Overcoming the Hurdles: A Comprehensive Guide to Challenges in Heterologous Expression of Biosynthetic Gene Clusters
Heterologous expression of biosynthetic gene clusters (BGCs) is a powerful strategy for discovering novel natural products with pharmaceutical potential, yet it is fraught with technical challenges that limit its success...
Overcoming the Hurdles: A Comprehensive Guide to Challenges in Heterologous Expression of Biosynthetic Gene Clusters
Abstract
Heterologous expression of biosynthetic gene clusters (BGCs) is a powerful strategy for discovering novel natural products with pharmaceutical potential, yet it is fraught with technical challenges that limit its success rate to 11-32%. This article provides a systematic analysis for researchers and drug development professionals, covering the foundational principles of BGC activation, advanced methodological approaches for cloning and host selection, practical strategies for troubleshooting and optimization, and rigorous validation techniques. By synthesizing recent scientific advances, we offer a actionable framework to navigate the complexities of expressing silent gene clusters, from initial cluster prioritization to the final isolation of novel compounds, thereby accelerating the pipeline for new drug discovery.
The Silent Majority: Unlocking the Potential of Cryptic Biosynthetic Gene Clusters
The quest for novel bioactive natural products, crucial for developing new antibiotics, anticancer agents, and other therapeutics, increasingly turns to microbial biosynthetic gene clusters (BGCs). These clusters are sets of co-localized genes that coordinate the production of secondary metabolites. However, the field faces a fundamental challenge: the vast majority of microbial diversity, and its associated biosynthetic potential, remains inaccessible through traditional laboratory cultivation techniques [1]. This "uncultured microbial diversity" represents a massive reservoir of uncharacterized BGCs. Compounding this issue, even in culturable microorganisms, many BGCs are "silent" or "cryptic," meaning they are not expressed under standard laboratory conditions, hiding their functional products [1] [2]. This article examines the scale of these challenges and the sophisticated experimental protocols being developed to overcome them, with a specific focus on the implications for heterologous expression—the process of transferring and expressing BGCs in a tractable laboratory host.
Quantifying the Uncultured and Silent Majority
Recent comprehensive genomic studies have begun to quantify the immense biosynthetic potential hidden within microbial communities, particularly in underexplored environments like the human gut and subterranean ecosystems.
BGC Abundance in the Human Gut Microbiome
A systematic analysis of 4,744 human gut microbial genomes from the Unified Human Gastrointestinal Genome (UHGG) database revealed a staggering abundance and diversity of BGCs. The findings are summarized in the table below.
Table 1: Biosynthetic Gene Clusters in the Human Gut Microbiome [2]
| Metric | Finding | Implication |
|---|---|---|
| Total Genomes Analyzed | 4,744 | Establishes a broad baseline for human gut biosynthetic potential. |
| Dominant Genus | Paenibacillus | Identified as a key genus with exceptional biosynthetic capacity. |
| Key Discovery | Production of Leinamycin | A potent anticancer compound previously thought exclusive to Streptomyces. |
| BGC Classes Identified | Non-ribosomal peptide synthetases (NRPS), Polyketide synthases (PKS), Terpenoids, Bacteriocins | Highlights diversity of potential therapeutic compounds. |
This study underscores the human gut as a rich, largely untapped resource for novel drug discovery. The identification of Paenibacillus as a dominant biosynthetic genus, capable of producing potent compounds like the anticancer agent leinamycin, illustrates the potential rewards of effectively accessing this hidden diversity [2].
Microbial Adaptation in Oligotrophic Environments
Subterranean environments, such as caves and underground mines, represent another frontier for natural product discovery. These ecosystems are characterized by extreme oligotrophy (nutrient scarcity), complete darkness, and high humidity, which have driven microbial communities to develop unique adaptations [1]. The ability of these communities to withstand such conditions creates a unique reservoir of untapped biosynthetic potential, likely encoding for novel bioactive compounds with applications in medicine and biotechnology [1]. The historical use of cave substances like moonmilk for wound healing provides anecdotal evidence supporting the bioactivity of these underground metabolites [1].
The Technical Hurdles in Heterologous Expression
Heterologous expression is a cornerstone strategy for accessing silent BGCs and those from uncultured organisms. However, the process is fraught with technical challenges that can prevent the successful production of the target metabolite.
Promoter Compatibility and Transcriptional Regulation
A critical challenge is ensuring the heterologous host can properly transcribe the foreign BGC. A 2025 study on the heterologous expression of an 11 kb nitrogen-fixing (nif) gene cluster from Paenibacillus polymyxa CR1 in Bacillus subtilis 168 provides a clear example [3]. The researchers successfully integrated and confirmed transcription of the cluster, yet detected no nitrogenase activity. Only after replacing the native promoter with a host-derived constitutive promoter (Pveg) was active nitrogenase produced [3]. This demonstrates that systemic compatibility, particularly for complex metalloenzymes requiring precise cofactor assembly, is as crucial as raw transcriptional strength. Interestingly, stronger promoters (P43 and Ptp2) did not further enhance activity, underscoring the need for balanced expression [3].
Table 2: Key Experimental Steps for nif Cluster Expression in B. subtilis [3]
| Step | Method/Technology | Purpose/Outcome |
|---|---|---|
| Cluster Identification | Genomic analysis of P. polymyxa CR1 | Revealed an 11 kb nif gene cluster (from nifB to nifV). |
| Cluster Assembly | ExoCET (exonuclease combined with RecET recombination) | Modular assembly and cloning of the synthesized nif cluster. |
| Genome Integration | Double-exchange chromosomal recombination | Stable integration of the cluster into the genome of B. subtilis 168. |
| Transcription Check | RT-PCR | Verified transcription of the integrated nif cluster. |
| Activity Assay | Acetylene reduction assay | Detected functional nitrogenase activity only after promoter engineering. |
| Promoter Engineering | Native promoter replacement with Pveg | Enabled production of active nitrogenase, proving critical for functionality. |
Identification of Minimal Gene Sets
For many BGCs, especially large and complex ones, the minimal set of genes required for producing the core metabolite is unknown. The function of unclear genes is often inferred through time-consuming gene knockout studies in the native producer. A rapid combinatorial assembly method presents an alternative solution [4]. This synthetic biology approach involves assembling individual genes from a BGC into a collection of partial or complete clusters in a heterologous host. By then screening these strains using mass spectrometry, researchers can directly identify the minimum genes required for compound production. This method was successfully applied to resolve conflicting results regarding the glidobactin gene cluster and had the added advantage of generating strains that produce novel analogues of the target compound [4].
The Scientist's Toolkit: Essential Research Reagents and Methods
To address these challenges, researchers rely on a suite of specialized tools and protocols. The table below details key reagents and methodologies critical for heterologous expression studies.
Table 3: Research Reagent Solutions for Heterologous Expression
| Reagent / Method | Function / Application | Key Features / Examples |
|---|---|---|
| ExoCET Technology | Assembly of large DNA constructs like BGCs. | Used for modular assembly of the 11 kb nif cluster prior to integration [3]. |
| antiSMASH | In silico identification of BGCs in genomic data. | A state-of-the-art tool for BGC prediction; version 6.0 used to mine 4,744 gut genomes [2]. |
| Combinatorial Assembly | Rapid determination of minimal functional gene sets. | Enables parallel assembly of different gene combinations to find the core biosynthetic machinery [4]. |
| Heterologous Hosts | Tractable chassis for BGC expression. | Bacillus subtilis: PGPR with agronomic benefits [3]. E. coli: Well-understood genetics, but poor root colonizer [3]. |
| Promoter Systems | Driving transcription of heterologous genes. | Constitutive promoters (e.g., Pveg, P43) are often required to activate silent BGCs in a new host [3]. |
| Mass Spectrometry | Detection and characterization of synthesized metabolites. | Critical for screening combinatorial libraries and confirming successful heterologous production [4]. |
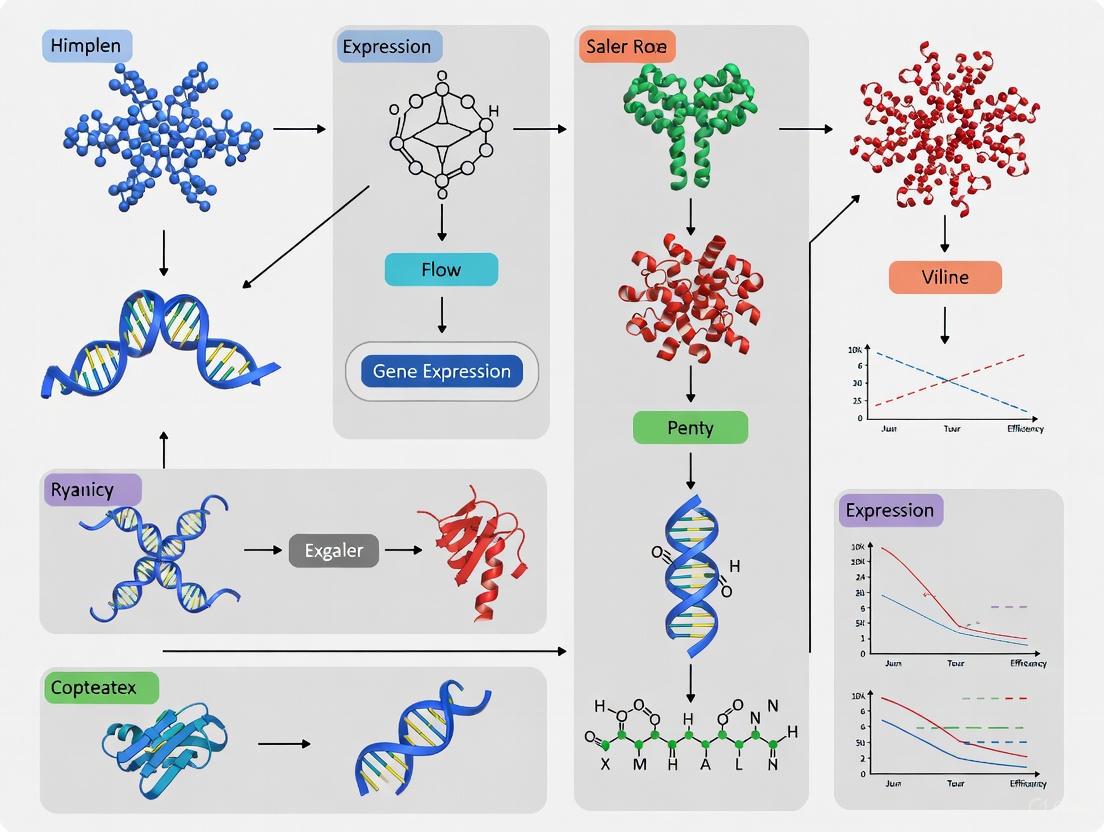
Visualizing the Heterologous Expression Workflow and Challenges
The following diagrams outline the general workflow for heterologous expression and the specific decision points involved in overcoming key challenges.
BGC Heterologous Expression Workflow
Overcoming Expression Challenges
Biosynthetic gene clusters (BGCs) represent a vast reservoir of potential natural products with applications in medicine and agriculture. However, the majority of these BGCs remain "silent" or "cryptic" under standard laboratory conditions, presenting a significant challenge for natural product discovery. This technical guide explores the multifaceted molecular and regulatory mechanisms underlying BGC silence in native hosts and examines how this fundamental biological challenge shapes subsequent efforts in heterologous expression. By integrating recent advances in genomics, molecular biology, and synthetic biology, this review provides a framework for understanding and overcoming the obstacles in accessing the hidden microbial metabolome.
Microbial natural products have historically been a prolific source of therapeutic agents, with over half of FDA-approved small molecule drugs originating from natural product scaffolds [5] [6]. Genomic sequencing has revealed that the biosynthetic potential of microorganisms far exceeds previously identified metabolites, with an estimated 97% of bacterial natural products remaining undiscovered [5]. This discrepancy stems from the prevalence of cryptic or silent BGCs—genomic regions encoding biosynthetic machinery for specialized metabolites that are not expressed under conventional laboratory conditions [6] [7].
The silence of these BGCs represents a fundamental bottleneck in natural product discovery. Understanding why these clusters remain inactive in their native hosts is crucial for developing strategies to activate them, either endogenously or through heterologous expression. This challenge exists within the broader context of overcoming technical hurdles in heterologous expression research, where fundamental biological questions directly inform practical experimental design [8] [9].
Mechanisms of BGC Silence in Native Hosts
Transcriptional Regulation and Silencing
The expression of BGCs is tightly controlled by complex regulatory networks that respond to environmental and physiological cues. In native hosts, this regulation often results in transcriptional silence under laboratory conditions.
Global and pathway-specific regulators: Many BGCs are controlled by pathway-specific transcription factors that respond to unknown environmental signals or are repressed by global regulatory systems [6] [10]. In Streptomyces, for instance, the global regulator DasR links nutrient stress to antibiotic production, and its activity can prevent BGC expression under non-inducing conditions [10].
Chromatin-level regulation: In fungi, the position of BGCs within heterochromatic regions near telomeres contributes to their silence through reduced DNA accessibility [11]. Histone modifications—including methylation, acetylation, and phosphorylation—govern chromatin structure and consequently BGC expression [7] [11]. DNA methylation can also epigenetically silence cluster expression.
Environmental Cues and Signaling
In natural environments, BGC expression is frequently triggered by specific ecological interactions that are absent in axenic laboratory cultures.
Quorum sensing and population density: Many bacteria employ cell-to-cell communication systems that activate secondary metabolism only at certain population densities, which may not be reached or synchronized in laboratory flasks [6].
Biotic interactions: Cross-talk with competing or symbiotic microorganisms often induces BGC expression through chemical signaling that is absent in pure cultures [10] [7]. For example, the synthesis of the cytotoxin malleicyprol in Burkholderia thailandensis is triggered by limited nucleotide pools, a condition that may arise during competition with other microbes [6].
Abiotic factors: Environmental parameters such as pH, temperature, oxygen availability, and nutrient composition in natural habitats differ significantly from standard laboratory media and can dramatically influence BGC expression [7] [11].
Genetic and Genomic Constraints
Structural and genetic features intrinsic to BGCs can also contribute to their silence.
Cluster boundary inaccuracy: Bioinformatic tools may misdefine the start and end points of BGCs, potentially excluding essential regulatory genes or biosynthetic components [5].
Dispersed genetic organization: Some BGCs are composed of multiple operons or genes located in disparate genomic loci, requiring coordinated regulation that may not occur in laboratory settings [6] [12].
Horizontal gene transfer: Recently acquired BGCs may lack appropriate integration into host regulatory networks or may be silenced as "foreign" DNA [11].
Table 1: Primary Mechanisms of BGC Silence in Native Hosts
| Mechanism Category | Specific Factors | Representative Examples |
|---|---|---|
| Transcriptional Regulation | Pathway-specific regulators, Global regulators, Chromatin structure | DasR nutrient regulation in Streptomyces [10], Histone modifications in fungi [7] [11] |
| Environmental Signaling | Quorum sensing, Microbial interactions, Abiotic factors | Malleicyprol induction by nucleotide limitation [6], Co-culture induction [7] |
| Genetic Organization | Cluster boundary issues, Dispersed genes, Horizontal transfer | Misidentified BGC boundaries [5], Multi-operon clusters [6] |
Experimental Approaches for Investigating Cryptic BGCs
Endogenous Activation Strategies
Endogenous approaches aim to activate silent BGCs within their native hosts, preserving native regulatory and biosynthetic contexts.
Culture-Based Methods:
- OSMAC (One Strain Many Compounds): Systematic variation of culture parameters including media composition, temperature, aeration, and cultivation time [7] [9]. This approach simulates environmental variation that may trigger BGC expression.
- Co-cultivation: Growing the target microorganism with other microbes to replicate ecological interactions that induce silent BGCs [10] [7].
- Small molecule elicitors: Addition of chemical inducers such as histone deacetylase inhibitors (e.g., suberoylanilide hydroxamic acid) or DNA methyltransferase inhibitors to disrupt epigenetic silencing [7].
Genetic Manipulation in Native Hosts:
- Promoter engineering: Replacement of native promoters with constitutive or inducible variants using CRISPR-Cas9 or homologous recombination [10].
- Regulator manipulation: Overexpression of pathway-specific activators or deletion of repressors [6] [10].
- Reporter-guided mutant selection (RGMS): Generation of random mutant libraries (via UV or transposon mutagenesis) coupled with reporter systems to identify mutants with activated BGCs [6] [10].
Heterologous Expression Strategies
Heterologous expression involves transferring BGCs to engineered host organisms optimized for natural product production, circumventing native regulatory constraints.
Host Selection and Engineering:
- Streptomyces chassis development: Creation of optimized hosts such as S. coelicolor M1152 and S. albus J1074 with deleted endogenous BGCs to reduce background interference and enhance precursor availability [13] [14].
- Escherichia coli platforms: Engineering of E. coli strains with optimized codon usage, precursor pathways, and specialized enzymes (e.g, phosphopantetheinyl transferases) for expression of certain BGC classes [8].
- Fungal expression systems: Development of Saccharomyces cerevisiae and Aspergillus nidulans as hosts for fungal BGCs [11].
BGC Capture and Refactoring:
- Direct cloning methods: Transformation-associated recombination (TAR) and exonuclease combined with RecET recombination (ExoCET) for capturing large BGCs (>100 kb) [8] [13].
- Pathway refactoring: Replacement of native regulatory elements with synthetic promoters, ribosome binding sites, and terminators to optimize expression in heterologous hosts [8].
- Multi-copy integration: Use of recombinase-mediated cassette exchange (RMCE) systems (e.g., Cre-lox, Vika-vox) to integrate multiple copies of BGCs into the host genome [13].
Table 2: Success Rates of Heterologous Expression in Large-Scale Studies
| BGC Source | BGCs Cloned | BGCs Expressed | Success Rate | New NP Families | Host(s) Used | Reference |
|---|---|---|---|---|---|---|
| Saccharothrix espanaensis | 17 | 4 | 11% | 2 | S. lividans DYA, S. albus J1074 | [5] |
| 14 Streptomyces spp., 3 Bacillus spp. | 43 | 7 | 16% | 5 | S. avermitilis SUKA17, S. lividans TK24, B. subtilis JH642 | [5] |
| 100 Streptomyces spp. | 58 | 15 | 24% | 3 | S. albus J1074, S. lividans RedStrep 1.7 | [5] |
| Multiple phyla | 83 | 27 | 32% | 3 | E. coli BL21 (DE3) | [5] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Cryptic BGC Investigation
| Reagent/Category | Specific Examples | Function/Application | Experimental Context |
|---|---|---|---|
| Bioinformatics Tools | antiSMASH, PRISM, RODEO | BGC identification, boundary prediction, product prediction | In silico analysis [5] [8] |
| Cloning Systems | TAR, ExoCET, BAC vectors | Capture and maintenance of large BGCs | Heterologous expression [8] [13] |
| Genetic Tools | CRISPR-Cas9, Red/ET recombineering | Precise genome editing, promoter replacement | Endogenous activation, refactoring [10] [13] |
| Epigenetic Modifiers | SAHA (Vorinostat), 5-azacytidine | Histone deacetylase inhibition, DNA methyltransferase inhibition | Chemical induction of silent BGCs [7] |
| Expression Hosts | S. coelicolor M1152, S. albus J1074, E. coli BAP1 | Optimized chassis for heterologous expression | Heterologous production [13] [14] |
| Analytical Platforms | HPLC-HRMS, NMR spectroscopy | Metabolite separation, detection, and structure elucidation | Compound identification [6] [11] |
Challenges in Heterologous Expression of Cryptic BGCs
The very mechanisms that maintain BGC silence in native hosts create specific technical challenges for heterologous expression efforts.
Molecular and Technical Hurdles
BGC capture and stability: Large BGC size (often >50 kb, up to 215 kb for polyketides), high GC content, and repetitive sequences complicate cloning and maintenance in heterologous systems [8]. Instability of repetitive sequences in E. coli conjugation systems can prevent successful transfer of intact BGCs [13].
Incomplete cluster identification: Incorrect prediction of BGC boundaries may exclude essential biosynthetic or regulatory genes. Additionally, genes outside the core cluster may be required for biosynthesis, such as those encoding precursor supply or transporter functions [5].
Incompatible regulation and expression: Heterologous hosts may lack specific transcription factors, post-translational modification systems, or chaperones required for proper enzyme folding and function [8]. Differences in codon usage, ribosomal binding sites, and promoter recognition between source and host organisms can prevent adequate expression [9].
Metabolic and Physiological Barriers
Precursor availability: Heterologous hosts may lack sufficient pools of required biosynthetic precursors (e.g., unusual acyl-CoAs, amino acids) or cofactors (e.g, NADPH, SAM) [8] [14].
Product toxicity: Expression of cryptic BGCs may produce compounds toxic to the heterologous host, limiting production yields or preventing detection [9].
Incompatible cellular environment: Differences in pH, redox potential, or subcellular compartmentalization between native and heterologous hosts can hinder proper biosynthesis [8] [9].
The silence of BGCs in native hosts represents both a challenge and an opportunity for natural product discovery. Understanding the complex regulatory networks, environmental cues, and genetic factors that maintain this silence provides crucial insights for developing activation strategies. Heterologous expression has emerged as a powerful approach for accessing cryptic metabolites, with recent advances in host engineering, DNA assembly, and pathway refactoring increasing success rates.
However, significant challenges remain, particularly in the functional expression of complex BGCs requiring specialized precursors, cofactors, or post-translational modifications. Future directions will likely involve the development of more specialized chassis hosts tailored to specific BGC classes, improved bioinformatic tools for predicting cluster boundaries and regulatory requirements, and integration of cell-free systems for rapid prototyping of BGC expression [12]. As these technologies mature, the gap between biosynthetic potential and characterized natural products will continue to narrow, unlocking new chemical space for therapeutic development and biological discovery.
Heterologous expression, the process of expressing a gene or set of genes from one organism in a different host species, is a fundamental technique in modern molecular biology, biotechnology, and drug development. It enables the production of recombinant proteins and natural products that are difficult to obtain from their native sources. Despite its widespread use, achieving efficient heterologous expression of biosynthetic gene clusters (BGCs) faces several core technical hurdles that can drastically reduce or even prevent the production of the target molecule.
This whitepaper provides an in-depth examination of three critical challenges in heterologous expression research: managing GC-content, optimizing codon usage, and handling large cluster sizes. We will explore the underlying principles of these hurdles, present quantitative data on their impacts, summarize proven experimental methodologies to overcome them, and visualize key workflows. Understanding and addressing these challenges is essential for researchers aiming to successfully express complex gene clusters for scientific and therapeutic applications, such as the production of novel marine natural products with pharmaceutical potential [9].
The GC-Content Challenge
Definition and Biological Significance
GC-content refers to the percentage of nitrogenous bases in a DNA molecule that are guanine (G) or cytosine (C). It is a critical factor in heterologous expression because it influences DNA stability, mRNA secondary structure, and the overall efficiency of transcription and translation. Every organism has a characteristic genomic GC-content, which can vary dramatically between species [15].
