Orthogonal Transcription Factor Systems: Engineering, Applications, and Validation for Advanced Genetic Control
This article provides a comprehensive evaluation of orthogonal transcription factor (TF) systems, a cutting-edge toolset in synthetic biology for decoupling genetic circuits from host regulatory networks.
Orthogonal Transcription Factor Systems: Engineering, Applications, and Validation for Advanced Genetic Control
Abstract
This article provides a comprehensive evaluation of orthogonal transcription factor (TF) systems, a cutting-edge toolset in synthetic biology for decoupling genetic circuits from host regulatory networks. Aimed at researchers, scientists, and drug development professionals, it explores the foundational principles of orthogonal TFs, including bacterial σ54 factors and phage-derived RNA polymerases. The scope covers methodological advances in their design and deployment, strategies for troubleshooting and optimizing system performance, and rigorous validation across diverse bacterial chassis. By synthesizing recent breakthroughs, this review serves as a critical resource for the application of these systems in programming complex cellular functions, from metabolic engineering to intelligent drug development.
Core Principles and the Expanding Universe of Orthogonal Transcription
In synthetic biology, orthogonality refers to the design of genetic systems that operate independently of the host cell's native regulatory networks. This decoupling is crucial for ensuring that engineered gene circuits function predictably and robustly without interference from host processes or unintended impact on host viability. The pursuit of orthogonality has become a central theme in advancing therapeutic applications, including gene and cell therapy, where precise control over genetic output is paramount [1]. This guide provides a comparative evaluation of current orthogonal transcription factor (TF) systems, detailing their performance metrics, experimental methodologies, and key reagent solutions for research and development.
Comparative Analysis of Orthogonal Genetic Systems
The table below summarizes the core characteristics and performance data of four major classes of orthogonal genetic systems, highlighting their key features and experimentally measured orthogonality.
Table 1: Comparison of Orthogonal Genetic Systems for Synthetic Biology
| System Type | Core Components | Mechanism of Orthogonality | Key Performance Metrics | Reported Advantages |
|---|---|---|---|---|
| σ54 Factor Variants [2] | Engineered σ54 (R456H/Y/L), cognate promoters, bEBPs | Rewired promoter recognition specificity; requires activation by bEBPs. | Ideal mutual orthogonality between variants; specific transcription demonstrated in 3+ non-model bacteria. | Eukaryotic-like regulation; low basal leakage; high fold change upon induction. |
| λ cI TF Variants [3] | Engineered λ cI repressor/activator variants, synthetic bidirectional promoters. | Modified protein-DNA binding specificity to engineered operator sites. | 12 orthogonal TFs operating on up to 270 synthetic promoters. | Flexible operation as activators/repressors; slots into existing projects. |
| Phage RNAP Mutators (OTM) [4] | Deaminase-MmP1/K1F/VP4 RNAP fusions, orthogonal phage promoters. | Phage polymerase specificity for its own promoter; targeted mutagenesis. | >1,500,000-fold increased mutation rates; high specificity (minimal off-target effects). | Accelerated protein evolution; broad-host-range functionality. |
| Prime TF Reporters [5] | Optimized synthetic DNA response elements, minimal core promoters. | Optimized TF-specific response elements with minimal cross-reactive motifs. | High sensitivity & specificity for 62 TFs; outperform available reporters in >80% of comparisons. | Direct, multiplexed TF activity measurement; covers diverse signaling pathways. |
Experimental Protocols for Validating Orthogonality
Protocol for Testing σ54 Orthogonality
This protocol is adapted from studies that expanded the σ54-dependent transcription system [2].
- Step 1: Construct Generation. Clone mutant σ54 genes (e.g., R456H, R456Y, R456L) and their partnered promoter sequences into separate expression plasmids. A broad-host-range plasmid vector (e.g., pBBR-derived) is recommended for transferability testing.
- Step 2: Host Strain Preparation. Generate a ΔrpoN knockout strain in E. coli using λ-red homologous recombination to eliminate the native σ54 factor, providing a clean background.
- Step 3: Reporter Assay. Co-transform the σ54 variant and its corresponding promoter driving a fluorescent reporter (e.g., GFP) into the ΔrpoN host. Include controls with non-cognate σ54-promoter pairs.
- Step 4: Specificity Measurement. Quantify fluorescence output to assess activation by the cognate pair. Measure the fold-change difference in output between cognate and non-cognate pairs to quantify orthogonality. A high signal from the cognate pair with minimal background from non-cognate pairs indicates strong mutual orthogonality.
- Step 5: Cross-Activation Test. Repeat the assay in multiple bacterial species (e.g., Klebsiella oxytoca, Pseudomonas fluorescens) to validate the transferability of the orthogonal system.
Protocol for Profiling TF Activity with Prime Reporters
This protocol outlines the use of massively parallel reporter assays (MPRAs) for evaluating orthogonal TF reporters [5].
- Step 1: Library Design and Cloning. Design a library of reporter constructs for each TF. Systematically vary key features, including the number of TF binding sites, spacer sequences and length between sites, and the core promoter sequence. Clone these constructs into a plasmid library, each associated with a unique DNA barcode.
- Step 2: Cell Transfection and Perturbation. Transfer the reporter library into the target cell line. Subject the cells to a wide array of TF perturbation conditions—including TF knockouts, overexpression, and pathway stimulation—to activate diverse TFs.
- Step 3: Barcode Sequencing and Analysis. After a set period, extract cellular RNA and sequence the barcode regions. The abundance of each barcode in the RNA pool serves as a direct measure of the transcriptional activity of its associated reporter.
- Step 4: Sensitivity and Specificity Calculation. For a given TF perturbation, the sensitivity of a reporter is its level of activation. Specificity is determined by calculating the ratio of its activity under its cognate TF perturbation versus its activity under all other non-cognate perturbations. The reporters with the highest sensitivity and specificity are designated "prime" reporters.
System Workflows and Mechanisms
The following diagrams illustrate the core architectures and functional workflows of two primary orthogonal systems.
σ54-Dependent Orthogonal Transcription
Diagram Title: σ54 Orthogonal System Activation
λ cI-Based Orthogonal Logic Gate
Diagram Title: λ cI Bidirectional Logic Gate
The Scientist's Toolkit: Essential Research Reagents
The table below catalogs key reagents and their functions essential for designing and testing orthogonal genetic systems.
Table 2: Key Research Reagent Solutions for Orthogonal System Development
| Reagent / Tool | Function in Research | Specific Example / Note |
|---|---|---|
| Engineered σ Factors [2] | Provides promoter recognition specificity orthogonal to native host σ factors. | σ54 variants (R456H, R456Y, R456L) with distinct, non-cross-reacting promoter preferences. |
| Bacterial Enhancer-Binding Proteins (bEBPs) [2] | Required activator for σ54-dependent transcription; enables AND-gate logic. | Proteins like NifA; can be regulated by environmental or chemical signals for inducible control. |
| Orthogonal Transcription Factors [3] | Engineered DNA-binding proteins that regulate synthetic promoters without affecting native genes. | λ cI variant TFs that can function as activators, repressors, or dual-function switches. |
| Synthetic Promoter Libraries [5] [3] | DNA sequences containing optimized binding sites for orthogonal TFs, driving expression of downstream genes. | Bidirectional promoters for λ cI; promoters with varied spacer sequences for tuning output strength. |
| Phage RNA Polymerases [4] | Provides orthogonal transcription and a platform for targeted, in vivo mutagenesis systems. | MmP1, K1F, VP4 RNAPs; can be fused to deaminases (e.g., PmCDA1) for continuous evolution. |
| Prime TF Reporters [5] | Optimized DNA reporter constructs to directly and sensitively measure the activity of specific TFs in living cells. | A collection of 62 highly specific reporters for TFs from pathways like MAPK, PI3K/AKT, and TGF-β. |
| Conditional Phage/Phagemid Systems [3] | A selection platform for evolving new orthogonal TF-promoter pairs inside host cells. | M13 phagemid system linking TF activity to essential phage gene (gVI) production for enrichment. |
In the pursuit of predictable and customizable genetic circuits in synthetic biology, the concept of orthogonality—where a system operates without crosstalk with the host's native processes—is paramount. Among the various molecular tools, bacterial sigma factors represent a primary mechanism for promoter recognition and transcription initiation. The σ54 factor, a specialized alternative sigma factor, occupies a unique regulatory niche distinct from the housekeeping σ70 factor. Its inherent biological characteristics, including stringent dependence on activator proteins and distinct promoter recognition sequences, provide a native and robust platform for orthogonal design [6] [7]. This guide objectively evaluates the performance of σ54-based orthogonal systems against other prevalent transcriptional regulators, providing a foundational resource for researchers and drug development professionals engaged in engineering complex biological systems.
Comparative Analysis of Orthogonal Transcriptional Systems
The table below provides a quantitative comparison of σ54-based systems against other common transcriptional regulators used in synthetic biology, highlighting key performance metrics.
Table 1: Performance Comparison of Orthogonal Transcriptional Systems
| System Feature | σ54-Dependent System | σ70-Dependent System (e.g., TetR, LacI) | AraC/PBAD System | CRISPRa (σ54-based) |
|---|---|---|---|---|
| Native Regulation | Stress responses, nitrogen metabolism [6] | Housekeeping & stress responses [6] | Carbon metabolism [8] | N/A (fully engineered) |
| Core Promoter Recognition | -24 / -12 (GG-N10-GC) [6] | -35 / -10 (TTGACA-N17-TATAAT) [6] | -35 / -10 site [8] | σ54-dependent promoter [9] |
| Activation Mechanism | ATP-dependent remodeling by bEBPs [6] | Spontaneous isomerization [6] | Conformational change in AraC [8] | Engineered dCas9-PspF fusion [9] |
| Typical Dynamic Range | > 1,000-fold [9] | ~10-100 fold | ~7-fold improvement possible [8] | > 1,000-fold [9] |
| Key Orthogonality Feature | Distinct promoter sequence & energy requirement [6] [10] | Operator sequence engineering | Operator sequence engineering | sgRNA-programmable UAS targeting [9] |
| Demonstrated Orthogonal Pairs | 3 (σ54-R456H, R456Y, R456L) [10] | Multiple (e.g., TetR, LacI) | Limited by host AraC | 1 (PspF-based) [9] |
Structural and Functional Basis for σ54 Orthogonality
Fundamental Mechanisms of σ54-Dependent Transcription
Unlike σ70-dependent transcription, which can often proceed spontaneously, σ54-dependent transcription is inherently locked in a closed complex. This complex requires ATP-dependent remodeling by specialized bacterial enhancer-binding proteins (bEBPs) to isomerize into an open complex capable of initiation [6]. This energy-dependent switch provides a fundamental layer of control not present in other systems. Structurally, σ54 achieves this inhibition through its Region I (RI) and an extra-long helix (ELH) in Region III, which physically block the DNA entry channel of RNA polymerase, preventing spontaneous DNA opening [6].
Orthogonal Engineering of the σ54-Promoter Interface
The primary strategy for creating orthogonal σ54 systems involves rewiring the specific interaction between the sigma factor and its target promoter. The RpoN domain within σ54's Region III is responsible for recognizing the conserved -24/-12 promoter element [6]. Research has demonstrated that targeted mutations in this domain, particularly at residue R456, can rewire promoter specificity. For instance, the mutations σ54-R456H, R456Y, and R456L were shown to create three mutually orthogonal sigma factor variants, each with distinct promoter preferences and minimal crosstalk with each other or the native σ54 [10]. This orthogonality was successfully transferred into three non-model bacteria, showcasing its robustness and broad-host applicability [10].
Table 2: Key Research Reagent Solutions for σ54 System Engineering
| Reagent / Method | Function in Research | Application Example |
|---|---|---|
| Bacterial Enhancer-Binding Proteins (bEBPs) | AAA+ ATPases that remodel σ54-RNAP closed complex; can be engineered for orthogonal control [6] [7] | Nla28 from M. xanthus used to activate natural product gene promoters [7] |
| Machine Learning (BT Model) | Computational tool to identify critical residue regions (CRRs) in transcription factors for engineering [11] | Narrowed down 669 residues in BmoR to 36 key residues for achieving strict signal molecule orthogonality [11] |
| Sort-Seq | Massively parallel reporter assay to map regulatory sequences at nucleotide resolution [8] | Used to characterize and improve arabinose (PBAD) and rhamnose (PRha) inducible promoters [8] |
| Orthogonal σ54 Variants (e.g., R456H/Y/L) | Engineered sigma factors with rewired promoter specificity for orthogonal gene circuits [10] | Used to orthogonalize complex biological pathways and genetic circuits in multiple bacterial hosts [10] |
| PspFΔHTH::λN22plus | A truncated, modular activation domain from bEBP PspF for synthetic systems [9] | Fused to RNA-binding peptides in eukaryote-like CRISPRa systems to activate σ54-dependent promoters [9] |
Experimental Protocols for Engineering and Validation
Protocol 1: Engineering Orthogonal σ54-Promoter Pairs
This protocol is adapted from the method used to create the orthogonal σ54-R456 mutants [10].
- Target Identification: Based on structural knowledge, target the RpoN domain, specifically the residues involved in -24/-12 promoter recognition (e.g., R456) [6] [10].
- Saturation Mutagenesis: Perform site-directed mutagenesis on the key residue (e.g., R456) to generate a library of σ54 variants.
- Promoter Library Design: In parallel, create a library of promoter variants with mutations in the -24/-12 region.
- Combinatorial Screening: Co-transform the σ54 variant library and promoter library into a reporter strain (e.g., carrying GFP) and use fluorescence-activated cell sorting (FACS) to isolate pairs that show strong activation only when matched.
- Orthogonality Validation: Test the top-performing σ54-promoter pairs against each other and the native system to confirm minimal cross-activation.
Protocol 2: Measuring Orthogonality and Dynamic Range
This protocol is used to quantitatively assess the performance of engineered systems [10] [9].
- Strain Construction: Create reporter strains where the expression of a fluorescent protein (e.g., GFP) is under the control of the orthogonal promoter.
- Transformation: Introduce the plasmid encoding the orthogonal σ54 variant and its cognate bEBP (if required) into the reporter strain.
- Culture and Induction: Grow bacterial cultures to mid-log phase and, if using an inducible bEBP, add the specific inducer.
- Flow Cytometry: Measure the fluorescence intensity of thousands of individual cells using a flow cytometer. The mean fluorescence of the induced population indicates the ON state.
- Data Analysis: Calculate the dynamic range as the ratio of the mean fluorescence in the fully induced (ON) state to the non-induced or non-cognate (OFF) state. Orthogonality ratio is calculated as the ON signal with the cognate factor divided by the ON signal with a non-cognate factor.
Diagram 1: Orthogonal System Engineering Workflow
Advanced Applications and Comparative Performance
σ54 in CRISPR-Based Activation Systems
The unique "eukaryote-like" properties of σ54 have been leveraged to create highly effective CRISPR activation (CRISPRa) systems. In one design, a truncated activation domain of the bEBP PspF (PspFΔHTH) was fused to an RNA-binding peptide (λN22plus) and recruited to a dCas9-sgRNA complex. This system, which targets σ54-dependent promoters, demonstrated a dynamic range exceeding 1000-fold and could be programmed for multi-input regulation due to the long-distance action of bEBPs [9]. This performance significantly surpasses that of early σ70-based CRISPRa systems, which were limited by the need for the dCas9 complex to be near the core promoter and often showed lower dynamic ranges [9].
Regulation of Natural Product Biosynthesis
σ54 systems are directly implicated in regulating bacterial natural product genes, which are crucial sources of therapeutics. In Myxococcus xanthus, σ54 promoters, activated by specific bEBPs like Nla28, control the expression of polyketide and non-ribosomal peptide gene clusters [7]. This regulatory link often ties natural product synthesis to changes in nutritional status, providing a native paradigm for using orthogonal σ54 systems to dynamically control the expression of biosynthetic pathways in response to engineered signals.
Diagram 2: σ54 Transcription Activation Pathway
The σ54 factor provides a uniquely powerful and native foundation for constructing orthogonal transcriptional systems in bacteria. Its inherent separation from σ70-driven housekeeping transcription, coupled with its mandatory requirement for ATP-dependent activation, offers layers of control that are difficult to achieve with other systems. Quantitative data confirms that well-engineered σ54 systems can achieve dynamic ranges exceeding 1000-fold, support multiple orthogonal channels within a single cell, and function across diverse bacterial species [10] [9]. While σ70-based systems and classical repressor-based switches (like TetR/LacI) remain useful for many applications, the σ54 paradigm is particularly superior for applications demanding ultra-low leakage, high-level expression, and complex, multi-input logic. As synthetic biology continues to move into non-model chassis and demand more sophisticated genetic circuitry, the σ54 system, especially when combined with modern tools like machine learning and CRISPR, is poised to be an indispensable component of the genetic engineer's toolkit.
Orthogonal transcription systems, which function independently of the host's native transcriptional machinery, are foundational tools in synthetic biology. These systems enable precise control over gene expression for applications ranging from fundamental research to industrial bioproduction and therapeutic development. Bacteriophage-derived RNA polymerases represent the most established and widely adopted orthogonal transcription systems. Among them, the T7 RNA polymerase (T7RNAP) system is considered the gold standard, prized for its high transcriptional activity and exceptional specificity for its cognate promoter. T7RNAP exhibits a transcriptional rate approximately five-fold higher than that of native Escherichia coli RNA polymerase [12]. Furthermore, its operation as a single-subunit enzyme—requiring no additional protein factors for function—simplifies its deployment across biological chassis [12].
The core principle of orthogonality ensures that the phage polymerase and its associated promoter sequence interact minimally with the host's regulatory networks. This allows synthetic genetic circuits to operate without unintended crosstalk, facilitating the predictable engineering of microbial cell factories (MCFs), advanced biosensors, and sophisticated gene expression controls. While T7RNAP has dominated the field for decades, recent research has significantly expanded the synthetic biology toolbox. The discovery and engineering of alternative phage polymerases, such as MmP1, K1F, and VP4, now provide a broader palette of orthogonal systems. These alternatives are vital for overcoming limitations of the T7 system, particularly its restricted host range and inefficient performance in non-model organisms, thereby unlocking new possibilities for genetic manipulation across diverse bacterial species [4].
Comparative Analysis of Phage Polymerases
This section provides a detailed, data-driven comparison of the performance characteristics of T7RNAP and other emerging orthogonal phage RNA polymerases, highlighting their specific advantages and suitable application contexts.
Table 1: Key Characteristics of Orthogonal Phage RNA Polymerases
| Polymerase | Primary Hosts | Transcription Rate | Key Advantages | Documented Limitations |
|---|---|---|---|---|
| T7 RNAP | E. coli, Eukaryotes (with engineering) | ~5x native E. coli RNAP [12] | High specificity and processivity; vast established toolkit (e.g., pET systems); enables dynamic control & biosensing [12]. | Inefficient in many non-model organisms [4]; uncapped transcripts in eukaryotes limit utility [13]. |
| MmP1 RNAP | H. bluephagenesis, E. coli, P. entomophila [4] | Efficient transcription in non-model hosts [4] | High orthogonality; functions in non-model organisms like Halomonas; enables mutagenesis in new chassis [4]. | Lower baseline recognition in scientific community; toolkit less mature than T7. |
| K1F RNAP | H. bluephagenesis, E. coli, C. testosteroni [4] | Efficient transcription in non-model hosts [4] | Broad-host-range capability; high orthogonality; part of a modular system with other phage RNAPs [4]. | Similar to MmP1, requires further characterization and adoption. |
| VP4 RNAP | H. bluephagenesis, E. coli, P. putida [4] | Efficient transcription in non-model hosts [4] | Broad-host-range capability; high orthogonality; enables new application spaces for in vivo evolution [4]. | Similar to MmP1 and K1F. |
Table 2: Quantitative Performance Metrics in Key Applications
| Application | Polymerase | Performance Metric | Result | Experimental Context |
|---|---|---|---|---|
| In Vitro mRNA Synthesis | T7RNAP (Wild Type) | mRNA Yield | 2-5 g L⁻¹ (standard); up to 12-14 g L⁻¹ (optimized) [14] | IVT reaction with optimized AT-rich downstream sequences [14]. |
| dsRNA Byproduct | Up to 30% reduction vs. wild-type promoter [14] | Using promoters with AT-rich insertions [14]. | ||
| In Vivo Mutagenesis | T7RNAP (MutaT7) | Mutation Frequency Increase | >80,000-fold vs. control [4] | C:G to T:A mutations in E. coli [4]. |
| MmP1 RNAP (pMT2-MmP1) | Mutation Frequency Increase | >80,000-fold vs. control [4] | C:G to T:A mutations in H. bluephagenesis; mutation rate of 2.9 x 10⁻⁵ s.p.b. [4]. | |
| Orthogonal Gene Expression in Eukaryotes | Evolved T7RNAP-Capping Enzyme Fusion | Protein Expression Increase | ~100x (two orders of magnitude) vs. wild-type T7RNAP [13] | Directed evolution in yeast; validated in mammalian cells [13]. |
The data reveal that T7RNAP remains unparalleled for applications in E. coli and cell-free systems, offering a combination of high yield, speed, and well-characterized behavior. Its use in cell-free protein synthesis can yield up to 2.3 mg/mL of a model protein like eGFP [12], and recent promoter engineering has pushed IVT mRNA yields to 14 g L⁻¹ [14]. However, a significant limitation of IVT with T7RNAP is the co-production of immunostimulatory double-stranded RNA (dsRNA) byproducts. Engineering efforts, including rational design of the polymerase and optimization of the promoter template, have successfully reduced these dsRNA levels by up to 30%, thereby improving the safety profile and purity of mRNA vaccines and therapeutics [12] [14].
For non-model organisms, the T7 system shows pronounced limitations. For example, in the industrially promising Halomonas bluephagenesis, T7RNAP failed to effectively transcribe genes downstream of its cognate promoter [4]. This critical shortcoming has driven the adoption of broader-host-range alternatives. The MmP1, K1F, and VP4 phage RNA polymerases have demonstrated high orthogonality and efficient transcription in Halomonas and Pseudomonas, effectively bypassing the host-range restriction of T7 [4]. When deployed for in vivo targeted mutagenesis, these systems achieved mutation frequencies more than 80,000-fold higher than controls, enabling rapid protein evolution in chassis previously intractable to such engineering [4].
In eukaryotic systems, the primary barrier for T7RNAP has been its inability to produce 5' methyl guanosine caps, which are essential for mRNA stability and translation. A groundbreaking solution involved fusing T7RNAP to a capping enzyme from the African swine fever virus and applying directed evolution in yeast. This engineered "Capping-T7" system resulted in variants exhibiting a 100-fold increase in protein expression compared to wild-type T7RNAP, finally enabling efficient, orthogonal, and programmable gene expression in both yeast and mammalian cells [13].
Experimental Protocols for Key Applications
Protocol: In Vitro Transcription (IVT) for High-Yield mRNA Production
This protocol, adapted from recent optimizations, details the production of mRNA using T7RNAP with a focus on maximizing yield while minimizing immunostimulatory byproducts [14].
- Template DNA Preparation: A linear DNA template is required, containing the T7 promoter sequence immediately upstream of the mRNA sequence to be transcribed. The mRNA sequence should include the 5' untranslated region (UTR), the open reading frame (ORF) for the antigen or protein of interest, the 3' UTR, and a poly(A) tail sequence. The template can be generated via PCR amplification or by linearizing a plasmid vector.
- Reaction Mixture Assembly: The IVT reaction is assembled in a nuclease-free tube on ice. A standard reaction mixture includes:
- Template DNA: 5-10 μg of purified linear DNA template.
- Nucleoside Triphosphates (NTPs): A final concentration of 5-10 mM of each NTP (ATP, CTP, GTP, UTP).
- T7 RNA Polymerase: Commercially available or purified, used according to manufacturer's specifications.
- Reaction Buffer: A proprietary buffer typically supplied with the enzyme, often containing components like Tris-HCl (pH 7.9), MgCl₂, spermidine, and dithiothreitol (DTT).
- Incubation and mRNA Synthesis: The reaction mixture is incubated at 37°C for 45 minutes to 2 hours. Recent optimizations show that high yields of up to 12-14 g L⁻¹ can be achieved within 45 minutes to 2 hours using promoters with specific AT-rich downstream sequences [14].
- DNase Treatment: After incubation, 1-2 units of DNase I (RNase-free) are added to the reaction to digest the template DNA, and the mixture is incubated for an additional 15 minutes at 37°C.
- mRNA Purification: The mRNA is purified from the reaction mixture using methods such as lithium chloride precipitation or chromatographic purification to remove enzymes, proteins, and short abortive transcripts. Critical attention is paid to removing dsRNA contaminants, which can be achieved with specialized purification kits or cellulose-based purification.
dot code for In Vitro Transcription (IVT) for High-Yield mRNA Production:
IVT Workflow for mRNA Production
Protocol: Targeted In Vivo Mutagenesis Using an Orthogonal Transcription System
This protocol describes the use of deaminase-fused phage RNAPs, such as the MmP1-based system, for directed evolution in non-model bacteria like H. bluephagenesis [4].
- Mutator Plasmid Construction: A plasmid is constructed to express a fusion protein comprising a cytosine deaminase (e.g., PmCDA1), a uracil glycosylase inhibitor (UGI), and a phage RNA polymerase (e.g., MmP1 RNAP). The fusion protein is expressed from an inducible promoter (e.g., IPTG-inducible Ptac). The UGI is included to prevent the repair of U:G mismatches, thereby increasing mutation efficiency.
- Target Plasmid Construction: The gene of interest (GOI) to be evolved is cloned into a separate plasmid under the control of the cognate phage promoter (e.g., PMmP1).
- Strain Transformation and Culture: The host strain (e.g., H. bluephagenesis) is co-transformed with both the mutator and target plasmids. The transformed cells are grown in an appropriate medium. Once the culture reaches the desired optical density, mutagenesis is induced by adding a defined concentration of IPTG (e.g., 0.5-1.0 mM).
- Mutation and Screening/Selection: The culture is allowed to grow for a specified mutagenesis period (e.g., 24 hours). During this time, the mutator protein is expressed, binds to the target promoter, and transcribes the GOI. The associated deaminase domain introduces point mutations (C:G to T:A) into the single-stranded DNA of the non-transcribed strand during transcription. Following mutagenesis, the population is subjected to a screening or selection process to identify clones with desired, improved phenotypes.
