Editing in the Drug Discovery Sector [https://www.synthego.com/blog/survey-report-crispr-therapeutics/]
- 54. AI-guided Cas9 engineering provides an effective strategy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12583830/]
- 55. Linking CRISPR–Cas9 double-strand break profiles to ... [https://www.nature.com/articles/s41587-024-02238-8]
- 56. Rationally engineered Staphylococcus aureus Cas9 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6800346/]
- 57. Unveiling the cut-and-repair cycle of designer nucleases in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12583642/]
- 58. GUIDE-Seq enables genome-wide profiling of off-target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4320685/]
- 59. Defining genome-wide CRISPR-Cas genome editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9331158/]
- 60. GUIDEseq: a bioconductor package to analyze GUIDE-Seq ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-3746-y]
- 61. Recent Advances in Improving Gene-Editing Specificity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9319960/]
- 62. Methods for detecting off-target effects of CRISPR/Cas9 [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002368]
- 63. Current Strategies for Increasing Knock-In Efficiency in ... [https://www.mdpi.com/1422-0067/25/5/2456]
- 64. HDR vs NHEJ: DNA Repair Pathway Comparison [https://invivobiosystems.com/crispr/hdr-vs-nhej/]
- 65. Efficient precise knockin with a double cut HDR donor after ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1164-8]
- 66. Knock-in of large reporter genes in human cells via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4872082/]
- 67. In search of an ideal template for therapeutic genome editing [https://pmc.ncbi.nlm.nih.gov/articles/PMC9992834/]
- 68. Effective control of large deletions after double-strand breaks ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02462-4]
- 69. Efficient high-precision homology-directed repair ... [https://www.nature.com/articles/s41592-023-01949-1]
- 70. Optimized CRISPR guide RNA design for two high-fidelity ... [https://www.nature.com/articles/s41467-019-12281-8]
- 71. Benchmarking of SpCas9 variants enables deeper base ... [https://www.nature.com/articles/s41467-022-28884-7]
- 72. DeepMEns: an ensemble model for predicting sgRNA on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11735754/]
- 73. High-fidelity Cas9-mediated targeting of KRAS driver ... [https://www.nature.com/articles/s41467-025-62350-4]
- 74. SpCas9-HF1 enhances accuracy of cell cycle-dependent ... [https://www.sciencedirect.com/science/article/pii/S2162253124000118]
- 75. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]