Impact on Heterologous Expression
Extreme GC-content (either too high or too low) in a heterologous gene can lead to numerous problems. High GC-content can promote the formation of stable secondary structures in mRNA, such as hairpins, which can impede the progression of the ribosome during translation [16]. It can also lead to increased recombination events in the host and create difficulties during PCR amplification and DNA sequencing, which are essential steps in clone verification. Conversely, low GC-content can affect codon usage patterns and potentially destabilize the DNA molecule [15].
Table 1: Impact of GC-Content Optimization on Gene Synthesis Success
| Gene Example | Original GC Content | Optimized GC Content | Experimental Outcome |
|---|---|---|---|
| Mouse Hoxa4 Gene [15] | 69.3% | 59.5% | Significant increase in chances of successful gene synthesis |
| General Recommendation [15] | >65% or <30% | ~60% | Optimal for stable heterologous expression and synthesis |
Experimental Protocol for GC-Content Analysis and Optimization
Protocol 1: In silico GC-Content Analysis and Optimization
- Sequence Analysis: Input your DNA sequence into a GC analysis tool, such as the GC Content Calculator integrated into VectorBuilder's platform [15]. This will determine the overall GC-content and can often provide a windowed analysis to show GC-distribution along the sequence.
- Identification of Problematic Regions: Identify regions with extreme GC-content (e.g., >65% or <30%) that may hinder expression or synthesis.
- Codon Optimization Tool: Use a codon optimization software (e.g., VectorBuilder's Codon Optimization tool, OPTIMIZER) [15] [17]. These tools allow you to set a target GC-content.
- Parameter Setting: Select the desired host organism and set the target GC-content to an optimal level (typically around 60% for most applications). The software will generate a new DNA sequence that encodes the same protein but uses synonymous codons to adjust the GC-content.
- Sequence Validation: Check the optimized sequence for the introduction or removal of restriction enzyme sites, repetitive elements, and other undesirable features.
The Codon Usage Bias Hurdle
Principles of Codon Usage and Bias
The genetic code is degenerate, meaning most amino acids are encoded by multiple triplets of nucleotides, known as codons. However, organisms do not use these synonymous codons with equal frequency; this preference is termed codon usage bias [15]. This bias exists because the abundance of transfer RNA (tRNA) molecules, which recognize codons and deliver the corresponding amino acids, varies within a cell. Highly expressed genes in an organism preferentially use codons that match the most abundant tRNA species, leading to efficient translation [17].
Consequences of Non-Optimal Codon Usage
When a heterologous gene contains a high frequency of codons that are rare in the expression host, translation can be slow, inefficient, or prone to errors. This is because the ribosome may stall waiting for a scarce tRNA, which can lead to a reduction in protein yield, premature translation termination, or misincorporation of amino acids [16] [18]. The Codon Adaptation Index (CAI) is a quantitative measure that predicts the expression level of a gene based on how well its codon usage matches that of the host's highly expressed genes. A CAI of 1.0 is ideal, while a value below 0.8 is often indicative of potential poor expression [15].
Table 2: Codon Optimization Tools and Their Key Features
| Tool Name | Key Features | Optimization Methods | Reference |
|---|---|---|---|
| VectorBuilder Tool [15] | Integrated with vector design; optimizes CAI, GC-content, and avoids restriction sites. | Proprietary algorithm balancing multiple factors. | [15] |
| OPTIMIZER [17] | Uses pre-computed tables of highly expressed genes or tRNA copy numbers for >150 prokaryotes. | One amino acid-one codon; Guided random; Customized one amino acid-one codon. | [17] |
| Deep Learning Models (e.g., BiLSTM-CRF) [18] | Learns complex codon distribution patterns from host genomes without relying on simple indices like CAI. | Sequence annotation via deep learning. | [18] |
Experimental Protocol for Codon Optimization
Protocol 2: Practical Codon Optimization for Heterologous Expression
- Determine the Host System: Identify the organism you will use for expression (e.g., E. coli, yeast, mammalian cells).
- Select a Reference Set: Choose a appropriate codon usage table for your host. For E. coli and other prokaryotes, tools like OPTIMIZER provide tables derived from highly expressed genes (e.g., ribosomal proteins), which are more reflective of translational efficiency than the genome-wide average [17].
- Choose an Optimization Method:
- One-to-One Replacement: Replaces all codons with the single most frequent synonymous codon in the host. This is simple but can cause tRNA imbalance and is not generally recommended [17] [18].
- Probabilistic/Harmonization Method: Redesigns the sequence so that the frequency of codon usage mirrors the natural distribution in the host's highly expressed genes. This is considered a superior strategy as it maintains a more natural translation rhythm, which can be important for correct protein folding [18].
- AI-Based Optimization: Employs deep learning models (e.g., BiLSTM-CRF) trained on the host genome to predict optimal codon sequences, potentially capturing complex patterns beyond traditional metrics [18].
- Generate and Analyze the Sequence: Use your selected tool (see Table 2) to generate the optimized sequence. Verify that the amino acid sequence is unchanged and check other parameters like GC-content.
- Gene Synthesis and Cloning: The optimized DNA sequence is typically synthesized de novo and cloned into an appropriate expression vector for the host.
Diagram 1: A generalized workflow for the computational codon optimization of a gene for expression in a heterologous host.
The Cluster Size and Complexity Obstacle
The Nature of Biosynthetic Gene Clusters (BGCs)
Many valuable natural products, such as antibiotics, antifungals, and anticancer agents, are synthesized by biosynthetic gene clusters (BGCs). These are sets of co-localized genes in a genome that encode the enzymes, regulators, and resistance mechanisms for a specific metabolic pathway [19] [9]. BGCs can be very large, spanning tens to over a hundred kilobases, and their genes are often organized in operons with complex regulation.
Challenges in Heterologous Cluster Expression
The large size of BGCs presents a primary technical challenge. Cloning large DNA fragments into standard vectors is difficult and often leads to instability in the host, with a high probability of rearrangements or deletions [9]. Furthermore, the heterologous host may lack the necessary precursors, co-factors, or specific post-translational modification enzymes required for the pathway to function. Many BGCs are also "silent" or "cryptic" under laboratory conditions, meaning their expression requires specific environmental or regulatory triggers that are not present in the new host [9].
Table 3: Challenges Associated with Expressing Large Gene Clusters
| Challenge | Description | Impact on Heterologous Expression |
|---|---|---|
| Large DNA Inserts | Clusters can be >100 kb in size. | Difficult to clone and maintain stably in a heterologous host; prone to recombination and deletion. |
| Complex Regulation | Native regulation may involve multiple, host-specific transcription factors. | The cluster may be silent in the heterologous host without the correct regulatory cues. |
| Missing Cofactors/Precursors | The pathway may require specific metabolic building blocks. | The final product may not be produced, or intermediates may accumulate. |
| Gene Toxicity | Expression of cluster genes may be toxic to the heterologous host. | Inhibits host growth, leading to low yields or selection for non-producing mutants. |
Experimental Protocol for BGC Heterologous Expression
Protocol 3: Strategy for Expressing Large Biosynthetic Gene Clusters
- Cluster Identification and Prioritization: Use genome mining tools like antiSMASH to identify and annotate BGCs in a source organism's genome [19] [9].
- Cluster Refactoring (if necessary): This involves replacing the native promoters and regulatory elements of the BGC with well-characterized, strong promoters that are functional in the heterologous host. This ensures all necessary genes are expressed and can awaken silent clusters [9].
- Host Selection:
- Closely Related Host: Choosing a host phylogenetically close to the native producer (e.g., another Streptomyces species for a Streptomyces BGC) can increase success due to similar cellular machinery [9].
- Standard Model Host: Using a well-established host like E. coli or S. cerevisiae is advantageous due to the availability of extensive genetic tools and fast growth, but it may lack necessary functionalities [20] [9].
- DNA Assembly and Transfer:
- For large clusters, use specialized techniques such as Transformation-Associated Recombination (TAR) in yeast, bacterial artificial chromosomes (BACs), or cosmids to capture and maintain the large DNA fragment [9].
- Alternatively, break the cluster into smaller parts and assemble them in the host using techniques like Gibson Assembly.
- Screening and Metabolite Analysis: Screen transformations for the production of the target compound using liquid chromatography-mass spectrometry (LC-MS) or other analytical methods. Further optimization may involve metabolic engineering of the host to supply pathway precursors [9].
Diagram 2: A multi-step experimental pipeline for the heterologous expression of a large biosynthetic gene cluster (BGC).
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successfully navigating the technical hurdles of heterologous expression requires a suite of specialized reagents and tools.
Table 4: Key Research Reagents for Overcoming Heterologous Expression Hurdles
| Reagent / Tool | Function & Application |
|---|---|
| Codon-Optimized Synthetic Genes | Custom DNA fragments designed in silico and synthesized to have optimal codon usage and GC-content for the target host, directly addressing the codon bias hurdle [15] [18]. |
| Specialized Expression Vectors (e.g., pET series, BACs, Cosmids) | Plasmids designed for specific hosts. pET vectors are standard for E. coli; BACs and cosmids are essential for stably maintaining large gene cluster inserts [9] [16]. |
| Engineered E. coli Strains (e.g., BL21(DE3), C41(DE3), C43(DE3)) | Host strains with mutated tRNA genes to accommodate rare codons (e.g., BL21-CodonPlus), or membranes to tolerate toxic protein expression (C41/C43) [20] [16]. |
| Disulfide Bond Helper Strains & Plasmids (e.g., CyDisCo system) | Systems that promote the correct formation of disulfide bonds in the E. coli cytoplasm, enabling the production of complex eukaryotic proteins [20]. |
| Cell-Free Protein Synthesis Systems | In vitro transcription/translation systems that bypass cellular growth and toxicity issues, useful for rapid screening of toxic proteins or pathways [20]. |
The heterologous expression of genes and biosynthetic pathways is fraught with technical challenges, of which GC-content, codon usage bias, and cluster size are among the most fundamental. These factors are not independent; for instance, codon optimization inherently affects GC-content, and the large size of BGCs compounds the difficulties of sequence optimization. A successful expression strategy requires a holistic and integrated approach, leveraging sophisticated computational tools for in silico design, a growing toolkit of specialized biological reagents and host strains, and robust experimental protocols for assembly and screening. As deep learning and synthetic biology continue to advance, they promise to provide even more powerful solutions, enabling researchers to more efficiently harness the vast potential of heterologous expression for drug discovery and bioproduction.
The exploration of microbial biosynthetic gene clusters (BGCs) represents a frontier in discovering novel bioactive natural products with therapeutic potential. These clusters, physically grouped genes encoding enzymatic machinery for natural product biosynthesis, offer immense promise for developing new antibiotics, antifungals, immunosuppressants, and anticancer agents [21] [22]. However, a significant challenge impedes this discovery pipeline: the majority of BGCs are silent or weakly expressed under standard laboratory conditions, and their native microbial hosts are often uncultivable or genetically intractable [9]. This fundamental problem necessitates heterologous expression—transferring BGCs into well-characterized host organisms for activation and production.
Heterologous expression, while powerful, introduces its own complex challenges. The process is laborious, time-consuming, and has a high failure rate. A major bottleneck is selecting which BGCs, from the thousands identifiable in genomic data, warrant the substantial investment required for heterologous expression experiments [21]. Bioinformatic prioritization has therefore become an indispensable first step in natural product discovery. By leveraging specialized tools and databases, researchers can strategically identify BGCs with the highest likelihood of yielding novel and bioactive compounds. This guide details how the integrated use of antiSMASH (antibiotics and Secondary Metabolite Analysis Shell) and the MIBiG (Minimum Information about a Biosynthetic Gene Cluster) repository addresses these challenges, creating a robust framework for BGC prioritization within heterologous expression research.
The Bioinformatics Toolkit for BGC Analysis
A typical BGC prioritization workflow utilizes several key bioinformatics resources, each with a distinct role. The table below summarizes these essential tools and databases.
Table 1: Key Bioinformatics Resources for BGC Prioritization
| Resource Name | Type | Primary Function | Role in Prioritization |
|---|---|---|---|
| antiSMASH [21] [22] | Analysis Pipeline | Identifies & annotates BGCs in genomic data. | Core analysis tool for initial BGC detection and structural prediction. |
| MIBiG [21] [22] | Curated Repository | Collection of experimentally characterized BGCs. | Gold standard for dereplication and novelty assessment. |
| antiSMASH DB [21] [22] | Public Database | Repository of pre-computed antiSMASH results. | Provides a vast dataset for comparative analysis. |
| BIG-FAM [22] | Classification Database | Groups BGCs into gene cluster families. | Enables evolutionary and structural relationship analysis. |
| GATOR-GC [22] | Targeted Mining Tool | Identifies BGCs based on user-defined proteins. | Facilitates targeted searches for specific natural product families. |
The Central Role of antiSMASH
antiSMASH is the cornerstone software for BGC detection. It uses profile Hidden Markov Models (pHMMs) to scan genomic data against manually curated rules for over 70 types of BGCs [21] [22]. Its analysis provides critical data for prioritization, including:
- BGC Delineation and Classification: Precise identification of cluster boundaries and assignment to a natural product class (e.g., nonribosomal peptide, polyketide, terpene).
- Core Scaffold Prediction: For certain classes like polyketides and nonribosomal peptides, antiSMASH predicts the chemical structure of the core molecular scaffold based on substrate specificity of enzymatic domains and assumed collinearity [21].
- Comparative Genomics: Integrated tools like ClusterBlast allow for immediate comparison of a query BGC against known clusters in databases such as MIBiG and antiSMASH DB [21].
The Dereplication Standard of MIBiG
The MIBiG database is a critical resource for preventing the rediscovery of known compounds. It houses detailed, curated information on over 1,900 experimentally validated BGCs and their associated secondary metabolites [21] [22]. By comparing putative BGCs against MIBiG, researchers can quickly assess the novelty of a cluster. A BGC with low similarity to any MIBiG entry represents a prime candidate for further investigation, as it likely codes for a previously uncharacterized natural product.
BGC Prioritization Strategies and Workflows
The integration of antiSMASH and MIBiG enables several powerful prioritization logics. The following workflow diagram illustrates the multi-step process from genomic data to a prioritized candidate for heterologous expression.
Key Prioritization Logics in Detail
Building on the workflow above, the following specific strategies have proven effective in selecting high-priority BGCs from large-scale genomic datasets [21].
Table 2: Strategic Approaches to BGC Prioritization
| Strategy | Rationale | Methodology | Exemplary Discovery |
|---|---|---|---|
| Resistance-Gene-Guided | BGCs often include self-resistance genes; their presence indicates expression and bioactivity [21]. | Identify genes (e.g., efflux pumps, drug-resistant enzyme variants) within or adjacent to BGCs. | Alkylpyrone-407 and pyxidicycline A, identified using pentapeptide repeat protein sequences [21]. |
| Phylogenomics-Guided | Evolutionary analysis can pinpoint BGCs unique to an understudied taxonomic branch. | Construct phylogenetic trees of housekeeping genes to identify strains, then analyze their BGCs for novelty. | Aspterric acid, discovered by focusing on a unique BGC in Aspergillus terreus [21]. |
| Structure-Guided | Targets BGCs predicted to produce specific, desirable chemical features. | Use substrate specificity predictions from antiSMASH or target specific tailoring enzymes (e.g., P450s, methyltransferases). | Novel FK506 analogs, found by searching for the lysine cyclodeaminase (KCDA) enzyme [22]. |
Experimental Protocol: From In Silico Hit to Heterologous Expression
Once a BGC is prioritized, the experimental journey begins. The process is complex, requiring careful decision-making at each stage, as visualized below.
Detailed Methodologies
Step A: Heterologous Host Selection The choice of host is critical. Key considerations include [9] [23]:
- Phylogenetic Proximity: Selecting a host closely related to the native producer (e.g., another Streptomyces for an actinomycete BGC) increases the likelihood that native promoters, regulatory factors, and ribosomal binding sites will function correctly, and that essential substrates will be available [9].
- Genetic & Physiological Suitability: The host must be genetically tractable and lack endogenous pathways that could interfere with the analysis or production of the target compound. Common model hosts include Streptomyces coelicolor, Mycobacterium smegmatis, and E. coli strains engineered for natural product expression [23].
Step B-D: BGC Cloning, Expression, and Compound Characterization
- BGC Cloning: Due to their large size (often >10 kb), cloning intact BGCs is a primary technical hurdle. Methods include cosmid/fosmid library construction, direct capture via Transformation-Associated Recombination (TAR) in yeast, or complete synthetic synthesis of the cluster [9] [23].
- Vector Assembly and Transformation: The cloned cluster is placed into a suitable expression vector, which may be equipped with strong, constitutive promoters to drive expression of silent clusters—a strategy known as "heterologous expression awakening" [9].
- Heterologous Expression and Analysis: Transformed hosts are cultured under conditions that induce BGC expression. Metabolite extracts are then analyzed using Liquid Chromatography-Mass Spectrometry (LC-MS) and compared to control strains to identify compounds unique to the BGC-containing host. Subsequent purification and nuclear magnetic resonance (NMR) spectroscopy are used for full structural elucidation [9].
The following table details key reagents, tools, and materials essential for executing the bioinformatic and experimental workflows described in this guide.
Table 3: Essential Research Reagents and Solutions for BGC Prioritization and Heterologous Expression
| Item Name | Function/Application | Technical Specifications & Alternatives |
|---|---|---|
| antiSMASH Software | Core BGC detection and annotation from genome sequences. | Available via web server or command-line. PRISM is an alternative for activity prediction [21]. |
| MIBiG Database | Reference for dereplication and assessment of BGC novelty. | Contains ~1,900 curated entries. The antiSMASH DB and IMG-ABC offer larger, non-curated datasets [21] [22]. |
| GATOR-GC Tool | For targeted mining of specific BGC families. | Allows user-defined required/optional protein searches. Manual BLAST analysis is an alternative [22]. |
| Cosmid/Fosmid Vectors | Cloning large (>30 kb) DNA fragments of BGCs from genomic DNA. | Essential for constructing genomic libraries for BGC capture [9]. |
| E. coli / S. cerevisiae Hosts | Intermediate hosts for vector propagation and assembly. | E. coli is standard; S. cerevisiae enables TAR cloning of very large clusters [9] [23]. |
| Specialized Heterologous Hosts | Final chassis for BGC expression and compound production. | Includes Streptomyces coelicolor, Mycobacterium smegmatis, and engineered E. coli strains [23]. |
| LC-HRMS Instrumentation | Critical for detecting and analyzing novel metabolites from heterologous hosts. | Used to compare metabolic profiles and identify target compounds [9]. |
The challenges inherent in heterologous expression of BGCs—from the silence of clusters in native strains to the inefficiency of the process itself—make strategic prioritization not merely beneficial, but essential. The integrated use of the bioinformatics toolkit centered on antiSMASH and MIBiG provides a powerful solution. This methodology enables researchers to move beyond random screening to a targeted, hypothesis-driven discovery process. By applying the prioritization strategies and experimental protocols outlined in this guide, scientists can systematically identify BGCs with the highest potential for yielding novel bioactive molecules, thereby accelerating the development of new therapeutic agents and unlocking the vast hidden potential of microbial genomes.
From Blueprint to Product: Methodologies for Cloning and Expressing Complex Gene Clusters
The exploration of microbial genomes has revealed a vast reservoir of uncharacterized biosynthetic gene clusters (BGCs) encoding potential novel therapeutics. However, the majority of these BGCs remain functionally inaccessible because they are either silent under laboratory conditions or poorly expressed in their native hosts [24]. Heterologous expression—the process of transferring and expressing BGCs in optimized surrogate hosts—has emerged as a pivotal strategy to overcome this limitation, enabling the activation of silent pathways and the production of bioactive natural products [25] [26]. This approach not only facilitates novel drug discovery but also allows for yield optimization through host and pathway engineering [27].
Despite its promise, heterologous expression faces significant technical hurdles, particularly during the initial cloning of BGCs. The direct cloning of large, GC-rich, and repetitive BGCs represents a critical bottleneck that this guide aims to address. These challenges stem from several intrinsic properties of BGCs: their large size (often exceeding 50 kb), high GC content (which can complicate sequencing and PCR amplification), and repetitive sequences (common in polyketide synthase and non-ribosomal peptide synthetase systems) that promote recombination events and vector instability [28] [26]. Successfully navigating this "cloning conundrum" requires a sophisticated toolkit of methods, vectors, and host strains, which we explore in detail below.
Understanding the Cloning Bottleneck: BGC Characteristics and Technical Hurdles
Biosynthetic gene clusters present unique challenges that differentiate them from standard genetic cloning projects. Their large size, often spanning 10 to over 100 kilobases, exceeds the capacity of conventional plasmids [28]. This is compounded by repetitive sequences that pose problems for both sequencing accuracy and genetic stability in cloning hosts. The high GC content (often >70%) typical of actinobacterial BGCs leads to secondary structures that hinder sequencing and PCR amplification, while also creating codon usage biases that must be addressed for successful expression in heterologous hosts [26].
A significant barrier arises from the transcriptional silencing of many BGCs in their native genomic context. Under standard laboratory conditions, an estimated 90% of native BGCs are not transcribed or are only partially expressed, necessitating not just physical cloning but also genetic refactoring to activate them [25]. Furthermore, when BGCs are successfully cloned into standard high-copy vectors, the metabolic burden on the host can lead to growth defects or vector instability, particularly when dealing with large inserts containing complex genetic elements [27].
Modern Cloning Strategies for Challenging BGCs
Recent methodological advances have significantly improved our ability to capture and manipulate large, complex BGCs. The table below summarizes the key characteristics of prominent contemporary cloning methods.
Table 1: Comparison of Modern BGC Cloning Strategies
| Method | Key Principle | Optimal Insert Size | Key Advantages | Primary Limitations |
|---|---|---|---|---|
| Transformation-Associated Recombination (TAR) | Homology-based assembly in yeast | 10 - 200+ kb | Captures very large clusters directly from gDNA; handles repetitive sequences well [28] [25] | Requires yeast handling expertise; may capture non-target regions |
| ExoCET (Exonuclease combined with RecET) | In vitro recombination with exonuclease treatment | 10 - 100+ kb | High efficiency; works with partial genome assemblies; direct cloning from gDNA [3] | Requires specialized enzyme mixtures |
| Cas9-Assisted Targeting of Chromosome Segments (CATCH) | CRISPR-Cas9 mediated linear fragment retrieval | 10 - 100+ kb | High specificity; uses Cas9 to cleave cluster boundaries [26] | Requires highly accurate genome sequence for gRNA design |
| Micro-HEP Platform | Combines E. coli recombineering with conjugation transfer | Varies with vector | Integrated system for modification and transfer; superior stability with repeats [29] | Multi-step process requiring multiple specialized strains |
Selecting the Appropriate Cloning Strategy
The choice of cloning method depends on several factors: the quality of available genomic data, BGC size, and the presence of repetitive elements. For poorly characterized systems where only draft genomes exist, TAR cloning and ExoCET offer particular advantages as they can be applied with incomplete genomic information [28]. When high-quality genome sequences are available, CATCH provides precise targeting using CRISPR-Cas9 guidance [26]. For projects requiring extensive refactoring, platforms like Micro-HEP that combine E. coli-based recombineering with conjugation offer an integrated solution from cloning to expression [29].