- Isolation and Characterization: Plasmid DNA is isolated from the selected clones, and the mutated GOI is sequenced to identify the causative mutations. The target plasmid can then be retransformed into a fresh host to confirm that the phenotype is linked to the GOI.
dot code for Targeted In Vivo Mutagenesis Using an Orthogonal Transcription System:
In Vivo Mutagenesis Workflow
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of orthogonal polymerase systems requires a suite of specialized reagents and genetic tools. The table below catalogues the essential components for designing and executing experiments with T7 and other phage polymerases.
Table 3: Essential Reagents for Phage Polymerase Research
| Reagent / Tool Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Polymerase Expression Systems | pET System (Chromosomal/plasmid-driven T7RNAP in E. coli) [12] | Provides controlled expression of T7RNAP for high-yield protein production. The classic system for recombinant expression. |
| Broad-Host-Range Mutator Plasmids (e.g., pSEVA241 with PmCDA1-UGI-MmP1) [4] | Enables targeted in vivo mutagenesis in non-model organisms like H. bluephagenesis and E. coli. | |
| Promoter & Template Variants | Wild-type T7 Promoter | The standard promoter sequence for initiating transcription with T7RNAP. |
| Engineered T7 Promoters (with AT-rich downstream insertions) [14] | Increase mRNA yields in IVT (up to 14 g L⁻¹) and reduce dsRNA byproduct formation by up to 30%. | |
| Orthologous Phage Promoters (PMmP1, PK1F, PVP4) [4] | Recognized by their respective orthogonal RNAPs (MmP1, K1F, VP4) for gene expression in broad hosts. | |
| Specialized Enzymes & Proteins | T7 RNA Polymerase (Wild-type and engineered variants) [12] [14] | The core catalyst for IVT and in vivo T7-based expression. Engineered variants reduce dsRNA byproducts. |
| Capping Enzyme-T7RNAP Fusion (Evolved variant) [13] | Enables efficient orthogonal gene expression in eukaryotic cells (yeast, mammalian) by producing capped mRNAs. | |
| Cytosine Deaminase-UGI-Phage RNAP Fusions (e.g., PmCDA1-UGI-MmP1) [4] | The core mutator protein for targeted in vivo evolution, introducing C:G to T:A transitions. | |
| Selection & Reporter Tools | Erythromycin Resistance Gene (ermC) with Inactivating Mutation [4] | A reporter gene for quantifying mutation frequency and efficiency in mutagenesis systems. |
| Fluorescent Proteins (sfGFP, EGFP) [14] [4] | Standard reporters for visualizing and quantifying gene expression and transcriptional activity. | |
| Critical Reaction Components | Nucleoside Triphosphates (NTPs), including modified NTPs (e.g., Pseudouridine) [12] | The building blocks for RNA synthesis. Modified NTPs are used to reduce immunogenicity of mRNA therapeutics. |
| In Vitro Transcription Buffer Systems | Provides optimal pH, ionic strength (Mg²⁺), and reducing agents (DTT) for T7RNAP activity. |
The landscape of orthogonal transcription systems has evolved from a single dominant platform, T7RNAP, into a rich ecosystem of complementary tools. T7RNAP continues to be the workhorse for applications within its effective host range, driven by continuous engineering that enhances its yield, fidelity, and controllability. The most significant advancements, however, lie in the expansion of this toolkit to overcome historical barriers. The development of broad-host-range phage polymerases like MmP1, K1F, and VP4 is a pivotal achievement, democratizing advanced synthetic biology techniques for non-model organisms with unique biotechnological value [4]. Simultaneously, the successful engineering of a T7-based system for eukaryotic orthogonality shatters a long-standing constraint, opening new avenues for therapeutic development and basic research in yeast and mammalian cells [13].
Future progress in this field will likely be driven by several key trends. The discovery and characterization of novel phage polymerases from environmental metagenomes will further expand the diversity of available systems. The engineering of polymerases with expanded substrate specificity to incorporate a wider range of non-canonical nucleotides will be crucial for advancing RNA therapeutics and creating new biomaterials. Furthermore, the integration of these orthogonal transcription engines with other powerful technologies, such as CRISPR-Cas for precise genome regulation, will continue to yield increasingly sophisticated and predictable genetic control systems [12]. As these tools become more refined, modular, and interconnected, they will profoundly accelerate the engineering of biological systems for drug discovery, sustainable manufacturing, and fundamental biological insight.
Predictable Operation, Low Basal Leakage, and High Inducibility
In the field of synthetic biology, the engineering of life is achieved through the rational design of genetic programs. A cornerstone of this endeavor is the development of orthogonal transcription factor systems, which are synthetic genetic controllers that operate independently of the host's native regulatory networks. The performance of these systems is critically evaluated based on key engineering metrics: predictable operation, which ensures consistent and reliable circuit performance; low basal leakage, which minimizes unintended gene expression in the absence of an inducer; and high inducibility, which provides a strong, clear signal upon activation. This guide objectively compares the performance of several advanced orthogonal systems, from bacterial sigma factors to mammalian synthetic receptors, providing researchers and drug development professionals with a data-driven framework for selecting the optimal system for their specific application, whether in bioproduction, therapeutic cell engineering, or fundamental biological research.
Performance Comparison of Orthogonal Systems
The quantitative performance of orthogonal systems varies significantly across different biological chassis and operational principles. The table below summarizes key performance data from recent studies to enable direct comparison.
Table 1: Performance Metrics of Selected Orthogonal Transcription Systems
| System Type | Organism | Key Inducer/Activator | Fold Induction (Dynamic Range) | Reported Basal Leakage | Key Performance Features |
|---|---|---|---|---|---|
| Orthogonal σ54 Factors [2] | E. coli | Bacterial Enhancer-Binding Proteins (bEBPs) | High (Precise data not given) | "Stringently regulated" and "strongly activated" | Excellent mutual orthogonality; transferable across bacterial species; AND-gate logic capability. |
| Small Molecule-Inducible Systems [15] | Mouse Embryonic Stem Cells (mESCs) | 4OHT, ABA, GZV | Strong (Precise data not given) | "Minimal leakage" and "low background activation" | Titratable control; discrete (ON/OFF) or continuous response modes; functional in pluripotent stem cells. |
| TcpPH-EMeRALD Sensor [16] | E. coli | Taurocholic Acid (TCA) | 84.92-fold | Low (enabling high dynamic range) | High sensitivity (EC50: 28.344 μM); uses transmembrane sensor for extracellular cues. |
| NatE MESA Cytokine Receptors [17] | Mammalian T Cells | Cytokines (e.g., VEGF) | Varied by receptor design | "Low ligand-independent signal" in optimal designs | Orthogonal signaling; customizable for therapeutic sense-and-respond programs; can be multiplexed for logic. |
Experimental Protocols for Key Systems
Characterization of Small Molecule-Inducible Systems in mESCs
The protocol for characterizing the GAL4-UAS based inducible systems in mouse Embryonic Stem Cells (mESCs) provides a robust template for testing in mammalian cells [15].
- 1. Cell Line Engineering: A reporter mESC line (UAS-BFP mESCs) is first created by lentivirally transducing a cassette containing a 5x tandem GAL4 Upstream Activating Sequence (UAS) driving expression of mTagBFP2 (BFP). A constitutively expressed mCitrine enables sorting of successfully transduced cells.
- 2. Transduction with Transcription Factors: The reporter line is subsequently transduced with lentiviral vectors encoding the synthetic transcription factors. These factors consist of the GAL4 DNA-binding domain fused to a transactivation domain (VP16/VP64) and a regulatory domain (Ert2, ABI/Pyl, or NS3). A constitutive mCherry marker facilitates sorting of double-positive cells.
- 3. Induction and Flow Cytometry: Engineered cells are cultured for 3 days in the presence of titrated concentrations of the specific small-molecule inducers (4-Hydroxytamoxifen for Ert2, Abscisic Acid for ABI/Pyl, and Grazoprevir for NS3).
- 4. Data Analysis: Cells are analyzed by flow cytometry to quantify BFP fluorescence. Performance metrics, including dose-response curves, fold induction, and population distribution dynamics (discrete ON/OFF vs. continuous), are calculated from this data.
Establishing Orthogonality for Bacterial σ54 Systems
The methodology for validating the orthogonality of engineered σ54 factors in bacteria involves a systematic approach [2].
- 1. Strain Generation: An E. coli ΔrpoN knockout strain is constructed using λ-red homologous recombination to eliminate the native σ54 factor, providing a clean background.
- 2. Library Construction and Screening: Mutant libraries of rpoN (the gene encoding σ54) are generated, focusing on mutations at the R456 residue. These mutant σ54 factors are co-expressed with cognate promoter libraries carrying mutations in the -24 element.
- 3. Orthogonality Validation: The mutant σ54 factors and their partner promoters are tested in the ΔrpoN strain. Their ability to drive expression of a reporter gene (e.g., GFP) is measured both individually and in combination to assess cross-talk.
- 4. Cross-Species Transfer: The functional orthogonal pairs are then cloned into broad-host-range plasmids and transformed into non-model bacteria (Klebsiella oxytoca, Pseudomonas fluorescens, Sinorhizobium meliloti) to demonstrate transferability.
- 5. Circuit Implementation: The orthogonal systems are integrated into more complex genetic circuits, such as layered logic gates or synthetic metabolic pathways (e.g., sucrose utilization), to test their performance under applied conditions.
Signaling Pathways and Workflows
The following diagrams illustrate the core operational mechanisms of two major classes of orthogonal systems discussed in this guide.
Mammalian Inducible Transcription System
This diagram visualizes the mechanism of small molecule-inducible synthetic transcription factors in mammalian cells [15].
Bacterial Orthogonal σ54 System
This diagram depicts the unique, multi-component mechanism of orthogonal σ54-dependent transcription in bacteria [2].
Research Reagent Solutions
A successful experiment relies on key, well-characterized reagents. The following table details essential tools and materials for working with the orthogonal systems described [15] [2] [16].
Table 2: Essential Research Reagents for Orthogonal Transcription Systems
| Reagent / Solution | Function / Description | Example or Source |
|---|---|---|
| Orthogonal Promoter | A synthetic DNA sequence recognized specifically by an orthogonal transcription factor or polymerase, minimizing host crosstalk. | GAL4 Upstream Activating Sequence (UAS) [15]; Engineered σ54-dependent promoters [2] |
| Engineered Transcription Factor | The core protein component that binds the orthogonal promoter and activates transcription in a controlled manner. | GAL4-DBD fused to Ert2/ABI/Pyl/NS3 [15]; Mutant σ54 factors (e.g., R456H) [2] |
| Specific Inducer Molecules | Small molecules or proteins that trigger the activation of the orthogonal transcription system. | 4-Hydroxytamoxifen, Abscisic Acid, Grazoprevir [15]; Bacterial Enhancer-Binding Proteins (bEBPs) [2] |
| Reporter Genes | Genes with easily measurable outputs (e.g., fluorescence) used to quantify system performance. | mTagBFP2, sfGFP, mCherry [15] [16] |
| Delivery Vectors | Plasmids or viral vectors for stable and efficient delivery of genetic constructs into the target cell type. | Lentiviral vectors (mammalian cells) [15]; Broad-host-range plasmids (bacteria) [2] |
| Selection Markers | Genes that confer resistance to antibiotics or other selection pressures, enabling enrichment of successfully engineered cells. | Antibiotic resistance genes (e.g., Kanamycin, Ampicillin) [2] |
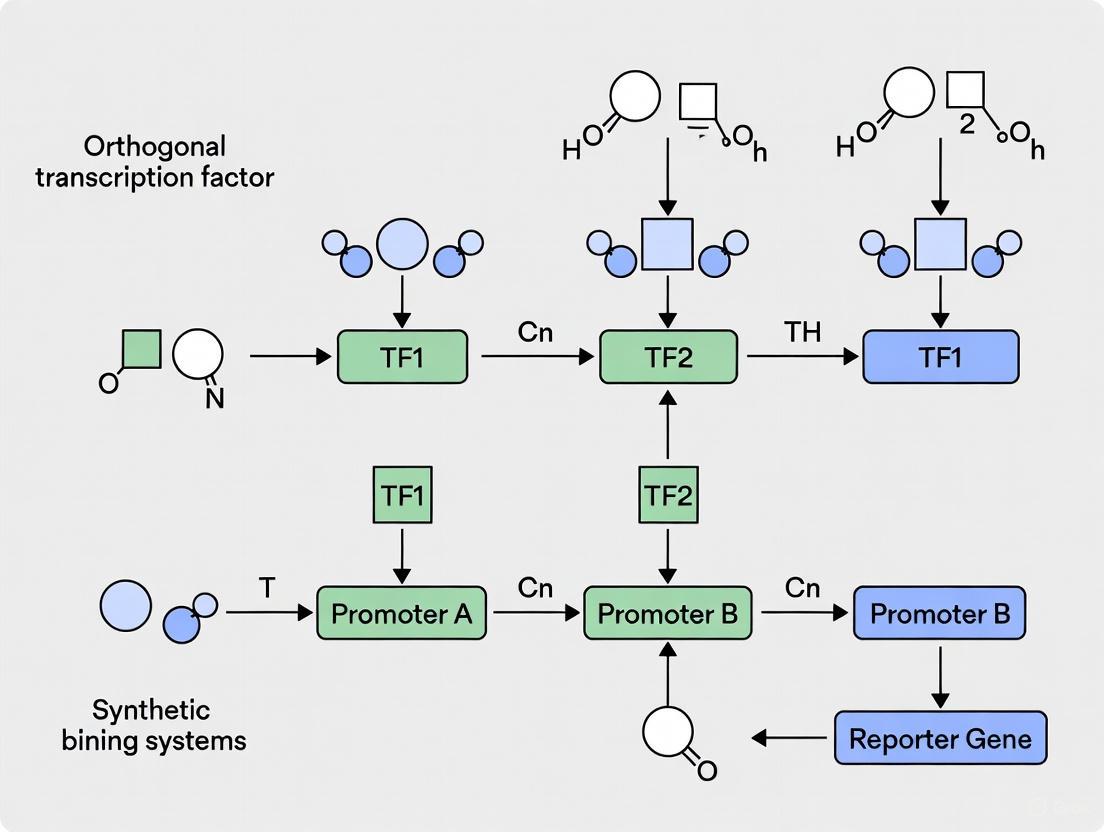
Bacterial enhancer-binding proteins (bEBPs) are a specialized class of AAA+ ATPases that function as transcriptional activators for genes dependent on the alternative sigma factor σ54 (also known as σN) [18]. Unlike the more common σ70-dependent transcription, σ54-dependent transcription exhibits a eukaryotic-like regulation mechanism where activator proteins are absolutely required for initiation [2] [18]. This dependency makes the σ54-bEBP partnership an attractive framework for engineering orthogonal genetic systems in synthetic biology [2] [10].
The σ54 factor itself recognizes distinct promoter sequences at the -12 (GG) and -24 (TGC) regions [18]. When bound to RNA polymerase (RNAP), σ54 forms a stable closed complex (RPc) that is transcriptionally inactive and cannot spontaneously isomerize to an open complex [18] [19]. This transition strictly requires the ATP-dependent remodeling activity of a bEBP, which interacts with the σ54-RNAP complex from binding sites typically located 100-150 base pairs upstream of the transcription start site [18] [19].
Structural and Mechanistic Basis of bEBP Function
Domain Architecture of bEBPs
bEBPs typically exhibit a conserved three-domain architecture:
- N-terminal regulatory domain (R): Often a receiver domain of a two-component system that senses environmental signals and regulates AAA+ domain activity.
- Central catalytic AAA+ domain (C): Responsible for ATP hydrolysis and mechano-chemical remodeling of the σ54-RNAP complex.
- C-terminal DNA binding domain (D): Contains a helix-turn-helix (HTH) motif that binds upstream activator sequences (UAS) [18].
These domains work cooperatively to ensure that transcription activation occurs only under appropriate environmental conditions sensed by the regulatory domain.
The AAA+ Domain and Conserved Motifs
The AAA+ domain of bEBPs belongs to the Helix-2-insert clade 6 of the AAA+ superfamily and contains several characteristic features [18]:
- Walker A and B motifs for nucleotide binding and hydrolysis
- Loop 1 (L1) containing the highly conserved GAFTGA motif essential for σ54 interaction
- Loop 2 (L2) that interacts with promoter DNA
The GAFTGA motif is particularly critical, as mutations in this sequence typically abolish transcription activation ability, either by impairing ATP hydrolysis, inter-subunit communication, or σ54 interaction [18].
Mechanism of Transcription Activation
Recent cryo-EM structures have captured snapshots of the conformational changes during σ54-mediated transcription initiation, revealing why bEBP activity is essential [18]. In the closed complex (RPc), σ54 regions I and III form a barrier that prevents DNA entry into the RNAP cleft [18].
bEBPs function as hexameric ring complexes that utilize ATP hydrolysis to remodel the σ54-RNAP complex through a mechanism involving:
- DNA looping facilitated by integration host factor (IHF), bringing distal bEBP binding sites proximal to the promoter
- Direct interaction between the bEBP AAA+ domain and σ54 region I
- Application of mechanical force via the GAFTGA-containing L1 loop to trigger σ54 conformational changes
- Promoter DNA melting and loading of the template strand into the RNAP active site
- Transition to open complex (RPo) competent for transcription initiation [18] [19]
This mechanism represents a sophisticated control system where transcription is tightly regulated at the isomerization step rather than RNAP binding, enabling stringent regulation and strong activation when environmental conditions dictate [2] [18].
bEBPs in Orthogonal Transcription System Engineering
Rationale for Orthogonality Engineering
The unique properties of σ54-dependent transcription make it particularly amenable to orthogonalization for synthetic biology applications. Key advantages include:
- Strict dependency on activator proteins prevents leaky expression
- Distinct promoter recognition patterns avoid crosstalk with host σ70 systems
- Strong activation potential enables high expression levels when required
- Natural stringency provides a framework for engineering predictable genetic circuits [2]
Orthogonal systems are defined by their ability to operate independently of host regulatory networks, enabling consistent and predictable performance of synthetic genetic circuits [2] [10].
Engineering Orthogonal σ54-bEBP Systems
Recent research has successfully engineered orthogonal σ54-bEBP systems through structure-guided rewiring of protein-DNA interaction interfaces. A key breakthrough involved modifying the RpoN box in σ54, which is responsible for recognizing the -24 promoter region [2] [10].
Liu et al. (2025) identified three orthogonal σ54 variants through knowledge-based screening and rational engineering:
Table 1: Orthogonal σ54 Variants and Their Properties
| σ54 Variant | Amino Acid Substitution | Promoter Preference | Orthogonality Performance |
|---|---|---|---|
| σ54-R456H | Arginine → Histidine | Altered -24 recognition | Ideal mutual orthogonality |
| σ54-R456Y | Arginine → Tyrosine | Altered -24 recognition | Ideal mutual orthogonality |
| σ54-R456L | Arginine → Leucine | Altered -24 recognition | Ideal mutual orthogonality |
These engineered σ54 factors exhibit distinct promoter preferences while maintaining strong mutual orthogonality toward each other and the native σ54 system [2] [10]. The orthogonality was demonstrated to be transferable across multiple bacterial species, including Klebsiella oxytoca, Pseudomonas fluorescens, and Sinorhizobium meliloti [2].
Applications in Genetic Circuit Design
The orthogonal σ54-bEBP systems have been successfully implemented in various synthetic biology applications:
- Layered logic gates with reduced crosstalk between circuit components
- Complex pathway orthogonalization to minimize metabolic burden and interference
- Environmental biosensing with customizable induction profiles
- Multi-input controllers for maintaining evolutionary longevity of synthetic circuits [2] [20]
When combined with different bEBPs, these orthogonal systems can control downstream outputs in response to environmental or chemical signals, enabling the construction of sophisticated genetic circuits with predictable computational performance [2].
Experimental Analysis of bEBP Systems
Key Experimental Methodologies
Research on bEBP structure and function employs several advanced techniques:
Table 2: Key Experimental Methods in bEBP Research
| Methodology | Application in bEBP Research | Key Insights Generated |
|---|---|---|
| Cryo-electron microscopy (cryo-EM) | Structural analysis of σ54-RNAP complexes at different activation states | Revealed conformational changes during closed-to-open complex transition [18] |
| High-throughput transcriptomics (HTTr) | Assessment of transcriptional activation potency and specificity | Enabled quantification of orthogonal system performance and detection of crosstalk [21] [22] |
| Genetic circuit characterization | Testing orthogonality and circuit performance in live cells | Demonstrated transferability of orthogonal systems across bacterial species [2] |
| Computational modeling & host-aware design | Predicting evolutionary longevity of synthetic circuits | Informed controller design to maintain circuit function despite mutation [20] |
Protocol for Orthogonal System Characterization
A standard protocol for assessing orthogonal σ54-bEBP system performance includes:
- Host strain preparation: Creation of ΔrpoN strains using λ-red homologous recombination to eliminate native σ54 background [2]
- Plasmid construction: Assembly of expression cassettes using Golden Gate assembly with orthogonal σ54 variants and cognate promoters
- Cross-activation testing: Transformation of σ54 variants with different promoter-reporter constructs to assess specificity
- Transcriptional output quantification: Measurement of reporter gene expression (e.g., GFP/RFP) under induced vs. uninduced conditions
- Orthogonality index calculation: Determination of activation specificity relative to non-cognate promoter pairs
- Host transfer validation: Testing orthogonal system performance in multiple non-model bacterial species [2]
This systematic approach enables comprehensive characterization of orthogonal system performance while identifying potential crosstalk between engineered components.
Comparative Performance of Orthogonal Systems
Quantitative Assessment of Orthogonal σ54 Variants
The performance of engineered orthogonal σ54-bEBP systems can be quantitatively evaluated across multiple metrics:
Table 3: Performance Metrics of Orthogonal σ54-bEBP Systems
| Performance Metric | σ54-R456H | σ54-R456Y | σ54-R456L | Native σ54 |
|---|---|---|---|---|
| Activation Fold-Change | High (comparable to native) | High (comparable to native) | Moderate to High | Reference level |
| Basal Expression | Low (stringent control) | Low (stringent control) | Low (stringent control) | Low (stringent control) |
| Promoter Specificity | High (altered -24 preference) | High (altered -24 preference) | High (altered -24 preference) | Native preference |
| Cross-talk with Native System | Minimal | Minimal | Minimal | N/A |
| Transferability Across Species | Demonstrated in 3 species | Tested in model systems | Tested in model systems | Species-dependent |
Advantages Over Alternative Orthogonal Systems
Compared to other orthogonal transcription systems like T7 RNA polymerase, σ54-bEBP systems offer distinct advantages:
- Lower cellular toxicity due to endogenous bacterial machinery
- Higher adjustability through bEBP regulation
- Natural stress response integration via bEBP sensing domains
- Flexible input control through diverse bEBP regulatory mechanisms [2]
However, potential limitations include the energy cost of AAA+ ATPase activity and the structural complexity of the multi-component activation mechanism.
Research Reagent Solutions Toolkit
Essential research tools for studying bEBPs and engineering orthogonal systems include:
Table 4: Essential Research Reagents for bEBP Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Bacterial Strains | E. coli ΔrpoN strains; Klebsiella oxytoca; Pseudomonas fluorescens; Sinorhizobium meliloti | Host organisms for orthogonal system testing and characterization [2] |
| Expression Vectors | pBBR-derived broad-host-range vectors; Golden Gate compatible plasmids | Genetic cargo delivery and modular circuit assembly [2] |
| Reporter Systems | GFP; RFP; metabolic markers (e.g., sucrose utilization cassettes) | Quantitative assessment of transcriptional activity and orthogonality [2] |
| bEBP Expression Constructs | NtrC; FleT; LasR; EsaR; engineered bEBP variants | Sources of activation function for σ54-dependent transcription [18] [23] |
| Promoter Libraries | Native σ54 promoters; engineered -24 variant promoters | Testing recognition specificity and orthogonal system performance [2] |
| Analytical Tools | Cryo-EM; HTTr; RNA-seq; growth rate assays | System characterization and mechanistic studies [18] [21] [22] |
Signaling Pathways and Experimental Workflows
bEBP Activation Mechanism
Orthogonal System Engineering Workflow
Future Directions and Applications
The engineering of orthogonal bEBP systems continues to evolve with several promising research directions:
- Expanded orthogonal sets through comprehensive mutagenesis of σ54-DNA interface
- Integration with CRISPR technologies for multi-layer genetic control
- Evolutionary stabilization of synthetic circuits using host-aware controller designs [20]
- Clinical and biotechnological applications in smart therapeutics and biosensing
- Cross-kingdom compatibility for broad-host-range genetic toolkits
The unique combination of stringency, strong activation potential, and engineerability positions σ54-bEBP systems as foundational tools for next-generation synthetic biology applications requiring precise transcriptional control.
Design, Assembly, and Functional Deployment in Synthetic Biology
The precise rewiring of protein-DNA interactions represents a frontier in synthetic biology, enabling the programming of custom gene regulatory networks for therapeutic and biotechnological applications. Central to this endeavor are orthogonal transcription systems—engineered biological components that function independently of the host's native machinery. These systems provide a powerful platform for directed evolution, allowing researchers to rapidly optimize protein-DNA binding specificities and affinities. The core principle involves creating a synthetic replication or transcription apparatus that operates in parallel to the cell's natural systems, facilitating targeted mutagenesis and selection without compromising host cell viability [24]. This approach has dramatically accelerated our ability to engineer novel transcription factors, DNA-binding proteins, and regulatory circuits with precision.
The emergence of tools like T7-ORACLE and the Orthogonal Transcription Mutation (OTM) system has transformed the protein engineering landscape. These systems address critical limitations of traditional directed evolution methods, which often involve laborious cycles of mutagenesis and screening. By harnessing error-prone viral polymerases and fusing them with deaminase enzymes, these platforms enable continuous hyper-mutation of target genes inside living cells, compressing evolutionary timelines from months to days while generating diverse mutational landscapes [24] [4]. This review comprehensively compares these pioneering technologies, their experimental performance, and their application in rewiring protein-DNA interactions for advanced research and therapeutic development.
Comparative Analysis of Orthogonal Mutagenesis Systems
The table below provides a systematic comparison of two leading orthogonal systems for evolving protein-DNA interactions, highlighting their distinct mechanisms and performance characteristics.
