LDBT vs DBTL: How Machine Learning is Redefining the Synthetic Biology Cycle
This article explores the paradigm shift from the traditional Design-Build-Test-Learn (DBTL) cycle to a new Learn-Design-Build-Test (LDBT) framework in synthetic biology.
LDBT vs DBTL: How Machine Learning is Redefining the Synthetic Biology Cycle
Abstract
This article explores the paradigm shift from the traditional Design-Build-Test-Learn (DBTL) cycle to a new Learn-Design-Build-Test (LDBT) framework in synthetic biology. Driven by advances in machine learning (ML) and artificial intelligence (AI), this reordering places data-driven learning at the forefront, enabling more predictive and precise biological design. We will examine the foundational principles of both cycles, detail the key ML technologies and high-throughput 'Build' and 'Test' methods that make LDBT possible, address critical troubleshooting and optimization challenges, and validate the approach through comparative analysis of its impact on efficiency and success rates. Aimed at researchers, scientists, and drug development professionals, this review synthesizes how LDBT accelerates therapeutic discovery, optimizes protein engineering, and paves the way for a more predictive engineering biology.
From DBTL to LDBT: Understanding the Foundational Shift in Engineering Biology
The Design-Build-Test-Learn (DBTL) cycle has long served as the foundational framework for systematic biological engineering, providing a structured approach to designing and optimizing biological systems. This iterative process begins with designing genetic constructs, building them in biological systems, testing their performance, and learning from the results to inform subsequent design iterations [1]. However, recent advances in machine learning (ML) and high-throughput testing platforms are fundamentally reshaping this paradigm. A new framework, dubbed "LDBT" (Learn-Design-Build-Test), proposes reordering the cycle to begin with machine learning, potentially accelerating biological design by leveraging predictive algorithms before physical construction [2]. This paradigm shift promises to transform synthetic biology from an empirical, trial-and-error discipline toward a more predictive engineering science.
The tension between traditional DBTL and the emerging LDBT framework represents a critical juncture for synthetic biology research and drug development. Where DBTL relies on empirical iteration to gain knowledge, LDBT leverages pre-trained machine learning models on vast biological datasets to generate initial designs, potentially reducing the number of physical cycles needed to achieve desired biological functions [2] [3]. This comparative analysis examines both frameworks through experimental data, methodological protocols, and practical implementations to guide researchers in selecting appropriate strategies for their biological engineering challenges.
Framework Fundamentals: DBTL vs. LDBT
The Traditional DBTL Cycle
The conventional DBTL cycle follows a sequential, iterative process. The Design phase involves defining objectives and designing genetic parts or systems using domain knowledge and computational modeling. The Build phase focuses on physical construction through DNA synthesis, assembly, and introduction into characterization systems (e.g., bacterial, mammalian, or cell-free systems). The Test phase experimentally measures the performance of engineered biological constructs. Finally, the Learn phase analyzes collected data to inform the next design iteration, repeating until desired functionality is achieved [2] [1].
This approach has proven effective but often requires multiple cycles to gain sufficient knowledge for optimal designs, with the Build-Test phases creating significant bottlenecks in timeline and resources [2]. The process is further constrained by the vast combinatorial space of biological sequences; for an average 300-residue protein, just three substitutions can yield approximately 3.1 × 10¹⁰ possible combinations, making exhaustive exploration impractical [4].
The Emerging LDBT Paradigm
The LDBT framework repositions "Learn" as the initial phase, leveraging pre-trained machine learning models on large biological datasets to generate initial designs. These models can capture complex patterns in high-dimensional spaces, enabling more efficient navigation of the biological design space before physical construction [2] [3]. Specifically, protein language models like ESM and ProGen, trained on millions of protein sequences, can perform zero-shot predictions of beneficial mutations and infer protein functions without additional training [2].
This learn-first approach is further enhanced by integrating cell-free transcription-translation (TX-TL) systems for rapid testing. These systems circumvent complexities of living cells, enabling swift assessment of genetic circuit performance within hours rather than days or weeks [3]. When coupled with machine learning predictions, they create a synergistic framework that accelerates validation while enriching training datasets for improved algorithmic learning [2] [3].