Experimental Design: A Step-by-Step Workflow for Successful BGC Cloning
Implementing a robust cloning strategy requires careful planning and execution. The following workflow outlines a comprehensive approach to BGC capture and refactoring, integrating the methods discussed above.
Detailed Protocol: ExoCET-Mediated BGC Cloning
The ExoCET method provides an efficient approach for direct BGC cloning, combining exonuclease treatment with RecET recombination. The following protocol has been successfully applied for capturing an 11 kb nitrogen-fixing gene cluster [3]:
Vector Preparation: Linearize your capture vector (e.g., pBR322-amp) using appropriate restriction enzymes. Purify the linearized vector using gel electrophoresis.
Genomic DNA Isolation: Extract high molecular weight genomic DNA from your source organism using a kit designed for Gram-negative bacteria (e.g., GenElute Bacterial Genomic DNA Kit). DNA quality is critical for success.
Fragment Preparation: Generate BGC-containing fragments from genomic DNA. This can be achieved through:
- PCR amplification of the target BGC using long-range polymerase
- Restriction enzyme digestion with enzymes that flank the BGC
- Synthesis of BGC fragments (as used for the nif cluster) [3]
ExoCET Recombination Reaction: Combine the following components:
- 300 ng of each purified DNA fragment (vector and BGC fragments)
- 0.13 μL T4 DNA polymerase
- 2 μL Reaction Buffer 2.1
- Nuclease-free water to 20 μL total volume
Incubate in a thermal cycler with the following program:
- 25°C for 60 minutes (recombination)
- 75°C for 20 minutes (enzyme inactivation)
- 50°C for 30 minutes (additional annealing)
- 4°C hold [3]
Transformation and Screening: Transform the reaction product into recombinase-proficient E. coli cells (e.g., GB05-dir). Select transformants on appropriate antibiotic plates. Validate positive clones through restriction analysis and sequencing.
Protocol: TAR Cloning from Oxford Nanopore Draft Genomes
For situations where only draft genome sequences are available, TAR cloning offers a powerful alternative:
Draft Genome Sequencing: Prepare genomic DNA libraries using the Rapid Barcoding Kit and sequence on Oxford Nanopore MinION flow cells. Assemble reads using Flye assembler and polish with Medaka [28].
BGC Identification: Use antiSMASH to identify BGCs of interest in the draft assembly.
TAR Vector Design: Design hooks (homology arms) of approximately 50 bp targeting the regions flanking the BGC. Incorporate these into a TAR vector containing yeast selection markers and origin of replication.
Yeast Transformation: Co-transform the TAR vector and genomic DNA into Saccharomyces cerevisiae. Select for transformants on appropriate dropout media.
Validation: Isolate yeast plasmids and transform into E. coli for amplification. Verify inserts by restriction digest and Sanger sequencing of cluster boundaries [28].
Successful BGC cloning requires specialized genetic tools and reagents. The following table catalogues essential components for establishing an effective cloning workflow.
Table 2: Essential Research Reagent Solutions for BGC Cloning
| Reagent/Resource | Function | Examples & Specifications |
|---|---|---|
| Cloning Vectors | BGC capture and maintenance | pCBA (low-copy, BAC-based), pSET152 (integration vector), pCAP-BAC, TAR vectors with yeast elements [27] |
| Engineering Strains | Recombination and conjugation | E. coli GB05-dir (direct cloning), GB05-red (recombineering), ET12567(pUZ8002) (conjugation) [3] [29] |
| Enzyme Systems | DNA manipulation and assembly | T4 DNA polymerase (ExoCET), RecET recombinase, Restriction enzymes (BamHI, Swal), Gibson assembly mix [3] |
| Heterologous Hosts | BGC expression and production | Streptomyces coelicolor M1152, Bacillus subtilis 168, Aspergillus oryzae, Engineered S. coelicolor A3(2)-2023 [3] [29] [30] |
| Promoter Libraries | BGC refactoring and activation | ermE*, kasOp, Pveg, P43, Ptp2 (strong, constitutive promoters) [3] [25] [27] |
| Selection Markers | Selection of successful clones | Ampicillin, kanamycin, spectinomycin, hygromycin resistance genes [3] [29] |
Refactoring and Optimization: From Cloned Cluster to Functional Expression
Successfully cloning a BGC is only the first step toward heterologous expression. Many BGCs, particularly silent ones, require refactoring to function in new host environments. Promoter replacement represents the most common and effective refactoring strategy, as demonstrated in the heterologous expression of the nitrogen-fixing gene cluster from Paenibacillus polymyxa in Bacillus subtilis. In this case, replacing the native promoter with the strong, constitutive Pveg promoter was necessary to achieve detectable nitrogenase activity [3].
The recent development of advanced genetic tools has dramatically improved our ability to refactor BGCs. CRISPR-Cas9 systems, particularly when applied in vitro, enable precise promoter replacements without leaving scar sequences or requiring multiple selection markers [27]. For instance, refactoring the oviedomycin BGC through promoter replacement of the ovm01 and ovmF genes resulted in a 20-fold increase in production titers [27].
Additional optimization strategies include:
Metabolic Engineering: Using genome-scale metabolic models (GEMs) to identify overexpression targets that enhance precursor supply. For oviedomycin production, overexpression of phosphoserine transaminase, methylenetetrahydrofolate dehydrogenase, and acetyl-CoA carboxylase significantly improved titers by enhancing malonyl-CoA and NADPH availability [27].
Multi-copy Integration: Employing recombinase-mediated cassette exchange (RMCE) systems to integrate multiple copies of BGCs into the host genome. Studies with the xiamenmycin BGC demonstrated that increasing copy number directly correlated with yield improvements [29].
Orthogonal Expression Systems: Implementing synthetic regulatory elements that function independently of host regulation. Completely randomized synthetic promoter libraries that include both promoter and ribosome binding site regions have shown success in achieving orthogonal expression in Streptomyces and other hosts [25].
The field of BGC cloning and heterologous expression has progressed dramatically from reliance on cosmic libraries to sophisticated direct cloning methods that can capture massive gene clusters with precision. As synthetic biology tools continue to advance, we are moving toward a future where accessing the vast hidden microbial metabolome becomes routine. Emerging technologies such as CRISPR-Cas12a systems for multiplexed editing, cell-free expression systems for rapid prototyping, and machine learning algorithms for predicting optimal refactoring strategies will further accelerate this field.
The strategies outlined in this guide provide a roadmap for researchers to navigate the complex landscape of BGC cloning. By selecting appropriate methods based on BGC characteristics, implementing systematic refactoring approaches, and leveraging the growing toolkit of genetic parts and optimized hosts, scientists can overcome the historical bottlenecks that have limited access to nature's chemical diversity. As these methodologies become more standardized and accessible, we anticipate a new wave of natural product discovery that will yield novel therapeutics and biochemical tools to address pressing challenges in medicine and biotechnology.
The successful production of recombinant proteins and natural products hinges on the strategic selection of an appropriate host chassis. Heterologous expression, the process of expressing genes or gene clusters in a non-native host, serves as a foundational strategy for engineering next-generation microbial agents for therapeutic, agricultural, and industrial applications [3]. However, this process is fraught with recurrent challenges, including the formation of insoluble protein aggregates, low product yield, genetic instability, and the failure to express biologically functional proteins [31]. These obstacles are often rooted in the complex interplay between the host's metabolic capacity, limitations of its cellular machineries, and the intrinsic characteristics of the foreign genetic material being introduced.
This technical guide provides a comprehensive landscape of the most prominent prokaryotic and eukaryotic chassis systems, from the workhorse E. coli to the complex Streptomyces, framing the discussion within the context of overcoming these universal challenges. By comparing their inherent advantages, limitations, and the specialized engineering strategies developed to optimize them, this document aims to equip researchers with the knowledge to make informed host selection decisions for their heterologous expression projects.
Core Challenges in Heterologous Expression
The functional expression of heterologous genes, particularly large biosynthetic gene clusters (BGCs), presents a consistent set of biological hurdles that must be addressed regardless of the host system.
- Improper Protein Folding and Solubility: Differences in cytoplasmic redox potential can interfere with disulfide bond formation essential for the stability of many eukaryotic proteins [31]. This often leads to the formation of inactive inclusion bodies [32].
- Incompatible Post-Translational Modifications: Prokaryotic hosts typically lack the machinery for eukaryotic post-translational modifications such as glycosylation, phosphorylation, and acetylation, which are critical for the activity of many therapeutic proteins [31]. It is estimated that over 50% of eukaryotic proteins are glycosylated [31].
- Host Toxicity and Reduced Viability: The expression of foreign proteins, especially membrane proteins, can be toxic to the host, leading to reduced cell viability and poor yields [31]. Unwanted byproducts from heterologous pathways can further exacerbate this issue [31].
- Codon Usage Bias: Discrepancies in codon preference between the native gene source and the expression host can lead to translational stalling, misincorporation of amino acids, and low yields [31].
- Proteolytic Degradation: Recombinant proteins can be recognized as foreign and subjected to degradation by the host's native protease systems [31].
- Silencing of Biosynthetic Gene Clusters (BGCs): In native hosts, especially Streptomyces, many BGCs are "silent" or "cryptic," meaning they are not expressed under standard laboratory conditions, making their products inaccessible without specialized activation strategies [33].
Prokaryotic Chassis Systems
Escherichia coli
E. coli remains the most prevalent and versatile prokaryotic expression host, with over 50% of recombinant proteins registered in the Protein Data Bank being produced in prokaryotic systems, predominantly E. coli [31].
Key Features:
- Well-Characterized System: Possesses the most extensively characterized genome, transcriptome, and translatome architectures [31].
- High Yield Potential: Can dedicate up to 40% of its dry cell weight to recombinant protein production in fed-batch cultures [31].
- Genetic Tractability: Benefits from a vast repertoire of synthetic biology tools, including libraries of promoters, RBSs, UTRs, and expression vectors [31].
Table 1: Engineered E. coli Strains for Specialized Expression Challenges
| Strain / System | Key Genetic Features | Primary Application | Mechanism of Action |
|---|---|---|---|
| Rosetta [31] | Overexpression of rare tRNA genes (AUA, AGG, AGA, CGG, CUA, CCC, GGA) | Expression of genes with divergent codon usage | Alleviates codon bias by supplementing tRNAs that are lowly expressed in standard E. coli strains. |
| Origami [31] | Mutations in thioredoxin reductase (ΔtrxB) and glutathione reductase (Δgor) pathways |
Production of disulfide-bonded proteins in the cytoplasm | Facilitates disulfide bond formation by creating an oxidizing cytoplasmic environment. |
| CyDisCo [31] | Introduction of eukaryotic thiol oxidase and disulfide isomerase | Cytoplasmic production of disulfide-bonded proteins | Encourages correct disulfide bond formation and isomerization within the E. coli cytoplasm. |
| C41(DE3)/C43(DE3) [31] | Derived from BL21(DE3); mutations conferring increased tolerance | Membrane protein production | Specific mutations reduce the toxicity associated with membrane protein overexpression, improving cell viability. |
| Lemo21(DE3) [31] | Tunable T7 RNA polymerase expression system | Membrane protein and toxic protein production | Fine-tuning of expression intensity prevents saturation of cellular machinery and mitigates toxicity. |
| Chaperone Co-expression [31] | Coordinated overexpression of GroEL/ES, DnaK/J/GrpE, etc. | Solubilization of aggregation-prone proteins | Molecular chaperones assist in the proper folding of recombinant proteins, reducing aggregate formation. |
Bacillus subtilis
As a Gram-positive bacterium, B. subtilis is a favored chassis for protein secretion due to its naturally high secretory capacity and generally recognized as safe (GRAS) status.
Key Features:
- Efficient Protein Secretion: Lacks an outer membrane, allowing direct secretion of proteins into the extracellular medium, which simplifies downstream purification [32].
- Industrial Robustness: Exhibits strong soil adaptability and is widely used as a plant-growth-promoting rhizobacterium (PGPR) [3].
- Well-Established Toolkits: Possesses a growing set of molecular tools, including promoters and transformation systems, for customized refactoring of heterologous pathways [3].
A landmark study demonstrated the heterologous expression of an 11 kb nitrogen-fixing (nif) gene cluster from Paenibacillus polymyxa in B. subtilis 168 [3]. Critical to success was the replacement of the native promoter with the host-derived constitutive promoter Pveg, highlighting the importance of promoter compatibility in achieving functional expression [3].
Streptomyces Species
Streptomyces are Gram-positive bacteria renowned for their innate capacity to produce a stunning array of natural products. They are the primary heterologous hosts for expressing complex bacterial BGCs.
Key Features:
- Native Proficiency: Naturally possess extensive biosynthetic precursor pools and secondary metabolic networks, making them ideal for producing complex natural products [29] [33].
- Secretory Capability: Efficiently secrete proteins into the culture medium, with Streptomyces lividans being a particularly noted host for recombinant protein production [34].
- Genetic Tools: Have well-established, though sometimes complex, genetic toolkits for manipulation [35].
Engineering Advanced Streptomyces Chassis: A prime example of chassis engineering is the development of S. coelicolor A3(2)-2023 [29]. This optimized chassis was created through:
- Genome Minimization: Deletion of four endogenous BGCs to minimize native metabolic interference and free up cellular resources [29].
- Integration Site Engineering: Introduction of multiple recombinase-mediated cassette exchange (RMCE) sites (
Cre-lox,Vika-vox,Dre-rox,phiBT1-attP) into the chromosome to enable stable, multi-copy integration of heterologous BGCs [29].
This system was validated by integrating two to four copies of the xiamenmycin BGC, demonstrating that increasing the gene copy number was associated with a higher yield of the final product [29].
Experimental Workflows for Heterologous Expression
The process of heterologously expressing a BGC involves a multi-step workflow, from cloning to final analysis.
The following diagram outlines the generalized steps for the heterologous expression of a biosynthetic gene cluster.
Cloning and Transfer of Large Gene Clusters
Capturing large and complex BGCs requires specialized methods beyond conventional cloning.
Key Methodologies:
- Transformation-Associated Recombination (TAR) Cloning: A powerful in vivo method that uses homologous recombination in yeast to directly clone large DNA fragments from genomic DNA. The linearized TAR vector contains homologous "hooks" to the target BGC, facilitating its capture [33].
- ExoCET (Exonuclease combined with RecET recombination): An in vitro method that employs T4 polymerase to facilitate annealing between linear target DNA and a vector. This method was successfully used to clone the intact 106 kb salinomycin BGC from S. albus [33].
- CATCH (Cas9-Assisted Targeting of CHromosome segments): An in vitro strategy that combines CRISPR-Cas9 with Gibson assembly. Cas9 is used to excise the target BGC from genomic DNA, which is then assembled into a vector [33].
- Bacterial Conjugation: A cornerstone method for transferring large BGC constructs from E. coli to Streptomyces. The system relies on the
oriTorigin of transfer and Tra proteins from IncP plasmids to mediate single-stranded DNA exchange [29]. Advanced systems, like the one in Micro-HEP, use engineered E. coli strains with superior stability for repeated sequences compared to traditional ET12567 (pUZ8002) strains [29].
Integration and Activation Strategies
Once transferred into the host, the BGC must be stably integrated and activated.
Recombinase-Mediated Cassette Exchange (RMCE): This versatile strategy allows for the precise, marker-less integration of a BGC into pre-defined chromosomal loci [29]. The process leverages orthogonal tyrosine recombinase systems (Cre-lox, Vika-vox, Dre-rox) which exhibit stringent substrate specificity with no cross-reactivity.
Diagram: RMCE Integration Mechanism
Protocol: Two-Step Recombineering in E. coli for BGC Modification [29]
- Step 1 - Plasmid Introduction: Electroporate the recombinase expression plasmid (e.g., pSC101-PRha-αβγA-PBAD-ccdA) into the E. coli strain harboring the BGC.
- Step 2 - First Recombination: Induce dual expression of the recombinase (Redα/Redβ/Redγ) and the CcdA anti-toxin with L-rhamnose and L-arabinose. Use a linear DNA cassette to replace the target gene with a selectable-counter-selectable marker (e.g.,
kan-rpsL). - Step 3 - Second Recombination: Induce only the recombinase to facilitate the replacement of the selectable-counter-selectable marker with the desired genetic modification (e.g., a new promoter).
- Step 4 - Plasmid Curing: Grow the cells at a non-permissive temperature to lose the temperature-sensitive recombinase plasmid, resulting in a marker-less, modified BGC.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents and Tools for Heterologous Expression
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| TAR Cloning Vector [33] | Captures large DNA fragments via homologous recombination in yeast. | Cloning intact BGCs directly from genomic DNA. |
| Red/ET Recombineering System [29] [33] | Enables precise DNA editing in E. coli using short homology arms (50 bp). | Refactoring BGCs (promoter replacements, gene deletions) prior to transfer. |
| PhiC31 Integrase System [33] | Mediates site-specific integration of DNA into the host chromosome. | Stable integration of BGCs into the attachment site (attB) of Streptomyces genomes. |
| RMCE Systems (Cre/lox, etc.) [29] | Enables precise, repeated, and marker-less cassette exchange at defined genomic loci. | Multi-copy integration of BGCs into a pre-engineered chassis like S. coelicolor A3(2)-2023. |
| Conjugative Plasmid (e.g., with oriT) [29] | Facilitates the transfer of large DNA constructs from E. coli to actinomycetes. | Moving large, refactored BGCs from the engineering host (E. coli) to the production host (Streptomyces). |
| Anti-SMASH Software [36] | A bioinformatics platform for genome mining to identify and analyze BGCs. | Predicting and annotating putative BGCs in a sequenced genome prior to cloning. |
The host selection landscape for heterologous expression is diverse, with no single chassis being universally optimal. The choice between the high-yield, tractable E. coli, the secretory-proficient B. subtilis, and the naturally gifted Streptomyces must be guided by the specific requirements of the target protein or natural product. The central challenge of reconciling the foreign genetic material with the host's innate biology is being met through sophisticated engineering strategies. These include genome streamlining, refactoring with compatible genetic parts, and the development of advanced cloning and integration platforms like Micro-HEP. As synthetic biology tools continue to advance, the trend towards creating specialized, minimal-genome chassis that are predictably programmed for specific expression tasks will undoubtedly accelerate, unlocking new possibilities in drug discovery and sustainable biomanufacturing.
The heterologous expression of biosynthetic gene clusters (BGCs) is a powerful strategy for discovering natural products and elucidating their biosynthetic pathways. This approach involves transferring genetic material from its native host into a well-characterized, tractable host system, enabling researchers to bypass the challenges of cultivating original source organisms and to more readily manipulate genetic elements. However, a central, persistent challenge in this field is the efficient cloning and faithful reconstruction of large, contiguous DNA sequences that constitute functional BGCs, which can span tens to hundreds of kilobases. Conventional cloning techniques often prove inadequate for handling such large DNA fragments, leading to fragmented or incomplete cluster recovery, which in turn results in failed expression or the production of incomplete metabolites.
This technical whitepaper explores advanced DNA assembly tools—specifically Transformation-Associated Recombination (TAR) cloning and related methodologies—designed to overcome these hurdles. By leveraging the highly efficient homologous recombination machinery of the yeast Saccharomyces cerevisiae, these techniques enable the precise capture, assembly, and maintenance of large DNA constructs. Framed within the context of heterologous expression challenges, this guide details the mechanisms, protocols, and applications of these tools, providing researchers and drug development professionals with the technical knowledge to advance their work in natural product discovery and genetic engineering.
The evolution of DNA assembly tools has been driven by the need to handle increasingly large and complex genetic constructs. Transformation-Associated Recombination (TAR) cloning stands out for its ability to selectively isolate large chromosomal segments from complex genomes. TAR cloning exploits the innate high frequency of homologous recombination in Saccharomyces cerevisiae to capture genomic regions of interest as circular Yeast Artificial Chromosome (YAC) or Bacterial Artificial Chromosome (BAC) molecules [37]. This method allows for the isolation of full-length genes and gene clusters, complete with their native regulatory elements, which is crucial for achieving physiologically relevant expression in heterologous hosts [37].
A significant advancement in this field is the CAPTURE (Cas9-Assisted Targeting of Chromosome Segments) system. While TAR cloning relies on in vivo homologous recombination in yeast, the CATCH method utilizes the RNA-guided Cas9 nuclease for in vitro cleavage of target DNA from a native chromosome, followed by ligation into a vector via Gibson assembly [37]. This method provides an alternative for isolating BGCs from individual microbial strains, though it has not been widely applied to complex environmental samples containing thousands of bacterial species [37].
The table below summarizes the core features of TAR cloning and its related techniques.
Table 1: Key Advanced DNA Assembly Tools for Gene Cluster Isolation
| Technology | Core Principle | Typical Insert Size | Key Advantage | Primary Application |
|---|---|---|---|---|
| TAR Cloning | In vivo homologous recombination in yeast [37] | Up to 300 kb [38] | Isolates intact genes with native regulatory elements; high efficiency (up to 35% for human genes) [37] | Functional studies, heterologous expression from complex genomes [37] |
| CAPTURE System | Cas9 in vitro cleavage + Gibson assembly [37] | Varies | Targeted isolation without the need for in vivo recombination | BGC isolation from individual microbial strains [37] |
| Combinatorial Assembly | In vitro or in vivo assembly of individual genes [39] | Defined by design | Rapid characterization of minimal gene sets and discovery of analogues [39] | Functional screening and pathway engineering [39] |
The TAR Cloning Workflow: Mechanism and Protocol
The TAR cloning process is a robust method for capturing large genomic regions. Its success hinges on a carefully designed vector and the preparation of genomic DNA containing the target region.
Mechanism of TAR Cloning
A TAR vector contains a YAC cassette (yeast selectable marker and centromere) for propagation in yeast and a BAC cassette (bacterial origin of replication and selectable marker) for subsequent propagation in E. coli [37]. The vector is engineered with two short, unique sequences ("hooks") homologous to the 5' and 3' flanks of the target genomic region. These hooks can be as short as 60 bp, though longer sequences can also be used [37] [38]. Before co-transformation with yeast, the vector is linearized between the hooks to expose these recombinogenic ends.
Upon co-transformation into competent yeast cells, the highly efficient homologous recombination system mediates the interaction between the hooks on the linearized vector and their complementary sequences on the target genomic DNA fragment. This recombination event circularizes the target fragment into a YAC/BAC molecule that can be selectively propagated in yeast and later shuttled to bacterial cells for amplification and analysis [37].
Detailed Experimental Protocol for TAR Cloning
Stage 1: Vector Preparation and Genomic DNA Isolation
- TAR Vector Design: Construct a TAR vector containing a YAC/BAC cassette and two targeting hooks (60+ bp each) specific to the flanks of your gene cluster of interest. The hooks must be unique within the source genome and cloned in the same orientation as they appear in the genome [37].
- Vector Linearization: Linearize the purified TAR vector using a restriction enzyme that cuts between the two targeting hooks [37].