Table 1: Comparison of Major Orthogonal Systems for Rewiring Protein-DNA Interactions
| Feature | T7-ORACLE System | Orthogonal Transcription Mutation (OTM) System |
|---|---|---|
| Core Mechanism | Orthogonal T7 replisome with error-prone DNA polymerase in E. coli [24] | Deaminase-phage RNA polymerase fusion proteins [4] |
| Mutation Types | Broad spectrum (unspecified) | Transition mutations: C:G to T:A and A:T to G:C [4] |
| Mutation Rate Increase | 100,000-fold above natural mutation rate [24] | >1,500,000-fold above natural mutation rate [4] |
| Evolution Timeframe | Days (versus months for traditional methods) [24] | Single day (shortest reported period) [4] |
| Host Organisms | Escherichia coli [24] | E. coli and non-model organisms (e.g., Halomonas bluephagenesis) [4] |
| Key Innovation | Continuous hypermutation without host genome damage [24] | Modular design with three different phage RNAPs (MmP1, K1F, VP4) [4] |
| Specificity | Targets only plasmid DNA, host genome untouched [24] | High specificity with minimal off-target effects (5-14 fold increase vs. 154-fold with suboptimal construct) [4] |
Experimental Protocols for System Validation
T7-ORACLE Workflow and Validation
The T7-ORACLE system employs a meticulously engineered experimental workflow to achieve accelerated evolution of target proteins:
- Step 1: System Construction – An artificial DNA replication system derived from bacteriophage T7 is engineered into E. coli host cells. The key component is an error-prone T7 DNA polymerase that replicates only specific plasmid DNA, leaving the host genome intact [24].
- Step 2: Target Gene Cloning – The gene of interest (e.g., an antibiotic resistance gene, transcription factor, or enzyme) is cloned into a special plasmid containing the T7 origin of replication, making it susceptible to the error-prone polymerase [24].
- Step 3: Continuous Evolution – The bacterial culture is propagated under standard laboratory conditions. With each cell division (approximately every 20 minutes), the target gene undergoes replication by the error-prone polymerase, introducing random mutations. This creates a continuously evolving gene library inside the cells [24].
- Step 4: Selection and Screening – Cells are subjected to selective pressure relevant to the desired protein function. In the validation experiment, cells harboring the TEM-1 β-lactamase gene were exposed to escalating doses of antibiotics. Variants with mutations conferring enhanced resistance survived and proliferated [24].
- Step 5: Outcome Analysis – In the validation study, this process yielded enzyme variants capable of resisting antibiotic levels 5,000 times higher than the original within one week. The identified mutations closely mirrored those found in clinical antibiotic resistance, confirming the system's biological relevance [24].
OTM System Workflow and Validation
The Orthogonal Transcription Mutation system utilizes a different mechanism based on transcriptional mutagenesis, with a protocol adaptable to multiple organisms:
- Step 1: Mutator Plasmid Construction – Plasmids are constructed to express fusion proteins. These fusions combine a deaminase enzyme (e.g., PmCDA1 for C→T mutations) with an orthogonal phage RNA polymerase (MmP1, K1F, or VP4) via a flexible XTEN linker [4].
- Step 2: Target Plasmid Design – The gene to be evolved is placed under the control of a promoter specific to the chosen phage RNA polymerase (e.g., PMmP1 for MmP1 RNAP) on a separate plasmid [4].
- Step 3: In Vivo Mutagenesis – Both plasmids are co-transformed into the host organism (E. coli or H. bluephagenesis). Upon induction, the fusion protein is expressed. It then transcribes the target gene and simultaneously introduces mutations into the nascent DNA transcript. Co-expression of uracil glycosylase inhibitor (UGI) is critical for enhancing C→T mutation efficiency by preventing repair mechanisms [4].
- Step 4: Functional Screening – The population of mutated cells is screened for desired phenotypes. The OTM system was validated by evolving fluorescent proteins, chromoproteins, and a dysfunctional erythromycin resistance gene (ErmC Y104S). A C-to-T reversion mutation at a specific site successfully restored antibiotic resistance, demonstrating the system's precision and effectiveness [4].
- Step 5: Efficiency Quantification – Mutation frequency and rates are calculated using a mutation-recovery method. The most efficient OTM construct (PmCDA1-UGI-MmP1) achieved an on-target mutation frequency of 2.5 × 10⁻², over 80,000 times higher than the control, with a calculated mutation rate of 2.9 × 10⁻⁵ substitutions per base [4].
Supporting Tools: Computational Prediction of Protein-DNA Interactions
Complementing experimental evolution, computational tools are vital for predicting and analyzing rewired protein-DNA interactions. The Interpretable protein-DNA Energy Associative (IDEA) model is a notable biophysical tool that predicts DNA recognition sites and binding affinities for DNA-binding proteins like transcription factors [25].
- Principle: IDEA integrates 3D structures and sequences of known protein-DNA complexes to learn an interpretable energy model. This model quantifies the physicochemical interactions between individual amino acids and nucleotides, effectively deciphering the "molecular grammar" of binding specificity [25].
- Application: In one benchmark, the IDEA model, using only the structure of the MAX transcription factor (PDB ID: 1HLO), achieved a Pearson correlation coefficient of 0.67 with experimentally measured binding affinities. Performance improved further when integrated with experimental SELEX data, providing a powerful hybrid approach for guiding and interpreting mutagenesis results [25].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of orthogonal evolution systems requires a suite of specialized reagents and tools. The following table details key components for establishing these platforms in a research setting.
Table 2: Essential Research Reagents for Orthogonal Evolution Studies
| Reagent/Tool Name | Function in Experimental Workflow | Example Application/Note |
|---|---|---|
| Error-Prone T7 DNA Polymerase | Drives continuous mutagenesis of target plasmid in T7-ORACLE [24] | Engineered variant with high error rate; core of the orthogonal replisome. |
| Deaminase-Phage RNAP Fusion | Introduces transition mutations during transcription in OTM [4] | e.g., PmCDA1-UGI-MmP1; modular for different mutation types and hosts. |
| Orthogonal Origin of Replication | Confines mutagenesis to target plasmid, sparing host genome [24] | Derived from bacteriophage (e.g., T7 origin). |
| Uracil Glycosylase Inhibitor (UGI) | Enhances C→T mutation yield by blocking DNA repair [4] | Co-expressed with cytosine deaminase fusions. |
| IDEA Model | Computationally predicts binding sites & affinities of DNA-binding proteins [25] | Provides interpretable, residue-level energy predictions. |
| Reporter Plasmids | Carry selectable or screenable markers for functional selection [24] [4] | e.g., TEM-1 β-lactamase (antibiotic resistance) or sfGFP (fluorescence). |
The direct comparison of T7-ORACLE and the Orthogonal Transcription Mutation system reveals a dynamic and rapidly advancing field. T7-ORACLE excels with its very high mutation rate and operation in the widely adopted E. coli chassis, making it a robust tool for broad protein engineering projects. In contrast, the OTM system offers unparalleled speed and modularity, with the distinct advantage of functioning in non-model organisms, thus expanding the scope of synthetic biology applications.
The integration of these advanced experimental evolution platforms with powerful computational predictors like the IDEA model creates a powerful feedback loop. Researchers can not only rapidly generate novel protein-DNA interfaces but also understand the biophysical principles governing their interactions. This synergy between laboratory evolution and computational design is fundamentally advancing our capacity to rewire biological systems, paving the way for breakthroughs in gene therapy, drug development, and cellular engineering.
Library Construction and High-Throughput Screening for Orthogonal Pairs
The engineering of biological systems requires genetic components that operate predictably and independently from the host's native regulatory networks. This principle, known as orthogonality, ensures that synthetic genetic circuits perform their intended functions without undesired crosstalk or interference. Orthogonal transcription systems, particularly those based on transcription factors and RNA polymerases, have become indispensable tools for programming cells with sophisticated capabilities. These systems enable controlled gene expression, pathway optimization, and the development of complex genetic circuits for therapeutic and industrial applications.
This guide provides a comparative evaluation of contemporary platforms for constructing orthogonal genetic systems and conducting high-throughput screening. We examine three pioneering approaches that demonstrate how strategic design of protein-DNA interactions, phage RNA polymerase engineering, and innovative selection mechanisms can overcome historical limitations in predictability, efficiency, and scalability. For each platform, we present quantitative performance data, detailed experimental protocols, and practical implementation guidelines to assist researchers in selecting the most appropriate technology for their specific applications.
Comparative Performance Analysis of Orthogonal Systems
Table 1: Performance Metrics of Orthogonal Systems
| Platform | Orthogonality Mechanism | Mutation Rate/ Efficiency | Key Applications | Host Organisms | Throughput Capacity |
|---|---|---|---|---|---|
| Orthogonal σ54 System | Engineered σ54-R456 mutations with modified promoter specificity | N/A (Transcriptional control) | Genetic circuit orthogonalization, metabolic engineering | E. coli, K. oxytoca, P. fluorescens, S. meliloti | Moderate (Library screening) |
| Orthogonal Transcription Mutator (OTM) | Deaminase-phage RNAP fusions generating transition mutations | >1,500,000-fold increase vs control; 2.9 × 10⁻⁵ substitutions per base [4] | Protein evolution, metabolic engineering | E. coli, H. bluephagenesis | High (>10¹¹ variants) |
| PANCS-Binders | Split RNA polymerase reconstitution upon target binding | 10⁶-fold amplification for high-affinity binders; 10¹⁵-fold relative enrichment [26] | Binder discovery, protein-protein interaction engineering | E. coli (with mammalian cell validation) | Very High (>10¹¹ protein-protein pairs) |
Table 2: Specificity and Selectivity Metrics
| Platform | On-target Efficiency | Off-target Effects | Orthogonality Between Components | Binding Affinity Range |
|---|---|---|---|---|
| Orthogonal σ54 System | Ideal mutual orthogonality between σ54-R456H, R456Y, R456L variants [2] | Minimal basal leakage due to bEBP requirement [2] | High (transferable across species) | N/A (Transcriptional tool) |
| pMT2-MmP1 Mutator | 80,000-fold increase over control; 7.4 × 10⁻⁴ mutation frequency [4] | 5-14 fold increase vs control (high specificity) [4] | High orthogonality between phage polymerases | N/A (Mutation generation) |
| PANCS-Binders | 55-72% hit rate for novel binders across 95 targets [26] | High-fidelity selection with low false positives | Specific phage-target pairing | 206 pM - 8.4 nM (after maturation) [26] |
Orthogonal σ54-Dependent Transcription System
The σ54-dependent orthogonal transcription system represents a novel approach to decoupling synthetic genetic circuits from native host regulation. Unlike the major σ70 factor in bacteria, σ54 recognizes distinct promoter sequences and requires activation by bacterial enhancer-binding proteins (bEBPs), providing an additional layer of regulatory control [2]. This dependency creates a stringent OFF state with minimal basal leakage, making it particularly valuable for applications requiring precise temporal control. Researchers engineered this system through knowledge-based screening and rewiring of the RpoN box in σ54, identifying three key mutations (R456H, R456Y, and R456L) that exhibit ideal mutual orthogonality and distinct promoter preferences while maintaining compatibility with native bEBP activation mechanisms [2].
Experimental Protocol
Library Construction Method:
- Strain Preparation: Generate ΔrpoN E. coli JM109 strain using λ-red homologous recombination with Gm-resistant gene flanked by 60 bp homologous arms [2].
- σ54 Mutant Library: Create random mutation libraries for rpoN R456/R457 sites using inverse PCR with plasmids carrying σ54 sequences as templates.
- Promoter Engineering: Introduce random mutations in the −24 elements of σ54-dependent promoters via inverse PCR.
- Vector Assembly: Assemble expression cassettes using Golden Gate assembly with BpiI and BsaI restriction sites.
- Host Transfer: Clone orthogonal systems into pBBR-derived broad-host-range vectors for testing in non-model bacteria (K. oxytoca, P. fluorescens, S. meliloti).
Screening Protocol:
- Primary Screening: Transform σ54 mutant library into ΔrpoN strain with GFP reporter under wild-type or modified σ54 promoters.
- Orthogonality Assessment: Co-transform different σ54 mutant-promoter pairs to assess cross-activation using fluorescent reporters (GFP/RFP).
- bEBP Integration: Co-express different bEBPs (KoNifA, RcNifA) under Ptet promoter to test activation specificity.
- Quantification: Measure fluorescence intensity via flow cytometry to calculate orthogonality coefficients and activation folds.
Key Research Reagents:
- Bacterial Strains: E. coli JM109, ΔrpoN strains, K. oxytoca, P. fluorescens, S. meliloti
- Vectors: pSEVA derivatives, pBBR-derived broad-host-range vectors
- Promoters: σ54-dependent native and engineered promoters
- Induction Systems: Ptet-regulated bEBP expression
- Reporters: GFP, RFP, sucrose utilization genes (cscA, cscB, cscK)
Orthogonal Transcription Mutation (OTM) System
The Orthogonal Transcription Mutation system represents a breakthrough in targeted in vivo mutagenesis by fusing deaminase enzymes with phage RNA polymerases to create hypermutation machinery. This platform addresses critical limitations of previous targeted evolution methods by achieving unprecedented mutation rates—over 1,500,000-fold increases compared to controls—while maintaining high specificity and minimal off-target effects [4]. The system employs three different phage RNA polymerases (MmP1, K1F, and VP4) that demonstrate high orthogonality, enabling simultaneous evolution of multiple genetic targets without cross-talk. Unlike traditional methods limited to model organisms, OTM functions effectively in non-model systems like Halomonas bluephagenesis, expanding directed evolution capabilities to industrially relevant chassis [4].
The mechanism involves fusion of cytosine deaminase (PmCDA1) or adenine deaminase (TadA8e) domains to phage RNAPs, creating mutator enzymes that introduce C:G to T:A and A:T to G:C transitions across target genes. When the deaminase-RNAP fusion binds to its specific promoter sequence, it locally unwinds DNA and exposes single-stranded regions for deamination, creating uracil or inosine bases that are processed into permanent transition mutations during subsequent replication cycles. The inclusion of uracil glycosylase inhibitor (UGI) domains significantly enhances mutation efficiency by preventing repair of uracil lesions [4].
Experimental Protocol
Mutator Library Construction:
- Plasmid Design: Clone PmCDA1 variants (PmCDA1, PmCDA1-UGI, evoPmCDA1-UGI) fused to N-terminus of MmP1, K1F, or VP4 RNAPs via XTEN linkers in high-copy-number pSEVA241 vector.
- Expression Control: Drive fusion expression with IPTG-inducible tac promoter (PTac).
- Target Plasmid Engineering: Clone sfGFP or inactivated ermC (Y104S mutation) under specific phage promoters (PMmP1, PK1F, PVP4) in pSEVA321 backbone.
Mutation Efficiency Assessment:
- Transcriptional Activity Verification: Transform mutator plasmids into H. bluephagenesis with sfGFP target plasmid, measure fluorescence intensity by flow cytometry.
- On-target Mutation Frequency: Use erythromycin resistance restoration assay with ErmC Y104S mutant, calculate mutation frequency as (EryR colonies)/(total colonies).
- Off-target Rate Evaluation: Measure rifampicin-resistant mutation frequency in genomic RNA polymerase beta subunit gene.
- Optimization: Titrate IPTG inducer concentration (0-1 mM) to balance mutation efficiency and cell viability.
High-Throughput Screening:
- Continuous Evolution: Co-culture cells containing mutator and target plasmids with appropriate antibiotic selection.
- Variant Isolation: Plate aliquots at time intervals on selective media to isolate functional mutants.
- Characterization: Sequence resistant colonies to identify mutation spectra and calculate mutation rates using maximum likelihood method.
Key Research Reagents:
- Phage RNA Polymerases: MmP1, K1F, VP4 RNAPs with cognate promoters
- Deaminase Enzymes: PmCDA1, evoPmCDA1, TadA8e
- Inhibitor Protein: Uracil glycosylase inhibitor (UGI)
- Vectors: pSEVA241 (high-copy), pSEVA321 (target)
- Selection Markers: Erythromycin resistance (ermC), rifampicin resistance (rpoB)
- Reporters: sfGFP, chromoproteins
PANCS-Binders Discovery Platform
PANCS-Binders (Phage-Assisted Noncontinuous Selection of Protein Binders) revolutionizes high-throughput binder discovery by linking the M13 phage life cycle to target protein binding through proximity-dependent split RNA polymerase biosensors. This platform enables comprehensive screening of protein-protein interaction pairs with unprecedented speed and scale—assessing over 10¹¹ variants against 95 separate targets in just two days [26]. The system achieves remarkable sensitivity, distinguishing binders with affinities as low as 206 pM and demonstrating 10¹⁵-fold relative enrichment for high-affinity interactions. By overcoming the sampling limitations of continuous evolution systems, PANCS-Binders successfully identifies de novo binders from extremely diverse libraries where active variants may be present at ratios below 1:10⁷ [26].
The molecular mechanism relies on engineered M13 phage encoding protein variant libraries tagged with one half of a split RNA polymerase (RNAPN). Host E. coli cells express the target protein of interest fused to the complementary RNAP fragment (RNAPC). When a phage-encoded protein variant binds to the target, the split RNAP reconstitutes and triggers expression of a required phage gene, allowing selective replication of binding clones. This direct coupling between binding and phage propagation creates a powerful selective pressure that efficiently enriches functional binders while eliminating non-functional variants from the population [26].
Experimental Protocol
Library and Strain Preparation:
- Phage Library Construction: Clone RNAPN-tagged protein variant libraries (affibodies, DARPins, etc.) into replication-deficient M13 phage vector. Achieve diversity of 10⁸-10¹⁰ unique variants.
- Selection Strain Engineering: Transform E. coli with plasmid expressing target protein fused to RNAPC under inducible promoter.
- Optimization: Validate split RNAP reconstitution efficiency with positive control binders.
PANCS Selection Protocol:
- Initial Infection: Mix phage library (10¹⁰-10¹¹ pfu) with selection cells (OD₆₀₀ ~0.5) at appropriate cell:phage ratio (optimized at 10:1).
- Primary Incubation: Incubate phage-cell mixture for 12 hours with shaking at 37°C to allow complete infection and binding-dependent replication.
- Serial Passaging: Transfer 5% of phage supernatant to fresh selection cells every 12 hours for 4-6 passages.
- Monitoring: Titer phage from each passage to track enrichment dynamics.
- Stringency Control: Adjust selection stringency using arabinose-inducible promoter strength for essential phage gene.
Hit Characterization:
- Phage Sequencing: Isolate individual phage clones from final passage, sequence variant regions.
- Affinity Measurement: Express and purify soluble binders, determine binding kinetics (K_D) via surface plasmon resonance or bio-layer interferometry.
- Functional Validation: Test binders in mammalian cells for target inhibition (Mdm2-p53) or degradation (KRAS-LC3B pathway).
Affinity Maturation:
- Secondary Library: Generate mutagenized library around initial hit sequences.
- Iterative PANCS: Perform additional selection rounds with increased stringency.
- Characterization: Identify matured binders with improved affinity (8.4 nM demonstrated from 200 nM starting binder) [26].
Key Research Reagents:
- Phage System: M13 phage derivatives, replication-deficient variants
- Split RNAP: RNAPN and RNAPC fragments with optimized split sites
- Selection Cells: E. coli with arabinose-inducible essential phage gene
- Protein Libraries: Affibody, DARPin, or other scaffold libraries
- Target Proteins: Diverse panel of 95 proteins for multiplex screening
- Mammalian Validation: HEK293T cells, degradation tag systems (LIR motifs)
Comparative Applications and Implementation Guidelines
System Selection Framework
Table 3: Application-Specific Platform Recommendations
| Research Goal | Recommended Platform | Implementation Timeline | Key Advantages | Technical Considerations |
|---|---|---|---|---|
| Genetic Circuit Orthogonalization | σ54 Transcription System | 2-3 weeks | Low basal leakage, bEBP regulation, transferable across species [2] | Requires bEBP co-expression, moderate throughput |
| Rapid Protein Evolution | Orthogonal Transcription Mutator | 1-2 days | Extreme mutation rates, broad host compatibility, uniform mutation distribution [4] | Limited to transition mutations, optimization needed for each host |
| De Novo Binder Discovery | PANCS-Binders | 2-7 days | Massive throughput (10¹¹ variants), pM affinities, direct functional validation [26] | Limited to E. coli initially, requires protein expression |
| Multiplexed Evolution | OTM with Multiple Phage RNAPs | 3-5 days | Orthogonal polymerases enable parallel evolution, high specificity [4] | Requires multiple selection markers, potential resource competition |
Technical Considerations and Optimization Strategies
Context Dependence and Host Interactions: Synthetic genetic systems operate within complex cellular environments where resource competition and growth feedback can significantly impact performance. Engineering orthogonal systems requires careful consideration of these circuit-host interactions:
Resource Competition: Synthetic constructs compete for limited transcriptional and translational resources, potentially causing unintended coupling between seemingly independent modules [27]. The σ54 system minimizes this issue through its specific recruitment mechanisms, while PANCS-Binders faces potential bottlenecks in RNAP availability during selection.
Growth Feedback: Circuit activity affects cellular growth rates, which in turn alters dilution rates of circuit components [27]. The OTM system addresses this through inducible expression control, while PANCS-Binders leverages the differential growth advantage of successful binders.
Burden Management: Implement resource-aware design principles by selecting appropriate copy number vectors, using tunable promoters, and incorporating feedback control to mitigate metabolic burden [27].
Troubleshooting Common Issues:
- Low Orthogonality: For σ54 systems, ensure complete knockout of native rpoN and optimize bEBP expression levels.
- Poor Mutation Efficiency: For OTM, verify deaminase-RNAP fusion integrity and optimize UGI fusion configuration.
- Limited Binder Enrichment: For PANCS-Binders, adjust cell:phage ratio and selection stringency, extend incubation time to 12 hours for complete library sampling.
The ongoing development of orthogonal biological systems represents a critical frontier in synthetic biology, enabling increasingly sophisticated genetic programming with enhanced predictability and reliability. The three platforms examined here—σ54-dependent transcription, orthogonal transcription mutation, and PANCS-Binders—demonstrate how strategic engineering of molecular interactions can overcome fundamental limitations in genetic circuit design and protein engineering.
When selecting an appropriate system, researchers should consider their specific application requirements: the σ54 system offers precise transcriptional control for metabolic engineering and circuit design; the OTM platform provides unprecedented mutagenesis capabilities for rapid protein evolution; and PANCS-Binders delivers exceptional throughput for binder discovery. As these technologies continue to mature, their integration with computational design and machine learning approaches will further accelerate the engineering of biological systems with novel functions, advancing both fundamental research and biotechnological applications.
In the field of synthetic biology, orthogonal gene expression systems represent a cornerstone technology for ensuring predictable operation of genetic circuits by decoupling them from host regulatory networks. The significance of orthogonal transcription lies in its ability to facilitate consistent and reliable performance of engineered genetic pathways, which is particularly crucial for applications in regenerative medicine, cancer therapy, and genetic disorders [28]. While systems like T7 RNA polymerase (T7 RNAP) have served as valuable tools, they face limitations including cellular toxicity and restricted promoter availability, creating a pressing need for expanded toolkits [2]. This review compares two innovative approaches that successfully generate multiple orthogonal systems from singular structural foundations: engineered σ54 factors and orthogonal transcription mutators (OTM) based on phage RNA polymerases. By evaluating their performance metrics, experimental methodologies, and practical applications, we provide researchers with a comprehensive framework for selecting appropriate orthogonal transcription systems for their specific experimental needs.
Comparative Analysis of Orthogonal System Performance
The development of orthogonal transcription systems has progressed significantly with two prominent approaches emerging: the engineering of bacterial σ54 factors and the creation of orthogonal transcription mutators using phage RNA polymerases. The table below summarizes key performance characteristics of these systems based on recent experimental findings.
Table 1: Performance Comparison of Orthogonal Transcription Systems
| System Characteristic | Engineered σ54 Factors [2] | Orthogonal Transcription Mutators (OTM) [4] |
|---|---|---|
| Base Scaffold | Native Escherichia coli σ54 factor | MmP1, K1F, and VP4 phage RNA polymerases |
| Engineering Approach | Knowledge-based screening and rewiring of RpoN box | Fusion of deaminases (PmCDA1, evoPmCDA1) with phage RNAPs |
| Orthogonal Variants Generated | 3 (σ54-R456H, R456Y, R456L) | 3 (based on MmP1, K1F, and VP4 RNAPs) |
| Key Functional Output | Transcriptional activation with specific promoter preferences | Targeted mutagenesis (C:G to T:A and A:T to G:C transitions) |
| Mutation Rate/Effect | Specific promoter recognition changes | >1,500,000-fold increased mutation rates over control |
| Host Organisms Demonstrated | E. coli, K. oxytoca, P. fluorescens, S. meliloti | E. coli and H. bluephagenesis |
| Activation Mechanism | Requires bacterial enhancer-binding proteins (bEBPs) | Inducible (IPTG-inducible Ptac promoter) |
| Primary Applications | Genetic circuits, pathway orthogonalization | Protein evolution, directed evolution |
Experimental Protocols and Methodologies
Engineering Orthogonal σ54 Systems
The development of orthogonal σ54 factors followed a systematic protein engineering approach with the following key methodological steps:
Library Construction: Created random mutation libraries focused on the RpoN box recognition region (specifically residues R456/R457) of σ54 using inverse PCR, with plasmids carrying native σ54 sequences as templates [2].
Screening Methodology: Employed a dual-reporter system with fluorescent proteins (GFP and RFP) to identify mutants with altered promoter specificity while maintaining transcriptional functionality.
Orthogonality Validation: Tested candidate σ54 mutants against a panel of engineered promoters containing variations in the -24 recognition element to identify orthogonal pairs with specific recognition patterns.
Host Transfer Verification: Validated orthogonal system functionality in non-model bacteria (Klebsiella oxytoca, Pseudomonas fluorescens, and Sinorhizobium meliloti) using broad-host-range plasmids with σ54 genes expressed from their native promoters.
Circuit Integration: Demonstrated application in genetic circuits by combining orthogonal σ54 factors with different bEBPs to create signal-responsive systems and implementing them in complex pathways such as sucrose utilization and nitrogen fixation [2].