Table 1: Core Conceptual Differences Between DBTL and LDBT Frameworks
| Aspect | Traditional DBTL | LDBT Paradigm |
|---|---|---|
| Starting Point | Design based on existing knowledge | Learning from pre-trained ML models on large datasets |
| Primary Driver | Empirical iteration | Predictive algorithms |
| Knowledge Acquisition | Gradual, through multiple cycles | Leveraged from foundational models at outset |
| Testing Approach | Often in vivo systems | Heavy utilization of rapid cell-free platforms |
| Cycle Goal | Converge through iteration | Achieve functionality in fewer cycles |
Experimental Comparisons and Performance Metrics
Case Study: Protein Engineering with DeepDE
A rigorous comparison of the frameworks emerges from protein engineering applications. Traditional directed evolution follows the DBTL approach, requiring labor-intensive screening of thousands of mutants over multiple rounds [4]. In contrast, the DeepDE algorithm exemplifies the LDBT approach, leveraging deep learning on a compact library of ~1,000 mutants as a training set [4].
When applied to GFP from Aequorea victoria, DeepDE achieved a 74.3-fold increase in activity over four rounds of evolution, far surpassing the benchmark superfolder GFP (40.2-fold increase) that required multi-year engineering efforts [4]. This demonstrates how machine-learning guided approaches can significantly accelerate optimization cycles while achieving superior results.
The algorithm employed a mutation radius of three (triple mutants), enabling exploration of a much greater sequence space compared to single or double mutants in each iteration. This approach explored a combinatorial library of approximately 1.5 × 10¹⁰ variants, a space impractical for traditional methods [4].
Case Study: Dopamine Production in E. coli
A knowledge-driven DBTL approach was used to optimize dopamine production in E. coli, demonstrating the traditional framework's capabilities when enhanced with mechanistic insights [5]. Researchers developed a high-throughput RBS engineering strategy to fine-tune expression levels of dopamine pathway enzymes.
The optimized strain achieved dopamine production of 69.03 ± 1.2 mg/L (equivalent to 34.34 ± 0.59 mg/g biomass), representing a 2.6 to 6.6-fold improvement over previous state-of-the-art in vivo production methods [5]. This success highlights how traditional DBTL cycles, when informed by upstream in vitro investigations and high-throughput engineering, can efficiently optimize complex metabolic pathways.
Table 2: Quantitative Performance Comparison of Engineering Approaches
| Engineering Approach | Target System | Performance Improvement | Screening Scale | Iterations/Timeframe |
|---|---|---|---|---|
| Traditional Directed Evolution (DBTL) | Various proteins | Variable, often requires extensive optimization | Thousands to millions of variants | Multiple rounds over months/years |
| DeepDE (LDBT) | avGFP | 74.3-fold increase in activity | ~1,000 mutants per round | 4 rounds [4] |
| Knowledge-Driven DBTL | Dopamine production in E. coli | 69.03 mg/L (2.6-6.6-fold improvement) | High-throughput RBS library | Not specified [5] |
| AI-Guided + Cell-Free (LDBT) | Antimicrobial peptides | 6 promising designs from 500,000 survey | 500 variants validated | Single round with computational pre-screening [2] |
Methodological Protocols
Protocol: DeepDE Implementation for Protein Engineering
The DeepDE algorithm exemplifies the LDBT approach through iterative deep learning-guided directed evolution [4]:
Training Data Curation: Compile a supervised training dataset of approximately 1,000 single or double mutants with associated fitness measurements. For avGFP, this dataset covered 219 of 238 sites.
Model Training: Implement three deep learning methods—unsupervised, weak-positive only, and supervised learning—using the curated dataset. Performance correlates with training dataset size, with Spearman's correlation coefficients increasing from 0.30 to 0.74 as dataset size grows from 24 to 2,000 mutants.
Mutant Prediction: Set a mutation radius of three for each evolution round. For the "mutagenesis by direct prediction" approach, compute all possible double mutants, identify top performers, calculate mutation frequency per site, and generate triple mutant combinations for prediction.
Experimental Validation: Synthesize and assay top-ranked triple mutants (e.g., top 10 predictions). For the "mutagenesis coupled with screening" approach, experimentally construct libraries of triple mutants for screening.
Iterative Cycling: Use best-performing mutants as templates for subsequent rounds, repeating the process for 4-5 rounds with the same training dataset before potentially transitioning to a different dataset.