- Genomic DNA (gDNA) Preparation: Extract high-molecular-weight gDNA from the source organism. For higher efficiency, the gDNA can be pre-treated with CRISPR/Cas9. Design gRNAs to cut near the 5' and 3' ends of the target region. Perform an overnight digestion of gDNA with Cas9-gRNA complexes to generate fragments with defined ends that facilitate recombination [37].
Stage 2: Yeast Transformation and Clone Selection
- Yeast Strain and Transformation: Use a highly transformable yeast strain (e.g., VL6-48N [40] or similar). Generate yeast spheroplasts to enhance DNA uptake [38].
- Co-transformation: Co-transform the spheroplasts with the linearized TAR vector and the prepared (and optionally Cas9-treated) gDNA using a standard polyethylene glycol (PEG) and calcium chloride transformation protocol [38] [37].
- Selection and Growth: Plate the transformed yeast cells onto synthetic dropout medium lacking a specific nutrient (e.g., without histidine or tryptophan, depending on the auxotrophic marker on the TAR vector) to select for yeast cells that have taken up and recombined the vector. Incubate plates at 30°C for 3-5 days until colonies form [40] [38].
Stage 3: Analysis and Validation of Positive Clones
- Initial Screening: Screen yeast colonies for the presence of the target insert using colony PCR with primers internal to the gene cluster or by analyzing yeast chromosome preparations via pulsed-field gel electrophoresis [40] [37].
- Shuttle to E. coli: Isolate YAC DNA from positive yeast clones and electroporate it into an appropriate E. coli strain (e.g., EPI300) for amplification. Select transformed bacteria using the appropriate antibiotic (e.g., chloramphenicol) [40].
- Validation: Isolate BAC DNA from bacterial cultures and validate the assembled construct using restriction enzyme digestion and gel electrophoresis, followed by next-generation sequencing (NGS) to confirm the fidelity of the entire captured sequence [40].
Diagram 1: TAR cloning workflow
A Case Study: Combinatorial Assembly for Glidobactin Analysis
Combinatorial assembly represents a powerful strategy for determining the minimal set of genes required for the production of a natural product, a common challenge in heterologous expression where superfluous genes can reduce titres.
Experimental Protocol for Combinatorial Assembly
A recent study on the glidobactin biosynthetic gene cluster (BGC) showcases this approach. The goal was to identify the core genes needed for glidobactin A production in E. coli BAP1, bypassing conflicting data from previous gene knockout studies [39].
- DNA Manipulation:
- Combinatorial Assembly:
- Use an in vitro DNA assembly method (e.g., NEBuilder HiFi DNA Assembly Master Mix) to assemble different combinations of the PCR-amplified genes into appropriate linearized plasmid vectors (e.g., pET28a, pACYCDuet-1) [39].
- The assembly strategy should generate a collection of plasmids containing partial or complete versions of the glidobactin BGC.
- Heterologous Expression and Analysis:
- Transform the assembled plasmids via electroporation into the heterologous host, E. coli BAP1 [39].
- Select single colonies and cultivate them in liquid medium for compound production.
- Analyze culture extracts using high-throughput mass spectrometry (LC-MS) to detect glidobactin A and its analogues, thereby correlating gene combinations with metabolic output [39].
This combinatorial approach confirmed that glbB, glbC, glbE, glbF, and glbG constitute the core biosynthetic genes for glidobactin production in this host, while also generating strains that produced potentially valuable analogues [39]. This method enables rapid functional characterization without preconceived biases.
Diagram 2: Combinatorial assembly workflow
Essential Reagents and Research Solutions
Successful implementation of advanced DNA assembly techniques requires a specific toolkit of reagents and vectors. The table below catalogs key materials used in the protocols cited in this guide.
Table 2: Research Reagent Solutions for Advanced DNA Assembly
| Reagent / Material | Function / Application | Specific Examples / Notes |
|---|---|---|
| TAR Vectors | Shuttle vectors for capture and propagation in yeast and bacteria. Contain YAC/BAC cassettes and cloning sites for homology hooks. | pCAP01, pCAP03 (for actinobacteria) [38]; pCAPB02 (for Bacillus subtilis) [38]. |
| Yeast Strain | Host for in vivo homologous recombination. | Saccharomyces cerevisiae VL6-48N (highly transformable, auxotrophic for selection) [40]. |
| High-Fidelity Polymerase | Accurate amplification of large gene fragments and vector components. | Phusion Polymerase [39]; Q5 Hot Start High-Fidelity 2X Master Mix [40]. |
| DNA Assembly Master Mix | In vitro assembly of multiple DNA fragments with overlapping homology. | NEBuilder HiFi DNA Assembly Master Mix [39]. |
| gDNA Isolation Kit | Preparation of high-quality, high-molecular-weight genomic DNA from source organisms. | DNeasy Blood & Tissue Kit (Qiagen) [40] [39]. |
| Heterologous Hosts | Expression chassis for cloned BGCs. | E. coli BAP1 [39]; Streptomyces coelicolor [38]; Pseudomonas putida [38] [39]. |
Advanced DNA assembly tools like TAR cloning and combinatorial assembly are overcoming the fundamental challenge of faithfully reconstructing large biosynthetic gene clusters for heterologous expression. By providing robust methods for capturing intact genomic regions and systematically defining functional core genes, these technologies are accelerating the discovery and engineering of natural products. As synthetic biology continues to evolve, the integration of these cloning strategies with other cutting-edge techniques like CRISPR/Cas and human artificial chromosome (HAC) systems promises to further expand their impact, paving the way for novel therapeutics and a deeper understanding of genetic function [37]. For researchers in drug development, mastering these tools is becoming indispensable for tapping into the vast genetic potential encoded within microbial and complex genomes.
The escalating crisis of antimicrobial resistance and the continuous need for novel bioactive compounds have intensified the search for new natural products. Polyketides (PKs) and non-ribosomal peptides (NRPs) represent two of the most therapeutically significant families of microbial natural products, boasting diverse structures and potent activities including antibiotic, antifungal, anticancer, and immunosuppressant properties [41]. While genome sequencing has revealed a vast and largely untapped reservoir of biosynthetic gene clusters (BGCs) encoding these compounds, a significant majority remain silent or "cryptic" under standard laboratory conditions, and many are sourced from genetically intractable or slow-growing native producers [42] [43]. Heterologous expression—the process of cloning and expressing BGCs in a genetically tractable host strain—has emerged as a powerful strategy to overcome these limitations. This approach provides a shortcut to pathway activation, modification, and optimization, facilitating the discovery, structural elucidation, and yield improvement of novel PKs and NRPs [42]. This case study explores seminal successes and the detailed methodologies that enabled them, framing these achievements within the broader context of overcoming persistent challenges in heterologous expression research.
Case Studies in Polyketide Production
Strategic Engineering ofE. colifor Oviedomycin Production
The heterologous production of type II polyketides in E. coli has historically been challenging due to difficulties in functionally expressing the ketosynthase heterodimer (KSα-KSβ), which often forms insoluble inclusion bodies, and the suboptimal codon usage between high-GC actinomycetes and E. coli [44]. A landmark study successfully engineered an E. coli strain for the overproduction of oviedomycin, a type II angucyclinone with cytotoxic activity, achieving a remarkable titer of 120 mg/L [44]. This success was underpinned by a multi-pronged engineering strategy.
Experimental Protocol & Key Findings: The research employed a systematic approach to overcome expression bottlenecks:
- Soluble Expression of Minimal PKS: The researchers tackled the critical bottleneck of KSα-KSβ insolubility by employing several parallel strategies:
- Fusion Partners: Attaching solubility-enhancing fusion partners (MBP, TrxA, NusA) to the OvmPKS proteins.
- Molecular Chaperones: Co-expressing chaperone systems (GroEL-GroES, DnaK-DnaJ-GrpE) to assist with proper protein folding.
- Rare tRNAs: Supplying a plasmid encoding rare tRNAs to mitigate codon bias issues. The most effective combination was found to be the NusA fusion partner co-expressed with the GroEL-GroES chaperone system [44].
- Scaffold Protein Fusion: To enhance the efficiency of the multi-enzyme complex, a synthetic scaffold protein was used to co-localize the oviedomycin biosynthetic enzymes (OvmPKS, OvmTAC, OvmO1). This strategy relieved the accumulation of biosynthetic intermediates and increased the final oviedomycin titer by approximately 2.5-fold [44].
- Efflux Transporter Engineering: To reduce potential intracellular toxicity from the accumulating product, an efflux transporter (RND family) was overexpressed. This further increased production by about 1.8-fold, likely by mitigating feedback inhibition [44].
- Precursor Pathway Engineering: Key metabolic genes involved in the supply of malonyl-CoA and NADPH—critical precursors for polyketide biosynthesis—were overexpressed based on genome-scale metabolic modeling predictions. This enhanced the metabolic flux toward the desired product [44].
This case demonstrates that overcoming the hurdles of heterologous PK production often requires an integrated approach that addresses protein expression, pathway efficiency, product toxicity, and precursor supply simultaneously.
2Burkholderiaspp. as Promising Hosts for Polyketide Expression
The choice of a phylogenetically proximate host can significantly enhance the success of heterologous expression. Burkholderia species are increasingly recognized as excellent chassis for expressing BGCs from the Betaproteobacteria class due to their intrinsic capacity to produce complex natural products and their shared metabolic and regulatory backgrounds with other members of the Burkholderiales order [42].
Experimental Protocol & Key Findings: Research utilizing Burkholderia thailandensis E264 exemplifies host development. The standard protocol involves:
- Host Engineering: The native BGC for thailandepsin (a cytotoxic PK-NRP) is knocked out (Δtdp::attB) to eliminate background and free up metabolic resources. Additional efflux pump mutants (ΔBAC::attB, ΔoprC::attB) can be created to enhance compound tolerance and secretion [42].
- DNA Transfer: Conjugation from E. coli is the primary method for introducing large BGC constructs.
- Expression System: The heterologous BGC is typically integrated into the chromosome using ϕC31 integrative vectors. Expression can be driven by strong constitutive promoters (e.g., Pgenta) or native autologous promoters from B. thailandensis [42].
- Achievement: This optimized system yielded an impressive 985 mg/L of a key FK228 (romidepsin) precursor, demonstrating the high production capacity of engineered Burkholderia hosts [42].
The table below summarizes and compares key engineered hosts for polyketide production.
Table 1: Engineered Heterologous Hosts for Polyketide and NRP Production
| Host Organism | Key Genetic Modifications | Biosynthetic Range Tested | Best Reported Titer | Primary Advantages |
|---|---|---|---|---|
| E. coli BAP1 | Integrated sfp (PPTase), chaperone co-expression, scaffold proteins, engineered efflux [44]. | Type II PKS (Oviedomycin) | 120 mg/L (Oviedomycin) [44] | Rapid growth, extensive genetic tools, FDA-approved, scalable fermentation. |
| Burkholderia thailandensis E264 | PK-NRP BGC knockout (Δtdp::attB), efflux pump deletions [42]. | PKs, PK-NRPs (from Betaproteobacteria, Myxococcia) | 985 mg/L (FK228 precursor) [42] | Phylogenetic proximity to many PK/NRP producers; native precursor pool. |
| Streptomyces albus J1074 | Minimized genome, high transformation efficiency [43] [45]. | NRPs, Depsipeptides, Type II PKS | High success rate for cryptic BGC activation [43] [45] | Native ability to express actinomycete BGCs; robust secondary metabolism. |
| Aspergillus oryzae | GRAS status; engineered precursor supply [46]. | Terpenoids, Polyketides, NRPs | Efficient producer of pleuromutilin & cephalosporin P1 [46] | Eukaryotic PTMs; superior protein secretion; high tolerance for acid/organics. |
Case Studies in Non-Ribosomal Peptide Production
Discovery of Jejumide via TAR Cloning inStreptomyces albus
The discovery of jejumide, a novel anti-inflammatory depsipeptide from marine Streptomyces sp. SNJ102, showcases a successful workflow for activating cryptic NRPS pathways. This case study highlights the power of transformation-associated recombination (TAR) cloning to capture large, complex BGCs directly from genomic DNA [43].
Experimental Protocol & Key Findings:
- BGC Identification & Capture: Genome mining of the producer strain using antiSMASH identified a cryptic, noncanonical NRPS BGC. A bacterial artificial chromosome (BAC) library was constructed, and the target BGC was captured directly in yeast using TAR cloning. The linearized pCB_Apr vector and DraI-digested BAC DNA were co-transformed into Saccharomyces cerevisiae, where homologous recombination assembled the complete BGC into the vector [43].
- Heterologous Expression: The constructed plasmid, pCB_102DP, was transferred into the heterologous host Streptomyces albus J1074 via intergeneric conjugation. S. albus is a preferred host due to its minimized genome and high propensity for expressing silent BGCs [43].
- Metabolite Analysis & Structure Elucidation: After cultivation in R5- medium for 7 days, metabolites were extracted from both the broth and mycelia. High-resolution LC-MS analysis confirmed the production of a new compound, which was isolated and purified using size-exclusion and reverse-phase chromatography. The structure of jejumide was elucidated using NMR spectroscopy, revealing a depsipeptide more similar to fungal metabolites than typical Streptomyces-derived compounds [43].
This end-to-end pipeline—from genome mining and TAR cloning to heterologous expression and structure elucidation—provides a robust blueprint for accessing novel NRPs from cryptic gene clusters.
Elucidating Trisubstituted Pyrazinone Biosynthesis
Research into the production of ichizinones A–C, rare trisubstituted pyrazinones from Streptomyces sp. LV45-129, demonstrates the critical role of gene deletion experiments in confirming the function of a heterologously expressed BGC and proposing a biosynthetic model [45].
Experimental Protocol & Key Findings:
- Cosmid Library Construction & Expression: A genomic library of the producer strain was constructed in cosmids. Cosmids harboring the putative NRPS BGC were conjugated into the heterologous host Streptomyces albus Del14 [45].
- Gene Inactivation via Red-ET Recombineering: To confirm the role of specific genes within the BGC, targeted inactivation was performed. Hygromycin resistance cassettes were amplified with primers containing homology arms and inserted into the cosmid-borne BGC to replace key genes (e.g., a cyclase, thioesterase, or PKS gene) using Red-ET recombineering in E. coli. The mutated cosmids were then introduced into S. albus [45].
- Results: HPLC-MS analysis of the mutant strains revealed abolished or altered production profiles of the ichizinones, directly linking the identified BGC to the production of these compounds and allowing researchers to propose a detailed biosynthetic pathway for these rare NRP-derived molecules [45].
The Scientist's Toolkit: Essential Reagents and Methods
Successful heterologous production relies on a suite of specialized reagents and methods. The table below details key solutions referenced in the case studies.
Table 2: Key Research Reagent Solutions for Heterologous Expression
| Reagent / Method | Function | Application Example |
|---|---|---|
| ϕC31 Integrative Vectors | Enables stable, single-copy integration of BGCs into the host chromosome. | Used in Burkholderia thailandensis and Streptomyces spp. for stable expression [42]. |
| Transformation-Associated Recombination (TAR) | Captures large, intact BGCs directly from genomic DNA in yeast. | Cloning the jejumide BGC from marine Streptomyces [43]. |
| Red-ET Recombineering | Permits precise gene knockouts or modifications directly on BACs or cosmids in E. coli. | Inactivating specific genes in the ichizinone BGC to confirm their function [45]. |
| MbtH-like Proteins (MLPs) | Small chaperone proteins essential for the proper folding and activity of NRPS adenylation (A) domains. | Co-purification with MLPs enabled the isolation of entire NRPS assembly lines for study [47]. |
| Phosphopantetheinyl Transferase (PPTase) | Activates carrier proteins (ACP in PKS, PCP in NRPS) by attaching the essential 4'-phosphopantetheine cofactor. | Essential for activity in all heterologous hosts; often encoded on a helper plasmid or engineered into the host genome (e.g., in E. coli BAP1) [44] [48]. |
Visualization of Workflows and Strategies
The following diagrams illustrate the core experimental workflows and engineering strategies discussed in this review.
General Workflow for Cryptic BGC Activation
This diagram outlines the standard pipeline for discovering novel natural products through heterologous expression.
Integrated Engineering Strategy for Oviedomycin in E. coli
This diagram details the multi-layered metabolic engineering strategy used to overcome production barriers in a heterologous host.
The case studies presented herein demonstrate that the heterologous production of polyketides and non-ribosomal peptides, while challenging, is a tractable and increasingly mature approach to natural product discovery and development. Key lessons for overcoming central challenges include: the critical importance of host selection (ranging from engineered E. coli to specialized hosts like Burkholderia and Streptomyces); the necessity of advanced genetic tools (TAR, recombineering) for BGC capture and manipulation; and the power of integrated metabolic engineering to address bottlenecks in protein expression, pathway flux, and product toxicity.
Future progress will be driven by several emerging trends. The development of cell-free protein synthesis (CFPS) systems offers a rapid platform for prototyping NRPS and PKS expression, bypassing cell viability constraints and accelerating design-build-test-learn cycles [48]. Furthermore, the refinement of CRISPR-based tools for genetic manipulation in non-model hosts like Aspergillus and Burkholderia will streamline host engineering efforts [42] [46]. Finally, the continued exploration and genomic minimization of non-traditional hosts will expand the phylogenetic range of BGCs that can be successfully expressed. By systematically applying and integrating these strategies, researchers can continue to unlock the vast potential of microbial genomes, paving the way for the next generation of therapeutic agents.
Optimizing for Success: Refactoring, Regulation, and Metabolic Engineering Solutions
Microbial natural products (NPs) are of paramount importance in human medicine, animal health, and plant crop protection due to their rich chemical diversity and bioactivity [25]. Large-scale genomic and metagenomic mining has revealed tremendous biosynthetic potential within microbial genomes, with a typical actinomycete containing an order of magnitude more biosynthetic gene clusters (BGCs) than are expressed under standard laboratory conditions [25] [26]. However, a significant challenge persists: approximately 90% of native BGCs are not expressed or are only partially transcribed under conventional fermentation conditions [25]. This "silent" majority represents an untapped reservoir of novel chemical entities with potential pharmaceutical applications.
The field faces three primary interconnected challenges in heterologous BGC expression. First, native regulatory networks are often poorly understood or incompatible with laboratory hosts, preventing cluster activation. Second, technical limitations in cloning, manipulating, and expressing large BGCs (particularly those encoding complex polyketide synthases and non-ribosomal peptide synthetases) hinder functional characterization. Third, host-pathway incompatibility can result in metabolic burden, toxicity, or missing essential cofactors [25] [26]. Pathway refactoring—the process of reconstructing genetic elements to optimize function and predictability—has emerged as a powerful synthetic biology approach to overcome these barriers by replacing native promoters and regulatory elements with well-characterized orthogonal parts [25].
Core Concepts: Principles and Components of Pathway Refactoring
Pathway refactoring involves the systematic redesign of BGCs using synthetic biology principles to enable reliable heterologous expression. This process typically includes codon optimization to match host preferences, elimination of native regulation, and standardization of genetic parts to create modular, predictable systems [26]. The primary goal is to disrupt complex native transcriptional networks that often maintain BGCs in a silent state and replace them with orthogonal regulatory elements that function reliably in the chosen heterologous host [25].
Table 1: Key Components in BGC Refactoring Strategies
| Component | Function | Examples | Considerations |
|---|---|---|---|
| Promoters | Initiate transcription | Constitutive (ermEp, kasOp), Inducible (tetracycline, thiostrepton) | Strength, regulatability, orthogonality [25] [26] |
| Ribosome Binding Sites (RBS) | Control translation initiation | Engineered sequences, degenerate libraries | Strength must be matched to gene product requirements [49] |
| Transcriptional Terminators | Prevent read-through | Well-characterized termination sequences | Transcriptional fidelity between adjacent genes [26] |
| Protein Degradation Tags | Modulate protein half-life | Various peptide tags | Fine-tuning metabolic flux [25] |
Central to this approach is promoter engineering, which involves replacing native promoters with constitutive or readily inducible promoters that function predictably in the heterologous host [25]. This strategy severs the BGC from its native regulatory context, potentially activating silent clusters and enabling high-level expression. Additional optimization of ribosomal binding sites (RBSs), terminators, and protein degradation tags further enhances pathway performance and predictability [25].
Next-Generation Transcriptional Regulatory Modules
Recent advances in synthetic biology have yielded sophisticated transcriptional regulatory systems that overcome limitations of earlier toolkits. Three innovative approaches exemplify this progress:
Orthogonal Regulatory Elements with Randomized Sequences
Ji et al. developed a novel design for synthetic promoter libraries in Streptomyces albus J1074 that randomizes sequences in both the promoter and RBS regions while partially fixing only the -10/-35 regions and Shine-Dalgarno sequence [25]. Using the blue pigment indigoidine as a reporter, they constructed a large pool of regulatory sequences with varying transcriptional activities. These elements demonstrated high orthogonality, enabling successful refactoring of the silent actinorhodin BGC from Streptomyces coelicolor by replacing seven native promoters with four strong regulatory cassettes, resulting in successful heterologous expression in S. albus J1074 [25].
Metagenomically-Mined Universal Promoters
To address the limited phylogenetic range of most promoter libraries, Johns et al. mined 184 microbial genomes to identify natural 5' regulatory sequences spanning Actinobacteria, Archaea, Bacteroidetes, Cyanobacteria, Firmicutes, Proteobacteria, and Spirochetes [25]. By systematically quantifying transcriptional and translational activities across different bacterial species and growth conditions, they identified a common subset of regulatory elements with varying sequence composition and orthogonal host ranges. This collection represents a rich resource for refactoring BGCs from underexplored bacterial taxa [25].
Copy Number-Insensitive Promoters
Segall-Shapiro et al. addressed the challenge of expression level variability using transcription-activator-like effectors (TALEs)-based incoherent feedforward loops (iFFLs) to create promoters with constant expression levels regardless of copy number in E. coli [25]. These stabilized promoters enabled consistent product titers when BGCs were transferred between plasmid and genomic locations and created metabolic pathways resistant to genomic mutations, growth conditions, and other stressors [25].
Novel BGC Refactoring Strategies and Methodologies
Several innovative methodologies have recently been developed to streamline and enhance BGC refactoring:
Plug-and-Play Pathway Refactoring Workflow
A highly modular, high-throughput refactoring system employs a two-tier Golden Gate assembly approach using BbsI and BsaI restriction enzymes [50]. This system utilizes helper plasmids containing promoters and terminators between which biosynthetic genes can be inserted, along with spacer plasmids containing random 20bp sequences to maintain proper assembly structure when fewer genes are used [50].
Figure 1: Two-Tier Golden Gate Assembly Workflow for Pathway Refactoring
The workflow achieves exceptional fidelity, with the first-tier reaction demonstrating 100% efficiency in cloning, and the final assembly showing 95-100% correct assembly rates [50]. This system facilitates rapid pathway construction, gene deletion studies, and combinatorial biosynthesis, enabling researchers to build 96 functional pathways for combinatorial carotenoid biosynthesis in just two days using polyclonal plasmids [50].