Development of Orthogonal Transcription Mutators
The OTM system was constructed and validated through the following experimental procedures:
Plasmid Construction: Assembled mutator plasmids in a high copy-number plasmid (pSEVA241) with PmCDA1 variants (PmCDA1, PmCDA1-UGI, and evoPmCDA1-UGI) fused to the N-terminus of MmP1 RNAP using an XTEN linker [4].
Transcriptional Activity Assessment: Evaluated mutator functionality using a target plasmid expressing sfGFP under the PMmP1 promoter, with fluorescence intensity measured via flow cytometry to confirm maintained transcriptional activity.
Mutation Rate Quantification: Employed an erythromycin resistance-based recovery assay where correction of an inactivating Y104S mutation in the ermC gene (changing TCT to TTT) restored resistance, enabling precise measurement of C:G to T:A mutation frequencies [4].
Specificity Evaluation: Measured off-target effects using rifampicin-resistant mutation frequency in the host genome and assessed cellular viability through colony-forming unit (CFU/mL) counts under mutator expression.
Orthogonality Testing: Demonstrated minimal cross-talk between different phage RNAP systems (MmP1, K1F, and VP4) when expressed simultaneously in the same host with their cognate promoters.
Protein Evolution Applications: Applied the system to evolve various proteins including fluorescent proteins, chromoproteins, cytoskeletal proteins, and metabolic exporters, evaluating functional improvements through relevant phenotypic assays [4].
Research Reagent Solutions
The following table details essential research reagents and their applications for researchers working with orthogonal transcription systems.
Table 2: Key Research Reagents for Orthogonal Transcription System Development
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Polymerase Scaffolds | σ54 factor, MmP1 RNAP, K1F RNAP, VP4 RNAP | Core components serving as foundation for engineering orthogonal systems |
| Deaminase Enzymes | PmCDA1, evoPmCDA1, rAPOBEC1, TadA8e | Generation of mutation signatures in OTM systems when fused to RNA polymerases |
| Inhibitor Proteins | Uracil glycosylase inhibitor (UGI) | Enhances mutation efficiency by preventing repair of deaminated bases |
| Reporter Systems | sfGFP, RFP, erythromycin resistance (ermC) | Functional assessment of orthogonal system performance and mutation rates |
| Selection Markers | Ampicillin, kanamycin, chloramphenicol, gentamycin resistance genes | Plasmid maintenance and selection in various host organisms |
| Induction Systems | IPTG-inducible Ptac promoter, tetracycline-inducible Ptet promoter | Controlled expression of orthogonal transcription components |
| Vector Backbones | pSEVA series, pBBR-derived broad-host-range vectors | Plasmid systems for cloning and expression across diverse bacterial hosts |
Signaling Pathways and System Architectures
σ54 Orthogonal System Engineering
Engineering workflow for creating orthogonal σ54 systems showing key stages from mutagenesis to application.
Orthogonal Transcription Mutator Mechanism
Functional mechanism of orthogonal transcription mutators showing how deaminase-RNAP fusions introduce targeted mutations.
Comparative Advantages and Limitations
σ54 Orthogonal Systems
Key Advantages:
- Native Integration: As engineered versions of endogenous bacterial transcription machinery, σ54-based systems demonstrate lower cellular toxicity compared to heterologous systems like T7 RNAP [2].
- Regulatory Precision: The requirement for bacterial enhancer-binding proteins (bEBPs) enables sophisticated regulation patterns, allowing construction of logic gates and signal-responsive circuits [2].
- Broad Host Compatibility: Engineered σ54 systems have demonstrated functionality across multiple bacterial species including non-model organisms, highlighting their transferability [2].
- Eukaryotic-like Regulation: The mechanism of transcription initiation resembles eukaryotic systems, providing unique engineering opportunities not available with σ70-type systems [2].
Notable Limitations:
- Complex Engineering: Creating functional orthogonal pairs requires coordinated engineering of both σ54 factor and cognate promoter elements.
- Dependency on Activators: Absolute requirement for bEBPs adds complexity to circuit design and implementation.
- Limited Mutant Spectrum: Current systems generate a relatively small number of orthogonal variants compared to phage polymerase-based systems.
Orthogonal Transcription Mutators
Key Advantages:
- High Mutagenesis Efficiency: OTM systems achieve exceptionally high mutation rates (>1,500,000-fold increase over background), enabling rapid protein evolution [4].
- Dual Mutation Capability: Engineered to introduce both C:G to T:A and A:T to G:C transition mutations, covering all possible transition mutations [4].
- Orthogonality Between Systems: Different phage RNAP-based mutators (MmP1, K1F, VP4) show minimal cross-talk, enabling parallel evolution of multiple genes [4].
- Application Versatility: Successfully applied to evolve diverse proteins including fluorescent proteins, structural proteins, and metabolic exporters [4].
Notable Limitations:
- Potential Off-target Effects: Although specificity is generally high, some constructs (particularly pMT2.1-MmP1) showed increased genome-wide mutation rates [4].
- Cellular Fitness Impact: Certain mutator constructs (pMT1-MmP1 and pMT2-MmP1) reduced colony-forming units, indicating potential fitness costs [4].
- Technical Complexity: Requires careful optimization of induction conditions to balance mutation efficiency with cell viability.
The development of multiple orthogonal systems from single scaffolds represents a significant advancement in synthetic biology toolkit expansion. Both σ54 engineering and orthogonal transcription mutator approaches demonstrate how strategic modification of core transcriptional components can generate families of orthogonal tools with distinct applications. The σ54 system excels in programmable genetic circuit implementation with minimal host interference, while OTM systems provide unprecedented capability for accelerated protein evolution. Future directions will likely focus on expanding the orthogonality landscape through additional scaffold engineering, enhancing specificity to minimize off-target effects, and broadening host compatibility to non-model organisms of industrial and therapeutic relevance. As these technologies mature, they will increasingly support complex synthetic biology applications requiring predictable, insulated genetic modules operating in parallel within the same cellular environment.
The engineering of orthogonal transcription systems represents a cornerstone of synthetic biology, enabling the construction of complex genetic circuits that function predictably and independently of host regulatory networks. These systems, which include orthogonal transcription factors, promoters, and RNA polymerases, provide the foundational components for building sophisticated biological computation devices, such as logic gates and layered logic circuits. The core principle of orthogonality involves creating biological parts that do not cross-react with each other or with the host's native systems, thereby ensuring consistent and predictable operation of synthetic genetic pathways [2]. This comparative guide evaluates the performance of several leading orthogonal transcription systems, focusing on their applications in constructing genetic logic gates and multi-layered circuits, providing researchers and drug development professionals with critical data for selecting appropriate systems for their specific applications.
Performance Comparison of Orthogonal Transcription Systems
The table below provides a comprehensive comparison of four major orthogonal transcription systems, highlighting their key characteristics, performance metrics, and demonstrated applications in genetic circuitry.
Table 1: Performance Comparison of Orthogonal Transcription Systems for Genetic Circuitry
| System Type | Key Components | Orthogonality Metrics | Demonstrated Logic Operations | Circuit Complexity Achieved | Host Organisms Tested |
|---|---|---|---|---|---|
| σ54 Mutants [2] | σ54-R456H/Y/L mutants, bEBPs, cognate promoters | Ideal mutual orthogonality; transferable across species | AND gates; Environmental signal-responsive logic | Orthogonalized complex pathways; Layered logic gates | E. coli, K. oxytoca, P. fluorescens, S. meliloti |
| Phage RNAP Mutators [29] | MmP1, K1F, VP4 RNAPs fused to deaminases | High orthogonality between phage polymerases | N/A (Targeted mutagenesis for evolution) | Mutation of protein pathways | E. coli, H. bluephagenesis |
| λ cI Variants [3] | 12 engineered cI TFs, synthetic bidirectional promoters | Orthogonal set of 12 TFs operating on 270 promoters | Activation, repression, dual activation-repression | Complex gene networks with combinatorial control | E. coli |
| LacI/GalR Chimeras [30] | 27 non-natural TFs with alternate DNA recognition | 201/210 non-cognate pairs unresponsive | AND, OR, NOT, NOR, half-AND | Combinatorial and non-canonical logical operations | E. coli |
Experimental Protocols for Key Systems
σ54-Dependent Orthogonal System Evaluation
The experimental protocol for characterizing σ54-dependent orthogonal systems involves several critical steps [2]:
- Library Construction: Random mutation libraries for rpoN R456/R457 sites and −24 elements of promoters were created using inverse PCR with plasmids carrying corresponding sequences as templates.
- Host Preparation: E. coli ΔrpoN strains were constructed using λ-red homologous recombination to eliminate native σ54 interference.
- Circuit Assembly: Expression cassettes were assembled using Golden Gate assembly with BpiI and BsaI restriction sites.
- Validation: Orthogonality was validated in multiple non-model bacteria (Klebsiella oxytoca, Pseudomonas fluorescens, and Sinorhizobium meliloti) using broad-host-range plasmids (pBBR-derived).
- Performance Assay: GFP and RFP reporters were used to characterize outputs of different orthogonal systems, while bEBP-dependent activation was tested using chemical or environmental signals.
Orthogonal Transcription Mutation System Protocol
The protocol for implementing the orthogonal transcription mutation system involves [29]:
- Mutator Construction: C:G to T:A mutator plasmids were constructed based on MmP1 phage RNAP and PmCDA1 variants in a high copy-number plasmid (pSEVA241).
- Fusion Protein Design: Cytosine deaminase PmCDA1, PmCDA1-UGI fusion, and evoPmCDA1-UGI fusion were fused to the N-terminus of MmP1 RNAP using an XTEN linker.
- Activity Assessment: Transcriptional activity of single mutators was evaluated using sfGFP under the PMmP1 promoter for induction studies and flow cytometry analysis.
- Mutation Rate Quantification: A mutation-recovery method based on the erythromycin resistance gene (ermC) was designed, where Y104S mutation demonstrated complete inactivation while Y104F mutation retained resistance.
- Specificity Validation: Off-target effects were evaluated using rifampicin-resistant mutation frequency, while cell viability was assessed by measuring colony-forming units (CFU/mL).
λ cI Transcription Factor Selection Protocol
The experimental workflow for selecting and characterizing orthogonal λ cI variants includes [3]:
- System Design: A phagemid-based selection platform was developed using conditional M13 bacteriophage replication.
- Component Separation: The system consists of three plasmids: helper phage plasmid (HP, providing phage components), accessory plasmid (AP, containing conditional gene circuit), and phagemid (PM, containing combinatorially-randomized TFs).
- Enrichment Assay: Enrichment of optimized cI mutants was tested from 10³-fold and 10⁶-fold excesses of phagemid expressing non-TF control protein (RFP).
- Promoter Engineering: Synthetic bidirectional promoters were designed by making symmetric variants of the consensus λ sequence with mutant operators differing by 2-4 base pair substitutions.
- Orthogonality Validation: Cross-reactivity of all selected cI variants and promoter pairs was systematically tested to ensure orthogonality before application in gene networks.
Signaling Pathways and Experimental Workflows
σ54-Dependent Transcription Activation Pathway
The diagram below illustrates the activation pathway of σ54-dependent transcription, highlighting the key components and regulatory mechanism that enable its use in orthogonal genetic circuits.
Phage RNAP Orthogonal Mutator Workflow
The following diagram outlines the experimental workflow for implementing the orthogonal transcription mutation system based on phage RNA polymerases.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Essential Research Reagents for Orthogonal Transcription System Engineering
| Reagent/Category | Specific Examples | Function/Application | Key Characteristics |
|---|---|---|---|
| Orthogonal σ Factors [2] | σ54-R456H, R456Y, R456L | Promoter recognition in orthogonal systems | Distinct promoter preferences; Require bEBPs for activation |
| Bacterial Enhancer-Binding Proteins [2] | KoNifA, RcNifA | Activate σ54-dependent transcription | Stringently regulated; Strong activation capability |
| Phage RNA Polymerases [29] | MmP1, K1F, VP4 RNAPs | Orthogonal transcription and mutagenesis | Broad host range; High efficiency in non-model organisms |
| Engineered Transcription Factors [3] [30] | λ cI variants, LacI/GalR chimeras | Transcriptional regulation in logic gates | Customizable DNA-binding; Orthogonal ligand response |
| Reporter Systems [2] [29] | GFP, RFP, sfGFP, 3WJdB | Circuit output measurement | Quantitative fluorescence; Real-time monitoring |
| Selection Plasmids [29] [3] | pSEVA241, pBBR-derived vectors | Maintain circuit components in hosts | Broad host range; Compatible assembly standards |
| Deaminase Fusion Proteins [29] | PmCDA1-MmP1, PmCDA1-UGI-MmP1 | Targeted mutagenesis for evolution | Introduce C:G to T:A mutations; High specificity |
Comparative Analysis and Research Applications
Performance Across Application Domains
Each orthogonal transcription system offers distinct advantages for specific research applications in genetic circuitry. The σ54-dependent system excels in environmental sensing applications due to its stringent requirement for bEBP activation, effectively creating natural AND gates where transcription only occurs when both the σ54 factor and its cognate bEBP are present [2]. This system has demonstrated remarkable transferability across diverse bacterial species, making it particularly valuable for applications requiring functionality in non-model organisms or environmental isolates. The phage RNAP-based systems provide unprecedented capabilities for directed evolution, achieving mutation rates exceeding 1,500,000-fold increases, enabling rapid protein engineering within complex pathways [29]. The λ cI variants and LacI/GalR chimeras offer the most extensive libraries of orthogonal components, with demonstrated capabilities for implementing complex Boolean logic and combinatorial control, making them ideal for constructing sophisticated computing elements in living cells [3] [30].
Considerations for Circuit Longevity and Stability
Recent research has highlighted the critical importance of evolutionary longevity in synthetic gene circuits, with studies showing that circuit function often degrades rapidly due to mutation and selection pressures [20]. The implementation of negative feedback controllers has been shown to significantly enhance circuit stability, with post-transcriptional control using small RNAs (sRNAs) generally outperforming transcriptional control via transcription factors [20]. When designing layered logic circuits, researchers should consider implementing such stabilization strategies, particularly for long-term applications in industrial biotechnology or therapeutic interventions where maintaining circuit function over extended periods is essential.
The expanding toolkit of orthogonal transcription systems provides researchers with versatile platforms for constructing increasingly sophisticated genetic circuitry. The selection of an appropriate system depends critically on the specific application requirements, including the desired logic operations, host organisms, circuit complexity, and required longevity. The σ54 systems offer unparalleled transferability across species and natural AND-gate functionality, while phage RNAP systems enable rapid directed evolution of circuit components. The extensive libraries of λ cI variants and LacI/GalR chimeras provide the component diversity needed for complex computing operations in single cells. As these technologies continue to mature, integrating orthogonal transcription systems with strategies for enhancing evolutionary longevity will be essential for realizing the full potential of synthetic genetic circuits in both basic research and applied biotechnology.
The precise engineering of cellular functions requires genetic tools that operate independently of the host's native regulatory networks. Orthogonal transcription systems, which use customized transcription factors and promoters that do not cross-react with the host's machinery, have emerged as powerful platforms for advanced synthetic biology applications. Within this landscape, Orthogonal Transcription Mutator (OTM) systems represent a groundbreaking advancement for in vivo protein evolution. These systems combine the precision of orthogonal transcription with targeted mutagenesis capabilities, enabling researchers to accelerate protein evolution directly within living cells under controlled conditions. This guide provides a comprehensive comparison of OTM systems, focusing on their performance against alternative protein evolution methods, with supporting experimental data and detailed protocols for implementation.
Fundamental Principles and Mechanism
OTM systems function through a elegant mechanism that links targeted transcription with directed mutagenesis. The core component is a fusion protein consisting of a phage-derived RNA polymerase (RNAP) coupled with a DNA deaminase enzyme. This fusion protein binds specifically to orthogonal promoters upstream of target genes, where it performs dual functions: initiating transcription of the target gene and introducing targeted mutations during the process [4].
The mutagenesis occurs through the catalytic activity of the deaminase domain, which directly converts cytosine to uracil (C→U) or adenine to inosine (A→I) in the DNA. During subsequent replication cycles, these modifications lead to transition mutations (C:G to T:A and A:T to G:C) throughout the target gene. The system's orthogonality stems from the specific pairing between the phage RNAP and its corresponding promoter sequence, which isn't recognized by the host's native transcription machinery [4] [31].
Table 1: Core Components of OTM Systems
| Component | Function | Examples |
|---|---|---|
| Phage RNA Polymerase | Provides orthogonal transcription capability; determines promoter specificity | T7 RNAP, MmP1 RNAP, K1F RNAP, VP4 RNAP |
| Deaminase Enzyme | Introduces targeted mutations by converting DNA bases | PmCDA1, evoPmCDA1, TadA8e |
| Orthogonal Promoter | Recognized specifically by the phage RNAP; drives target gene expression | PT7, PMmP1, PK1F, PVP4 |
| Linker Sequence | Connects deaminase and RNAP; affects fusion protein activity | XTEN linker |
| Inhibitor Protein | Enhances mutation efficiency by preventing repair | UGI (uracil glycosylase inhibitor) |
Comparative Advantages Over Traditional Methods
Compared to conventional protein evolution techniques, OTM systems offer several distinct advantages. Error-prone PCR, the traditional workhorse of directed evolution, requires iterative cycles of library construction, transformation, and screening, making it labor-intensive and limited by transformation efficiency [4]. CRISPR-based mutagenesis systems (EvolvR, CRISPR-X) offer better targeting but have narrow editing windows and require extensive gRNA design [4]. In contrast, OTM systems enable continuous in vivo mutagenesis of specific genes or pathways without the need for repeated transformations, dramatically accelerating the evolutionary timeline from months to days [4] [31].
The orthogonal nature of these systems minimizes cellular toxicity and off-target effects while allowing predictable operation of genetic circuits. This orthogonality is particularly valuable for evolving essential genes or complex pathways where maintaining cellular fitness is crucial [2] [4].
Performance Comparison: OTM vs. Alternative Protein Evolution Systems
Mutation Efficiency and Specificity
Recent studies have quantitatively demonstrated the superior performance characteristics of OTM systems. The latest OTM systems achieve remarkable mutation frequencies, with the PmCDA1-UGI-MmP1 fusion construct exhibiting >80,000-fold increased mutation frequency compared to controls in Halomonas bluephagenesis [4]. When applied in both E. coli and H. bluephagenesis, these systems have demonstrated >1,500,000-fold increased mutation rates while maintaining high specificity to target genes [4] [31].
Table 2: Performance Comparison of Protein Evolution Systems
| Method | Mutation Rate/Frequency | Mutation Types | Key Advantages | Key Limitations |
|---|---|---|---|---|
| OTM Systems | >1,500,000-fold increase vs control; 2.9×10⁻⁵ substitutions per base [4] | C:G→T:A and A:T→G:C transitions | Continuous in vivo evolution; broad host compatibility; minimal off-target effects | Limited to transition mutations; requires optimization for new hosts |
| Error-prone PCR | Limited by transformation efficiency | All mutation types | Well-established; random mutations throughout gene | Labor-intensive; limited library diversity; requires repeated transformations |
| CRISPR-Based Methods (EvolvR, CRISPR-X) | Varies by system; typically lower than OTM | Depends on editor used | Precise targeting; various mutation types | Narrow editing window; requires multiple gRNAs; lower efficiency in non-model organisms |
| OrthoRep (Yeast) | 100,000-fold increased mutation rates [4] | All mutations in target plasmid | Continuous evolution; specialized for yeast | Restricted to linear plasmids in specific host systems |
| MutaT7/T7RNAP-based | ~566-fold increase in Pseudomonas putida [4] | C→T or A→G depending on deaminase | Gene-specific targeting | Limited efficiency in non-model organisms; single polymerase type |
Applications and Host Compatibility
The versatility of OTM systems has been demonstrated through successful protein evolution across diverse biological targets. Researchers have applied these systems to rapidly evolve fluorescent proteins, chromoproteins, cytoskeletal proteins, cell division-related proteins, global sigma factors, and metabolite exporters such as the LysE transporter [4] [31]. Remarkably, this evolution has been accomplished within a single day of mutagenesis process, highlighting the unprecedented speed of OTM systems [4].
A critical advantage of modern OTM systems is their broad host compatibility. While earlier T7RNAP-based systems were largely confined to model organisms like E. coli, newer systems incorporating MmP1, K1F, and VP4 phage RNAPs function effectively in non-model organisms including Halomonas bluephagenesis, Pseudomonas entomophila, and Comamonas testosteroni [4]. This significantly expands the potential application of directed evolution to industrially relevant microbes that were previously difficult to engineer.
Experimental Protocols for OTM Implementation
System Construction and Validation
Plasmid Design and Assembly:
- Vector Selection: Use a high-copy-number plasmid backbone (e.g., pSEVA241) for mutator expression [4].
- Fusion Protein Construction: Fuse the deaminase domain (e.g., PmCDA1 variant) to the N-terminus of the phage RNAP (e.g., MmP1 RNAP) using an XTEN linker. Include UGI (uracil glycosylase inhibitor) for cytosine deaminase fusions to prevent repair of U:G mismatches [4].
- Promoter Selection: Place the fusion protein under an inducible promoter (e.g., IPTG-inducible PTac) for controlled expression [4].
- Target Plasmid Construction: Clone the gene of interest under the corresponding orthogonal promoter (e.g., PMmP1 for MmP1 RNAP) on a separate plasmid [4].
Validation Steps:
- Transcriptional Activity Assessment: Transform the mutator plasmid and a target plasmid expressing a fluorescent reporter (e.g., sfGFP) into the host. Induce mutator expression and measure fluorescence via flow cytometry to confirm functional transcription [4].
- Mutation Efficiency Quantification: Use a mutation-recovery assay with an inactivated antibiotic resistance gene (e.g., erythromycin resistance gene ermC with Y104S mutation). Measure the frequency of restored resistance after mutator induction [4].
- Off-Target Evaluation: Assess genome-wide mutations by measuring rifampicin-resistant mutation frequency in the presence of the mutator system [4].
Directed Evolution Workflow Using OTM Systems
The following diagram illustrates the complete experimental workflow for protein evolution using OTM systems:
Key Optimization Parameters:
- Inducer Concentration: Titrate inducer (e.g., IPTG) to balance mutation efficiency and cell viability [4].
- Temporal Control: Limit mutator activity to specific growth phases to manage mutational load.
- Selection Pressure: Apply appropriate selective conditions based on desired protein function.
- Host Strain: Consider using repair-deficient strains to enhance mutation retention, though this must be balanced against potential toxicity.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of OTM systems requires carefully selected genetic components and molecular tools. The table below details essential research reagents for establishing these systems:
Table 3: Essential Research Reagents for OTM Systems
| Reagent/Category | Specific Examples | Function/Purpose | Key Characteristics |
|---|---|---|---|
| Phage RNA Polymerases | MmP1 RNAP, K1F RNAP, VP4 RNAP, T7 RNAP [4] | Orthogonal transcription initiation | Determines promoter specificity; varies in efficiency across hosts |
| Deaminase Enzymes | PmCDA1, evoPmCDA1, TadA8e [4] | Introduction of targeted point mutations | Specificity for cytosine or adenine; varying activity levels |
| Orthogonal Promoters | PMmP1, PK1F, PVP4, PT7 [4] | Specific recognition by corresponding phage RNAP | Minimal cross-talk with host promoters; tunable strength |
| Expression Vectors | pSEVA series, pET derivatives [4] | Plasmid backbone for system components | Appropriate copy number; compatible origin and selection markers |
| Linker Sequences | XTEN linkers [4] | Connection of deaminase and RNAP domains | Affects fusion protein stability and activity |
| Enhancer Proteins | UGI (uracil glycosylase inhibitor) [4] | Increases mutation efficiency | Prevents repair of deamination products; critical for C→T mutations |
| Reporter Systems | sfGFP, RFP, antibiotic resistance genes [4] | Assessment of transcriptional activity and mutation efficiency | Enables quantitative measurement of system performance |
| Induction Systems | IPTG-inducible PTac, arabinose-inducible systems [4] | Controlled expression of mutator components | Enables temporal control over mutagenesis process |
Orthogonality and Circuit Integration
A defining feature of advanced OTM systems is their high degree of orthogonality, enabling simultaneous evolution of multiple genes or pathways. Studies have demonstrated that systems based on different phage RNAPs (MmP1, K1F, VP4) exhibit minimal cross-reactivity - each polymerase exclusively transcribes its cognate promoter without activating others [4]. This mutual orthogonality enables sophisticated synthetic biology applications where multiple genes can be independently mutated or controlled.
The orthogonal nature of these systems also facilitates their integration with complex genetic circuits. When combined with different bacterial enhancer-binding proteins (bEBPs), OTM systems can be designed to respond to environmental or chemical signals, creating dynamically controlled evolution platforms [2]. This programmability allows researchers to establish layered logic gates and conditional mutagenesis schemes that activate only under specific physiological conditions or in response to predetermined cues [2].
Orthogonal Transcription Mutator systems represent a significant leap forward in protein evolution technology, offering unprecedented speed, specificity, and programmability. The quantitative data presented in this guide demonstrates their clear advantages over traditional methods, particularly for applications requiring rapid evolution of specific proteins in diverse host organisms.
As synthetic biology continues to expand into non-model organisms with industrial and therapeutic relevance, the demand for robust, portable genetic tools will only increase. Future developments in OTM technology will likely focus on expanding the mutagenesis repertoire beyond transition mutations, improving orthogonality in diverse hosts, and enhancing temporal control over the evolutionary process. For researchers and drug development professionals, these systems offer a versatile platform to tackle complex protein engineering challenges that were previously impractical or impossible with conventional methodologies.
A Guide to Orthogonal Transcription Systems
In synthetic biology, the precise control of cellular output in response to specific signals is foundational for applications in therapeutics, biomanufacturing, and fundamental research. Orthogonal transcription factor systems, which operate independently of the host's native machinery, are key to achieving this precise control. This guide objectively compares the performance of several engineered systems that respond to environmental and chemical signals, providing a direct evaluation of their mechanisms, outputs, and experimental efficacy.