Protocol: Knowledge-Driven DBTL for Metabolic Engineering
The dopamine production case study demonstrates an enhanced DBTL approach [5]:
Upstream In Vitro Investigation: Conduct cell lysate studies to assess enzyme expression levels and pathway interactions before in vivo implementation. This mechanistic understanding informs initial design decisions.
Host Strain Engineering: Develop a high-production host (e.g., E. coli FUS4.T2 for dopamine) with precursor enhancement (e.g., l-tyrosine overproduction through genomic modifications like TyrR depletion and feedback inhibition mutation).
Pathway Optimization: Implement high-throughput RBS engineering to fine-tune relative expression of pathway enzymes. Modulate Shine-Dalgarno sequences without interfering secondary structures to predictably control translation initiation rates.
Automated Strain Construction: Utilize automated molecular cloning and cultivation processes to accelerate the Build and Test phases.
Multi-Omics Analysis: Integrate transcriptomic, proteomic, and metabolomic data during the Learn phase to identify bottlenecks and inform subsequent design iterations.
Visualization Frameworks
DBTL Cycle Workflow
LDBT Paradigm Workflow
Research Reagent Solutions
Table 3: Essential Research Tools for DBTL and LDBT Implementation
| Reagent/Platform | Function | Framework Application |
|---|---|---|
| Cell-Free TX-TL Systems | Rapid protein expression without living cells | LDBT: Enables high-throughput testing of ML predictions in hours [2] [3] |
| Protein Language Models | Predict protein structure-function relationships | LDBT: Pre-trained models (ESM, ProGen) enable zero-shot design [2] |
| UTR Designer Tools | Modulate RBS sequences for expression tuning | DBTL: Fine-tune metabolic pathway enzymes in iterative optimization [5] |
| Droplet Microfluidics | Ultra-high-throughput screening platform | Both: Enables screening of >100,000 picoliter-scale reactions [2] |
| Automated Biofoundries | Integrated robotic assembly and testing | Both: Full automation of DBTL/LDBT cycles for scaling [2] |
| Deep Learning Algorithms | Navigate vast protein sequence spaces | LDBT: Tools like DeepDE, ProteinMPNN predict optimized variants [2] [4] |
The choice between DBTL and LDBT frameworks depends on specific research contexts and available resources. The traditional DBTL cycle remains valuable when limited training data exists for machine learning models, when engineering well-characterized biological systems, and when working with constrained computational resources. The knowledge-driven DBTL approach, incorporating upstream in vitro investigations, demonstrates how traditional cycles can be enhanced for efficient optimization [5].
The emerging LDBT paradigm offers distinct advantages for exploring vast design spaces, engineering poorly characterized systems, and accelerating development timelines. Its strength lies in leveraging pre-existing biological knowledge embedded in foundational models, potentially achieving functionality in fewer physical cycles [2] [4]. For drug development professionals, LDBT approaches show particular promise for antibody engineering, enzyme optimization, and metabolic pathway design where large sequence datasets enable robust model training.
The future of biological engineering likely involves hybrid approaches that leverage the strengths of both frameworks. As the field advances, the distinction may blur into adaptive cycles that dynamically reorder phases based on available knowledge and resources. What remains clear is that the integration of machine learning and rapid experimental platforms is fundamentally transforming biological engineering from an empirical art toward a predictive science.
Synthetic biology operates on a core engineering mantra known as the Design-Build-Test-Learn (DBTL) cycle, a systematic framework intended to streamline the engineering of biological systems [6]. In this paradigm, researchers Design biological parts with desired functions, Build DNA constructs and introduce them into living systems, Test the resulting constructs to measure performance, and Learn from the data to inform the next design iteration [2]. This approach has enabled remarkable achievements over the past two decades, from basic genetic oscillators to microbial production of therapeutic compounds [6]. However, as the field advances toward more complex challenges, the traditional DBTL framework is revealing significant limitations in its ability to efficiently navigate biological complexity.
The fundamental weakness lies in what might be termed "the learning bottleneck" – the cycle's inability to effectively extract predictive knowledge from the growing volumes of biological data [6]. While synthetic biologists can now generate draft blueprints of desired biological systems, many still resort to top-down approaches based on likelihoods and trial-and-error to determine optimal designs [6]. This deviation from synthetic biology's aspiration of rational design stems from the fact that biological processes in cells are often highly dynamic and inscrutable "black boxes" [6]. As a result, even with massive improvements in DNA synthesis and testing capabilities, the learning phase has failed to keep pace, creating a critical bottleneck that limits the entire engineering process.