CRISPR-Enabled Multiplex Promoter Replacement
Advanced CRISPR-based methods have been developed for multiplexed promoter engineering, including:
- mCRISTAR (multiplexed CRISPR-based Transformation-Associated Recombination): Enables simultaneous replacement of multiple native promoters with synthetic counterparts [25]
- miCRISTAR (multiplexed in vitro CRISPR-based TAR): An in vitro version for enhanced precision and efficiency [25]
- mpCRISTAR (multiple plasmid-based CRISPR-based TAR): Facilitates complex multi-plasmid assemblies [25]
These techniques were successfully applied to activate a silent BGC, leading to the discovery of two antitumor sesterterpenes, atolypene A and B [25].
Algorithm-Guided Library Optimization
The RedLibs algorithm addresses the challenge of combinatorial explosion in library design by identifying optimal degenerate RBS sequences that uniformly sample the entire accessible translation level space while maintaining manageable library sizes [49]. This approach calculates translation initiation rate (TIR) distributions for all possible degenerate RBSs and selects those that most closely match a target distribution, enabling the creation of "smart" libraries that maximize coverage with minimal experimental effort [49].
Table 2: Comparison of BGC Refactoring Techniques
| Method | Key Features | Efficiency/Throughput | Applications |
|---|---|---|---|
| Plug-and-Play Golden Gate | Modular, uses helper/spacer plasmids, two-tier assembly | 95-100% assembly fidelity; 96 pathways in 2 days | Combinatorial biosynthesis, gene deletion studies [50] |
| CRISTAR Platforms | CRISPR-based, multiplexed promoter replacement | Simultaneous replacement of up to 8 promoters | Activation of silent BGCs, natural product discovery [25] |
| RedLibs Algorithm | Computationally optimized RBS libraries, uniform TIR coverage | Drastic reduction in library size while maintaining diversity | Metabolic flux optimization, pathway balancing [49] |
| Classical Cosmid/BAC Libraries | Large insert capacity, established methodology | Lower throughput, labor-intensive | Initial BGC capture from diverse sources [26] |
Heterologous Host Systems and Engineering Strategies
Streptomyces as Versatile Chassis
Streptomyces species have emerged as the most widely used heterologous hosts for bacterial and fungal natural product BGCs, with over 450 peer-reviewed studies published between 2004-2024 [26]. Quantitative analysis reveals a clear upward trajectory in successful heterologous expression events, reflecting technological advances and growing expertise in host engineering.
Table 3: Key Heterologous Host Platforms for BGC Expression
| Host Organism | Advantages | Limitations | Suitable BGC Types |
|---|---|---|---|
| Streptomyces spp. | High GC compatibility, native precursor supply, experienced with complex metabolites | Slower growth, more complex genetics | Actinobacterial PKS, NRPS, hybrid clusters [26] |
| Escherichia coli | Fast growth, extensive genetic tools, high-throughput capabilities | Limited precursor supply, may lack essential cofactors | Type II PKS, simple NRPS, terpenes [25] |
| Saccharomyces cerevisiae | Eukaryotic PTMs, compartmentalization, strong genetic tools | Different codon usage, may lack prokaryotic cofactors | Fungal PKS, NRPS, eukaryotic metabolites [50] |
| Myxococcus xanthus | Tolerant of cytotoxic compounds, proficient secretor | Specialized growth requirements, limited tools | Myxobacterial metabolites, cytotoxic compounds [25] |
The preference for Streptomyces hosts stems from several intrinsic advantages: (1) genomic compatibility with high-GC BGCs from actinomycetes, reducing the need for extensive codon optimization; (2) proven metabolic capacity for complex polyketides and non-ribosomal peptides; (3) advanced regulatory systems that can be co-opted for heterologous expression; (4) tolerant physiology that withstands cytotoxic compounds; and (5) established scalability for industrial fermentation [26].
Host Engineering for Enhanced BGC Expression
Strategic engineering of heterologous hosts has significantly expanded the range of expressible BGCs. Key approaches include:
- Precursor pathway engineering to enhance supply of essential building blocks
- Cofactor balancing to meet demands of heterologous enzymes
- Protease deletion to improve heterologous protein stability
- Efflux pump expression to enhance toxin resistance
- Global regulator manipulation to activate silent capacity [26]
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 4: Key Research Reagent Solutions for Pathway Refactoring
| Reagent/Resource | Function | Application Examples | Key Characteristics |
|---|---|---|---|
| Golden Gate Assembly System | Modular DNA assembly | Zeaxanthin pathway refactoring [50] | BbsI/BsaI enzymes, high fidelity (95-100%) |
| Orthogonal Promoter Libraries | Transcriptional control | Actinorhodin BGC activation [25] | Randomized sequences, varying strengths |
| RBS Calculator & Prediction Tools | Translation rate prediction | RedLibs algorithm implementation [49] | Biophysical modeling, TIR prediction |
| TAR/CATCH Systems | Direct BGC capture from genomes | Large PKS/NRPS cluster capture [26] | Homologous recombination-based |
| CRISPR-Cas9 Tools | Genome editing, promoter replacement | mCRISTAR/miCRISTAR platforms [25] | Multiplex editing capabilities |
| Specialized Host Strains | Heterologous expression | Streptomyces albus J1074, M. xanthus DK1622 [25] [26] | Optimized for BGC expression |
Pathway refactoring through promoter and regulatory element replacement has transformed the landscape of natural product discovery and optimization. The integration of sophisticated synthetic biology tools—including CRISPR-enabled genome editing, modular DNA assembly systems, and computational library design—has addressed fundamental challenges in heterologous BGC expression. These advances enable researchers to bypass native regulatory constraints, optimize pathway flux, and unlock the vast chemical diversity encoded in silent biosynthetic gene clusters.
The field continues to evolve toward more predictable, high-throughput platforms that minimize empirical optimization while maximizing success rates. Future developments will likely focus on expanding the repertoire of orthogonal regulatory parts, enhancing host chassis capabilities through systems-level engineering, and integrating machine learning approaches to predict optimal refactoring strategies. As these technologies mature, pathway refactoring will play an increasingly central role in accessing microbial chemical diversity for pharmaceutical applications, ultimately accelerating the discovery of novel therapeutic agents to address pressing medical needs.
Overcoming Cellular Toxicity and Inclusion Body Formation in Bacterial Hosts
The heterologous expression of gene clusters in bacterial systems is a cornerstone of modern biotechnology, enabling the production of therapeutic proteins, enzymes, and complex natural products. However, this process is frequently hampered by two major cellular phenomena: cellular toxicity and inclusion body formation [20]. When a recombinant protein disrupts the host's normal physiological processes, it can inhibit growth and lead to cell death, significantly reducing yields [16]. Simultaneously, the aggregation of overexpressed proteins into dense, amorphous cytoplasmic structures known as inclusion bodies (IBs) represents a significant bottleneck, often necessitating complex and inefficient refolding procedures to recover active protein [51]. This technical guide examines the underlying mechanisms of these challenges and details advanced strategies to overcome them, providing a framework for improving the success of heterologous expression projects.
Understanding Protein Toxicity: Mechanisms and Mitigation
Mechanisms of Cellular Toxicity
Protein toxicity in bacterial hosts, primarily Escherichia coli, arises when the heterologous protein interferes with essential host cell functions. Common toxic proteins include ribonucleases that degrade bacterial mRNA, proteases that cleave essential host proteins, membrane proteins that disrupt ion gradients or membrane integrity, and enzymes that deplete critical metabolites or cofactors [16]. Even proteins without overtly destructive functions can be toxic simply by overburdening the host's transcription and translation machinery or sequestering essential chaperones [20].
Strategic Solutions for Toxic Proteins
Dual Transcriptional-Translational Control
A primary strategy for expressing toxic proteins involves using inducible systems with tight regulatory control to prevent basal expression during early growth phases. While standard inducible systems (e.g., T7/lac-based systems) offer transcriptional control, they often suffer from leakage expression. For highly toxic proteins, a more effective approach employs dual transcriptional-translational control [20]. This can be achieved through:
- Incorporation of unnatural amino acids: Rendering translation dependent on the presence of a non-standard amino acid not found in natural proteins.
- Riboswitches and ribozymes: RNA elements that regulate translation initiation or mRNA stability in response to specific ligands.
- Antisense RNA: Sequences that bind complementary mRNA targets to block translation.
Table 1: Comparison of Dual Control Systems for Toxic Protein Expression
| Control Mechanism | Principle of Action | Inducing Agent | Advantages |
|---|---|---|---|
| Unnatural Amino Acid Incorporation | Translation requires synthetic amino acid | Unnatural amino acid (e.g., Azidophenylalanine) | Extremely low background; residue-specific control |
| Riboswitches | Conformational change in mRNA leader sequence regulates translation | Metabolites (e.g., theophylline) | Small molecule inducers; modular design |
| Antisense RNA | Complementary RNA binds target mRNA to block ribosome access | IPTG or temperature shift (to inactivate antisense RNA) | High specificity; tunable repression |
Alternative Solutions
- Fusion Tags: Fusion partners such as Maltose-Binding Protein (MBP) or Thioredoxin (Trx) can enhance solubility and reduce toxicity by improving folding and shielding disruptive surfaces [20].
- Specialized Bacterial Strains: Strains like C41(DE3) and C43(DE3) are derived from BL21(DE3) and selected for their ability to tolerate toxic membrane proteins [16].
- Cell-Free Expression Systems: Bypass cellular metabolism altogether by using E. coli extracts for in vitro protein synthesis, allowing direct control of the reaction environment [20].
Inclusion Body Formation: Challenge and Opportunity
The Nature and Structure of Inclusion Bodies
Inclusion bodies are submicron proteinaceous particles (typically 50-800 nm) observed in recombinant bacteria during high-level expression [52]. Traditionally viewed as amorphous aggregates of misfolded protein, IBs are now recognized as structurally complex. They exhibit amyloid-like properties, including binding to dyes like Congo red and thioflavin T, and contain a significant amount of cross-β sheet structure as revealed by X-ray diffraction [51]. The modern paradigm views IBs as a porous, hydrated matrix where correctly folded and functional proteins can coexist with misfolded and amyloid-like fibrillar species that form a structural scaffold [52].
Diagram 1: Architecture of a bacterial inclusion body showing multi-component nature.
Strategies for Managing Inclusion Bodies
Promoting Soluble Expression
- Lowered Growth Temperatures: Shifting cultures to 20-25°C post-induction slows protein synthesis, allowing more time for proper folding and reducing aggregation.
- Cytoplasmic Disulfide Bond Formation (CyDisCo): For proteins requiring disulfide bonds, the CyDisCo system co-expresses enzymes for disulfide formation and isomerization in the E. coli cytoplasm, enabling production of complex disulfide-bonded proteins [20].
- Fusion Tags and Molecular Chaperones: Co-expression of chaperones (GroEL/GroES, DnaK/DnaJ) or fusion with solubility-enhancing tags can guide proper folding.
Working with Inclusion Bodies: Refolding and Active IBs
When soluble expression proves intractable, IBs can be exploited as a source of active protein or used directly.
- Traditional Refolding: IBs are isolated, solubilized with denaturants (e.g., urea, guanidine hydrochloride), and the target protein is refolded by removing the denaturant through dilution or dialysis. This process is often low-yielding and requires extensive optimization [20].
- Non-Denaturing Solubilization: The discovery of native-like structure and activity within IBs has led to protocols using mild detergents or low concentrations of denaturants to extract functional proteins without complete unfolding [20] [52].
- Active IBs as Functional Materials: For some applications, IBs can be used directly as robust, immobilized biocatalysts or as slow-release protein depots in tissue engineering, leveraging their inherent mechanical stability and biological activity [52].
Table 2: Comparison of Inclusion Body Utilization Strategies
| Strategy | Methodology | Key Advantage | Typical Yield/Activity |
|---|---|---|---|
| Traditional Refolding | Denaturation → Renaturation | Can produce pure, soluble protein | Highly variable; often low |
| Non-Denaturing Solubilization | Mild detergent (e.g., Sarkosyl) extraction | Preserves native structure; simpler | Moderate to high specific activity |
| Active IB Application | Use of washed IBs directly | No solubilization needed; highly stable | Lower than soluble enzyme, but reusable |
Integrated Experimental Workflow
A systematic approach is required to troubleshoot and overcome toxicity and aggregation. The following workflow outlines a recommended pathway.
Diagram 2: Decision workflow for addressing toxicity and inclusion body formation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Overcoming Expression Challenges
| Reagent / Tool | Function / Principle | Application Context |
|---|---|---|
| pET Series Vectors | High-copy number plasmids with strong, inducible T7 promoter | Standard workhorse for high-level protein expression in BL21(DE3) |
| BL21(DE3) E. coli Strain | B-strain lacking Lon and OmpT proteases, carries T7 RNA polymerase gene | Standard host for pET system; reduces protein degradation |
| C41(DE3) & C43(DE3) Strains | Evolved mutants of BL21(DE3) with enhanced membrane integrity | Expression of toxic proteins, especially membrane proteins |
| CyDisCo System | Co-expression of sulfhydryl oxidase and disulfide isomerase | Production of proteins requiring disulfide bonds in the cytoplasm |
| Fusion Tags (MBP, Trx) | Enhances solubility and correct folding of fused passenger protein | Reducing aggregation and toxicity of difficult-to-express proteins |
| Molecular Chaperone Plasmids | Vectors for co-expressing GroEL/GroES or DnaK/DnaJ/GrpE | Assisting de novo folding of recombinant proteins to prevent IB formation |
| Site-Specific Unnatural Amino Acids | Incorporates synthetic amino acid via engineered tRNA/synthetase | Provides tight, translational-level control for toxic protein expression |
Cellular toxicity and inclusion body formation represent significant, yet not insurmountable, challenges in the heterologous expression of gene clusters. A deep understanding of their underlying mechanisms—where toxicity stems from interference with host physiology and IBs are structured aggregates with functional potential—enables the deployment of sophisticated strategies. Success hinges on selecting the appropriate combination of tightly regulated expression systems, specialized bacterial strains, and solubility-enhancing tools, while also considering the potential utility of inclusion bodies themselves as functional materials. As the field advances, the integration of high-throughput screening and synthetic biology approaches, such as designing genome-reduced hosts, promises to further expand the boundaries of what is possible to produce in bacterial systems, driving innovation in both basic research and industrial bioproduction.
The heterologous expression of biosynthetic gene clusters (BGCs) is a cornerstone strategy in modern natural product research and drug development, enabling the discovery and production of valuable compounds from genetically intractable or slow-growing organisms [53] [54]. This approach involves transferring genetic material responsible for the biosynthesis of a target compound into a well-characterized and genetically tractable host organism, such as Escherichia coli or Streptomyces species [13]. Despite its transformative potential, this field faces significant technical hurdles that limit its efficiency and broader application.
A primary challenge lies in the low production titers of target compounds, often resulting from inadequate precursor and energy supply in the heterologous host [53]. Native producers have evolved sophisticated regulatory mechanisms to balance primary and secondary metabolism, whereas heterologous hosts frequently lack such specialized infrastructure. Furthermore, the incorrect folding and assembly of complex enzymes, particularly those requiring specific metallo-cofactors or post-translational modifications, often leads to non-functional pathways in the new cellular environment [3]. The genetic instability of large BGCs and inefficient horizontal gene transfer between distantly related species further complicate expression efforts [13]. These challenges collectively underscore the critical need for sophisticated metabolic engineering strategies to optimize heterologous production systems.
This technical guide addresses two fundamental metabolic engineering approaches—precursor supplementation and co-factor balancing—to overcome these bottlenecks. By systematically designing host metabolism to support heterologous pathways, researchers can significantly enhance the production of valuable natural products for therapeutic applications.
Precursor Supplementation Strategies
Rationale and Core Principles
Precursor supplementation focuses on ensuring that the heterologous host produces adequate amounts of the essential building blocks required for biosynthetic pathways. When a BGC is introduced into a new host, the demand for specific precursors may exceed the host's native supply capacity, creating a bottleneck that limits final product yield. The core objective is to reinforce native metabolic pathways or introduce artificial pathways to generate sufficient precursor molecules.
This strategy typically follows a "push-pull-block" paradigm: "push" strategies enhance the flux through precursor-supplying pathways, "pull" strategies increase the consumption of precursors toward the desired product, and "block" strategies minimize diversion of precursors into competing metabolic pathways [55]. For instance, in cyanobacteria, which are prolific producers of bioactive natural products, the structural complexity of compounds like dolastatin 10 and curacin A places significant demand on malonyl-CoA and other polyketide precursors [53]. In such cases, merely introducing the BGC is insufficient without concurrently engineering the host's central carbon metabolism to meet these new metabolic demands.
Key Methodologies and Experimental Protocols
Central Carbon Pathway Optimization
Protocol: Enhancing Malonyl-CoA Supply for Polyketide Production
- Genetic Modifications: Overexpress the accABCD operon encoding acetyl-CoA carboxylase, which catalyzes the conversion of acetyl-CoA to malonyl-CoA. Use a strong, inducible promoter (e.g., Ptrc or PBAD) to control expression and avoid fitness burdens [53].
- Competitive Pathway Knockout: Delete genes encoding enzymes that compete for acetyl-CoA, such as phosphate acetyltransferase (pta) and lactate dehydrogenase (ldhA), to redirect flux toward malonyl-CoA synthesis.
- Precursor Supplementation: In cultivation media, supplement with precursors like aspartate or oleic acid that can enhance intracellular acetyl-CoA pools through β-oxidation or anaplerotic reactions.
- Analytical Validation: Quantify intracellular malonyl-CoA levels using LC-MS/MS and correlate with product titers to confirm flux enhancement.
MEP Pathway Enhancement for Isoprenoid Production
Protocol: Amplifying Isopentenyl Pyrophosphate (IPP) and Dimethylallyl Pyrophosphate (DMAPP) Supply
- Rate-Limiting Enzyme Overexpression: Overexpress 1-deoxy-D-xylulose-5-phosphate synthase (DXS), the first enzyme in the methylerythritol phosphate (MEP) pathway. In Phaeodactylum tricornutum, DXS overexpression resulted in a 2.4-fold increase in fucoxanthin production [55].
- Feedback Inhibition Relief: Engineer feedback-resistant versions of DXS and other key enzymes to avoid pathway regulation by downstream isoprenoids.
- Cultivation Strategy: Supplement cultures with glycerol and glycine, which enter the MEP pathway upstream, to boost carbon flux without genetic modifications.
Table 1: Quantitative Impact of Precursor Engineering Strategies
| Target Compound | Host Organism | Precursor Engineering Strategy | Yield Improvement | Reference |
|---|---|---|---|---|
| Fucoxanthin | Phaeodactylum tricornutum | DXS overexpression | 2.4-fold increase | [55] |
| Polyhydroxybutyrate (PHB) | Cupriavidus necator | Optimization of central carbon flux to acetyl-CoA | >80% cell dry weight | [56] |
| Riboflavin | Escherichia coli | Reinforced PPP flux & deregulated purine biosynthesis | Increased GTP availability | [57] |
| 2,4-Dihydroxybutyric Acid | Escherichia coli | Aspartate/malate-insensitive PEP carboxylase expression | Enhanced oxaloacetate supply | [58] |
The following diagram illustrates the strategic integration of precursor supplementation within a metabolic network to support heterologous production, highlighting key "push," "pull," and "block" interventions.
Diagram 1: Metabolic engineering strategies for precursor supplementation. "Push" strategies enhance precursor supply, "Pull" strategies increase BGC consumption, and "Block" strategies knock out competing pathways.
Co-factor Balancing Strategies
Rationale and Core Principles
Cofactor balancing addresses the critical need for appropriate ratios and availability of energy carriers and reducing equivalents, primarily NADPH/NADP+ and NADH/NAD+, which are essential for powering biosynthetic enzymes. Heterologous pathways often impose unnatural redox demands on the host, creating inefficiencies and limiting yields. Under aerobic conditions, the [NADPH]/[NADP+] ratio in E. coli is approximately 60, while the [NADH]/[NAD+] ratio is only 0.03, making NADPH-dependent reactions thermodynamically more favorable for reductive biosynthesis [58].
A common challenge in heterologous expression is the mismatch between the cofactor specificity of the introduced enzymes and the host's native cofactor balance. For instance, an enzyme requiring NADPH might be introduced into a host compartment where NADH is more abundant, leading to kinetic limitations. Successful cofactor engineering involves either modifying the host's cofactor metabolism to match the pathway's needs or engineering the enzymes themselves to alter their cofactor preference, thereby creating a more harmonious system.
Key Methodologies and Experimental Protocols
Enhancing NADPH Supply
Protocol: Engineering Pentose Phosphate Pathway (PPP) Flux
- Genetic Modifications: Overexpress the zwf gene encoding glucose-6-phosphate dehydrogenase, the first and rate-limiting enzyme of the PPP. This direct intervention increases NADPH generation.
- Cofactor System Swapping: Replace native NADH-dependent enzymes in central metabolism with NADPH-dependent alternatives. For example, introduce a NADP+-dependent glyceraldehyde-3-phosphate dehydrogenase (GapN) from Streptococcus mutans.
- Transhydrogenase Overexpression: Overexpress the membrane-bound transhydrogenase complex (pntAB), which reversibly converts NADH and NADP+ to NAD+ and NADPH, allowing interconversion of reducing equivalents [58].
Enzyme Engineering for Cofactor Specificity
Protocol: Switching Cofactor Preference from NADH to NADPH
- Identify Cofactor-Binding Pocket: Use crystal structure data or homology modeling to identify the amino acid residues responsible for coordinating the adenosine phosphate moiety of the cofactor. This region typically contains conserved Rossmann fold motifs.
- Rational Design/Site-Directed Mutagenesis: Introduce mutations that alter the binding pocket's electrostatic potential and size to favor NADPH. For example, in the engineering of a NADPH-dependent 2-oxo-4-hydroxybutyrate (OHB) reductase, the introduction of two point mutations (D34G and I35R) increased specificity for NADPH by more than three orders of magnitude [58].
- Screening and Validation: Express mutant libraries in a cofactor-auxotrophic strain or use high-throughput colorimetric assays to identify variants with the desired cofactor specificity.
Table 2: Cofactor Engineering Approaches and Outcomes
| Engineering Approach | Specific Method | Host Organism | Impact on Production |
|---|---|---|---|
| NADPH Supply Enhancement | pntAB overexpression | Escherichia coli | 50% increased yield of 2,4-dihydroxybutyric acid [58] |
| Enzyme Cofactor Specificity | D34G, I35R mutations in OHB reductase | Escherichia coli | Switched cofactor preference from NADH to NADPH [58] |
| Cofactor System Swapping | Introduction of NADP+-dependent GapN | Escherichia coli | Increased NADPH availability for product synthesis |
| PPP Flux Enhancement | zwf gene overexpression | Escherichia coli | Reinforced riboflavin biosynthesis [57] |
The workflow for implementing a cofactor balancing strategy, from identifying the need to validating the final strain, is outlined below.