System Performance Comparison
The following table summarizes the key performance characteristics and experimental outputs of different systems designed to control downstream gene expression.
| System Name / Type | Input Signals | Output Measured | Key Mechanism | Reported Performance / Experimental Data |
|---|---|---|---|---|
| AND Gate (T7 RNAP/supD) [32] | Arabinose, Salicylate (or Mg²⁺, AI-1) | GFP Fluorescence, Mammalian Cell Invasion | T7 RNA polymerase gene with amber stop codons translated only with suppressor tRNA supD from a second promoter. |
Near-digital AND-gate behavior; 5-fold gain in fluorescence with both inducers; successful activation of invasin gene for cell invasion. |
| Orthogonal Transcription Mutation System [31] | N/A (Continuous mutagenesis) | Protein Evolution (Fluorescent proteins, sigma factor, etc.) | Fusion of deaminases with phage RNA polymerases for targeted, high-frequency mutagenesis. | >1,500,000-fold increased mutation rates; achieved in E. coli and H. bluephagenesis; protein evolution within a single day. |
| Biophysical Prediction (motifDiff) [33] | Genetic Variants | Predicted TF Binding Affinity | Uses Position Weight Mateworks (PWMs) to quantify the effect of DNA sequence variants on TF binding. | High scalability (millions of variants in minutes); evaluated on gold-standard datasets (e.g., ADASTRA for allele-specific binding). |
| Chemical Signaling (Abiotic Stress in Plants) [34] | Stress (Cold, Salinity, Drought) | Expression of Stress-Response Genes | Cytosolic Ca²⁺ increase sensed by proteins (e.g., Calmodulin, CDPKs), triggering a phosphorylation cascade. | Ca²⁺ concentration shifts from 200 nM to micromolar levels; leads to downstream gene regulation and stress adaptation. |
Detailed Experimental Protocols
Protocol for a Synthetic AND Gate
This protocol details the construction and validation of a modular AND gate in E. coli that activates a downstream output only when two input promoters are active [32].
1. Circuit Construction:
- Input 1 Plasmid: Place the T7 RNA polymerase gene (
T7ptag), containing two internal amber stop codons, under the control of an inducible promoter (e.g., PBAD, induced by arabinose). - Input 2 Plasmid: Place the amber suppressor tRNA gene
supDunder the control of a different inducible promoter (e.g., Psal, induced by salicylate). - Output Plasmid: Place a reporter gene (e.g., Green Fluorescent Protein,
GFP, or invasin) under the control of a T7 promoter.
- Input 1 Plasmid: Place the T7 RNA polymerase gene (
2. Tuning and Transformation:
- The basal expression level of the
T7ptagcomponent is critical. To achieve proper AND-gate behavior, the ribosome binding site (RBS) preceding theT7ptaggene may need to be rationally designed or mutagenized to tune its translation efficiency [32]. - Co-transform all three plasmids into the host E. coli strain.
- The basal expression level of the
3. Induction and Measurement:
- Grow bacterial cultures and expose them to different combinations of inducers: no inducer, arabinose only, salicylate only, and both.
- Measure the output by quantifying GFP fluorescence using a fluorimeter or flow cytometer. For a phenotypic output like invasion, perform a mammalian cell invasion assay.
4. Data Analysis:
- Calculate the fold-change in output (e.g., fluorescence) for the condition with both inducers compared to all control conditions. A successful AND gate will show a high output only when both inputs are present [32].
Protocol for an Orthogonal Transcription Mutation System
This protocol describes the use of a phage polymerase-deaminase fusion system for in vivo continuous evolution to generate novel protein functions [31].
1. System Assembly:
- Create a fusion gene encoding a phage RNA polymerase (e.g., T7 RNAP) and a cytidine or adenosine deaminase.
- Place this fusion gene under a constitutive or inducible promoter on a plasmid.
2. Target Gene Cloning:
- Clone the gene of interest (GOI) you wish to evolve (e.g., a fluorescent protein, a metabolic enzyme) into a separate plasmid, downstream of the corresponding phage promoter (e.g., T7 promoter).
3. HyperMutation:
- Co-transform the system plasmid and the target gene plasmid into the desired host organism (E. coli or Halomonas bluephagenesis).
- Culture the transformed cells to allow the hypermutation system to operate. The fused RNA polymerase will transcribe the GOI, and the deaminase domain will introduce mutations (C→T and A→G) during transcription.
4. Selection and Screening:
- Apply the appropriate selective pressure for the desired function (e.g., antibiotic resistance for stability, fluorescence-activated cell sorting for enhanced fluorescence).
- Isolate improved variants and sequence the target gene to identify the beneficial mutations.
Signaling and Workflow Pathways
Synthetic AND Gate Logic
The following diagram illustrates the genetic logic and component interaction within a synthetic AND gate.
Cellular Chemical Signal Transduction
This diagram visualizes the generalized pathway for chemical signal transduction in cells, such as the calcium-mediated response to abiotic stress in plants.
Research Reagent Solutions
The table below lists key reagents and their functions for constructing and testing orthogonal genetic systems.
| Reagent / Component | Function in Experiment |
|---|---|
| T7 RNA Polymerase (T7ptag) [32] | Engineered transcriptional activator; the core component of the AND gate, requiring suppression for activity. |
| Amber Suppressor tRNA (supD) [32] | Decodes amber stop codons as serine; enables translation of T7ptag only when its promoter is active. |
| Inducible Promoters (PBAD, Psal) [32] | Provide well-characterized, external control (via arabinose/salicylate) over the two input signals. |
| Phage Polymerase-Deaminase Fusion [31] | Drives targeted hypermutation by introducing point mutations (transitions) during transcription of target genes. |
| Position Weight Matrix (PWM) [33] | A biophysical model representing TF binding specificity; used computationally to predict the impact of genetic variants. |
| Calcium Sensors (Calmodulin, CDPK) [34] | Native biological components that bind Ca²⁺ and transduce stress signals into downstream cellular responses. |
Overcoming Specificity, Toxicity, and Efficiency Challenges
{{ // Define colors from the specified palette const blue = "#4285F4" const red = "#EA4335" const yellow = "#FBBC05" const green = "#34A853" const white = "#FFFFFF" const lightGray = "#F1F3F4" const darkGray = "#202124" const mediumGray = "#5F6368" }}
Addressing Cellular Toxicity and Fitness Costs of Heterologous Systems
The engineering of heterologous systems, where organisms are modified to express foreign genetic elements, has become a cornerstone of modern biotechnology and therapeutic development. These systems enable the production of valuable compounds, proteins, and biomaterials that would otherwise be inaccessible. However, the introduction of non-native genetic material often imposes significant cellular toxicity and fitness costs on host organisms, limiting their efficiency and scalability. For researchers and drug development professionals, understanding and mitigating these challenges is crucial for advancing orthogonal transcription factor systems and other synthetic biology approaches. This guide provides a comprehensive comparison of current strategies, supported by experimental data and detailed methodologies, to help researchers navigate the complex landscape of heterologous system optimization.
Comparative Analysis of Mitigation Strategies
The table below summarizes the primary approaches currently employed to address cellular toxicity and fitness costs in heterologous systems, comparing their key features and effectiveness.
Table 1: Comparison of Strategies for Mitigating Heterologous System Challenges
| Strategy | Core Principle | Applicable Hosts | Key Advantages | Quantified Efficacy | Major Limitations |
|---|---|---|---|---|---|
| CRISPR Activation (CRISPRa) [35] | Uses deactivated Cas9 fused to transcriptional activators to upregulate endogenous genes. | E. coli, Eukaryotic cells | Targeted approach; can multiplex; reversible. | Established proof of concept for boosting recombinamer yields in E. coli [35]. | Requires further optimization for industrial-scale outputs; delivery efficiency can be low [35]. |
| DNA Repair Pathway Enhancement [36] | Overexpression of DNA repair enzymes (e.g., Smug1) to mitigate toxicity from nucleic acid-incorporating agents. | Mammalian cells (oocytes, early embryos), NIH 3T3 cells | Directly addresses a specific mechanism of genotoxicity. | Increased oocyte maturation rate and embryo development; reduced 5-FUrd in RNA by LC-MS/MS [36]. | Highly specific to certain toxins; potential for unintended metabolic shifts. |
| Systems Biology & Computational Modeling [37] | Employs bioinformatics tools (e.g., MAnorm2) to analyze high-throughput data and identify stress signatures. | Universal application across hosts | Data-driven; enables predictive modeling of host-responses early in design. | MAnorm2 reliably identifies differential ChIP/ATAC-seq signals between sample groups, even with high variability [37]. | Requires high-quality omics data; computational complexity. |
| Promoter & Expression Optimization | Engineering of inducible or synthetic promoters to fine-tune heterologous gene expression. | Yeast, E. coli, Mammalian cells | Reduces metabolic burden; controls expression timing. | Yeast systems allow for stable expression of large gene clusters [38]. | Can be context-dependent; requires extensive characterization. |
| Host Engineering | Direct modification of the host genome to improve tolerance and productivity. | Yeast, E. coli | Creates a specialized chassis; can confer robust, generalizable fitness. | Engineered yeast strains successfully express full gene clusters for compound production [38]. | Can be technically challenging and time-consuming. |
Detailed Experimental Protocols
To ensure the reproducibility of critical findings in this field, below are detailed methodologies for key experiments cited in the comparative analysis.
Protocol for Assessing CRISPRa Efficacy in Bacterial Systems
This protocol is adapted from studies using CRISPRa to enhance the expression of protein-based biomaterials like elastin-like recombinamers (ELRs) in E. coli [35].
- Key Reagents: dCas9 transcriptional activator fusion protein, guide RNA (gRNA) plasmids targeting promoter regions of genes of interest, E. coli expression strain (e.g., BL21), ELR expression construct, selective media (e.g., LB with appropriate antibiotics).
- Procedure:
- gRNA Design: Design and clone gRNAs targeting the upstream regulatory regions of host genes involved in metabolic pathways that support heterologous protein expression (e.g., chaperones, tRNA synthetases).
- Strain Transformation: Co-transform the E. coli host strain with the plasmid expressing the dCas9-activator and the gRNA plasmid.
- Control Preparation: Prepare a control strain transformed with a non-targeting gRNA plasmid.
- Cultivation and Induction: Grow both experimental and control strains in selective media to mid-log phase and induce CRISPRa system and ELR expression with appropriate inducers (e.g., IPTG, arabinose).
- Sample Analysis:
- Biomass Measurement: Monitor cell density (OD600) over time to assess fitness costs.
- Product Quantification: Harvest cells after a set induction period. Lyse cells and quantify ELR yield using SDS-PAGE with densitometry analysis or HPLC.
- Transcriptional Validation: Perform RT-qPCR on the targeted host genes to confirm successful upregulation.
Protocol for Evaluating DNA Repair Enzyme-Mediated Toxicity Relief
This protocol is based on research demonstrating that Smug1 alleviates the reproductive toxicity of 5-fluorouracil (5-FU) in a mouse model [36].
- Key Reagents: Experimental animal model (e.g., mice), toxicant (5-FU), reagents for mRNA synthesis (in vitro transcription kit), microinjection system, antibodies for Smug1 and Dkc1, LC-MS/MS system.
- Procedure:
- Animal Grouping and Exposure: Divide mice into three groups: Control (vehicle), Toxicant-Exposed (5-FU), and Recovery (5-FU with a recovery period).
- Functional Rescue:
- Clone the Smug1 gene into an expression vector suitable for in vitro transcription.
- Microinject Smug1 mRNA into GV-stage oocytes or early embryos collected from donor animals.
- Expose these injected cells to 5-FU in vitro.
- Phenotypic Assessment:
- For oocytes: Score maturation rates after culture.
- For embryos: Record development rates to 4-cell, morula, and blastocyst stages.
- Molecular Analysis:
- RNA Maturation Assay: Extract total RNA from 4-cell embryos. Use RT-qPCR to measure the levels of immature (47S) and mature (18S, 28S) rRNAs.
- Protein Localization: Perform immunofluorescence staining on embryos for Smug1 and Dkc1 to confirm their co-localization in the nucleus.
- Toxin Incorporation Quantification: Use LC-MS/MS to measure levels of 5-FUrd (a metabolite of 5-FU in RNA) in control and Smug1-overexpressing cells.
Protocol for Computational Analysis of Epigenetic Changes
This protocol utilizes the MAnorm2 model for comparing groups of ChIP-seq or ATAC-seq samples to understand how heterologous expression alters the host's epigenomic landscape [37].
- Key Reagents: Cultured cells with and without heterologous system expression, reagents for ChIP-seq or ATAC-seq library preparation, high-throughput sequencer.
- Procedure:
- Sample Preparation and Sequencing: Perform ChIP-seq (for a specific histone mark like H3K27ac) or ATAC-seq on your experimental and control cell groups. Include biological replicates (recommended minimum n=3).
- Data Preprocessing: Map sequencing reads to the reference genome using tools like Bowtie2 or BWA. Call peaks for each sample using specialized callers (e.g., MACS2 for ChIP-seq).
- Running MAnorm2 Analysis:
- Normalization: Use MAnorm2's hierarchical scaling strategy to normalize read counts across all samples, accounting for different group structures.
- Differential Analysis: Apply the MAnorm2 empirical Bayes framework to statistically compare normalized signals between groups at each genomic region. The model will automatically estimate and correct for differences in within-group variability (e.g., between normal and more heterogeneous test groups).
- Output Interpretation: The output will provide a list of genomic regions with statistically significant differential signals (e.g., gained or lost H3K27ac marks), which can be linked to nearby genes to hypothesize about the host's transcriptional response to the heterologous system.
Visualizing Experimental Workflows and Pathways
The following diagrams, generated with Graphviz, illustrate the logical flow of the key experimental and biological processes discussed.
CRISPRa Efficacy Workflow
Smug1 Toxicity Relief Pathway
The Scientist's Toolkit: Essential Research Reagents
Successful research into heterologous system toxicity requires a specific set of reagents and tools. The following table details key solutions for designing and executing these studies.
Table 2: Essential Research Reagents for Investigating Heterologous System Toxicity
| Reagent / Solution | Critical Function | Example Application | Key Considerations |
|---|---|---|---|
| Orthogonal Transcription Factor Systems | Provides a tunable, insulated gene expression circuit that minimizes interference with native host networks. | Controlled expression of heterologous gene clusters in yeast [38]. | Select for high dynamic range and low basal expression. |
| CRISPRa Toolkit (dCas9-Activator + gRNAs) | Enables targeted upregulation of endogenous host genes to bolster metabolic capacity or stress responses. | Enhancing host machinery for elastin-like recombinamer production in E. coli [35]. | gRNA specificity is paramount to avoid off-target effects. |
| Specialized Expression Hosts | Engineered strains (e.g., yeast, E. coli) with reduced protease activity or enhanced secretory pathways. | Stable expression of large gene clusters for compound production [38]. | Match host capabilities (e.g., post-translational modifications) to product needs. |
| Dual-Luciferase Reporter Assay Kits | Quantifies transcriptional activity and cellular toxicity simultaneously by measuring firefly and Renilla luciferase [39]. | Validating transcriptional activation of a target gene by a novel transcription factor. | Normalize experimental reporter activity to control reporter for accuracy. |
| ChIP-seq & ATAC-seq Kits | Genome-wide mapping of transcription factor binding (ChIP-seq) or chromatin accessibility (ATAC-seq). | Identifying global changes in the host epigenome upon introduction of a heterologous system [37]. | Requires high-quality antibodies for ChIP; requires fresh cells for ATAC. |
| MAnorm2 Software | A computational tool for the quantitative comparison of groups of ChIP-seq or ATAC-seq samples [37]. | Statistically identifying differential histone marks between cells with/without a heterologous system. | Requires bioinformatics expertise and properly formatted input files (BAM, BED). |
| Yeast One-Hybrid System | Screens for transcription factors that bind to a specific DNA sequence (cis-element) [39]. | Identifying host TFs that might interact with the heterologous system's genetic parts. | Can yield false positives that require secondary validation. |
Effectively addressing the cellular toxicity and fitness costs associated with heterologous systems is a multifaceted challenge that requires an integrated approach. As this guide illustrates, strategies range from CRISPRa-mediated host engineering and DNA repair pathway enhancement to sophisticated computational modeling with tools like MAnorm2. The optimal path forward often involves combining these strategies, such as using computational analyses to identify key bottlenecks and then employing CRISPRa or host engineering to alleviate them. For researchers in drug development and synthetic biology, a deep understanding of these comparative tools and methods is essential for designing robust, high-yielding production platforms that can advance therapeutic innovations from the bench to the clinic.
Enhancing Specificity and Minimizing Off-Target Effects
In the engineering of biological systems, the concept of orthogonality—creating components that function independently of the host's native machinery—has become a cornerstone for achieving predictable and reliable outcomes. Orthogonal transcription factor (TF) systems, derived from bacteriophages, prokaryotes, or engineered variants of host factors, provide synthetic biologists with programmable tools to control gene expression without interfering with endogenous regulatory networks. However, the development of these systems is fundamentally constrained by a dual challenge: maximizing their specific activity on intended target sequences while minimizing unintended interactions with host genomes or non-target pathways. This balance is not merely a technical consideration but a prerequisite for applications ranging from fundamental biological research to therapeutic development, where off-target effects can compromise experimental validity or clinical safety.
This guide provides a comparative analysis of contemporary orthogonal transcription systems, focusing on their documented specificity and off-target profiles. By synthesizing experimental data and methodologies from recent studies, we aim to equip researchers with the criteria necessary to select and optimize these powerful tools for their specific applications, with a particular emphasis on strategies for enhancing specificity and quantifying off-target effects.
Comparative Analysis of Orthogonal Transcription Systems
The performance of orthogonal transcription systems can vary significantly in their specificity and off-target rates. The table below summarizes key metrics for several recently developed systems.
Table 1: Performance Comparison of Orthogonal Transcription Systems
| System Name | Core Components | Reported Mutation Rate or Activity | Specificity (On-target Efficiency) | Off-target Rate (Genome-wide) | Host Organisms Validated |
|---|---|---|---|---|---|
| Orthogonal Transcription Mutator (OTM) [31] [4] | PmCDA1-UGI-MmP1 RNAP fusion | >1,500,000-fold increase in mutation rate; 2.9 × 10⁻⁵ s.p.b. [4] | High (uniform mutations across target genes) [31] | Rifampicin resistance assay: 5-fold increase over control [4] | E. coli, H. bluephagenesis [31] [4] |
| Orthogonal σ54 Systems [2] | Engineered σ54-R456H/Y/L variants with partner promoters | Demonstrated high orthogonality and specific output [2] | High mutual orthogonality between variants and to native σ54 [2] | Implied low off-targeting due to orthogonal design [2] | E. coli, K. oxytoca, P. fluorescens, S. meliloti [2] |
| dCas9-based synTFs [40] | dCas9 fused to effector domains, guided by sgRNA | Wide range of expression outputs, induction factors up to 400 [40] | High specificity guided by sgRNA; minimal background expression [40] | Dependent on sgRNA design and specificity [40] | S. cerevisiae [40] |
As the data illustrates, the OTM system excels in achieving hypermutation of target genes, a property valuable for directed protein evolution. In contrast, the orthogonal σ54 systems and dCas9-based synthetic TFs provide precise transcriptional control for gene circuit applications, with the σ54 system demonstrating particular utility in non-model bacterial hosts.
Experimental Protocols for Validating Specificity
Rigorous validation is critical for establishing the specificity of any orthogonal system. Below are detailed protocols for key experiments used to generate the data in the comparison table.
Protocol: Mutation-Rate Analysis via Recovery of Antibiotic Resistance
This method, used to characterize the OTM system, quantifies on-target mutation efficiency by measuring the reversion of an inactivated antibiotic resistance gene [4].
- Vector Construction: Clone a target gene, such as the erythromycin resistance gene (
ermC), into a plasmid under the control of the orthogonal promoter (e.g., PMmP1). Introduce a specific missense mutation (e.g., Y104S) that completely inactivates the protein. - Transformation and Culture: Co-transform the target plasmid alongside the plasmid expressing the orthogonal mutator (e.g., pMT2-MmP1) into the host strain. Include controls (e.g., a non-functional mutator) in parallel.
- Selection and Calculation: Plate the transformed cells on media containing the antibiotic (e.g., erythromycin). The mutation frequency is calculated as the number of resistant colonies divided by the total number of viable cells plated. The mutation rate can be further refined using a maximum-likelihood method [4].
Protocol: Assessing Genomic Off-Target Effects via Rifampicin Resistance
This assay measures genome-wide mutation rates by quantifying the emergence of resistance to the antibiotic rifampicin, which arises from mutations in the bacterial rpoB gene [4].
- Mutagenesis Phase: Grow cells harboring the orthogonal mutator system under inducing conditions for a specified period to allow for potential mutagenesis across the genome.
- Selection Phase: Plate the cultured cells onto agar plates containing rifampicin.
- Quantification: Count the resulting rifampicin-resistant colonies and compare the frequency to that of the control strain. A significant fold-increase indicates a higher genome-wide mutation rate and potential off-target activity of the system [4].
Protocol: Profiling TF-TF Interactions with CAP-SELEX
This high-throughput method maps cooperative binding between transcription factors and identifies specific composite DNA motifs, providing a deep understanding of specificity determinants [41].
- Library and Protein Preparation: Generate a comprehensive library of double-stranded DNA oligonucleotides with random sequences. Express and purify TF pairs of interest.
- Successive Rounds of Selection: Incubate the TF pair with the DNA library. First, capture TF-DNA complexes using an affinity tag on one TF. In a second step, use a tag on the partner TF to further purify complexes where both TFs are bound cooperatively to the same DNA molecule.
- High-Throughput Sequencing and Analysis: Isolate the selected DNA, amplify it, and subject it to high-throughput sequencing. Use specialized algorithms (e.g., based on mutual information or k-mer enrichment) to identify overrepresented composite motifs and preferred spacing/orientations for the TF pair [41].
Visualization of Mechanisms and Workflows
The following diagrams illustrate the core mechanisms of orthogonal transcription systems and the key experimental workflow for assessing their specificity.
Orthogonal Transcription and Mutagenesis Mechanisms
Specificity and Off-Target Validation Workflow
Successful implementation of orthogonal transcription systems requires a suite of specialized reagents and databases.
Table 2: Key Research Reagent Solutions for Orthogonal Transcription Research
| Tool / Reagent | Function / Application | Example / Source |
|---|---|---|
| Phage RNA Polymerases | Core component of orthogonal systems; provides promoter specificity. | T7 RNAP, MmP1 RNAP, K1F RNAP, VP4 RNAP [31] [4] |
| Engineered σ Factors | Enables orthogonal promoter recognition in prokaryotes. | σ54-R456H, R456Y, R456L variants [2] |
| Programmable synTFs | Provides customizable DNA-binding specificity for targeted regulation. | dCas9-effector fusions, TALE-TFs [40] |
| CAP-SELEX Platform | High-throughput mapping of TF-TF interactions and composite motifs. | In vitro screening of >58,000 TF pairs [41] |
| CollecTF Database | Repository of experimentally validated TF binding sites. | Curated data on naturally occurring TF-binding sites [42] |
| Motif Analysis Tools | Predicts and analyzes TF binding motifs from sequence data. | JASPAR, PROMO, RcisTarget, MEIRLOP, monaLisa [43] [44] |
| Mutation Reporter Plasmids | Quantifies on-target mutation efficiency in vivo. | Plasmid with inactivated ermC (Y104S) gene [4] |
Optimizing Promoter Context and Transcription Factor Binding Units (TFBUs)
In synthetic biology, the predictable engineering of cellular functions requires precise control over gene expression. This control is fundamentally governed by the interaction between transcription factors (TFs) and the regulatory DNA sequences they bind. While transcription factor binding sites (TFBSs) have long been recognized as crucial elements, emerging research highlights that the surrounding context sequences significantly influence TF binding efficacy and subsequent transcriptional activity [45]. This comprehensive understanding has led to the conceptualization of the transcription factor binding unit (TFBU), defined as a modular entity comprising both the core TFBS and its surrounding context sequence (TFBS-context) [45].
The optimization of TFBUs is particularly critical for advancing orthogonal transcription factor systems, which are designed to function independently of host cellular networks and of each other. The development of such systems enables the construction of complex, multi-input genetic circuits for applications ranging from fundamental biological research to therapeutic drug development. This guide provides a comparative analysis of current technologies and methodologies for TFBU optimization, presenting structured experimental data and protocols to inform research and development efforts in this rapidly evolving field.
Comparative Analysis of Orthogonal Transcription Factor Systems
The engineering of orthogonal TF systems has expanded the toolkit available for synthetic biology. The table below compares the key characteristics of several prominent systems documented in recent literature.
Table 1: Comparison of Orthogonal Transcription Factor Systems
| System Name / Type | Core Components | Key Features & Applications | Orthogonality Assessment | Experimental Evidence |
|---|---|---|---|---|
| λ cI Variant Toolkit [3] | Engineered λ cI repressor/activator variants and synthetic bidirectional promoters (O1, O2, O3 operators). | 12 TFs operating as activators, repressors, or dual activator-repressors; operates on ~270 synthetic promoters; useful for complex logic gates. | Selected via M13 phagemid system to eliminate cross-reactivity; high orthogonality confirmed. | Phage enrichment assays; GFP/mCherry reporter characterization in E. coli. |
| T-Pro (Transcriptional Programming) [46] | Synthetic repressors/anti-repressors (e.g., CelR, LasR scaffolds) responsive to ligands (IPTG, cellobiose, D-ribose) and cognate synthetic promoters. | Enables circuit "compression" for 3-input Boolean logic (256 operations) with minimal genetic footprint; reduces metabolic burden. | Implicit in design via orthogonal ligand responsiveness and alternate DNA recognition (ADR) domains. | Fluorescence-activated cell sorting (FACS) of anti-repressor libraries; quantitative characterization of logic circuits. |
| LuxR-Type QS Systems [23] | EsaI/EsaR and LasI/LasR quorum sensing systems from E. coli. | Applied in population-density-dependent gene expression; requires crosstalk elimination for simultaneous use. | Demonstrated low-level crosstalk (LasR activating EsaR promoter; LasR responding to EsaI signal). | Promoter engineering (nucleotide change in EsaR binding site); rational mutant LasR(P117S) screened for reduced crosstalk. |
| TF Recognition Element (RE) Arrays [47] | Plasmids containing long, repetitive arrays of TF recognition elements (up to 256 REs). | Sequesters TFs to tune gene expression and direct cell fate; alternative to high-burden overexpression. | Orthogonality depends on the specific TF's binding specificity used to build the array. | Proof-of-concept in mammalian cell lines with TetR and dCas9; measurement of gene expression alteration. |
Quantitative Analysis of TFBU Optimization
Impact of Context Sequence Design
The DeepTFBU toolkit demonstrates that manipulating the context sequence within a TFBU can dramatically alter enhancer activity. Experimental measurements on designed sequences reveal the significant potential of this approach.