The Learning Bottleneck: Where DBTL Stumbles
Fundamental Limitations in Handling Biological Complexity
The DBTL cycle struggles particularly with the multidimensional complexity inherent to biological systems. Three key factors contribute to this learning bottleneck:
System Heterogeneity and Component Interactions: Biological systems exhibit extraordinary complexity and heterogeneity, with numerous interacting components that create emergent properties not easily predicted from individual parts [6]. The traditional DBTL approach often oversimplifies these interactions, leading to designs that fail when scaled from individual parts to systems.
Data Interpretation Challenges: The "Learn" phase faces difficulties due to "variations in experimental setups" and the challenge of integrating multi-omics data [6]. Without standardized approaches to data generation and analysis, knowledge gained from one cycle often fails to transfer effectively to the next.
Trial-and-Error Inefficiency: The current paradigm frequently deviates into "top-down approaches based on likelihoods and trial-and-error" [6]. This empirical approach contrasts with the foundational vision of synthetic biology as a discipline built on rational design principles.
The Throughput Mismatch: Building and Testing Outpacing Learning
Technical advancements have dramatically accelerated the Build and Test phases while leaving Learning behind. DNA sequencing costs have plummeted from approximately $10 million per human genome in 2007 to around $600 today [6]. This cost reduction has enabled the accumulation of vast genomic databases, while innovations in DNA synthesis and assembly methodologies allow researchers to rapidly construct complex genetic systems [6].
The establishment of biofoundries worldwide has further accelerated this process through high-throughput automated assembly and screening methods [6]. These facilities can generate enormous amounts of multi-omics data at single-cell resolution, creating a deluge of information that outpaces traditional analytical approaches [6]. The result is a fundamental mismatch between data generation capacity and knowledge extraction capabilities – the core of the learning bottleneck.
Table: Throughput Comparison Across DBTL Stages
| DBTL Stage | Traditional Approach | Modern Capabilities | Limitations |
|---|---|---|---|
| Design | Manual, experience-based | Computational modeling | Limited by biological understanding |
| Build | Manual cloning | Automated DNA synthesis & assembly | Cost-effective but limited by design quality |
| Test | Low-throughput assays | High-throughput multi-omics | Data volume exceeds analysis capacity |
| Learn | Manual data interpretation | Basic statistical analysis | Inability to extract complex patterns |
LDBT: A Paradigm Shift to Overcome the Bottleneck
Reordering the Cycle: Learning Before Design
A paradigm shift is emerging in synthetic biology that directly addresses the learning bottleneck: the LDBT framework, which repositions "Learning" at the beginning of the cycle [2]. This approach leverages machine learning (ML) models trained on vast biological datasets to make predictive designs before any building or testing occurs. Rather than relying on iterative experimental cycles to accumulate knowledge, LDBT starts with knowledge embedded in pre-trained models capable of "zero-shot" predictions – generating functional designs without additional training [2].
This reorientation represents more than a simple procedural change; it fundamentally alters the relationship between data generation and knowledge application. As researchers note, "the data that would be 'learned' by Build-Test phases may already be inherent in machine learning algorithms" [2]. This approach brings synthetic biology closer to established engineering disciplines like civil engineering, which rely on first principles to create functional designs without extensive iterative testing [2].
Machine Learning and Foundational Models in LDBT
The LDBT paradigm is enabled by specialized machine learning approaches trained on biological data:
Protein Language Models: Sequence-based models like ESM and ProGen are trained on evolutionary relationships between protein sequences across phylogeny [2]. These models can predict beneficial mutations and infer protein function, enabling zero-shot prediction of diverse antibody sequences and other protein engineering tasks [2].
Structure-Based Design Tools: Approaches like ProteinMPNN use deep learning to design protein sequences that fold into specific backbone structures [2]. When combined with structure-assessment tools like AlphaFold, these methods have demonstrated "nearly 10-fold increase in design success rates" compared to traditional methods [2].