Diagram 2: A workflow for cofactor balancing, showing parallel strategies of engineering host metabolism or pathway enzymes.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Metabolic Engineering
| Reagent/Material | Function | Example Application |
|---|---|---|
| ExoCET (Exonuclease combined with RecET) | Cloning and assembly of large BGCs (>10 kb) from genomic DNA [3] [13]. | Assembled an 11 kb nitrogen-fixing (nif) gene cluster from Paenibacillus polymyxa for heterologous expression [3]. |
| Redα/Redβ/Redγ Recombineering System | λ phage-derived recombinases enabling precise DNA editing in E. coli using short (50 bp) homology arms [13]. | Facilitated markerless integration of RMCE cassettes into BGC-containing plasmids within the Micro-HEP platform [13]. |
| RMCE (Recombinase-Mediated Cassette Exchange) Systems | Orthogonal tyrosine recombinase systems (Cre-lox, Vika-vox, Dre-rox) for precise, multi-copy BGC integration into defined chromosomal loci [13]. | Enabled integration of 2-4 copies of the xiamenmycin BGC, increasing product yield proportionally with copy number [13]. |
| Constitutive Promoters (e.g., Pveg, P43) | Drive consistent gene expression in heterologous hosts; critical for optimizing transcription of refactored BGCs [3]. | Replacing the native promoter of a nitrogen-fixing gene cluster with Pveg enabled functional expression in Bacillus subtilis [3]. |
| Membrane-Bound Transhydrogenase (pntAB) | Enzyme complex that converts NADH + NADP+ to NAD+ + NADPH, balancing intracellular redox cofactors [58]. | Overexpression in E. coli improved NADPH supply and increased the yield of 2,4-dihydroxybutyric acid by 50% [58]. |
Integrated Case Studies in Natural Product Discovery
Cyanobacterial Natural Products and Drug Discovery
Cyanobacteria produce structurally complex natural products with potent biological activities, such as dolastatin 10, curacin A, and apratoxin A, which are promising leads for anticancer therapies [53]. However, their heterologous production is notoriously challenging due to low titers (typically 0.1–0.2% dry weight) and slow growth of native cyanobacterial hosts. Metabolic engineering in more tractable hosts like E. coli or Streptomyces is essential.
Integrated Engineering Approach:
- Precursor Strategy: The biosynthesis of dolastatin 10 involves hybrid polyketide-nonribosomal peptide assembly, requiring malonyl-CoA-extended amino acids. Engineering central carbon metabolism to supply both methylmalonyl-CoA and the unique amino acid precursors is critical.
- Cofactor Strategy: The integrated PKS/NRPS machinery consumes substantial NADPH for reductive steps. Balancing the NADPH pool via PPP enhancement or pntAB overexpression is necessary to support high-level production.
Automated High-Throughput Strain Engineering
The FAST-NPS (Functional Analysis of Synthetic Transcripts for Natural Products Screening) platform represents a cutting-edge integration of these metabolic engineering principles within an automated workflow [59]. This system addresses the dual challenges of identifying bioactive compounds and scaling up their production.
Key Workflow Steps:
- Bioinformatic Prioritization: Uses self-resistance genes within BGCs as markers to predict bioactivity, ensuring focus on the most promising clusters.
- Automated Cloning: The CAPTURE method is automated on the iBioFAB platform to clone hundreds of BGCs in parallel with 95% success rate, eliminating manual bottlenecks.
- Heterologous Expression: Cloned BGCs are transferred into optimized Streptomyces chassis strains for expression.
- Metabolic Engineering: Successful expression strains can subsequently be engineered using the precursor and cofactor strategies outlined in this guide to enhance titers to commercially viable levels.
Precursor supplementation and cofactor balancing are not standalone solutions but are deeply interconnected strategies that form the foundation of efficient heterologous production systems. The success of expressing complex BGCs from cyanobacteria and other organisms in engineered chassis like E. coli and Streptomyces hinges on the simultaneous optimization of both building block supply and the cellular energy currency [53] [13]. As the field advances, the integration of these rational metabolic engineering strategies with automated high-throughput platforms like FAST-NPS and sophisticated bioinformatics tools will dramatically accelerate the discovery and development of novel therapeutic agents, ultimately overcoming the persistent challenges in heterologous expression research.
Advanced Codon Optimization and Algorithms for Predicting Expression Success
The heterologous expression of gene clusters is a foundational strategy in synthetic biology, enabling the production of valuable natural products, therapeutic proteins, and industrial enzymes in tractable host organisms. However, a persistent and central challenge in this field is the frequent failure or suboptimal yield of the target molecule when its biosynthetic pathway is transferred from its native organism into a heterologous chassis. A critical factor underpinning this challenge is the incompatibility between the genetic language of the donor gene cluster and the translational machinery of the host organism.
The genetic code is degenerate, meaning most amino acids are encoded by multiple synonymous codons. Different organisms have evolved distinct preferences for which codons they use most frequently, a phenomenon known as codon usage bias. This bias exists because the cellular pools of transfer RNAs (tRNAs) are adapted to match the codon preferences of the organism's highly expressed genes. When a heterologous gene is introduced, its non-optimal codon composition can lead to ribosomal stalling, translation errors, reduced protein yields, and even incorrect protein folding [60]. Consequently, codon optimization—the computational redesign of a gene's nucleotide sequence to match the codon preferences of the host without altering the amino acid sequence—has become an indispensable tool for successful heterologous expression.
This whitepaper provides an in-depth technical guide to advanced codon optimization algorithms and their critical role in predicting and ensuring expression success. Framed within the broader challenges of heterologous expression research, it examines the limitations of traditional methods, explores next-generation data-driven approaches, and provides detailed experimental protocols for validating optimized sequences.
Beyond Single Metrics: The Evolution of Codon Optimization Algorithms
Early codon optimization tools relied heavily on simple metrics like the Codon Adaptation Index (CAI), which measures the similarity of a gene's codon usage to the usage of highly expressed genes in the target host [60]. While improving upon native sequences, these single-metric approaches often yield inconsistent results because they oversimplify the complex biological reality of gene expression.
The Shift to Multi-Parameter Optimization
Modern algorithms have evolved to perform multi-parameter optimization, simultaneously balancing numerous sequence features that influence transcriptional and translational efficiency [61]. The key parameters considered by advanced platforms like the GeneOptimizer algorithm are summarized in the table below.
Table 1: Key Parameters in Multi-Parameter Codon Optimization
| Level of Regulation | Parameter | Influence on Expression |
|---|---|---|
| Transcriptional | GC Content | Influences mRNA stability; optimal range is host-specific [60]. |
| Cryptic Splice Sites, TATA Boxes | Can cause aberrant transcription and must be eliminated [61]. | |
| mRNA Stability | RNA Instability Motifs (e.g., AU-rich elements) | Trigger rapid mRNA degradation [61]. |
| mRNA Secondary Structure (ΔG) | Stable 5' secondary structures can inhibit ribosome binding and scanning [62] [60]. | |
| Translational | Codon Usage / CAI | Matches codon frequencies to host tRNA pools for efficient elongation [60] [61]. |
| Codon Pair Bias (CPB) | Optimizes adjacent codon pairs to prevent ribosomal stalling [60]. | |
| Ribosomal Entry Sites | Ensures the Shine-Dalgarno sequence (in prokaryotes) or Kozak sequence (in eukaryotes) is optimal. |
This holistic approach is exemplified by commercial and academic platforms. For instance, MNDL Bio's AI-driven platform co-optimizes coding and non-coding regions and models translation dynamics to prevent protein misfolding, reportedly achieving up to 20-fold increases in protein production yield [63]. Similarly, Thermo Fisher's GeneOptimizer software processes a wide array of these parameters in a single operation, with documented success in significantly boosting expression for challenging protein classes like kinases and membrane proteins [61].
The Rise of AI and Deep Learning
The most significant recent advancement is the integration of deep learning, which moves beyond predefined rules to learn the complex relationships between mRNA sequence and expression output directly from experimental data.
A leading example is RiboDecode, a deep learning framework that predicts translation levels by training on large-scale ribosome profiling (Ribo-seq) data [62]. This method provides a genome-wide snapshot of ribosome positions, directly measuring translational efficiency. RiboDecode's architecture integrates three components:
- A translation prediction model trained on over 320 paired Ribo-seq and RNA-seq datasets from 24 human tissues and cell lines.
- An MFE prediction model implemented as a differentiable deep neural network to evaluate mRNA secondary structure.
- A codon optimizer that uses gradient ascent to iteratively explore the vast sequence space and generate codon sequences with improved predicted fitness (translation and/or stability) [62].
This data-driven, context-aware approach allows RiboDecode to account for the influence of specific cellular environments. In vitro and in vivo validation has demonstrated its superiority, showing substantial improvements in protein expression and enabling dramatic dose-reduction in mRNA therapeutic applications [62].
Table 2: Comparative Analysis of Codon Optimization Tool Strategies
| Tool / Platform | Core Methodology | Key Features | Reported Outcome |
|---|---|---|---|
| JCat, OPTIMIZER, ATGme [60] | Heuristic, rule-based | Strong alignment with host-specific codon usage (high CAI); considers GC content and CPB. | Effective for standard applications; performance varies. |
| GeneOptimizer [61] | Multi-parameter algorithmic optimization | Simultaneously balances >20 parameters related to transcription, mRNA stability, and translation. | Up to 15-fold increase in protein yield; 86% of optimized genes showed increased expression. |
| MNDL Bio [63] | AI-driven deep learning | Models hidden genetic information, vector stability, and non-uniform translation dynamics. | Up to 20-fold yield increase; successful expression of notoriously difficult targets (e.g., human G6PD in E. coli). |
| RiboDecode [62] | Deep learning on Ribo-seq data | Directly learns from translational data; context-aware; generative sequence exploration. | ~10x stronger antibody responses in vivo; equivalent efficacy at 1/5th the mRNA dose. |
Experimental Protocols for Validation
Optimized in silico sequences must be rigorously validated experimentally. The following are detailed protocols for key validation assays cited in the literature.
Acetylene Reduction Assay for Nitrogenase Activity
This protocol was used to validate the functional heterologous expression of a nitrogen-fixing (nif) gene cluster in Bacillus subtilis [3].
- Objective: To detect and quantify nitrogenase activity by measuring its ability to reduce acetylene to ethylene.
- Materials:
- Engineered B. subtilis strain 168::CR1nif and an appropriate control strain.
- Nitrogen-limiting medium: KH₂PO₄ (3.4 g/L), Na₂HPO₄ (26.3 g/L), biotin (10 μg/L), MgSO₄ (30 mg/L), p-aminobenzoic acid (10 μg/L), CaCl₂·2H₂O (26 mg/L), ferric citrate (36 mg/L), MnSO₄·H₂O (0.33 mg/L), Na₂MoO₄·2H₂O (7.6 mg/L), glucose (4 g/L) [3].
- Acetylene gas.
- Gas-tight sealed vials (e.g., 50 mL serum bottles).
- Gas chromatograph (GC) equipped with a flame ionization detector and a Porapak N column.
- Methodology:
- Culture Preparation: Grow the engineered and control strains in nitrogen-limiting medium under micro-aerobic or anaerobic conditions optimal for nitrogenase function.
- Acetylene Injection: Transfer log-phase cultures to gas-tight vials. Inject acetylene to a concentration of 10% (v/v) of the headspace.
- Incubation: Incubate the vials at the growth temperature with shaking for a predetermined time (e.g., 2-4 hours).
- Gas Sampling: Remove a defined volume (e.g., 100 μL) from the headspace of each vial.
- Chromatographic Analysis: Inject the gas sample into the GC. Quantify the amount of ethylene produced by comparing peak areas to a standard curve of known ethylene concentrations.
- Normalization: Normalize the ethylene production rate to cell dry weight or protein content.
Comparative Expression Analysis via Western Blot
This is a standard method for quantifying relative protein expression levels between wild-type and optimized genes, as used in validation studies for GeneOptimizer and other tools [61].
- Objective: To compare the recombinant protein expression yields from wild-type and optimized gene sequences.
- Materials:
- Expression vectors containing wild-type and optimized genes (e.g., transfected into HEK293T cells).
- Lysis buffer (e.g., RIPA buffer with protease inhibitors).
- SDS-PAGE gel and Western blotting apparatus.
- Primary antibody against the target protein or an affinity tag (e.g., His-tag).
- HRP-conjugated secondary antibody.
- Chemiluminescent substrate and a digital imager.
- Methodology:
- Transfection & Culture: Transfect host cells in triplicate with wild-type, optimized, and empty vector (negative control) constructs. Culture for an optimal duration.
- Protein Extraction: Lyse cells and quantify total protein concentration of the lysates.
- SDS-PAGE & Transfer: Separate equal amounts of total protein by SDS-PAGE and transfer to a nitrocellulose or PVDF membrane.
- Immunoblotting: Block the membrane, then incubate with primary antibody, followed by HRP-conjugated secondary antibody.
- Detection & Quantification: Develop the blot with chemiluminescent substrate and capture the image. Quantify the band intensities using image analysis software (e.g., ImageJ).
- Data Analysis: Calculate the fold-increase in expression for the optimized construct relative to the wild-type construct, using the mean of the independent replicates [61].
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and tools essential for conducting research in heterologous expression and codon optimization validation.
Table 3: Research Reagent Solutions for Heterologous Expression
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| ExoCET (Exonuclease combined with RecET) [3] [13] | Cloning and assembly of large DNA fragments like biosynthetic gene clusters (BGCs). | Used to assemble an 11 kb nif gene cluster from Paenibacillus polymyxa for integration into B. subtilis [3]. |
| Red/ET Recombineering System [13] | Precise genetic engineering in E. coli using homologous recombination with short arms (~50 bp). | Essential for modifying BGCs in intermediary hosts like E. coli GB05-dir or GB05-red strains prior to conjugation [3] [13]. |
| RMCE Cassettes (Cre-lox, Vika-vox, Dre-rox) [13] | Enables precise, marker-less integration of heterologous DNA into specific chromosomal loci of the host. | Used in the Micro-HEP platform for stable, multi-copy integration of BGCs into Streptomyces chassis genomes [13]. |
| Ribo-seq (Ribosome Profiling) [62] | Provides a genome-wide snapshot of translating ribosomes, allowing direct measurement of translation efficiency. | Generates the primary data for training deep learning models like RiboDecode to predict and optimize translation levels [62]. |
| E. coli ET12567 (pUZ8002) [13] | A conjugation donor strain for transferring DNA from E. coli to actinomycetes like Streptomyces. | Standard tool for inter-species transfer of large BGCs, though newer systems seek to improve its limitations [13]. |
Visualizing Workflows and Relationships
Next-Generation Codon Optimization with RiboDecode
The following diagram illustrates the generative deep learning workflow of the RiboDecode framework.
Integrated Heterologous Expression Pipeline
This workflow maps the multi-stage process from gene cluster identification to heterologous expression validation, integrating codon optimization as a critical step.
The field of codon optimization has evolved decisively from simplistic, rule-based algorithms to sophisticated, multi-parameter, and data-driven approaches. The integration of deep learning and direct learning from ribosome profiling data represents a paradigm shift, enabling context-aware optimization that can dynamically adapt to specific cellular environments and therapeutic formats [62]. These advanced methods are proving critical for overcoming the persistent challenges in heterologous expression, such as the inefficient translation of complex gene clusters from phylogenetically distant organisms.
Future developments will likely focus on the holistic optimization of entire metabolic pathways, balancing the codon usage of multiple genes to avoid resource competition and metabolic burden [64]. Furthermore, as the delivery of gene therapies advances, the optimization of sequences for specific tissue and cell types using context-aware AI models will become increasingly important. The successful application of these advanced codon optimization strategies will be a cornerstone in accelerating the development of high-value biopharmaceuticals, sustainable biomaterials, and robust microbial cell factories.
Measuring Success: Validation Techniques and Comparative Host Performance
The discovery of novel bioactive compounds from natural sources is a cornerstone of drug development. A significant challenge in this field lies in the fact that a vast majority of biosynthetic gene clusters (BGCs)—the genetic blueprints for natural product synthesis—are silent or poorly expressed under standard laboratory conditions [9] [65]. Heterologous expression, the process of transferring these BGCs into a surrogate host organism, has emerged as a powerful strategy to activate these cryptic pathways and access their chemical products [9] [8]. However, the success of this approach is contingent upon robust analytical workflows capable of detecting, profiling, and identifying the novel compounds produced, often in minute quantities amidst a complex metabolic background.
Metabolomic profiling, defined as the comprehensive analysis of all low-molecular-weight metabolites in a biological system, provides this essential analytical link [66] [67]. It serves as the direct readout of BGC activity, enabling researchers to differentiate between known and potentially novel compounds. Among the available analytical platforms, mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy have become the techniques of choice due to their complementary strengths and abilities to survey complex chemical mixtures [65] [68] [69]. This technical guide outlines integrated MS/NMR workflows for novel compound identification, specifically framed within the challenges of heterologous expression research.
The Central Challenge: Silent Biosynthetic Gene Clusters and Heterologous Expression
The primary motivation for employing heterologous expression is the inability to access the chemical potential of most microorganisms through conventional cultivation.
- The Silent Majority: Genomic sequencing reveals that in normal culture conditions, around 90% of BGCs are cryptic and remain uncharacterized [8]. Furthermore, as many as 99% of environmental microbes are uncultivable in the lab, making heterologous expression the only viable route to explore their biosynthetic capabilities [9] [8].
- The Heterologous Expression Solution: This strategy involves cloning and transferring a BGC from its native host into a well-characterized "chassis" organism, such as E. coli, S. albus, or S. cerevisiae [9] [8]. This bypasses the native regulatory mechanisms that may suppress expression and allows for production in a host with a simpler metabolic background that is more amenable to genetic manipulation and fermentation.
- Persistent Hurdles: Despite its promise, heterologous expression is fraught with challenges. These include the technical difficulty of cloning large, often GC-rich BGCs; ensuring functional expression of all enzymes in the foreign host; and providing necessary precursors and cofactors that may be absent in the new cellular environment [8]. The success of this entire endeavor hinges on sensitive analytical methods to confirm production and identify the target compound.
Analytical Platforms: MS and NMR as Complementary Tools
The metabolome's chemical diversity necessitates analytical platforms with broad detection capabilities. MS and NMR serve as the two pillars of metabolomics, each with distinct advantages and limitations that make them highly complementary [68] [69].
Table 1: Strengths and Weaknesses of NMR and MS in Metabolomics
| Characteristic | NMR Spectroscopy | Mass Spectrometry (MS) |
|---|---|---|
| Strengths | Highly reproducible and quantitative; enables structural elucidation; non-destructive; requires no separation; analyzes intact samples. | Exceptional sensitivity; detects thousands of metabolites; high mass accuracy; can be coupled with separation techniques. |
| Weaknesses | Relatively low sensitivity (Limit of Detection ~1 μM); limited spectral resolution; detects fewer metabolites compared to MS. | Less reproducible; destructive to samples; requires internal standards for quantitation; difficulty analyzing salty samples. |
Mass Spectrometry Workflows
MS-based metabolomics involves ionizing metabolites and measuring their mass-to-charge ratios (m/z). It is typically coupled with a separation technique like liquid chromatography (LC) to reduce sample complexity.
Key Technical Descriptors for Metabolite Characterization: Modern MS platforms provide a suite of analytical descriptors that aid in metabolite identification [65]:
- Mass Accuracy: The deviation of the experimentally determined m/z from the true m/z. With mass errors of under 1 ppm, an exact elemental composition can be determined for compounds under 600 Da [65].
- Isotopic Modeling: The analysis of the isotopic envelope abundances can rapidly indicate the presence and quantity of heteroatoms like sulfur or chlorine [65].
- Chromatographic Retention Time: Provides a separation dimension orthogonal to mass, based on metabolite hydrophobicity or other chemical properties.
- Fragmentation (Tandem MS): Using collision-induced dissociation (CID) or other activation methods, metabolites are fragmented to provide structural information and differentiate between isomers [65] [67].
Experimental Protocol: LC-HRMS/MS for Metabolite Profiling
- Sample Preparation: Precipitate proteins from the culture broth or cell lysate using cold methanol or acetonitrile. Centrifuge and collect the supernatant for analysis [68].
- Chromatographic Separation: Employ a reversed-phase C18 column with a binary solvent system (e.g., water and acetonitrile, both modified with 0.1% formic acid) using a gradient elution over 10-20 minutes [67].
- Mass Spectrometry Analysis:
- Ionization: Use electrospray ionization (ESI) in both positive and negative modes to maximize metabolite coverage.
- Data Acquisition: Run in data-dependent acquisition (DDA) mode. A full high-resolution MS scan (e.g., 70,000 resolution) is followed by MS/MS scans on the most intense ions. Alternatively, data-independent acquisition (DIA) fragments all ions within a selected m/z range, providing a more complete fragmentation map [65] [67].
- Data Processing: Use software (e.g., XCMS, MS-DIAL) for peak picking, alignment, and deconvolution to generate a list of metabolite features (m/z, retention time, intensity).
Nuclear Magnetic Resonance Spectroscopy Workflows
NMR spectroscopy exploits the magnetic properties of atomic nuclei, providing unparalleled structural information in a quantitative and non-destructive manner.
Key Strengths for Novel Compound Identification:
- Structural Elucidation: A combination of 1D and 2D NMR experiments (e.g., COSY, HSQC, HMBC) allows for the determination of atomic connectivity and the full structure of unknown metabolites, a task that is challenging for MS alone [68].
- Quantitative Accuracy: The NMR signal intensity is directly proportional to the concentration of the nucleus, allowing for absolute quantitation with a single internal standard [68].
- Non-Targeted and Non-Destructive: NMR can profile intact biofluids or crude extracts with minimal preparation, and the sample can be recovered for further analysis [67] [68].
Experimental Protocol: 1H-NMR Metabolite Profiling of Culture Extracts
- Sample Preparation: Lyophilize the culture supernatant or cell extract and reconstitute it in a deuterated buffer (e.g., D2O phosphate buffer, pH 7.4). Include a known concentration of a chemical reference standard, such as TSP (trimethylsilylpropanoic acid) or DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid), for chemical shift referencing and quantitation [70] [68].
- Data Acquisition:
- Acquire a standard 1D 1H-NMR spectrum with water suppression (e.g., presaturation).
- For complex mixtures, acquire 2D spectra such as 1H-1H COSY (Correlation Spectroscopy) and 1H-13C HSQC (Heteronuclear Single Quantum Coherence) to resolve overlapping signals and identify coupled spins [68].
- Data Processing and Analysis: Fourier transform the free induction decay (FID) data after applying exponential line broadening. Phase and baseline correct the spectra. Use chemometric software to perform multivariate statistical analysis (e.g., PCA, OPLS-DA) to identify significant differences between control and expression samples [70].
Integrated MS/NMR Workflow for Novel Compound Identification
The most powerful approach for de-orphaning silent BGCs involves the sequential and integrated use of MS and NMR. The workflow below outlines this process from heterologous expression to novel compound identification.
Detailed Methodologies for Key Workflow Steps
Step 3: Data Processing and Dereplication Dereplication is the critical process of comparing analytical data against databases of known compounds to avoid rediscovery [67].
- MS Dereplication: Use the high-resolution m/z value to search natural product databases (e.g., CMNPD, AntiBase). Confirm potential matches by comparing MS/MS fragmentation patterns and LC retention times, if available [9] [67].