Table 2: Quantitative Impact of TFBU Context Sequence Optimization on Enhancer Activity [45]
| Transcription Factor (TF) | Cell Line | Average Activity Increase | Maximum Achieved activity Increase | Notes |
|---|---|---|---|---|
| ELF1 | HepG2 | >20-fold | Not Specified | Effect observed for 82.9% (97/117) of TFs tested. |
| HNF1A | HepG2 | >20-fold | Not Specified | Single TFBU design. |
| HNF4A | HepG2 | >20-fold | Not Specified | No introduction of other obvious TFBSs. |
| General Application | HepG2 | >30-fold | Not Specified | For tandem repeats of TFBSs. |
| Cell Type-Specific Design | Specific vs. Non-Specific | Up to 60-fold | 60-fold | Achieved cell type-specific enhancer activity. |
Performance of Optimized Enhancers
The TFBU-based optimization strategy has been successfully applied to enhance the activity of existing, strong enhancers, showcasing its practical utility.
Table 3: Performance of Enhancers Optimized via TFBU-Based Design [45]
| Enhancer / Design Target | Optimization Method | Resulting Activity Change | Key Experimental Method |
|---|---|---|---|
| Cytomegalovirus (CMV) Enhancer | Decoupling enhancer effect into individual TFBUs and optimizing context sequences with a few mutations. | +60% increase | Massively Parallel Reporter Assay (MPRA) |
| Synthetic Enhancer (Tandem TFBS) | Joint optimization of context sequences for multiple TFBSs within the TFBU framework. | >30-fold increase | MPRA |
Essential Experimental Protocols
Protocol 1: Orthogonality Assessment of Dual Transcription Factor Systems
This protocol is adapted from studies assessing crosstalk between LuxR-type quorum sensing systems [23].
- Clone Reporter Constructs: Insert the promoter sequence for each TF (e.g., Pe for EsaR, Pl for LasR) upstream of a fluorescent reporter gene (e.g., GFP, mCherry) in separate plasmids.
- Express TFs Individually: Co-transform the reporter strains with plasmids expressing a single TF (e.g., EsaR or LasR).
- Measure Basal Activity: For each TF-reporter pair, measure fluorescence in the absence of its cognate signal (acyl-homoserine lactone, AHL) to establish baseline activity.
- Measure Cross-Activation: Expose each TF-reporter pair to the non-cognate AHL signal. For example, expose the LasR reporter strain to the EsaI-produced 3OC6-HSL. A significant increase in fluorescence indicates signal crosstalk.
- Measure Promoter Crosstalk: In a strain expressing one TF (e.g., LasR), measure the activity of the other TF's reporter (e.g., the EsaR promoter). Increased activity indicates promoter crosstalk.
- Mitigation Strategies:
- Promoter Engineering: Introduce nucleotide substitutions into the core TFBS of the affected promoter (e.g., Pe) to reduce affinity for the non-cognate TF.
- TF Engineering: Create mutant TF libraries (e.g., via site-directed mutagenesis or error-prone PCR) and screen for variants that retain response to their cognate signal but show reduced response to non-cognate signals and reduced activation of non-cognate promoters.
Protocol 2: Measuring Enhancer Activity of Designed TFBUs Using MPRA
This protocol is based on the experimental validation performed by the DeepTFBU toolkit developers [45].
- Library Design: Synthesize a library of DNA sequences containing the designed TFBU variants. This includes sequences with optimized TFBS-contexts, scrambled controls, and reference enhancers.
- Plasmid Library Construction: Clone the oligonucleotide library into a specific location in a reporter plasmid, typically downstream of a minimal promoter and a barcoded open reading frame (e.g., for a fluorescent protein or a surface marker).
- Cell Transfection & Culturing: Transfect the plasmid library into the target cell line (e.g., HepG2). Use a high multiplicity of infection to ensure each cell receives only one plasmid variant. Culture cells for a set period to allow for gene expression.
- Sample Harvesting and Sequencing:
- DNA Sequencing: Harvest a sample of cells immediately after transfection to sequence the plasmid DNA. This establishes the initial representation of each barcode in the library.
- RNA Sequencing: After the expression period, harvest cells and extract mRNA. Reverse transcribe the mRNA and sequence the resulting cDNA to quantify the abundance of each barcode.
- Data Analysis: For each TFBU variant, calculate its enhancer activity as the ratio of its barcode abundance in the RNA pool to its abundance in the DNA pool. Normalize these ratios to a control enhancer included in the library.
Visualization of Key Concepts and Workflows
Architecture of a Transcription Factor Binding Unit (TFBU)
Diagram 1: Modular structure of a TFBU, integrating the core TFBS and its functional context sequence.
Workflow for Orthogonality Assessment and Engineering
Diagram 2: Iterative process for assessing and engineering orthogonality in TF systems.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagent Solutions for TFBU and Orthogonal TF Research
| Reagent / Resource | Function/Description | Example Source / System |
|---|---|---|
| Engineered λ cI Variant Toolkit | A set of 12 orthogonal TFs that can function as activators, repressors, or dual switches on a library of ~270 synthetic promoters. | [3] |
| T-Pro Synthetic Transcription Factors | Ligand-responsive synthetic repressors and anti-repressors (e.g., based on CelR, LasR scaffolds) for building compressed genetic circuits. | [46] |
| Orthogonal Quorum Sensing Systems | Characterized pairs of signal synthase and receptor (e.g., EsaI/EsaR, LasI/LasR) for cell-cell communication and multi-input sensing. | [23] |
| TF Recognition Element (RE) Arrays | Plasmid-based arrays of repeated TF binding sites (up to 256x REs) for tuning gene expression by sequestering native or synthetic TFs. | [47] |
| DeepTFBU Computational Toolkit | A deep learning-based toolkit for predicting and optimizing the impact of context sequences on TF binding and enhancer activity. | [45] |
| Massively Parallel Reporter Assay (MPRA) | A high-throughput experimental method for quantitatively measuring the activity of thousands of designed DNA sequences in parallel. | [45] |
| M13 Phagemid Selection System | A directed evolution platform for selecting functional TF-promoter pairs with high orthogonality from combinatorial libraries. | [3] |
In the engineering of biological systems, the precise control of gene expression is a fundamental requirement for achieving predictable and optimal performance. Orthogonal transcription factor systems have emerged as powerful tools in synthetic biology, enabling researchers to decouple synthetic genetic circuits from the host's native regulatory networks [2]. The performance and applicability of these systems are critically dependent on two interconnected factors: the careful titration of inducer concentrations and the strategic balancing of expression levels within the host chassis. This guide provides a comparative evaluation of contemporary orthogonal transcription systems, analyzing their operational parameters and performance characteristics to inform selection and implementation strategies for research and therapeutic development.
Orthogonal Transcription Systems: A Comparative Analysis
The development of orthogonal transcription systems has diversified, offering distinct mechanisms to achieve specific, tunable control of gene expression. The table below compares the core characteristics, performance metrics, and optimal use cases for four prominent classes of these systems.
Table 1: Comparative Analysis of Orthogonal Transcription Systems
| System Type | Core Mechanism | Key Performance Metrics | Reported Performance Data | Ideal Use Cases |
|---|---|---|---|---|
| Phage RNAP-based Mutagenesis (OTM) [4] | Fusion of phage RNA polymerase (e.g., MmP1) with deaminases (e.g., PmCDA1) for targeted in vivo mutagenesis. | Mutation frequency, specificity (on-target vs. off-target), cell viability. | >80,000-fold increase in on-target mutation frequency; Off-target rate only 5-14x above control [4]. | Continuous protein evolution; Generating diverse variant libraries in non-model organisms. |
| σ54-Dependent Orthogonal Systems [2] | Engineered bacterial σ54 factors (e.g., R456H/Y/L) with rewired promoter recognition, requiring bEBP activation. | Orthogonality (cross-talk), transferability across hosts, activation fold. | High mutual orthogonality between variants; Functional in multiple non-model bacteria [2]. | Multi-gene pathway regulation; Complex genetic circuits in diverse bacterial hosts. |
| Allosteric Transcription Factor (aTF) Biosensors [48] | Engineered ligand-binding domains (e.g., TtgR) that undergo conformational change to regulate transcription. | Dynamic range (F-score), ligand specificity, sensitivity. | F-scores >1 with high dynamic range for non-native ligands (e.g., naltrexone, quinine) [48]. | Metabolic engineering; Biosensing for small molecules; Diagnostic cell-free systems. |
| CRISPRi-Aided Genetic Switches [49] | Integration of TF-based biosensors with FndCas12a, which processes crRNAs from sensor transcripts for repression. | Dynamic range, leakiness (basal expression), tunability. | Reduced basal transcription; Enhanced dynamic range via terminator filters [49]. | Dynamic pathway repression; Signal amplification circuits; Metabolic flux balancing. |
Experimental Protocols for System Characterization
A critical step in deploying any orthogonal system is the empirical determination of its operational parameters within a specific host context. The following protocols outline key methodologies for characterizing system performance, with a focus on the relationship between inducer concentration and expression output.
Protocol for Titrating Inducer Concentration and Assessing Mutagenesis Efficiency
This protocol, adapted from studies on the Orthogonal Transcription Mutation (OTM) system, is designed to quantify how inducer concentration modulates system activity and cellular impact [4].
- Strain Transformation and Culture: Transform the host strain (e.g., E. coli or Halomonas bluephagenesis) with two plasmids: the mutator plasmid (e.g., pMT2-MmP1 expressing PmCDA1-UGI-MmP1) and a target plasmid containing a reporter gene (e.g., sfGFP) and a mutation-recovery cassette (e.g., inactivated ermC).
- Inducer Gradient Setup: Inoculate cultures and grow them to mid-log phase. Add the inducer (e.g., IPTG) across a range of concentrations (e.g., 0 μM to 1 mM) to separate culture aliquots.
- Flow Cytometry Analysis: After a set induction period, analyze cells from each inducer concentration via flow cytometry to measure fluorescence distribution. This assesses the transcriptional activity and the proportion of cells with inactivated reporter genes.
- Mutation Frequency Assay: Plate induced cells on media containing a selective agent (e.g., erythromycin) to count colonies where mutation has restored function. Calculate the mutation frequency for each inducer concentration by dividing the number of resistant colonies by the total viable count.
- Off-Target and Viability Assessment: In parallel, plate cells on media containing rifampicin to assess genome-wide mutation rates. Perform colony-forming unit (CFU) counts to correlate inducer concentration with cell viability.
Protocol for Profiling aTF Biosensor Response Using Sensor-Seq
Sensor-seq is a high-throughput method for quantifying the dose-response of thousands of aTF variants to ligands, linking transcriptional output to deep sequencing [48].
- Library Construction and Barcoding: Clone a diverse library of aTF variants (e.g., TtgR mutants) into a screening construct. Each variant is placed in cis with a unique randomized RNA barcode, which is transcribed as part of the reporter output.
- Ligand Exposure and RNA Sequencing: Grow the pooled library and divide it into aliquots. Treat each aliquot with a different ligand or a vehicle control. Harvest cells during log phase and extract total RNA for sequencing.
- Genotype-Phenotype Linking: From the same culture, also extract plasmid DNA. Perform a Golden Gate Assembly to link each aTF variant's sequence to its associated barcode for short-read sequencing.
- F-score Calculation: For each variant, calculate the F-score—a normalized measure of activity—using the formula:
F-score = (cDNA_count_ligand / cDNA_count_control) / (plasmid_DNA_count_ligand / plasmid_DNA_count_control). This quantifies the ligand-induced fold-change in reporter expression, corrected for cellular plasmid abundance.
System Workflows and Signaling Pathways
The functional principles of these systems can be visualized as standardized workflows. The diagram below illustrates the generalized experimental pipeline for tuning and evaluating an orthogonal transcription system.
Diagram 1: System Tuning Workflow
The molecular logic of a CRISPRi-aided genetic switch demonstrates how signal responsiveness is engineered by integrating transcription factors with RNA-processing Cas proteins.
Diagram 2: CRISPRi Switch Mechanism
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of orthogonal transcription systems relies on a core set of reagents and tools. The following table details essential components for setting up and characterizing these systems.
Table 2: Key Research Reagent Solutions for Orthogonal Transcription Systems
| Reagent / Tool | Function | Example(s) / Notes |
|---|---|---|
| Phage RNAP & Promoter Systems | Drives orthogonal transcription initiation, often with high specificity and strength. | MmP1, K1F, and VP4 RNAPs; T7 RNAP. Broader host range than T7 in some non-model organisms [4]. |
| Engineered σ54 Factors | Provides orthogonal promoter recognition in bacteria, requires activation by bEBPs. | σ54-R456H, R456Y, R456L mutants for orthogonal pathways in E. coli and non-model hosts [2]. |
| Allosteric Transcription Factor (aTF) Scaffolds | Serves as a programmable scaffold for designing biosensors that respond to new ligands. | TtgR variant library; chosen for its large, promiscuous binding pocket (1500 ų) [48]. |
| CRISPR-Cas Regulatory Proteins | Enables programmable transcriptional repression (CRISPRi) or activation. | Nuclease-deficient FndCas12a (dFnCas12a); features innate RNase activity for crRNA processing [49]. |
| Reporter Genes | Quantifies system performance, dynamic range, and leakiness. | Fluorescent proteins (sfGFP, mCherry), chromoproteins, or antibiotic resistance genes (ermC) [4] [49]. |
| High-Throughput Screening Platforms | Maps genotype to phenotype for thousands of variants in a single experiment. | Sensor-seq: uses RNA barcoding and deep sequencing to calculate F-scores for aTF libraries [48]. |
Strategies for Transferring Orthogonality to Non-Model Organisms
The transfer of orthogonal biological systems to non-model organisms represents a significant frontier in synthetic biology, enabling precise genetic control in industrially and environmentally relevant species that often lack sophisticated genetic toolkits. Orthogonal systems, which operate independently of host cellular machinery, provide predictable and insulated function essential for reliable synthetic circuit operation, metabolic engineering, and controlled gene expression. However, achieving true orthogonality in non-model organisms presents unique challenges, including incompatible transcriptional/translational machinery, differing cellular environments, and limited characterization of host interference factors. This review comparatively analyzes two primary strategic frameworks—orthogonal transcription systems based on phage RNA polymerases and engineered bacterial sigma factors—for establishing orthogonal control in non-model hosts, evaluating their performance characteristics, implementation requirements, and applicability across diverse biological systems.
Comparative Analysis of Orthogonal System Performance
Table 1: Quantitative Performance Comparison of Orthogonal Systems in Non-Model Organisms
| Performance Metric | Phage RNAP System (OTM) | Engineered σ54 System |
|---|---|---|
| Mutation Rate Increase | >1,500,000-fold vs. control [4] | Not Applicable (Transcriptional Control) |
| Mutation Types | C:G to T:A and A:T to G:C transitions [4] | N/A |
| Orthogonality Between Variants | High orthogonality between MmP1, K1F, and VP4 RNAPs [4] | Ideal mutual orthogonality between σ54-R456H, R456Y, R456L [2] |
| Host Range Demonstrated | Halomonas bluephagenesis, E. coli [4] | Klebsiella oxytoca, Pseudomonas fluorescens, Sinorhizobium meliloti [2] |
| Key Applications | Protein evolution (fluorescent proteins, chromoproteins, exporters) [4] | Genetic circuits, pathway orthogonalization, nitrogen fixation control [2] |
| Activation Requirement | Constitutive or inducible expression | Bacterial enhancer-binding proteins (bEBPs) [2] |
| System Specificity | High specificity with minimal off-target effects [4] | Stringent regulation with low basal leakage [2] |
Table 2: Experimental Validation and Methodological Requirements
| Experimental Aspect | Phage RNAP System | Engineered σ54 System |
|---|---|---|
| Validation Timeframe | Single-day mutagenesis process [4] | Varies with host and application |
| Primary Readout Methods | Flow cytometry, erythromycin resistance restoration, rifampicin resistance testing [4] | Fluorescent reporters (GFP/RFP), phenotypic assays (sucrose utilization) [2] |
| Key Molecular Components | PmCDA1-UGI-MmP1 fusion, evoPmCDA1-UGI fusion, target plasmids with phage promoters [4] | σ54 mutants, orthogonal promoters, bEBPs (RcNifA, KoNifA) [2] |
| Host Engineering Requirements | Expression of phage RNAP-deaminase fusions, target genes with cognate promoters [4] | Modified σ54 recognition, cognate promoter engineering, bEBP expression [2] |
| Critical Controls | Empty vector controls, RNAP-only constructs, off-target mutation assessment [4] | Wild-type σ54 controls, promoter specificity tests, cross-talk evaluation [2] |
Orthogonal Transcription Mutation Systems: Phage RNA Polymerase Approach
System Architecture and Mechanism
The Orthogonal Transcription Mutation (OTM) system represents a breakthrough in targeted protein evolution, employing phage RNA polymerase-deaminase fusions to enable continuous, targeted mutagenesis of specific genes in vivo. This system addresses the critical limitation of previous T7RNAP-based systems, which showed inefficient transcription in many non-model organisms such as Halomonas bluephagenesis and Pseudomonas species [4]. By fusing cytosine deaminases (PmCDA1) and adenine deaminases (TadA8e) with broad-host-range phage RNA polymerases (MmP1, K1F, VP4), the system achieves uniform transition mutations across target genes while maintaining high specificity and minimal off-target effects [4].
The system's orthogonal nature derives from the use of phage-derived transcriptional machinery that operates independently of host RNA polymerases. Each phage RNAP recognizes only its specific promoter sequence, enabling simultaneous but independent operation of multiple mutagenesis systems within the same cell. This orthogonality was demonstrated through the successful use of three different phage RNAPs (MmP1, K1F, and VP4) that showed minimal cross-talk while maintaining high mutation efficiency [4].
Experimental Implementation and Validation
The experimental protocol for implementing the OTM system involves constructing fusion proteins with deaminases linked to phage RNAPs via XTEN linkers, followed by comprehensive evaluation of mutation efficiency and specificity [4]. Key methodological steps include:
Plasmid Construction: High copy-number plasmids (pSEVA241) carrying PmCDA1 variant-phage RNAP fusions under IPTG-inducible tac promoter control [4].
Mutation Rate Quantification: Employing an erythromycin resistance-based mutation-recovery assay where specific C-to-T mutations restore antibiotic resistance, enabling precise measurement of on-target mutation frequencies [4].
Specificity Assessment: Evaluating off-target effects through rifampicin-resistant mutation frequency in the host genome and measuring impacts on cell viability via colony-forming units [4].
Application Testing: Demonstrating system utility by evolving diverse proteins including fluorescent proteins, chromoproteins, cytoskeletal proteins, cell division-related proteins, global sigma factors, and exporters [4].
Performance optimization revealed that inclusion of uracil glycosylase inhibitor (UGI) significantly enhanced mutation activity by preventing repair of deaminated bases, with the PmCDA1-UGI-MmP1 construct (pMT2-MmP1) exhibiting the highest mutation frequency—over 80,000-fold greater than controls [4]. The system maintained high specificity with only 5-14-fold increase in genomic off-target mutations for the most efficient constructs while achieving remarkable mutation rates up to 2.9 × 10⁻⁵ substitutions per base [4].
Engineered Sigma Factor Systems: Endogenous Transcription Rewiring
System Architecture and Mechanism
As an alternative to phage-derived systems, researchers have developed orthogonal transcription systems based on engineered bacterial sigma factors, particularly focusing on σ54 due to its distinct recognition properties and eukaryotic-like regulation mechanisms. Unlike the major σ70 factor, σ54 recognizes conserved promoter sequences at -24 and -12 elements and requires bacterial enhancer-binding proteins (bEBPs) for transcription initiation, creating a naturally modular system amenable to orthogonal engineering [2].
Through knowledge-based screening and rewiring of the RpoN box in σ54, researchers identified three orthogonal variants (σ54-R456H, R456Y, and R456L) with different promoter preferences and ideal mutual orthogonality [2]. These engineered systems maintain the stringent bEBP-dependence of native σ54 transcription while operating independently of both native σ54 and each other, enabling multiplexed orthogonal control within single cells.
Experimental Implementation and Validation
Implementation of orthogonal σ54 systems involves coordinated engineering of both the sigma factor and its cognate promoter sequences, followed by validation across multiple host organisms:
Sigma Factor Engineering: Targeted mutagenesis of key recognition residues (R456) in the RpoN box to alter promoter specificity while maintaining structural integrity and bEBP responsiveness [2].
Promoter Library Construction: Creating variant libraries of -24 elements to identify cognate promoters for each orthogonal σ54 variant through selection and screening [2].
Cross-Talk Testing: Comprehensive evaluation of mutual orthogonality between wild-type σ54 and engineered variants (R456H, R456Y, R456L) through fluorescent reporter assays in ΔrpoN backgrounds [2].
Host Transfer Validation: Demonstrating system functionality in non-model organisms including Klebsiella oxytoca, Pseudomonas fluorescens, and Sinorhizobium meliloti using broad-host-range plasmids and species-adapted components [2].
The orthogonal σ54 system successfully controlled complex biological pathways, including sucrose utilization and nitrogen fixation, and enabled construction of sophisticated genetic circuits with minimal host interference [2]. The system maintained the naturally low basal expression and high inducibility of σ54-dependent transcription, making it particularly valuable for applications requiring tight regulation and minimal metabolic burden.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Implementing Orthogonal Systems
| Reagent/Component | Function | Example Sources/References |
|---|---|---|
| Phage RNA Polymerases | Orthogonal transcription initiation | MmP1, K1F, VP4 RNAPs [4] |
| Deaminase Enzymes | Targeted base conversion | PmCDA1, evoPmCDA1, TadA8e [4] |
| Uracil Glycosylase Inhibitor | Prevents repair of deaminated bases | UGI protein [4] |
| Engineered Sigma Factors | Altered promoter recognition | σ54-R456H, R456Y, R456L variants [2] |
| Bacterial Enhancer-Binding Proteins | ATP-dependent activation of σ54 systems | RcNifA, KoNifA [2] |
| Broad-Host-Range Plasmids | Genetic system transfer across species | pSEVA vectors, pBBR-derived plasmids [4] [2] |
| Orthogonal Promoters | Specific recognition by engineered systems | Modified -24/-12 promoters for σ54; phage-specific promoters [4] [2] |
| Reporter Systems | Quantitative measurement of orthogonality | Fluorescent proteins, antibiotic resistance genes [4] [2] |
The selection between phage RNAP-based and engineered sigma factor systems for transferring orthogonality to non-model organisms depends critically on the specific application requirements and host characteristics. Phage RNAP systems excel in applications requiring high mutation rates and continuous evolution capabilities, particularly for protein engineering in industrially relevant hosts where traditional T7 systems fail. The demonstrated success in Halomonas bluephagenesis—a chassis for Next Generation Industrial Biotechnology—highlights their practical value for metabolic engineering and industrial biotechnology [4].
Conversely, engineered σ54 systems offer advantages for applications requiring precise regulatory control, minimal metabolic burden, and predictable circuit behavior. Their modular activation through bEBPs enables sophisticated genetic computing and environmental sensing while maintaining orthogonality from native host regulation [2]. The transferability of these systems across diverse bacterial species without redesign demonstrates their robustness as platform technologies.
Future developments will likely combine strengths from both approaches, potentially creating hybrid systems that leverage the mutagenesis capability of deaminase fusions with the precise control of engineered transcription factors. As synthetic biology expands into increasingly diverse non-model hosts, these orthogonalization strategies will become essential tools for predictable biological engineering across the tree of life.
Benchmarking Performance and Cross-Species Validation
Orthogonal transcription factor (TF) systems are genetically encoded tools that enable precise control of gene expression in synthetic biology. They function independently of the host's native regulatory networks, allowing researchers to program cellular behavior for applications in therapeutic drug development, metabolic engineering, and fundamental biological research. The performance of these systems is quantitatively assessed by three critical metrics: induction level (the maximum expression achieved in the "ON" state), dynamic range (the fold-difference between ON and OFF states), and leakiness (undesired basal expression in the OFF state). Evaluating these metrics provides crucial insights for selecting appropriate TFs for specific applications, where high dynamic range and minimal leakiness are often paramount. This guide objectively compares the performance of current orthogonal TF systems, presenting experimental data and methodologies to inform researcher selection for specific scientific and industrial applications.
Performance Comparison of Orthogonal TF Systems
The field of orthogonal transcription factors has expanded beyond traditional TF-based systems to include advanced CRISPR-based technologies. The quantitative performance of these systems varies significantly based on their design and mechanism of action.
Table 1: Performance Comparison of Orthogonal Transcription Factor Systems
| System Type | Key Components | Reported Induction Level | Reported Dynamic Range | Reported Leakiness | Primary Applications |
|---|---|---|---|---|---|
| CRISPRi-Aided Genetic Switch [50] | FnCas12a, TF-based biosensors, terminator filters | High (Precise repression >90%) | ~50-fold increase over baseline | Significantly reduced via terminator filters | Metabolic pathway reprogramming, high-precision genetic circuits |
| Deaminase-Phage RNAP Systems [4] | PmCDA1-UGI fused to MmP1/K1F/VP4 RNAPs | High transcriptional activity | >80,000-fold mutation frequency increase | Moderate (addressed via inducer concentration tuning) | Continuous in vivo protein evolution, mutagenesis |
| TF-TF Composite Systems [41] | Cooperating TF pairs (e.g., HOXB13-MEIS1) | Enhanced via cooperative binding | Dependent on specific TF pair and spacing | Not explicitly quantified | Decoding complex gene regulation, developmental biology |
Experimental Protocols for Key Systems
Protocol for Evaluating CRISPRi-Aided Genetic Switches
The CRISPRi-aided genetic switch platform integrates transcription factor-based biosensors with the FnCas12a CRISPR system to achieve signal-responsive transcriptional regulation [50].