- NMR Dereplication: Compare the 1H-NMR spectrum (chemical shifts, coupling constants) of the crude extract or fraction with spectral databases to identify known compounds.
Step 6: Bioassay-Guided Fractionation If a bioactivity assay is available (e.g., antimicrobial, enzyme inhibition), this step ensures the isolation of the bioactive constituent.
- The crude extract is fractionated using preparative HPLC or MPLC (Medium-Pressure Liquid Chromatography).
- All fractions are tested for the desired bioactivity.
- The active fraction(s) are subjected to further purification steps (e.g., analytical HPLC with different chromatographic phases) until a pure compound is obtained [67].
Step 7: NMR Analysis for Structural Elucidation With a pure compound in hand, a full suite of NMR experiments is conducted to solve its structure.
- 1D Experiments: 1H and 13C NMR for initial structural overview.
- 2D Experiments:
- COSY: Identifies 1H-1H coupling networks (through-bond correlations, typically over 2-3 bonds).
- HSQC: Identifies direct 1H-13C correlations, defining protonated carbons.
- HMBC: Identifies long-range 1H-13C correlations (typically 2-4 bonds), crucial for connecting structural units across heteroatoms or quaternary carbons.
- The combined data from these experiments allows for the assembly of the molecular structure [68].
Data Fusion: Integrating MS and NMR Data
To fully leverage the complementary nature of MS and NMR, data fusion strategies are increasingly employed. These strategies integrate datasets from both platforms to build more robust and informative models [69].
Table 2: Data Fusion Strategies for MS and NMR Metabolomics
| Fusion Level | Description | Methodologies | Advantages and Challenges |
|---|---|---|---|
| Low-Level | Direct concatenation of raw or pre-processed data matrices from NMR and MS. | PCA, PLS on the fused matrix. | Advantage: Retains all original information.Challenge: High dimensionality; requires careful data scaling. |
| Mid-Level | Fusion of features extracted from each dataset separately. | PCA scores from NMR and MS data are merged into a new matrix for analysis. | Advantage: Reduces data dimensionality; focuses on most relevant features.Challenge: Risk of losing subtle but important information during feature extraction. |
| High-Level | Combination of the final predictions or classifications from models built on each dataset. | Bayesian consensus, majority voting on model outputs. | Advantage: Uses optimized model for each platform; flexible.Challenge: Most complex; requires building separate models first. |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Metabolomic Profiling Workflows
| Item | Function/Brief Explanation |
|---|---|
| Methanol & Acetonitrile (LC-MS Grade) | High-purity solvents for protein precipitation during sample preparation and as mobile phases for LC-MS to minimize background interference. |
| Deuterated Solvents (e.g., D2O, CD3OD) | NMR solvents that allow for signal locking and do not produce large interfering signals in the 1H-NMR spectrum. |
| Internal Standards (for NMR: TSP, DSS; for MS: isotope-labeled amino acids) | Reference compounds added in known concentrations for quantitative NMR and for correcting instrument variability in MS. |
| Silica-based C18 LC Columns | The workhorse stationary phase for reversed-phase chromatographic separation of complex natural extracts. |
| Culture Media for Heterologous Hosts | Defined media (e.g., LB for E. coli, R5 for Streptomyces) optimized for the growth and production of secondary metabolites in the chassis organism. |
| Cloning Kits for Large DNA Fragments | Specialized kits (e.g., utilizing Gibson Assembly, Transformation-Associated Recombination - TAR) are essential for capturing large, GC-rich BGCs [8]. |
The integration of advanced metabolomic profiling with heterologous expression represents a powerful pipeline for modern natural product discovery. While the challenges of activating silent BGCs are significant, the combination of sensitive MS-based detection for dereplication and prioritization, followed by definitive NMR-based structural elucidation, provides a robust solution. Furthermore, the emerging paradigm of data fusion promises to more deeply exploit the complementary nature of MS and NMR, offering a more holistic view of the metabolome. As both analytical technologies and genetic engineering techniques continue to advance, this integrated workflow will undoubtedly accelerate the discovery of novel compounds with potential applications in drug development and beyond.
Heterologous expression, the process of expressing a gene or gene cluster in a host organism different from its origin, is a cornerstone of modern biotechnology. It enables the production of valuable proteins, enzymes, and natural products for applications ranging from therapeutic development to industrial biocatalysis. However, the path from gene insertion to high-yield product formation is fraught with challenges. Unpredictable expression levels, improper protein folding, host toxicity, and suboptimal post-translational modifications frequently result in disappointingly low success rates and titers. This whitepaper synthesizes insights from recent large-scale studies to quantify these success rates, analyze the factors governing them, and present optimized experimental protocols designed to overcome these pervasive challenges.
Quantifying Success: Performance Metrics Across Diverse Systems
The success of heterologous expression is measured by key performance indicators including protein yield, enzyme activity, and the successful production of complex natural products. The data summarized in Table 1 reveals how strategic host and engineering choices directly impact these outcomes.
Table 1: Quantitative Outcomes from Heterologous Expression Systems
| Host System | Target Product(s) | Engineering Strategy | Key Performance Metrics | Citation |
|---|---|---|---|---|
| Aspergillus niger (Chassis AnN2) | Four diverse proteins (e.g., glucose oxidase, pectate lyase) | Deletion of 13/20 native glucoamylase genes & major protease gene (PepA); Site-specific integration | Yields: 110.8 - 416.8 mg/L in shake-flasks; Enzyme activities: ~1276 - 1907 U/mL | [71] |
| Streptomyces aureofaciens (Chassis2.0) | Oxytetracycline, Actinorhodin, Flavokermesic acid | Deletion of two endogenous polyketide gene clusters to eliminate precursor competition | 370% increase in oxytetracycline production vs. commercial strains; High-efficiency synthesis of tri-ring T2PKs | [72] |
| Komagataella phaffii | Recombinant Acidocin 4356 (rACD) | Codon optimization; Response Surface Methodology for fermentation | 34.12% yield increase; 58.29% reduction in P. aeruginosa growth at 150 µg/mL | [73] |
| Corynebacterium glutamicum, E. coli, Pseudomonas putida | Engineered Type I Polyketide Synthase (T1PKS) | Systematic testing of 11 different codon optimization strategies | ≥50-fold increase in PKS protein levels enabling unnatural polyketide production | [74] |
| Salmonella enterica serovar Typhimurium | manA and ova genes | Codon optimization using COSEM model (OCTOPOS software) | 3-fold increase in protein yield vs. wildtype and commercially optimized sequences | [75] |
The data demonstrates that success is not monolithic but highly dependent on the synergy between the target product and the host. Prokaryotic hosts like Streptomyces and E. coli excel in producing bacterial enzymes and natural products [72] [74], whereas eukaryotic hosts like A. niger and K. phaffii are superior for complex eukaryotic proteins requiring specific post-translational modifications [71] [73]. A critical finding across studies is that "one-size-fits-all" approaches are ineffective. Instead, maximizing success rates requires a tailored, multi-factorial strategy integrating genomic, transcriptional, and metabolic engineering.
Detailed Experimental Protocols for Enhanced Success
To translate the quantitative insights from Table 1 into practical outcomes, researchers require robust and reproducible experimental workflows. The following sections detail two of the most effective protocols from recent literature.
This protocol creates a clean background host strain (chassis) that minimizes native protein interference and provides defined, high-expression loci for gene integration.
Step 1: Parent Strain Selection
- Begin with an industrial glucoamylase-producing strain, such as A. niger AnN1, which possesses robust native secretion machinery and multiple copies of the TeGlaA gene.
Step 2: CRISPR/Cas9-Mediated Genomic Streamlining
- Design gRNAs: Design guide RNAs targeting the promoter and terminator regions of the tandemly repeated native TeGlaA gene.
- Co-transformation: Co-transform the parent strain with a plasmid expressing Cas9 and the designed gRNAs, along with a donor DNA template for homology-directed repair.
- Marker Recycling: Use a recyclable selection marker to sequentially delete 13 out of the 20 TeGlaA gene copies. This drastically reduces the background of secreted native proteins.
- Protease Disruption: In the same strain, use CRISPR/Cas9 to disrupt the gene encoding the major extracellular protease PepA, thereby minimizing degradation of the target heterologous protein.
- Outcome Validation: The resulting chassis strain, AnN2, shows a 61% reduction in total extracellular protein and significantly reduced glucoamylase activity, providing a low-noise background for expression.
Step 3: Site-Specific Gene Integration
- Vector Design: Construct a modular donor plasmid containing the target gene flanked by the native AAmy promoter and AnGlaA terminator. These act as homology arms for integration into the loci previously occupied by TeGlaA genes.
- Transformation and Screening: Introduce the donor plasmid into the AnN2 chassis and screen for successful site-specific integration. This strategy leverages native, high-transcription loci to drive strong expression of the heterologous gene.
Step 4: Secretory Pathway Engineering (Optional Enhancement)
- To further boost yields, overexpress key components of the secretory machinery, such as the COPI vesicle trafficking component Cvc2. This has been shown to enhance production of a pectate lyase (MtPlyA) by 18% [71].
This platform facilitates the cloning, modification, and expression of large Biosynthetic Gene Clusters (BGCs) for natural product discovery.
Step 1: In vivo Modification in an Optimized E. coli Host
- Host Strain: Use engineered E. coli strains (e.g., GB2005, GB2006) equipped with a rhamnose-inducible Redαβγ recombination system and a arabinose-inducible CcdA counter-selection system.
- Plasmid Assembly: Clone the target BGC into a suitable vector. Use the Redαβγ system to perform precise, markerless modifications, such as inserting Recombinase-Mediated Cassette Exchange (RMCE) cassettes (Cre-lox, Vika-vox, Dre-rox, phiBT1-attP).
- Conjugation Readiness: Ensure the final plasmid contains the transfer origin (oriT) for subsequent bacterial conjugation.
Step 2: Conjugative Transfer to a Streptomyces Chassis
- Chassis Strain: Use a genetically streamlined Streptomyces chassis, such as S. coelicolor A3(2)-2023, in which four endogenous BGCs have been deleted to minimize metabolic competition and background interference.
- Conjugation: Mate the donor E. coli strain with the Streptomyces chassis. The E. coli Tra proteins will mobilize the plasmid containing the BGC into Streptomyces cells via single-stranded DNA transfer.
Step 3: Genomic Integration via RMCE
- Site-Specific Integration: The RMCE cassette on the plasmid recombines with pre-engineered, matching RMCE sites on the Streptomyces chromosome. This process cleanly integrates the BGC without co-integrating the plasmid backbone.
- Multi-Copy Integration: Utilize heterospecific recombination sites (e.g., lox5171 and lox2272) to integrate multiple copies of the BGC. Studies show that increasing the copy number of the xiamenmycin BGC from two to four leads to a corresponding increase in final product titer [13].
Step 4: Fermentation and Product Analysis
- Culture the exconjugants under optimized fermentation conditions and screen for the production of the target natural product using analytical techniques like HPLC or LC-MS.
Workflow Visualization
The following diagram illustrates the logical relationship and workflow of the two key experimental protocols described above:
The Scientist's Toolkit: Essential Research Reagent Solutions
The successful implementation of the protocols above relies on a suite of specialized reagents and genetic tools. Table 2 catalogs these key components and their critical functions in heterologous expression workflows.
Table 2: Essential Research Reagents for Heterologous Expression
| Reagent / Tool Name | Function / Application | Key Feature / Benefit | |
|---|---|---|---|
| CRISPR/Cas9 System | Targeted gene knockout and genomic deletions in fungal and bacterial hosts. | Enables precise, multiplexed genome editing without leaving marker scars. | [71] |
| Redαβγ Recombineering System | In vivo genetic engineering of large DNA constructs (e.g., BGCs) in E. coli. | Uses short homology arms (50 bp) for efficient recombination; essential for BAC modification. | [13] |
| RMCE Cassettes (Cre-lox, Vika-vox, Dre-rox, phiBT1-attP) | Precise, backbone-free integration of gene clusters into specific chromosomal loci. | Orthogonal systems allow for multiple, sequential integrations in a single chassis. | [13] |
| Codon Optimization Tools (e.g., DNA Chisel, BaseBuddy, OCTOPOS) | Algorithmic adaptation of gene sequences to match the codon bias of the host organism. | Can improve protein yields by >50-fold; some tools model ribosome dynamics. | [74] [75] |
| Biparental Conjugation System (e.g., E. coli ET12567/pUZ8002) | Transfer of large, non-mobilizable plasmids from E. coli to Streptomyces and other actinomycetes. | Bypasses the need for direct transformation of difficult-to-transform hosts. | [13] |
| Streamlined Chassis Strains (e.g., A. niger AnN2, S. coelicolor A3(2)-2023) | Optimized host backgrounds with reduced native interference for heterologous expression. | Deletion of native proteases and competing BGCs enhances target product yield and detection. | [71] [13] |
Visualization of a Key Concept: Modeling Translation Dynamics
A significant challenge in heterologous expression is understanding how codon choice impacts translation efficiency. The Codon-Specific Elongation Model (COSEM) provides a mechanistic framework for this, moving beyond simple codon frequency analysis. The following diagram illustrates the core principles and dynamics of this model.
The COSEM model illustrates that optimal protein yield is not achieved by simply using the most frequent codons. Instead, it requires a balance where the initiation rate and elongation rates (which are codon-specific) are synchronized to avoid ribosome traffic jams, thereby maximizing the ribosomal current and protein output [75]. Tools like OCTOPOS leverage this understanding for gene optimization, leading to significant yield improvements—up to a 3-fold increase—compared to traditional methods [75].
Large-scale heterologous expression studies consistently demonstrate that high success rates and titers are achievable through integrated, systematic approaches. Quantifiable data confirms that strategies such as employing genomically streamlined chassis strains, implementing sophisticated codon optimization based on translation dynamics, and leveraging precise genome editing tools can lead to yield improvements of over 50-fold for proteins and nearly 4-fold for natural products. The protocols and toolkits detailed herein provide a actionable roadmap for researchers to overcome the historical challenges in the field. By moving beyond heuristic, one-gene-at-a-time experimentation and adopting these holistic, platform-based methodologies, scientists can significantly de-risk and accelerate the development of bioproduction processes for novel therapeutics, enzymes, and specialty chemicals.
The discovery of microbial natural products has been revolutionized by next-generation sequencing, revealing an extensive hidden reservoir of biosynthetic gene clusters (BGCs) encoding potential novel bioactive compounds [76]. This genomic potential vastly outstrips the number of compounds successfully characterized through traditional cultivation approaches. For instance, computational predictions estimate that Streptomyces bacteria alone may have the capability to produce 150,000 chemically distinct antimicrobial agents, while only a fraction have been identified [76]. This discrepancy represents both a tremendous opportunity and a fundamental challenge for natural product discovery.
The field faces a critical bottleneck: while bioinformatic identification of BGCs has become routine through tools like antiSMASH, the majority of these clusters remain "cryptic" or "silent" under standard laboratory conditions [76] [77]. Heterologous expression—the transfer of BGCs into genetically tractable host organisms—has emerged as a pivotal strategy for activating these silent clusters. However, this approach is fraught with technical challenges, including inefficient cluster assembly, host compatibility issues, and the inability to predict which of the countless BGCs merit the substantial investment required for expression attempts [13].
This technical guide frames comparative genomics and phylogenetic analysis as essential prioritization frameworks to overcome these challenges. By understanding the evolutionary distribution patterns of BGCs across bacterial taxa, researchers can make informed decisions about which clusters represent the most promising candidates for heterologous expression, thereby focusing resources on genetically distinctive BGCs with the highest likelihood of yielding novel chemistry.
Core Principles: Linking Phylogeny to Biosynthetic Potential
Phylogenetic Distribution Patterns of BGCs
Biosynthetic gene clusters evolve through both vertical inheritance and horizontal gene transfer, creating distinct phylogenetic distribution patterns that can inform prioritization strategies. Comparative genomics studies have revealed that BGC distribution often correlates with phylogeny, indicating that vertical gene transfer plays a major role in the evolution of secondary metabolite gene clusters [78]. However, the vast majority of BGCs are derived from clusters unique to specific strains, highlighting the simultaneous importance of horizontal acquisition and rapid evolution [78].
In Amycolatopsis species, phylogenetic characterization together with pan-genome analysis distinguished four major lineages that differed significantly in their potential to produce secondary metabolites [78]. This lineage-specific distribution was particularly pronounced in Streptomyces, where a specific monophyletic group characterized by the formation of rugose-ornamented spores was found to possess exceptional biosynthetic potential, containing an average of 50 BGCs per genome compared to the genus average of 33 BGCs [77]. These strains also had the largest genomes, averaging 11.5 Mb in size [77].
Table 1: BGC Abundance Across Streptomyces Phylogenetic Groups
| Phylogenetic Group | Average Genome Size (Mb) | Average BGC Abundance | Notable Characteristics |
|---|---|---|---|
| General Streptomyces | 8.5 | 33 BGCs | Representative of most strains |
| Group F (Rugose-spored) | 11.5 | 50 BGCs | Largest genomes, highest BGC count |
| Other Major Clades | 7.8-8.7 | 20-45 BGCs | Wide variation between lineages |
Genetic Locus and Cluster Stability
The genomic context of BGCs provides crucial insights into their evolutionary history and functional stability. Studies on Amycolatopsis have revealed that BGCs acquired by horizontal gene transfer tend to be incorporated into non-conserved regions of the genome, allowing researchers to distinguish between core and hypervariable genomic regions [78]. This localization has practical implications for heterologous expression, as clusters in conserved regions may represent more stable, ancestral traits with better-established regulatory networks in the native host.
Marine bacteria studies further demonstrate the dynamic nature of BGC architecture. In vibrioferrin-producing BGCs, while core biosynthetic genes remained conserved, the accessory genes exhibited high genetic variability [79]. This structural plasticity may influence functional properties such as iron-chelation efficiency and microbial interactions [79]. For heterologous expression, this suggests that prioritizing clusters with conserved core architectures may increase the likelihood of successful functional reconstitution.
Methodological Framework: From Genomes to Candidate BGCs
Phylogenetic Analysis and Tree Construction
Robust phylogenetic reconstruction provides the essential framework for correlating biosynthetic potential with evolutionary relationships. A comprehensive phylogenetic study of Streptomyces utilized full-length 16S rRNA sequences from 615 type strains to establish an evolutionary blueprint of the genus, grouping strains into 130 distinct clades based on statistically significant evolutionary relatedness [77]. For finer resolution, Multi-Locus Sequence Analysis (MLSA) using concatenated housekeeping genes (e.g., atpD, clpB, gapA, gyrB, nuoD, pyrH, rpoB) provides enhanced discriminatory power compared to single-gene analyses [78].
Experimental Protocol: Multi-Locus Sequence Analysis
- Gene Selection: Select 5-7 housekeeping genes with appropriate evolutionary rates
- PCR Amplification: Amplify target genes from strain collection using conserved primers
- Sequence Alignment: Perform multiple sequence alignment for each gene locus
- Concatenation: Concatenate aligned sequences into a single supermatrix
- Tree Construction: Generate maximum likelihood phylogeny with bootstrap support (1000 replicates)
- Topology Validation: Compare with 16S rRNA phylogeny to identify discrepancies
For marine bacteria, the rpoB gene has proven particularly valuable as a phylogenetic marker due to its relatively conserved nature, allowing for accurate reconstruction of evolutionary relationships among diverse bacterial strains [79].
BGC Identification and Classification
The identification and classification of BGCs relies on specialized bioinformatics tools that detect signature biosynthetic domains and architectures.
Experimental Protocol: BGC Identification and Analysis
- Genome Assembly: Assemble high-quality draft or complete genomes (preferably <100 contigs to avoid BGC fragmentation)
- Cluster Prediction: Use antiSMASH 7.0 with default detection settings, enabling KnownClusterBlast, ClusterBlast, and SubClusterBlast [79]
- Manual Curation: Verify putative BGC boundaries through comparative analysis with known clusters
- Cluster Classification: Categorize BGCs by type (NRPS, PKS, RiPP, terpene, etc.) and novelty
- Similarity Networking: Use BiG-SCAPE to group BGCs into Gene Cluster Families (GCFs) based on domain sequence similarity [79]
Table 2: Bioinformatics Tools for BGC Analysis
| Tool | Primary Function | Key Features | Application in Workflow |
|---|---|---|---|
| antiSMASH | BGC prediction and annotation | Identifies core biosynthetic machinery, compares to known clusters | Initial BGC discovery and classification |
| BiG-SCAPE | BGC similarity networking | Groups BGCs into Gene Cluster Families (GCFs) based on sequence similarity | Clustering and prioritization of BGCs |
| MIBiG | Reference database of known BGCs | Provides standardized annotation and metadata for characterized clusters | Novelty assessment and functional prediction |
| PRISM | Chemical structure prediction | Predicts chemical scaffolds from genetic sequences | Prioritization based on predicted chemistry |
Correlation Analysis: Linking Phylogeny to BGC Distribution
The core analytical step involves determining the relationship between phylogenetic position and biosynthetic potential to identify lineages with elevated or distinctive BGC content.
Experimental Protocol: Phylogeny-BGC Correlation
- BGC Abundance Mapping: Map BGC abundance and diversity data onto phylogenetic trees
- Statistical Analysis: Determine correlation between phylogenetic distance and BGC similarity
- Lineage-Specific Enrichment: Identify phylogenetic groups with statistically significant enrichment of specific BGC types
- Novelty Assessment: Compare identified BGCs against MIBiG database to determine sequence novelty
- Venn Diagram Construction: Visualize BGC distribution across phylogenetic groups to identify shared, group-specific, and strain-specific clusters
In practice, this approach revealed that within the high-potential Streptomyces group F, the majority of BGCs were either subgroup-specific (rare) or strain-specific (unique), highlighting the value of targeting these lineages for novel natural product discovery [77].
Experimental Workflow: From Candidate BGC to Heterologous Expression
The transition from bioinformatic identification to functional expression requires a coordinated series of molecular biology techniques. The following workflow diagrams the complete process from genomic DNA to compound production in a heterologous host.
BGC Heterologous Expression Workflow
Cluster Assembly and Engineering
Capturing and engineering target BGCs requires specialized molecular techniques capable of handling large DNA fragments with repetitive sequences.
Experimental Protocol: ExoCET Assembly
- Cluster Fragmentation: Divide large BGCs into 4-6 overlapping fragments (e.g., 3-5 kb each)
- Vector Preparation: Linearize receiving vector via restriction enzyme digestion
- ExoCET Reaction: Combine fragments with T4 DNA polymerase in appropriate buffer
- In Vitro Recombination: Incubate at 25°C for 1 hour, 75°C for 20 minutes, 50°C for 30 minutes
- Transformation: Transfer reaction product into competent E. coli GB05-dir cells
- Validation: Screen positive recombinants through restriction analysis and sequencing [3]
For combinatorial assembly of partial clusters to identify minimal gene sets, researchers have developed rapid methods to assemble individual genes involved in biosynthesis into collections of partial or complete clusters in heterologous hosts [4]. This approach simultaneously generates multiple strains that produce potentially desirable analogues in addition to the target compound.
Promoter Engineering and Regulatory Optimization
A critical challenge in heterologous expression is achieving appropriate expression of all necessary biosynthetic genes. Native promoters often fail to function optimally in new host contexts.
Experimental Protocol: Promoter Replacement
- Promoter Selection: Choose constitutive or inducible promoters compatible with the host system (e.g., Pveg, P43, Ptp2 for Bacillus subtilis)
- Fusion PCR: Amplify promoter and antibiotic resistance fragments, then fuse via overlap extension PCR
- Recombineering: Co-electroporate fusion fragments with cluster-containing plasmid into recombination-competent E. coli
- Screening: Select on appropriate antibiotics, verify promoter insertion by colony PCR [3]
Studies demonstrate that promoter selection requires balancing transcriptional strength with systemic compatibility, particularly for complex metalloenzymes demanding precise cofactor assembly [3]. While promoter replacement (e.g., replacing native promoters with Pveg) can enable heterologous hosts to produce active enzyme, stronger promoters do not necessarily further enhance activity [3].
Heterologous Expression Platforms and Chassis Engineering
Advanced Expression Systems
Specialized expression platforms have been developed to streamline the process of BGC transfer and expression in heterologous hosts.
Experimental Protocol: Micro-HEP Platform
- BGC Modification: Use E. coli strains with rhamnose-inducible redαβγ recombination system for precise insertion of RMCE cassettes
- Vector Mobilization: Incorporate oriT transfer origin for conjugation into Streptomyces
- Conjugative Transfer: Mobilize BGC as single-stranded DNA into chassis strain via Tra protein
- Site-Specific Integration: Integrate BGC into pre-engineered chromosomal loci using recombinase-mediated cassette exchange (RMCE)
- Multi-Copy Integration: Introduce additional attB sites for chromosomal amplification of heterologous BGCs [13]
The Micro-HEP platform demonstrates how systematic engineering of both transfer systems and chassis strains can overcome historical bottlenecks in heterologous expression. This system successfully expressed BGCs for the anti-fibrotic compound xiamenmycin and griseorhodins, with increasing copy number associated with increasing yield of the target compound [13].
Chassis Strain Optimization
Engineered host strains with reduced genetic complexity and enhanced precursor supply provide superior platforms for heterologous expression.
Experimental Protocol: Chassis Development
- BGC Deletion: Remove endogenous BGCs to minimize metabolic competition and background interference
- Integration Site Introduction: Incorporate multiple orthogonal recombinase-mediated cassette exchange sites (Cre-lox, Vika-vox, Dre-rox, phiBT1-attP)
- Precursor Pathway Enhancement: Modify primary metabolic pathways to supply essential cofactors and building blocks
- Regulatory Network Engineering: Modify global regulators to favor secondary metabolite production [13]
The chassis strain S. coelicolor A3(2)-2023 was generated by deleting four endogenous BGCs followed by introducing multiple RMCE sites in the chromosome, creating a clean background optimized for heterologous expression [13].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for BGC Heterologous Expression
| Reagent/Category | Specific Examples | Function in Workflow | Technical Considerations |
|---|---|---|---|
| Cloning Systems | ExoCET, TAR, Gibson Assembly | Large DNA fragment assembly | ExoCET enables direct cloning from genomic DNA without intermediate hosts |
| Expression Vectors | p15A-ha-spec, pBR322-amp | BGC delivery and maintenance | Must include appropriate selectable markers and integration elements |
| Host Strains | B. subtilis 168, S. coelicolor A3(2)-2023 | BGC expression and compound production | Engineered chassis strains with deleted endogenous BGCs improve success |
| Conjugation Systems | E. coli ET12567 (pUZ8002), engineered E. coli GB2005 | Inter-species DNA transfer | Improved systems address instability of repeated sequences in large BGCs |
| Recombinase Systems | Cre-lox, Vika-vox, Dre-rox, PhiC31-int | Site-specific genomic integration | Orthogonal systems enable multiple integrations in single chassis |
| Inducible Promoters | Pveg, P43, Ptp2 | Drive expression of BGC genes in heterologous hosts | Strength must be balanced with host compatibility |
The integration of comparative genomics and phylogenetic analysis with advanced heterologous expression platforms represents a powerful paradigm for overcoming the fundamental challenges in natural product discovery. By applying the methodologies outlined in this technical guide, researchers can systematically prioritize the most promising BGCs from the vast genomic landscape, focusing experimental resources on clusters with the highest likelihood of yielding novel bioactive compounds. As synthetic biology tools continue to advance, the marriage of evolutionary insight with engineering precision will undoubtedly accelerate the discovery and development of next-generation therapeutic agents from microbial sources.
The heterologous expression of biosynthetic gene clusters (BGCs) in engineered host organisms represents a powerful strategy for discovering novel natural products and elucidating their biosynthetic pathways. This approach is particularly valuable for studying marine microorganisms and other recalcitrant species where a significant proportion—often around 90% of BGCs—remain silent or weakly expressed under standard laboratory conditions [9] [8]. Furthermore, as many as 99% of environmental microbes resist cultivation, making direct genetic manipulation impossible and heterologous expression essential for accessing their biosynthetic potential [8].
A central challenge in this field lies in conclusively demonstrating that a heterologously expressed BGC is responsible for producing a specific compound and deciphering the function of individual genes within the cluster. Simply observing product formation in a heterologous host is insufficient; rigorous functional validation requires genetic evidence. Site-directed mutagenesis (SDM) provides this critical link by enabling researchers to make precise, targeted changes to a BGC and observe the resulting phenotypic changes in the metabolic output, thereby confirming the cluster's function and the role of its constituent genes [80] [81].
This technical guide outlines the primary challenges in heterologous BGC expression and details how mutagenesis serves as an indispensable tool for confirming BGC function and elucidating product structure, providing detailed protocols and resources for implementation.
Key Challenges in Heterologous Expression of Gene Clusters
Functional expression of BGCs in a heterologous host is fraught with difficulties, which can be categorized into several areas, as detailed in the table below.
Table 1: Major Challenges in Heterologous Expression of Biosynthetic Gene Clusters
| Challenge Category | Specific Obstacles | Impact on Functional Expression |
|---|---|---|
| Cluster Cloning & Integrity | Large cluster size (>100 kb), high GC content, repetitive sequences [8] | Difficult to capture intact, functional clusters; time-consuming cloning processes |
| Host Compatibility | Lack of essential substrates/precursors, incompatible transcription/translation machinery, improper post-translational modifications, toxicity of intermediates/products [9] [8] | Silent clusters, low product titers, or formation of incorrect or incomplete products |
| Regulatory Control | Complex native regulation lost in new host; cryptic promoters and ribosomal binding sites not recognized [9] [8] | Failure to activate the pathway or suboptimal expression of biosynthetic genes |
| Enzyme Functionality | Misfolding of large, complex enzymes (e.g., PKS, NRPS); insufficient cofactors; inadequate post-translational activation [8] | Inactive biosynthetic pathways or the production of aberrant compounds |
Overcoming these challenges often requires a combination of specialized cloning techniques, careful host selection, and pathway refactoring—replacing native regulatory elements with well-characterized, constitutive parts to optimize expression in the new host [8]. However, even after successful production is achieved, mutagenesis remains the definitive method for confirming the pathway-product relationship.
Site-Directed Mutagenesis as a Validation Tool
Fundamental Principles and Applications
Site-directed mutagenesis is a molecular biology method used to make specific and intentional changes to a DNA sequence, including point mutations, insertions, or deletions [80]. In the context of BGC validation, its principle is straightforward: a designed mutation is introduced into a putative biosynthetic gene, and the heterologous host is re-screened for changes in metabolite production. The absence, reduction, or structural alteration of the target compound provides direct genetic evidence linking the modified gene to the biosynthetic pathway [80] [81].
Key applications of SDM in BGC functional analysis include:
- Gene Inactivation: Introducing premature stop codons or deleting critical domains to knockout the function of a specific gene and observe the resulting loss of product formation.
- Active Site Probing: Mutating key catalytic residues (e.g., substituting a critical serine with alanine) to study enzyme mechanism and confirm its role in a specific biosynthetic step [80].
- Pathway Engineering: Altering substrate specificity or re-routing biosynthetic pathways to generate novel analogues, simultaneously confirming hypotheses about enzyme function [81].
Advanced Methodologies for Large BGCs
Conventional PCR-based mutagenesis methods often struggle with the large size and repetitive nature of BGCs, particularly those encoding modular polyketide synthases (PKS) and nonribosomal peptide synthetases (NRPS) [81]. Advanced methods that combine oligonucleotide recombineering and CRISPR/Cas9 counter-selection have been developed to address this.
Recombineering uses single-stranded oligonucleotides and bacterial phage proteins to enable precise, markerless alteration of DNA sequences, even on large bacterial artificial chromosomes (BACs) [81]. When coupled with CRISPR/Cas9, which introduces double-strand breaks in unmutated, wild-type sequences, the system powerfully selects for successfully edited clones, dramatically improving efficiency.
Table 2: Essential Research Reagents for CRISPR/Recombineering-Based BGC Mutagenesis
| Reagent / Tool | Function in the Mutagenesis Workflow |
|---|---|
| Shuttle Vector (e.g., pCAP-BAC) | Carries the large BGC for cloning, manipulation in E. coli, and transfer to heterologous hosts [81] |
| Recombineering Strain (e.g., E. coli HME68) | Supplies inducible Red genes (exo, bet, gam) for efficient oligonucleotide recombination [81] |
| Targeting Oligonucleotide | Single-stranded DNA designed with the desired mutation; incorporated into the BGC via recombineering [81] |
| CRISPR/Cas9 Plasmid (e.g., pJZ002) | Provides Cas9 and a guide RNA (protospacer) for counter-selection against the unmutated parent sequence, enriching for mutants [81] |
| Mismatch Repair (MMR) Deficiency | Enhances recombineering efficiency by preventing the cellular machinery from rejecting the oligonucleotide-template mismatch [80] [81] |
Experimental Protocol: CRISPR/Recombineering-Mediated Mutagenesis of a BGC
The following protocol details the key steps for performing site-directed mutagenesis on a large BGC cloned in a shuttle vector, based on the validated method from [81].
Stage 1: Preparation
- Clone Target BGC: Clone the BGC of interest into a suitable shuttle vector (e.g., pCAP-BAC) to create the parent construct [81].
- Design Mutagenic Oligo: Design a single-stranded oligonucleotide (typically 70 nucleotides) complementary to the lagging strand with the desired mutation centrally located. To evade mismatch repair, include at least 5 consecutive base changes or silent mutations near the target site [81].
- Design CRISPR Protospacer: Design a 20-30 nt protospacer sequence adjacent to a PAM site (e.g., "AGG" for S. pyogenes Cas9) that is unique to the unmutated parent sequence. The desired mutation should disrupt this PAM or protospacer to prevent Cas9 from cleaving successfully mutated plasmids [81].
- Transform Parental Construct: Transform the parent BGC construct into a recombineering-proficient E. coli strain (e.g., HME68) that carries an inducible λ Red system and is MMR-deficient (ΔmutS) [81].
Stage 2: Recombineering and Counter-Selection
- Induce Recombineering Proteins: Grow the transformed culture and induce the expression of the Red proteins (Exo, Bet, Gam) to make the cells competent for recombination.
- Electroporation: Electroporate the mixture of the mutagenic oligonucleotide and the CRISPR/Cas9 plasmid (pJZ002::vioX) into the induced cells [81].
- Selection and Screening: Plate the cells on media containing antibiotics that select for both the BGC vector and the CRISPR plasmid. The presence of Cas9 will kill cells retaining the unmutated BGC, dramatically enriching the population for successfully recombined clones. Screen resulting colonies (e.g., by PCR or phenotypic assay) to identify potential mutants.
Stage 3: Validation
- Sequence Verification: Isolate plasmid DNA from candidate clones and perform Sanger sequencing across the targeted region to confirm the incorporation of the desired mutation and the absence of unintended errors.
- Functional Validation in Heterologous Host: Transfer the validated mutant BGC construct into the chosen heterologous production host (e.g., Pseudomonas putida, Streptomyces albus). Analyze the metabolic profile using LC-MS or other analytical methods and compare it to the profile of the wild-type BGC to determine the impact of the mutation on product structure and yield [81].
The following diagram visualizes the core workflow and mechanism of this combined CRISPR/recombineering method.
Case Study: Mutagenesis of Thalassospiramide BGCs
A practical application of this methodology involved the genetic dissection of two massive hybrid PKS-NRPS BGCs (ttc and ttm) from marine bacteria, which produce immunosuppressive thalassospiramides [81].
- Objective: To probe the function of non-canonical domains within the ~25.5 kb ttc and ~19.1 kb ttm assembly lines.
- Method: Researchers used the combined CRISPR/recombineering system in E. coli HME68 to perform site-directed mutagenesis on the BGCs, which were cloned in the pCAP-BAC shuttle vector.
- Implementation: A total of 12 point mutations were successfully introduced into key catalytic domains of the megasynthase genes to inactivate them.
- Outcome and Validation: The mutant BGCs were transferred to Pseudomonas putida for heterologous expression. Analysis of the metabolic output by LC-MS revealed either the absence of thalassospiramides or the accumulation of novel, earlier-stage intermediates. This directly linked the inactivated domains to specific biochemical steps in the biosynthesis, confirming their predicted functions and validating the overall BGC assignment [81].
Mutagenesis remains a cornerstone of functional genetics, providing the critical, causal evidence needed to connect a BGC to its metabolic product. The challenges of heterologous expression—from cloning massive gene clusters to achieving functional enzyme production in a foreign host—can be formidable. However, as demonstrated by advanced techniques like CRISPR/recombineering, precise genetic manipulation is now possible even for the largest and most complex BGCs. By systematically inactivating or altering genes and observing the resulting chemical phenotypes, researchers can definitively confirm BGC function, elucidate biosynthetic pathways, and engineer new natural product analogues, thereby fully unlocking the genetic potential encoded within microbial genomes.
Conclusion
The heterologous expression of biosynthetic gene clusters remains a challenging yet indispensable frontier in natural product discovery. While significant hurdles—from cloning large GC-rich sequences to ensuring proper regulation and folding in a foreign host—result in a low overall success rate, the integration of advanced bioinformatics, sophisticated DNA assembly techniques, and systematic host engineering provides a clear path forward. Future progress will depend on developing more universal and genetically tractable chassis organisms, creating smarter, AI-driven refactoring tools, and deepening our understanding of the complex regulatory networks that govern secondary metabolism. For biomedical and clinical research, mastering these challenges is paramount to unlocking the vast reservoir of unseen chemical diversity encoded in microbial genomes, offering a promising pipeline for the next generation of therapeutic agents against drug-resistant pathogens and other pressing human diseases.
References
- 1. Subterranean marvels: microbial communities in caves and ... [https://pubs.rsc.org/en/content/articlehtml/2025/np/d4np00055b]
- 2. A comprehensive analysis of human gut microbial ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12282132/]
- 3. Heterologous Expression of the Nitrogen-Fixing Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12195393/]
- 4. A rapid combinatorial assembly method for gene cluster ... [https://www.sciencedirect.com/science/article/pii/S2405805X25001516]
- 5. Bacterial natural product discovery by heterologous expression [https://pmc.ncbi.nlm.nih.gov/articles/PMC10727000/]
- 6. A Natural Product Chemist's Guide to Unlocking Silent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9148385/]
- 7. Epigenetic Activation of Silent Biosynthetic Gene Clusters ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.815008/full]
- 8. Challenges of functional expression of complex polyketide ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166920301920]
- 9. Recent Advances in the Heterologous Expression of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9225448/]
- 10. Recent advances in activating silent biosynthetic gene ... [https://www.sciencedirect.com/science/article/abs/pii/S1369527418300572]
- 11. Strategies for Natural Product Discovery by Unlocking ... [https://www.mdpi.com/2297-8739/10/6/333]
- 12. Cell-free synthetic biology for natural product biosynthesis and ... [https://pubs.rsc.org/en/content/articlehtml/2025/cs/d4cs01198h]
- 13. A highly efficient heterologous expression platform to facilitate ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02722-z]
- 14. Discovery, characterization, and engineering of an ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02416-y]
- 15. Codon Optimization Tool [https://en.vectorbuilder.com/tool/codon-optimization.html]
- 16. Strategies to overcome the challenges of low or no ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975024001113]
- 17. OPTIMIZER: a web server for optimizing the codon usage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1933141/]
- 18. Codon optimization with deep learning to enhance protein ... [https://www.nature.com/articles/s41598-020-74091-z]
- 19. Natural Products and the Gene Cluster Revolution - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5123934/]
- 20. Heterologous Expression of Difficult to Produce Proteins in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10815505/]
- 21. Targeted Large-Scale Genome Mining and Candidate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9231227/]
- 22. Targeted genome mining for natural product discovery [https://www.sciencedirect.com/science/article/abs/pii/S0076687925001624]
- 23. Methods and options for the heterologous production of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9896020/]
- 24. Recent advances in the direct cloning of large natural ... [https://www.sciencedirect.com/science/article/pii/S2667370323000176]
- 25. Refactoring biosynthetic gene clusters for heterologous ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8238852/]
- 26. Streptomyces as a versatile host platform for heterologous ... [https://pubs.rsc.org/en/content/articlehtml/2025/np/d5np00036j]
- 27. Heterologous overproduction of oviedomycin by refactoring ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-023-02218-8]
- 28. Enabling Access to Novel Bacterial Biosynthetic Potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11876861/]
- 29. A highly efficient heterologous expression platform to facilitate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12076808/]
- 30. Recent Advances in Heterologous Protein Expression and ... [https://www.mdpi.com/2309-608X/11/7/534]
- 31. Engineering Biology to Construct Microbial Chassis for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7037952/]
- 32. Challenges and advances in the heterologous expression of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4212100/]
- 33. Recent Advances in Silent Gene Cluster Activation ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2021.632230/full]
- 34. Advancing recombinant protein production by bacteria [https://www.sciencedirect.com/science/article/pii/S2590262825000735]
- 35. A flexible, modular and versatile functional part assembly ... [https://www.sciencedirect.com/science/article/pii/S2405805X23001102]
- 36. Bioprospecting Through Cloning of Whole Natural Product ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2020.00526/full]
- 37. Transformation-associated recombination (TAR) cloning ... [https://www.oncotarget.com/article/28546/text/]
- 38. Direct cloning and heterologous expression of natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6555405/]
- 39. A rapid combinatorial assembly method for gene cluster ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492648/]
- 40. Assembly of ascovirus HvAV-3h long DNA fragment using the ... [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/s12896-025-00964-8]
- 41. Application of Synthetic Biology to the Biosynthesis ... [https://www.sciepublish.com/article/pii/248]
- 42. The development of Burkholderia bacteria as heterologous ... [https://pubs.rsc.org/en/content/articlehtml/2025/np/d5np00024f]
- 43. Transformation-associated recombination and ... [https://link.springer.com/article/10.1007/s42995-025-00296-8]
- 44. Strategic engineering for overproduction of oviedomycin, a ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717625000485]
- 45. Identification and heterologous expression of an NRPS ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02753-6]
- 46. Recent Advances in Heterologous Protein Expression and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12299000/]
- 47. In-Situ Purification of Non-Ribosomal Peptide Synthetases ... [https://www.mdpi.com/1422-0067/26/4/1750]
- 48. Cell-free protein synthesis for nonribosomal peptide ... [https://www.frontiersin.org/journals/natural-products/articles/10.3389/fntpr.2024.1353362/full]
- 49. Rationally reduced libraries for combinatorial pathway ... [https://www.nature.com/articles/ncomms11163]
- 50. A plug-and-play pathway refactoring workflow for natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5500230/]
- 51. Towards revealing the structure of bacterial inclusion bodies [https://pmc.ncbi.nlm.nih.gov/articles/PMC2802778/]
- 52. Bacterial Inclusion Bodies: Discovering Their Better Half [https://www.sciencedirect.com/science/article/abs/pii/S0968000417300269]
- 53. Green genes from blue greens: challenges and solutions to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12415834/]
- 54. Heterologous expression facilitates the discovery and ... [https://www.sciencedirect.com/science/article/pii/S2667370323000693]
- 55. Metabolic engineering and cultivation strategies for ... [https://link.springer.com/article/10.1007/s00253-025-13441-1]
- 56. Metabolic Engineering Strategies for Enhanced ... [https://www.mdpi.com/2073-4360/17/15/2104]
- 57. Recent advances in metabolic engineering of Escherichia ... [https://pubmed.ncbi.nlm.nih.gov/40999135/]
- 58. Cofactor engineering for improved production of 2,4- ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1504785/full]
- 59. New automated method increases the efficiency of bioactive ... [https://www.igb.illinois.edu/leakey/article/new-automated-method-increases-efficiency-bioactive-natural-product-discovery]
- 60. Comparative Analysis of Codon Optimization Tools [https://pmc.ncbi.nlm.nih.gov/articles/PMC12010093/]
- 61. GeneOptimizer Process for Successful Gene Optimization [https://www.thermofisher.com/us/en/home/life-science/cloning/gene-synthesis/geneoptimizer.html]
- 62. Deep generative optimization of mRNA codon sequences ... [https://www.nature.com/articles/s41467-025-64894-x]
- 63. MNDL Bio | Optimization of Gene Expression [https://www.mndl.bio/]
- 64. Codon Optimization Tools: What to Look for in 2025 [https://synapse.patsnap.com/article/codon-optimization-tools-what-to-look-for-in-2025]
- 65. Comparative mass spectrometry-based metabolomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5214543/]
- 66. Challenges and Opportunities of Metabolomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6309313/]
- 67. Current approaches and challenges for the metabolite ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967314017038]
- 68. Can NMR solve some significant challenges in metabolomics? [https://pmc.ncbi.nlm.nih.gov/articles/PMC4646661/]
- 69. Integrating NMR and MS for Improved Metabolomic Analysis [https://www.mdpi.com/1420-3049/30/12/2624]
- 70. 1H-NMR-based metabolomic profiling and proteomic ... [https://www.sciencedirect.com/science/article/pii/S2405844024134974]
- 71. Development of an efficient heterologous protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12236015/]
- 72. Development of a versatile chassis for the efficient ... [https://www.nature.com/articles/s41467-025-62659-0]
- 73. Heterologous expression and optimization of the antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12535540/]
- 74. Maximizing Heterologous Expression of Engineered Type I ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10661030/]
- 75. Optimizing the dynamics of protein expression [https://www.nature.com/articles/s41598-019-43857-5]
- 76. Activation of microbial secondary metabolic pathways [https://pmc.ncbi.nlm.nih.gov/articles/PMC6190515/]
- 77. Comparative Genomics Reveals a Remarkable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8407293/]
- 78. Comparative genomics reveals phylogenetic distribution ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-4809-4]
- 79. Genomic insights into biosynthetic gene cluster diversity ... [https://www.nature.com/articles/s41598-025-21523-3]
- 80. Site-directed mutagenesis [https://en.wikipedia.org/wiki/Site-directed_mutagenesis]
- 81. Site-Directed Mutagenesis of Large Biosynthetic Gene Clusters via... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8142379/]