- Plasmid Construction: Clone a nuclease-deficient FndCas12a (D917A) into an appropriate expression backbone (e.g., pFnSECRVi). The crRNA array, targeting the gene of interest, is expressed from a separate plasmid under a biosensor-responsive promoter.
- Terminator Filter Integration: To mitigate leakiness, incorporate transcriptional terminator filters (e.g., strong terminators like T1 or T7) between the promoter and the crRNA coding sequence. This step is critical for reducing basal transcription.
- Cell Culture and Transformation: Transform the constructed plasmids into the desired host strain (e.g., E. coli DH5α). Select transformants using the appropriate antibiotics.
- Induction and Measurement: Grow cultures and add the specific inducer ligand. Monitor cell growth (OD₆₀₀) and output (e.g., fluorescence for a reporter gene) over time using a microplate reader.
- Data Analysis:
- Dynamic Range: Calculate the ratio of output signal (e.g., fluorescence) in the fully induced state (ON) to the signal in the uninduced state (OFF).
- Leakiness: Quantify the residual output signal in the uninduced state (OFF), normalized to cell density.
Protocol for Testing Deaminase-Phage RNAP Mutator Systems
This protocol evaluates the mutation efficiency of orthogonal transcription mutators (OTM) in vivo, which is a direct reflection of their transcriptional activity and targeting capability [4].
- Mutator Plasmid Construction: Fuse a cytosine deaminase (e.g., PmCDA1) and UGI to the N-terminus of a phage RNA polymerase (e.g., MmP1 RNAP) via a flexible linker (e.g., XTEN). This construct is cloned into a plasmid under an inducible promoter (e.g., PTac).
- Target Plasmid Design: Clone a reporter gene (e.g., sfGFP) or a disabled antibiotic resistance gene (e.g., ErmC Y104S) under the corresponding phage promoter (e.g., PMmP1) on a separate plasmid.
- Evaluation of Mutation Frequency:
- Co-transform both plasmids into the host organism (e.g., E. coli or H. bluephagenesis).
- Induce the mutator system with IPTG and allow for growth.
- Plate the cells on antibiotic-containing media (e.g., erythromycin) to select for clones where a C:G to T:A mutation has restored gene function.
- Calculate the mutation frequency as the ratio of antibiotic-resistant colonies to the total number of viable cells.
- Assessment of Orthogonality: Test the specificity of the system by measuring off-target mutation rates in the host genome, for example, by assessing rifampicin-resistant mutation frequencies.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of orthogonal TF systems requires a suite of specialized reagents. The table below details key solutions for constructing and evaluating these systems.
Table 2: Key Research Reagent Solutions for Orthogonal TF Research
| Reagent / Solution | Function | Examples & Specifications |
|---|---|---|
| Expression Vectors | Provides backbone for TF or CRISPR component expression. | pSEVA series plasmids; pET vectors for protein expression [51] [4]. |
| Chromatin Profiling Kits | Identifies active regulatory elements for TF binding site analysis. | H3K27ac ChIP-seq kits; ATAC-seq kits [52]. |
| Fluorescent Reporters | Quantifies induction level, dynamic range, and leakiness. | sfGFP, mCherry; codon-optimized for host organism [50]. |
| Affinity Purification Kits | Purifies recombinant TFs for in vitro binding studies. | Ni-NTA resin for His-tagged proteins (e.g., GATA4 DBD) [51]. |
| Motif Analysis Software | Predicts TF binding sites and analyzes sequence specificity. | RcisTarget, MEIRLOP, monaLisa; uses PWMs/iPWMs [53] [52]. |
| Synthetic Oligonucleotides | Creates DNA probes, mutant constructs, and gRNA spacers. | IR700-labeled dsDNA for EMSA; variant-containing sequences [51]. |
System Workflows and Regulatory Pathways
The functional principles of advanced orthogonal systems can be visualized as streamlined workflows. The following diagram illustrates the operational logic of a CRISPRi-aided genetic switch.
Diagram 1: CRISPRi-aided genetic switch workflow.
The next diagram depicts the mechanism of a deaminase-phage RNAP fusion system, highlighting its core mutagenesis function.
Diagram 2: Deaminase-phage RNAP mutator system mechanism.
Orthogonality is a foundational concept in synthetic biology, describing engineered biological systems that operate independently of each other and the host's native regulatory networks. For transcription factor (TF) systems, this independence encompasses both mutual compatibility (non-interaction between multiple synthetic systems) and host compatibility (minimal crosstalk with host cellular processes). The pursuit of orthogonal systems is driven by the need for predictable, robust, and complex genetic circuits in applications ranging from basic research to therapeutic drug development. This guide objectively compares the performance of contemporary orthogonal TF systems, providing a detailed analysis of their design principles, experimental validation, and suitability for various biotechnological applications.
Comparative Analysis of Orthogonal Transcription Factor Systems
The table below summarizes the key performance metrics and characteristics of four recently developed orthogonal TF systems, highlighting their specific strategies for achieving mutual and host compatibility.
Table 1: Comparison of Orthogonal Transcription Factor Systems
| System Name / Type | Core Orthogonality Mechanism | Key Orthogonality Metrics | Host Organisms Demonstrated | Primary Applications |
|---|---|---|---|---|
| Orthogonal σ54 System [2] | Knowledge-based rewiring of RpoN box in σ54 factor and its partnered promoter. | "Ideal mutual orthogonality" demonstrated between three mutant σ54 factors (R456H, R456Y, R456L) and native σ54. [2] | E. coli, Klebsiella oxytoca, Pseudomonas fluorescens, Sinorhizobium meliloti. [2] | Control of orthogonal downstream outputs in response to environmental/chemical signals; complex pathway orthogonalization. [2] |
| Orthogonal Transcription Mutators (OTM) [4] | Use of three distinct phage RNA polymerases (MmP1, K1F, VP4) fused to deaminases. | "High orthogonality between phage polymerases" with minimal off-target effects (e.g., 5-14 fold increase in off-target rates for best performer). [4] | E. coli, Halomonas bluephagenesis (a non-model organism). [4] | In vivo hypermutation for accelerated protein evolution; targeted mutagenesis of specific genes or pathways. [4] |
| Engineered λ cI Variants [3] | Directed evolution of bacteriophage λ cI protein for new DNA-binding specificities using a phagemid-based selection system. | A toolkit of 12 TFs operating on up to 270 synthetic promoters with minimal cross-reactivity. [3] | E. coli. [3] | Construction of dual activator-repressor switches and complex logic gates in synthetic gene circuits. [3] |
| Engineered Quorum Sensing Systems [23] | Rational mutagenesis to eliminate crosstalk between LasI/LasR and EsaI/EsaR systems. | Solved promoter crosstalk (LasR interacting with EsaR promoter) and signal crosstalk (LasR responding to EsaI autoinducers) via a LasR(P117S) mutant. [23] | Escherichia coli. [23] | Simultaneous, independent use of multiple quorum sensing systems within the same cell for complex cell-cell communication circuits. [23] |
Experimental Protocols for Orthogonality Assessment
Assessing orthogonality requires rigorous experimental designs to quantify both mutual and host compatibility. The following protocols are derived from the cited studies and represent standard methodologies in the field.
Protocol for Measuring Mutual Orthogonality
This protocol, as used in the characterization of the orthogonal σ54 systems, tests for non-interaction between multiple, co-existing TF-promoter pairs. [2]
- Strain Construction: Generate a set of bacterial strains, each harboring a single test TF gene and a reporter plasmid carrying its cognate synthetic promoter driving a measurable output (e.g., GFP, RFP). Additionally, construct strains containing combinations of multiple TF genes and their respective, uniquely tagged reporter plasmids.
- Cross-Activation Assay: For each TF, measure the reporter output from every non-cognate promoter in the set. This identifies unwanted activation (crosstalk).
- Co-habitation Assay: In strains containing multiple TF-promoter pairs, induce expression and measure the output of each reporter simultaneously. Compare these outputs to the levels measured in the single-pair strains.
- Data Analysis: Calculate the fold-induction for each TF on its cognate versus non-cognate promoters. Orthogonality is demonstrated by high fold-induction on the cognate promoter and minimal activity on all others. Ideal mutual orthogonality is achieved when the performance of each pair is unaffected by the presence of others.
Protocol for Assessing Host Compatibility
This protocol evaluates the interaction between an engineered orthogonal system and the host's native regulatory networks, as critical for systems like the orthogonal transcription mutators. [4]
- Control Strain Preparation: Construct a control strain containing the reporter gene but lacking the orthogonal TF system.
- Genomic Off-Target Measurement: Use a mutation-reporter gene, such as the rifampicin resistance gene (rpsL), to quantify the mutation rate across the host genome in the presence of the active orthogonal system. An orthogonal system should not significantly increase the genomic mutation rate beyond the background level.
- Transcriptomic Analysis: Perform RNA sequencing (RNA-seq) on host cells expressing the orthogonal TF system and compare the transcriptomic profile to that of the control strain.
- Data Analysis: The off-target mutation frequency is calculated and compared to the control. A high-specificity system will show a major increase in mutation rate only on the target plasmid, with a minimal increase in the rifampicin resistance mutation rate. [4] For transcriptomics, differential gene expression analysis identifies any significant unintended perturbations to host gene expression. A highly host-compatible system will show minimal significant changes in host gene expression.
Signaling Pathways and System Workflows
The functionality of orthogonal systems often relies on sophisticated molecular pathways. The diagrams below, defined using the DOT language, illustrate the core mechanisms of two key systems.
σ54-Dependent Orthogonal Transcription
The σ54 system's orthogonality and tight regulation depend on a unique two-step activation mechanism involving a bacterial enhancer-binding protein (bEBP). [2]
Orthogonal Mutator System Workflow
The Orthogonal Transcription Mutation (OTM) system uses phage polymerases targeted by deaminase fusions to introduce mutations specifically into genes of interest. [4]
The Scientist's Toolkit: Key Research Reagents
Successful implementation of orthogonal TF systems relies on a standardized set of genetic parts and experimental tools. The following table catalogs essential reagents as featured in the compared studies.
Table 2: Essential Research Reagents for Orthogonal System Development
| Reagent / Solution | Function | Specific Examples |
|---|---|---|
| Orthogonal Polymerases | Engineered enzymes that recognize unique promoter sequences to decouple transcription from host RNA polymerase. | MmP1, K1F, and VP4 phage RNAPs; [4] T7 RNAP; engineered σ54 factors. [2] |
| Synthetic Promoters | Custom DNA sequences designed to be recognized exclusively by their cognate orthogonal TFs and not by host TFs. | Promoters for orthogonal σ54 (e.g., with rewired RpoN box); [2] Phage promoters (PMmP1, PK1F, PVP4); [4] Engineered λ PR/PRM variants. [3] |
| Reporter Genes | Genes with easily quantifiable outputs (fluorescence, luminescence, antibiotic resistance) used to measure TF activity and specificity. | GFP, RFP, sfGFP for real-time monitoring; [2] [4] Erythromycin resistance gene (ermC) for mutation-recovery assays. [4] |
| Selection Systems | Tools for enriching functional TF-promoter pairs from combinatorial libraries or for assessing mutation efficiency. | M13 phagemid system for directed evolution of TFs; [3] Antibiotic resistance restoration assays. [4] |
| Modular Cloning Systems | Standardized DNA assembly frameworks for rapid and reliable construction of multi-part genetic circuits. | Golden Gate Assembly; [2] MoClo (Modular Cloning) framework. [54] |
The reliability of genetic circuits across different bacterial species is a fundamental challenge in synthetic biology. Orthogonal transcription systems, which operate independently of the host's native regulatory networks, provide a promising solution to this challenge. The σ54-dependent transcription system, with its unique requirement for bacterial enhancer-binding proteins (bEBPs) and distinct promoter recognition mechanism, presents an ideal candidate for such orthogonal applications [2]. Unlike the σ70-dependent housekeeping system, σ54-dependent transcription requires activation by bEBPs that hydrolyze ATP to remodel the closed promoter complex, enabling stringent regulation and strong activation outputs [2]. This complex mechanism offers unique advantages for engineering predictable genetic circuits that can function reliably across diverse bacterial species.
This guide objectively compares the performance of orthogonal σ54-factor systems across three bacterial chassis: Escherichia coli, Pseudomonas fluorescens, and Sinorhizobium meliloti. By examining experimental data from recent studies, we provide a comprehensive analysis of the transferability, functionality, and application potential of these engineered systems in both model and non-model organisms, with particular emphasis on their use in synthetic biology and metabolic engineering applications.
Orthogonal σ54 Systems: Engineering and Mechanism
Engineering Orthogonality Through RpoN Box Rewiring
The engineering of orthogonal σ54 systems centered on targeted modifications to the RpoN box, the key region responsible for promoter recognition. Researchers employed knowledge-based screening and rewiring of this critical domain, specifically modifying residue R456 of σ54 to create mutant variants with altered promoter specificities [2]. Three primary mutant systems were identified and characterized: σ54-R456H, σ54-R456Y, and σ54-R456L. Each variant demonstrated distinct promoter preferences while maintaining ideal mutual orthogonality toward each other and the native σ54 system [2].
The orthogonal systems preserve the fundamental regulatory mechanism of native σ54 transcription, including the essential requirement for bEBP-mediated activation. This conservation ensures that the engineered systems maintain the desirable properties of low basal leakage and high fold-change upon induction that characterize native σ54-dependent transcription [2]. The retention of bEBP dependence provides an additional layer of regulatory control, enabling these systems to respond to environmental or chemical signals through the appropriate bEBP partners.
Molecular Architecture and Transcriptional Activation
Figure 1: Mechanism of σ54-Dependent Transcription Activation. The RNAP-σ54 holoenzyme forms a closed complex at the promoter region, which remains stable until a bacterial enhancer-binding protein (bEBP) is recruited. The bEBP hydrolyzes ATP to remodel the complex into the open conformation, initiating transcription [2].
Experimental Protocols for Cross-Species Validation
Strain Construction and Genetic Engineering
The validation of orthogonal σ54 systems across multiple bacterial species required meticulous strain construction and genetic engineering. For initial testing in E. coli, researchers constructed an ΔrpoN knockout strain using the λ-red homologous recombination method [2]. This involved transforming the temperature-sensitive plasmid pKD46 carrying red recombinase into E. coli JM109, inducing recombinase expression with arabinose, and electroporating with a linear DNA fragment containing a gentamycin-resistant gene flanked by 60 bp homologous arms to the rpoN gene [2]. Successful knockouts were screened by PCR and confirmed by DNA sequencing.
For expression in non-model bacteria, the orthogonal σ54 components were cloned into pBBR-derived broad-host-range vectors to ensure compatibility and maintenance across diverse species [2]. The σ54 mutants (σ54-R456H, R456Y, and R456L) were constitutively expressed using the Pbla2 promoter, while partner bEBPs such as KoNifA and RcNifA were expressed under the control of the Ptet promoter [2]. In P. fluorescens and S. meliloti, codon-optimized versions of key components were employed to enhance expression efficiency, with nifA from Azotobacter vinelandii used in Pseudomonas and native nifA from S. meliloti driven by the Pcat promoter in rhizobial species [2].
Functional Assessment and Phenotypic Characterization
Functional validation of the orthogonal systems employed multiple reporter genes and phenotypic assays. GFP and RFP served as primary reporters for quantitative characterization of promoter activity and orthogonality assessments [2]. Flow cytometry and fluorescence measurements provided precise quantification of transcriptional outputs.
For phenotypic characterization in S. meliloti, researchers utilized nitrogenase activity assays conducted in KPM minimal medium with varying nitrogen levels [2]. These assays measured the system's ability to activate symbiotic nitrogen fixation pathways in response to bEBP inputs. Additionally, sucrose utilization pathways provided a selective growth-based readout, with genes cscA, cscB, and cscK (encoding sucrose metabolic enzymes) serving as reporters for functional transcription system performance [2].
Comparative Performance Across Bacterial Chassis
Orthogonality and Transferability Metrics
Table 1: Performance Comparison of Orthogonal σ54 Systems Across Bacterial Species
| Parameter | E. coli JM109 | P. fluorescens Pf-5 | S. meliloti Sm1021 |
|---|---|---|---|
| σ54-R456H Specific Transcription | Demonstrated | Demonstrated | Demonstrated |
| Mutual Orthogonality | Ideal between mutants and native σ54 | Preserved | Preserved |
| bEBP Dependency | Maintained | Maintained | Maintained |
| Signal Response Capability | Environmental and chemical signals | Environmental and chemical signals | Nitrogen level signals |
| Genetic Circuit Operation | Functional AND/NAND gates | Compatible | Compatible |
| Host Regulatory Interference | Minimal | Minimal | Minimal |
The orthogonal σ54 systems demonstrated remarkable transferability across the three bacterial species, with specific transcription via σ54-R456H confirmed in all tested chassis [2]. The mutual orthogonality – the ability of each mutant system to operate independently without cross-talk – remained ideal not only in E. coli but also in the non-model organisms [2]. This preservation of orthogonality highlights the robustness of the engineering approach and suggests that the fundamental mechanism of σ54-promoter recognition is sufficiently conserved across these species to maintain the engineered specificities while being sufficiently flexible to accommodate the designed modifications.
A critical finding was the maintenance of bEBP dependency in all tested species, enabling the systems to control orthogonal downstream outputs in response to environmental or chemical signals [2]. This preserved regulatory feature allows researchers to leverage the native signaling pathways of each chassis while maintaining orthogonal expression of synthetic circuits.
Applications in Metabolic Pathway Control
Table 2: Metabolic Engineering Applications of Orthogonal σ54 Systems
| Application | E. coli | Pseudomonas | Sinorhizobium |
|---|---|---|---|
| Sucrose Utilization Pathway | Functional cscABK expression | Not reported | Not reported |
| Nitrogen Fixation Control | Not applicable | Not applicable | RcNifA-mediated in KPM medium |
| Layered Genetic Circuits | AND/NAND gates demonstrated | Compatible | Compatible |
| Chemical Production | Compatible | Compatible | Compatible |
The orthogonal σ54 systems enabled precise control of metabolic pathways in a species-appropriate manner. In E. coli, the systems successfully controlled a heterologous sucrose utilization pathway (cscA, cscB, cscK genes), allowing growth on sucrose as a carbon source [2]. In S. meliloti, the systems responded to RcNifA activation under low nitrogen conditions in KPM minimal medium, demonstrating their potential for controlling nitrogen fixation processes relevant to symbiosis [2].
The systems proved capable of orthogonalizing complex biological pathways and genetic circuits in all three species, facilitating the implementation of layered logic operations [2]. This capability is particularly valuable for metabolic engineering applications where balanced expression of multiple pathway enzymes is required to optimize flux toward desired compounds while minimizing metabolic burden.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Orthogonal Transcription System Validation
| Reagent/Solution | Function | Example Application |
|---|---|---|
| pBBR-derived Vectors | Broad-host-range cloning | System transfer to non-model bacteria |
| Pbla2 Promoter | Constitutive expression | σ54 mutant expression |
| Ptet Promoter | Inducible expression | bEBP expression control |
| GFP/RFP Reporters | Quantitative output measurement | Orthogonality assessment |
| cscABK Genes | Sucrose utilization phenotype | Metabolic pathway control |
| KPM Minimal Medium | Defined growth conditions | Nitrogenase activity assays |
| LB/TY Media | Standard cultivation | E. coli and Sinorhizobium growth |
Implementation Workflow for Cross-Species Validation
Figure 2: Experimental Workflow for Cross-Species Validation of Orthogonal Transcription Systems. The process begins with strain construction in E. coli, followed by system transfer to target species using broad-host-range vectors, functional validation through reporter assays, and finally application-specific testing in each chassis [2].
The validation of orthogonal σ54-dependent transcription systems across E. coli, P. fluorescens, and S. meliloti demonstrates the robust transferability of these engineered genetic tools beyond model organisms. The consistent performance of the σ54-R456H, R456Y, and R456L variants across diverse bacterial chassis highlights their potential for reliable synthetic biology applications in both industrial and environmental settings.
The preservation of bEBP dependency across species provides a critical advantage for applications requiring precise temporal and signal-responsive control of gene expression. This feature enables researchers to leverage native regulatory signals while maintaining orthogonal operation of synthetic circuits—a crucial capability for metabolic engineering, bioremediation, and biomedication production.
Future development of orthogonal transcription systems will likely focus on expanding the toolkit of orthogonal σ factors and bEBPs, enhancing the systems' compatibility with additional non-model bacteria, and refining the dynamic range and sensitivity of these systems for precise metabolic control. The successful cross-species validation outlined in this guide establishes a foundation for these advances, providing researchers with proven methodologies for transferring sophisticated genetic control systems across diverse bacterial chassis.
Orthogonal transcription systems are indispensable tools in synthetic biology, enabling the decoupling of engineered genetic circuits from the host's native regulatory networks. Among these, bacterial σ54-dependent systems and phage-derived RNA polymerase (RNAP) systems represent two powerful, yet mechanistically distinct, approaches to achieving precise gene expression control. This guide provides a comparative analysis of these systems, focusing on their fundamental operating principles, performance characteristics, and practical research applications. The evaluation is framed within the context of advancing genetic circuit design, metabolic engineering, and therapeutic development, providing researchers with the data necessary to select the appropriate system for their specific experimental needs.
Fundamental Characteristics and Mechanisms
The σ54-Dependent Transcription System
The σ54 system is an endogenous bacterial transcription machinery component that requires activation by specialized bacterial enhancer-binding proteins (bEBPs). Its key distinguishing feature is a mechanism that tightly locks transcription in an "off" state until a specific activation signal is received [55]. The RNA polymerase holoenzyme formed by the core RNAP and the σ54 factor binds to conserved promoter sequences at the -12 and -24 regions, forming a stable closed complex. However, unlike the major σ70 factor, this closed complex cannot spontaneously isomerize into an open complex [2]. Transcription initiation absolutely requires the intervention of a bEBP, which is typically an ATP-dependent AAA+ ATPase. The bEBP binds to upstream enhancer sequences, often located 80-150 bp from the promoter, and uses the energy from ATP hydrolysis to remodel the closed complex, enabling DNA melting and transcription initiation [55] [7]. This requirement for a remote activator and ATP hydrolysis makes the σ54 system uniquely suited for constructing complex genetic logic gates and achieving very low basal expression with high dynamic range [2].
Phage Polymerase-Based Transcription Systems
Phage polymerase systems, such as the well-characterized T7 RNAP system, employ a virally-encoded, single-subunit RNA polymerase that recognizes a specific phage promoter sequence. This system is fundamentally orthogonal to the host's multi-subunit RNAP because it operates independently of the host's transcription machinery [56]. The T7 RNAP and its cognate promoters form a self-contained transcription module that is highly specific and active. Engineered versions of phage systems have been developed for sophisticated applications, such phage infection-induced gene expression. In one implementation, an engineered M13 phage carries the T7 RNAP gene, which is only expressed upon infection of the host cell. This delivered T7 RNAP then activates a reporter or target gene under the control of a T7 promoter, ensuring that gene expression is confined only to infected cells [56]. This spatial and temporal control is a key advantage over traditional, constitutively active phage systems.
Table 1: Core Characteristics of Orthogonal Transcription Systems
| Feature | σ54-Dependent System | Phage Polymerase-Based System |
|---|---|---|
| Origin | Endogenous bacterial factor [55] | Bacteriophage (e.g., T7) [56] |
| Core Components | Core RNAP, σ54 factor, bEBP [55] | Single-subunit RNAP (e.g., T7 RNAP) [56] |
| Promoter Recognition | σ54 factor (-12/-24 boxes) [55] | Phage RNAP (e.g., T7 promoter) |
| Activation Mechanism | bEBP-dependent ATP hydrolysis & DNA looping [55] | Polymerase expression or delivery (e.g., via phage infection) [56] |
| Key Regulatory Feature | Enhancer elements & bEBP control [2] | Phage-host interaction & polymerase specificity [56] |
| Basal Expression | Very low (locked closed complex) [55] | Varies; can be designed for low leakage [56] |
Performance and Experimental Data
Orthogonality and Tunability
Both systems offer a high degree of orthogonality, but achieve it through different strategies. The σ54 system's orthogonality stems from its unique promoter recognition and absolute requirement for a specific bEBP partner. Recent advances have further expanded the σ54 toolbox through protein engineering. By rewiring the RpoN box in σ54, researchers have created mutant variants (e.g., σ54-R456H, R456Y, R456L) with distinct promoter preferences. These mutants exhibit ideal mutual orthogonality toward each other and the native σ54 system, enabling multiple independent transcription channels within a single cell [2]. This orthogonality has been successfully transferred to non-model bacteria, including Klebsiella oxytoca, Pseudomonas fluorescens, and Sinorhizobium meliloti [2].
Phage polymerase systems, by contrast, derive their orthogonality from the fundamental incompatibility between the phage-derived polymerase and host promoters. The specificity is engineered at the system delivery level. For instance, in the Spatial Phage-Assisted Continuous Evolution (SPACE) system, the gene for T7 RNAP is carried by an engineered M13 phage. The expression of genes under T7 promoter control is therefore strictly contingent upon phage infection, allowing for targeted induction in specific strains within a mixed bacterial community [56]. This provides a form of spatial orthogonality that is difficult to achieve with small-molecule inducers.
Applications in Complex Genetic Circuits
The unique properties of each system make them suitable for different classes of genetic circuits. The σ54 system, with its requirement for ATP-hydrolysis and DNA looping, is inherently suited for constructing layered logic gates and amplifiers. Its "AND"-gate-like behavior—requiring both the σ54-RNAP holoenzyme and an activated bEBP—allows for the construction of complex decision-making circuits [2]. Furthermore, because bEBPs can be controlled by diverse signal transduction pathways (e.g., phosphorylation cascades), the σ54 system can be wired to respond to a wide array of environmental and chemical signals [2] [55].
Phage polymerase systems excel in applications requiring compartmentalized or population-level control. The SPACE system, which uses phage-delivered T7 RNAP, is a powerful example of this applied to directed evolution. It leverages the spatial expansion of motile bacteria in soft agar to provide fresh host cells for iterative phage infection. The phage infection-induced expression of a mutagenesis module ensures that genetic diversity is generated specifically in host cells that have been infected and are carrying the gene of interest to be evolved [56]. This spatial and conditional control remarkably simplifies the operational setup for continuous evolution experiments.
Table 2: Comparative Performance Metrics
| Metric | σ54-Dependent System | Phage Polymerase (T7) System |
|---|---|---|
| Dynamic Range | High (strong activation, low basal) [2] | High [56] |
| Orthogonal Variants | Multiple (σ54-R456H/Y/L) [2] | Limited (promoter/polymerase pairs) |
| Transferability Across Species | Demonstrated in diverse bacteria [2] | Requires phage host range & delivery |
| Toxicity to Host | Low (native, low-leakage system) [2] | Can be high if overexpressed [2] |
| Suitability for Logic Gates | Excellent (AND-gate behavior) [2] | Moderate |
| Spatial/Temporal Control | Via bEBP regulation | Via phage infection [56] |
Experimental Protocols
Protocol: Establishing an Orthogonal σ54 System
This protocol outlines the steps for implementing a mutant σ54 orthogonal system in a new host, based on the methodology described in [2].
Host Strain Preparation (if necessary):
- For a clean background, construct an rpoN (σ54) knockout strain using λ-Red homologous recombination. The linear DNA fragment for knockout should consist of a gentamycin-resistant (GmR) gene flanked by 60 bp homologous arms targeting the rpoN gene.
- Transform the temperature-sensitive plasmid pKD46 (carrying Red recombinase) into the host (e.g., E. coli JM109).
- Induce Red recombinase expression with arabinose and electroporate the linear knockout DNA fragment.
- Plate on Gm plates and incubate at 37°C overnight. Screen colonies by PCR and DNA sequencing to confirm the knockout.
Plasmid Assembly:
- Use Golden Gate assembly to construct the expression vector. The essential components include:
- A constitutive promoter (e.g., Pbla2) to express the orthogonal σ54 factor (e.g., σ54-R456H).
- The gene for the corresponding enhancer-binding protein (EBP) under a regulated promoter (e.g., Ptet for inducible expression).
- A reporter gene (e.g., gfp or rfp) under the control of the cognate orthogonal σ54 promoter.
- For broad-host-range applications, clone the assembled cassette into a suitable vector, such as a pBBR-derived plasmid.
- Use Golden Gate assembly to construct the expression vector. The essential components include:
Validation and Characterization:
- Transform the assembled plasmid into the prepared host strain.
- Induce the EBP expression with the relevant signal (e.g., anhydrotetracycline for Ptet).
- Measure output (e.g., fluorescence) over time to quantify the dynamic range, orthogonality against the native σ54 system, and leakiness. Flow cytometry can be used for single-cell resolution.
Protocol: Phage Infection-Induced Gene Expression
This protocol details the setup for a phage-delivered T7 RNAP system for conditional gene expression, as used in SPACE [56].
Reporter and Accessory Plasmid Construction:
- Reporter Plasmid (RP): Clone the gene of interest (e.g., GFP) downstream of a T7 promoter on a medium-copy plasmid (e.g., pUC origin) with appropriate antibiotic resistance (e.g., Chloramphenicol, Chl).
- Accessory Plasmid (AP): Construct a plasmid carrying the T7 RNAP gene under a promoter that is activated by phage infection components. Alternatively, this plasmid can provide factors necessary for phage propagation (e.g., gIII for M13 phage).
Engineered Activator Phage Preparation:
- Engineer an M13 phage genome by replacing the gIII gene with the T7 RNAP gene. This creates a selection linkage where the phage's ability to propagate is tied to the functional expression of T7 RNAP.
- Produce high-titer stocks of this engineered "Activator Phage" (e.g., AP1-SPT7).
Host Cell Preparation and Infection:
- Transform the host bacteria (e.g., E. coli) with both the Reporter Plasmid and the necessary Accessory Plasmid.
- Grow the transformed cells to mid-log phase.
- Infect the culture with the engineered Activator Phage at the desired multiplicity of infection (MOI). A low MOI (<1) can be used to induce gene expression in only a subpopulation of cells.
- Incubate with shaking to allow for phage infection, T7 RNAP expression, and subsequent activation of the gene of interest on the Reporter Plasmid.
Analysis:
- Monitor gene expression output (e.g., fluorescence) over time.
- Use flow cytometry or microscopy to confirm that expression is correlated with phage infection events.
System Diagrams and Workflows
The diagrams below illustrate the core mechanisms and a key application for each orthogonal transcription system.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Orthogonal Transcription Research
| Reagent / Material | Function / Description | Example Use Case |
|---|---|---|
| Orthogonal σ54 Plasmids | Expression vectors for σ54 mutants (R456H/Y/L) and cognate promoters [2]. | Establishing multiple orthogonal transcription channels in a single host. |
| bEBP Expression Constructs | Plasmids for inducible expression of enhancer-binding proteins (e.g., under Ptet) [2]. | Providing the essential activation signal for σ54-dependent transcription. |
| Engineered Activator Phages | Modified bacteriophages (e.g., M13-AP1-SPT7) carrying polymerase genes [56]. | Delivering transcriptional activators via infection for spatial/temporal control. |
| Phage Shock Protein (psp) Reporter | Plasmid with GFP downstream of pspA-E promoter [56]. | Reporting on phage infection events or related stress responses. |
| Mutagenesis Plasmid (e.g., pLM1) | Plasmid carrying mutator genes (dnaQ926) under phage-inducible control [56]. | Targeted evolution experiments (e.g., in SPACE) to generate diversity. |
| λ-Red Recombination System | Plasmid (pKD46) for efficient gene knockout/integration in E. coli [2]. | Creating clean genetic backgrounds (e.g., ΔrpoN strains). |
| Broad-Host-Range Vectors | Plasmids (e.g., pBBR-derived) for system transfer to non-model bacteria [2]. | Deploying orthogonal systems in clinically or industrially relevant strains. |
The choice between σ54-dependent systems and phage polymerase-based systems is not a matter of superiority, but of strategic application. The σ54 system offers unparalleled precision and programmability for internal regulatory circuits within a bacterial population. Its low basal expression, high activation range, and the availability of mutually orthogonal variants make it ideal for constructing complex synthetic networks, biosensors, and finely-tuned metabolic pathways. In contrast, phage polymerase systems excel in inter-cellular and population-level control, where the key variable is which specific cell within a population expresses a gene. Their strength lies in coupling gene expression to the physical event of phage infection, enabling powerful applications in directed evolution, bacterial community engineering, and potentially, antimicrobial strategies that require precise targeting. A deep understanding of their distinct operational principles, as provided in this guide, empowers researchers to deploy these systems effectively, pushing the boundaries of synthetic biology and therapeutic development.
In synthetic biology, the functional validation of complex metabolic pathways is often complicated by crosstalk with the host's native regulatory networks. Orthogonal transcription factor systems, which operate independently of host machinery, provide a powerful solution to this challenge. These systems enable precise control and measurement of pathway activity without confounding host interactions, making them particularly valuable for evaluating multifaceted processes like nitrogen fixation and sucrose utilization. This guide compares the performance of current orthogonal tools and their application in dissecting these essential biological pathways, providing researchers with a framework for selecting appropriate validation strategies.
The emerging significance of σ54-dependent transcription presents a particularly promising orthogonal platform. Unlike the more common σ70 factor, σ54-dependent promoters require activation by bacterial enhancer-binding proteins (bEBPs), creating a multi-layer control system that offers low basal leakage and high inducibility [2]. This "eukaryotic-like" regulation mechanism provides a valuable tool for pathway analysis where precise, low-noise control is essential. Furthermore, the development of orthogonal σ54 factors with mutated RpoN boxes (R456H, R456Y, R456L) has expanded this toolkit, enabling simultaneous analysis of multiple pathway components without interference [2].
Comparative Analysis of Orthogonal Validation Systems
Table 1: Performance Comparison of Orthogonal Transcription Systems
| System Type | Key Components | Mutation Rate/Induction Fold | Target Pathways | Host Organisms | Key Advantages |
|---|---|---|---|---|---|
| Orthogonal Transcription Mutators (OTM) | Phage RNAPs (MmP1, K1F, VP4) fused to deaminases | >1,500,000-fold increased mutation rates; Uniform C:G to T:A and A:T to G:C transitions | Fluorescent proteins, chromoproteins, metabolic enzymes | E. coli, H. bluephagenesis | High specificity, minimal off-target effects, single-day mutagenesis |
| σ54-Dependent Orthogonal Systems | σ54 mutants (R456H, R456Y, R456L) with bEBPs | Strong activation with minimal basal expression; Tunable via bEBP input | Nitrogen fixation, sucrose utilization | E. coli, K. oxytoca, P. fluorescens, S. meliloti | Low basal leakage, eukaryotic-like regulation, transferable across species |
| Deaminase-T7RNAP Fusion Systems | rAPOBEC1, PmCDA1, or TadA8e fused to T7RNAP | Varies by deaminase; C→T or A→G transitions | cis-aconitate decarboxylase, various enzymes | Mammals, yeast, plants | Gene-specific mutagenesis, proven in eukaryotes |
Table 2: Quantitative Performance Metrics in Pathway Engineering
| Pathway | Engineering Approach | Performance Improvement | Key Measured Parameters | Validation Method |
|---|---|---|---|---|
| Sucrose Utilization (Csc) in E. coli | Adaptive Laboratory Evolution (ALE) | 1.72x growth rate, 1.40x sucrose uptake rate [57] | Growth rate, substrate consumption, genomic mutations | Whole genome sequencing, fermentation analysis |
| Energy-Conserving Sucrose Utilization in B. amyloliquefaciens | Heterologous pathway (CscB + SucP) | 49.4% more sucrose consumed, 38.5% more γ-PGA produced [58] | Product yield, substrate utilization, ATP consumption | HPLC, yield calculations, fermentation kinetics |
| Nitrogen Fixation in R. palustris | Suppressor mutations (Fer1, AadN) | Restored growth under nitrogen-fixing conditions in ΔfixC strain [59] | Growth under N-limiting conditions, electron transfer | Genetic complementation, enzyme activity assays |
| Soil Sucrose Impact on Legume BNF | Sucrose soil amendment | %Ndfa increased from 83% to 96%; 3x biomass in C. juncea [60] | %Ndfa, dry matter production, nitrogen accumulation | Isotopic ratio mass spectrometry, biomass measurement |
Experimental Protocols for Key Validation Approaches
Orthogonal Mutagenesis for Pathway Optimization
The Orthogonal Transcription Mutation (OTM) system enables rapid in vivo protein evolution for pathway components. The following protocol, adapted from the OTM system [4], allows comprehensive mutagenesis of target pathways:
Construct Mutator Plasmids: Fuse cytosine deaminase (PmCDA1) and adenine deaminase (TadA8e) variants with phage RNA polymerases (MmP1, K1F, VP4) using XTEN linkers in a high-copy-number plasmid (e.g., pSEVA241).
Clone Target Pathway: Insert genes of interest under control of corresponding phage promoters (PMmP1, PK1F, PVP4) in a separate reporter plasmid.
Transformation and Induction: Co-transform mutator and target plasmids into host strain (E. coli or H. bluephagenesis). Induce mutator expression with IPTG (optimize concentration between 0.1-1.0 mM).
Mutation Generation: Grow cultures for 12-24 hours to accumulate mutations. The system introduces uniform C:G to T:A and A:T to G:C transitions across target genes.
Screening and Selection: Plate cells on selective media or use fluorescence-activated cell sorting (FACS) for high-throughput screening of desired phenotypes.
Validation: Sequence target genes to verify mutation profiles and measure off-target effects using rifampicin resistance frequency assays.
This system achieves >1,500,000-fold increased mutation rates with minimal off-target effects, enabling comprehensive pathway optimization within a single day [4].
σ54-Dependent Orthogonal Pathway Expression
For precise control and measurement of pathway activity without host interference, the σ54-dependent orthogonal system provides stringent regulation [2]:
Strain Preparation: Create ΔrpoN host strain using λ-red homologous recombination with Gm-resistant gene cassette.
Orthogonal σ54 Mutant Expression: Clone orthogonal σ54 mutants (R456H, R456Y, R456L) under constitutive promoters (e.g., Pbla2) in appropriate vectors.
Promoter Engineering: Modify target pathway genes (nif, csc) with orthogonal σ54-dependent promoters containing specific -24 region mutations (e.g., GG-GAAC for σ54-R456H).
bEBP Integration: Co-express compatible bEBPs (NifA, CbrA) under inducible promoters (Ptet, Pcat) for pathway activation control.
Pathway Assembly: Assemble complete pathway with orthogonal transcription components using Golden Gate assembly for modular construction.
Cross-Species Validation: Transfer constructs to broad-host-range vectors (pBBR-derived) for validation in non-model organisms (K. oxytoca, P. fluorescens, S. meliloti).
This system enables discrete analysis of pathway components with minimal basal expression and strong inducibility (>1000-fold induction in some configurations) [2].
Diagram Title: σ54 Orthogonal Transcription System
Pathway-Specific Validation Methodologies
Nitrogen Fixation Pathway Analysis
The nitrogen fixation pathway presents particular challenges for functional validation due to its oxygen sensitivity, complex metalloenzyme requirements, and multi-component electron transfer system. The following specialized approaches enable accurate functional assessment:
Electron Transfer Pathway Validation [59]:
- Engineer suppressor strains by introducing specific mutations in ferredoxin (Fer1) and NAD+-dependent Fd:NADPH oxidoreductase (AadN)
- Measure nitrogenase activity through acetylene reduction assays under anaerobic conditions
- Quantify electron flux using Fd reduction assays and monitor H2 production as obligate nitrogenase byproduct
- Validate pathway specificity through genetic complementation in ΔfixC backgrounds
Nitrogenase Variant Identification [61]:
- Utilize comprehensive databases (NFixDB) with Hidden Markov Models for nifHDK, vnfHDK, and anfHDK identification
- Apply E-value cutoff <9.9×10-10 and bitscore >50 for sequence validation
- Verify gene cluster integrity (proximity of H, D, K genes) to distinguish functional nitrogenases from pseudo-nitrogenases
- Link nitrogenase genes to ribosomal RNA operons (16S-5S-23S) for phylogenetic analysis
Environmental Interaction Studies [60] [62]:
- Assess nitrogen fixation efficiency through 15N isotopic dilution methods
- Calculate %Ndfa (percentage of nitrogen derived from atmosphere) using mass spectrometry
- Measure nitrogenase gene expression (nifH transcript levels) under varying soil conditions
- Correlate functional gene abundance (nifH, anfH, vnfH) with process rates using qPCR
Table 3: Nitrogen Fixation Functional Assays
| Assay Type | Key Reagents/Methods | Measured Parameters | Applications | Advantages/Limitations |
|---|---|---|---|---|
| Acetylene Reduction Assay | Acetylene gas, gas chromatography | Ethylene production, nitrogenase activity | Laboratory and field measurements | High sensitivity, but indirect measurement |
| Isotopic Methods | 15N2 gas, mass spectrometry | Ndfa, nitrogen flux, incorporation rates | Quantitative fixation measurement | Direct but technically complex |
| nifH Expression Analysis | RNA extraction, RT-qPCR, RNA-Seq | Transcript abundance, regulation | Gene expression studies | Correlative, not direct activity |
| Functional Gene Arrays | DNA microarrays, metagenomics | Gene abundance, diversity | Community analysis | Comprehensive but expensive |
Sucrose Utilization Pathway Engineering
Sucrose utilization pathways vary significantly in their energy efficiency and regulatory complexity, requiring distinct validation approaches:
Pathway Efficiency Analysis [58] [57]:
- Compare ATP consumption between PTS (2 ATP/sucrose) and non-PTS (1 ATP/sucrose) pathways
- Measure growth rates and biomass yields in minimal sucrose media
- Quantify sucrose uptake rates using HPLC analysis of extracellular sucrose depletion
- Calculate carbon conversion efficiency through metabolic flux analysis
Adaptive Laboratory Evolution (ALE) Protocol [57]:
- Initialize evolution with engineered strains containing integrated cscBKA genes
- Culture in M9 minimal media with 20 g/L sucrose as sole carbon source
- Perform serial passages during exponential growth phase using automated platform
- Monitor population dynamics through regular OD600 measurements
- Sequence evolved clones to identify causal mutations (rpoB, rpoC, pyrE, cscR)
- Validate mutations through reverse engineering in ancestral strains
Orthogonal Pathway Expression [2]:
- Implement σ54-dependent expression of cscA, cscB, and cscK genes
- Assess orthogonality through growth comparison in sucrose vs. glucose media
- Measure pathway specificity through transcript analysis of native sugar utilization genes
- Validate cross-species functionality in non-model organisms
Diagram Title: Sucrose Utilization Pathways Comparison
Research Reagent Solutions Toolkit
Table 4: Essential Research Reagents for Pathway Validation
| Reagent/Category | Specific Examples | Function/Application | Key Features/Benefits |
|---|---|---|---|
| Orthogonal σ54 Factors | σ54-R456H, R456Y, R456L mutants | Pathway component expression | Mutual orthogonality, transferable across species |
| Phage RNA Polymerases | MmP1, K1F, VP4 RNAPs | Orthogonal transcription mutation | Broad host range, high efficiency in non-model organisms |
| Deaminase Fusion Enzymes | PmCDA1-UGI, evoPmCDA1, TadA8e | Targeted mutagenesis | Specific transition mutations, reduced off-target effects |
| Bacterial Enhancer Proteins | NifA, CbrA, HrpR/S | σ54-dependent activation | Stringent regulation, environmental responsiveness |
| Reporter Systems | sfGFP, RFP, ErmC resistance | Pathway output measurement | Quantitative readouts, selection capability |
| Sucrose Pathway Components | CscB (permease), CscA (hydrolase), SucP (phosphorylase) | Sucrose utilization engineering | Energy-conserving options, heterologous expression |
| Nitrogenase Components | nifHDK, vnfHDK, anfHDK clusters | Nitrogen fixation analysis | Metal cofactor specificity, phylogenetic distribution |
| Database Resources | NFixDB, GTDB, FunGene (historical) | Sequence analysis and identification | HMM-based search, rRNA operon linkage |
Functional validation of nitrogen fixation and sucrose utilization pathways requires careful selection of orthogonal systems matched to specific research goals. For nitrogen fixation studies where low background expression is critical, σ54-dependent systems provide exceptional stringency and are transferable across diverse microbial hosts. For rapid optimization of pathway components, orthogonal mutagenesis systems offer unparalleled speed in generating diversity. The strategic integration of these tools, combined with the experimental protocols and reagents outlined in this guide, enables researchers to overcome traditional challenges in complex pathway validation.
When designing validation experiments, consider the energy efficiency of sucrose pathways, the metal cofactor requirements of nitrogenase variants, and the orthogonality of expression systems relative to your host organism. The quantitative data and comparative tables provided here serve as benchmarks for evaluating your system performance. As orthogonal tools continue to evolve, these foundational approaches will enable increasingly sophisticated engineering of complex biological pathways for both basic research and biotechnological applications.
Conclusion
Orthogonal transcription factor systems represent a paradigm shift in synthetic biology, offering unprecedented control over gene expression by operating independently of host machinery. The foundational work on σ54 and phage polymerases has blossomed into a versatile toolkit, enabling the creation of multiple, mutually orthogonal systems for programming complex biological functions. Methodological advances allow for the rational design and tuning of these systems, while robust troubleshooting frameworks address critical challenges like specificity and toxicity. Successful validation across a range of bacterial species underscores their broad applicability. Future directions will focus on refining these systems for therapeutic applications, including cell-based therapies and intelligent drug delivery, and expanding their functionality in eukaryotic cells. The continued expansion of this orthogonal toolkit promises to unlock new frontiers in biomedical research, metabolic engineering, and clinical interventions.
References
- 1. Gene regulation technologies for gene and cell therapy [https://www.sciencedirect.com/science/article/pii/S1525001625002783]
- 2. Expanding the σ54-dependent transcription process with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12096077/]
- 3. Engineering orthogonal dual transcription factors for multi ... [https://www.nature.com/articles/ncomms13858]
- 4. An orthogonal transcription mutation system generating all ... [https://www.nature.com/articles/s41467-025-61354-4]
- 5. Optimized reporters for multiplexed detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11667439/]
- 6. Mechanisms of σ54-Dependent Transcription Initiation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7057263/]
- 7. The σ 54 system directly regulates bacterial natural product ... [https://www.nature.com/articles/s41598-021-84057-4]
- 8. Nucleotide-level characterization and improvement of l ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11983282/]
- 9. Engineered CRISPRa enables programmable eukaryote ... [https://www.nature.com/articles/s41467-019-11479-0]
- 10. Expanding the σ54-dependent transcription process with ... [https://pubmed.ncbi.nlm.nih.gov/40401558/]
- 11. Design of strictly orthogonal biosensors for maximizing ... [https://www.sciencedirect.com/science/article/pii/S2090123225006988]
- 12. Innovations, Challenges and Future Directions of T7RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12090346/]
- 13. Directed evolution of an orthogonal transcription engine for ... [https://www.sciencedirect.com/science/article/pii/S2589004224027688]
- 14. Comprehensive evaluation of T7 promoter for enhanced ... [https://www.nature.com/articles/s41598-024-59978-5]
- 15. Small molecule- and cell contact-inducible systems for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12188245/]
- 16. BNDS-China - iGEM 2025 [https://2025.igem.wiki/bnds-china/design]
- 17. Conversion of natural cytokine receptors into orthogonal ... [https://www.nature.com/articles/s41589-025-01986-1]
- 18. Bacterial Enhancer Binding Proteins—AAA + Proteins in ... [https://www.mdpi.com/2218-273X/10/3/351]
- 19. Is Enhancer Function Driven by Protein–Protein Interactions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12101050/]
- 20. Genetic controllers for enhancing the evolutionary ... [https://www.nature.com/articles/s41467-025-63627-4]
- 21. High-throughput transcriptomics toxicity assessment of ... [https://www.sciencedirect.com/science/article/pii/S0269749124015410]
- 22. High-throughput transcriptomics analysis of equipotent and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12408750/]
- 23. Characterization and orthogonality assessment of two ... [https://www.sciencedirect.com/science/article/pii/S1871678425000780]
- 24. Revolutionizing therapeutic protein design with synthetic ... [https://www.news-medical.net/news/20250808/Revolutionizing-therapeutic-protein-design-with-synthetic-biology.aspx]
- 25. Interpretable protein-DNA interactions captured by ... [https://elifesciences.org/articles/105565]
- 26. PANCS-Binders: a rapid, high-throughput binder discovery ... [https://www.nature.com/articles/s41592-025-02740-0]
- 27. Context-Dependent Redesign of Robust Synthetic Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11223972/]
- 28. Advances in transcription factor delivery: Target selection, ... [https://www.sciencedirect.com/science/article/pii/S016836592500505X]
- 29. An orthogonal transcription mutation system generating all ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12218169/]
- 30. Transcriptional programming using engineered systems of ... [https://www.nature.com/articles/s41467-019-12706-4]
- 31. An orthogonal transcription mutation system generating all ... [https://pubmed.ncbi.nlm.nih.gov/40593783/]
- 32. Environmental signal integration by a modular AND gate [https://pmc.ncbi.nlm.nih.gov/articles/PMC1964800/]
- 33. Ultra-fast variant effect prediction using biophysical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12507518/]
- 34. Chemical signaling under abiotic stress environment in plants [https://pmc.ncbi.nlm.nih.gov/articles/PMC2634487/]
- 35. Assessing the feasibility of CRISPRa approaches to ... [https://www.sciencedirect.com/science/article/pii/S2590006425002790]
- 36. Smug1 竟能缓解5-FU 生殖毒性,为抗癌治疗开辟新思路 [https://www.ebiotrade.com/newsf/2025-2/20250218050928590.htm]
- 37. 上海营养与健康研究所建立用于多样本ChIP-seq数据分组 ... [https://shb.cas.cn/kydt2024/kjjz2024/202011/t20201130_5804142.html]
- 38. CN110268057B - 用于鉴定和表达基因簇的系统和方法 [https://patents.google.com/patent/CN110268057B/zh]
- 39. 一文详解转录因子研究策略(上) [https://www.bestofbest.top/news_1/141.html]
- 40. Frontiers | Synthetic Promoters and Transcription for... Factors [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2017.00063/full]
- 41. DNA-guided transcription factor interactions extend human ... [https://www.nature.com/articles/s41586-025-08844-z]
- 42. CollecTF: a database of experimentally validated transcription ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3965012/]
- 43. A Comprehensive Guide to Transcription Factor Research ... [https://www.pronetbio.com/News/172.html]
- 44. Benchmarking tools for transcription factor prioritization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11137382/]
- 45. Modeling and designing enhancers by introducing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11807178/]
- 46. Engineering wetware and software for the predictive ... [https://www.nature.com/articles/s41467-025-64457-0]
- 47. Engineered transcription factor-binding arrays for DNA- ... [https://www.sciencedirect.com/science/article/pii/S0167779925001726]
- 48. Highly multiplexed design of an allosteric transcription ... [https://www.nature.com/articles/s41467-024-54260-8]
- 49. Modular and signal-responsive transcriptional regulation ... [https://jbioleng.biomedcentral.com/articles/10.1186/s13036-025-00526-8]
- 50. Modular and signal-responsive transcriptional regulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12142916/]
- 51. Protocol for evaluating the impact of non-coding variants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12169763/]
- 52. Benchmarking tools for transcription factor prioritization [https://www.sciencedirect.com/science/article/pii/S2001037024001648]
- 53. Discovery and validation of information theory-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5389469/]
- 54. Orthogonal control of gene expression in plants using ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-022-00867-1]
- 55. The Bacterial Enhancer-Dependent ς54 (ςN) Transcription ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC101881/]
- 56. Engineered Phage Enables Efficient Control of Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11720261/]
- 57. Generation of an E. coli platform strain for improved sucrose ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-019-1165-2]
- 58. Construction of energy-conserving sucrose utilization ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-017-0712-y]
- 59. Evolving a New Electron Transfer Pathway for Nitrogen ... [https://pubmed.ncbi.nlm.nih.gov/36645294/]
- 60. Effects of Soil Sucrose Application on Biological Nitrogen ... [https://www.mdpi.com/2504-3129/5/3/50]
- 61. NFixDB (Nitrogen Fixation DataBase) [https://pmc.ncbi.nlm.nih.gov/articles/PMC11155484/]
- 62. Microbial functional genes involved in nitrogen fixation, ... [https://www.sciencedirect.com/science/article/abs/pii/S0038071714001163]