Functional Prediction Models: Specialized models focus on predicting key protein properties like thermostability (Prethermut, Stability Oracle) and solubility (DeepSol) [2]. These tools help eliminate potentially problematic designs before the Build phase.
Table: Machine Learning Approaches in LDBT
| ML Approach | Training Data | Key Capabilities | Demonstrated Applications |
|---|---|---|---|
| Protein Language Models (ESM, ProGen) | Millions of protein sequences | Predict beneficial mutations, infer function | Antibody sequence prediction, enzyme engineering |
| Structure-Based Design (ProteinMPNN) | Experimentally determined structures | Design sequences for specific folds | TEV protease optimization (increased activity) |
| Stability Prediction (Stability Oracle) | Protein stability data with structures | Predict ΔΔG of mutations | Enzyme stabilization for industrial applications |
| Hybrid Approaches | Multiple data types combined | Enhanced predictive power | PET hydrolase engineering with improved performance |
Comparative Analysis: DBTL vs. LDBT in Practice
Experimental Workflows and Methodologies
The fundamental difference between DBTL and LDBT approaches becomes evident in their experimental workflows:
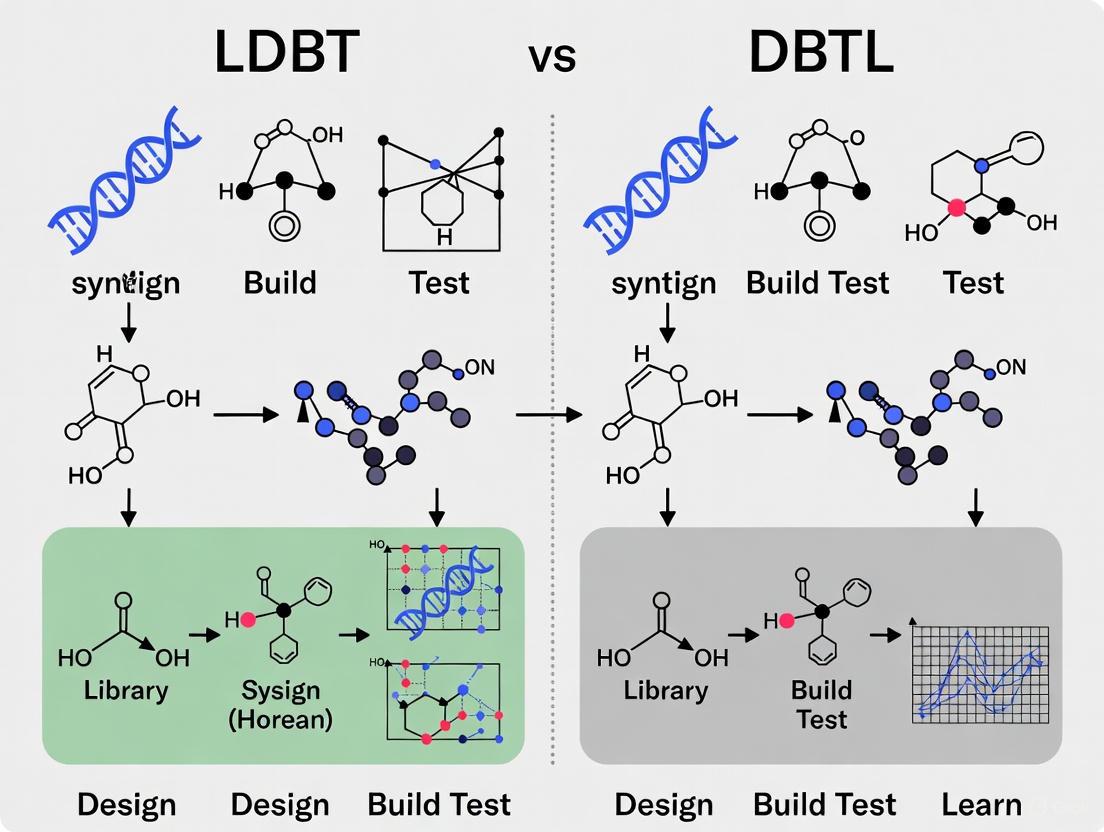
The traditional DBTL cycle (top) follows a sequential, iterative process where learning occurs only after building and testing. In contrast, the LDBT paradigm (bottom) begins with learning through machine learning analysis of existing biological data, enabling more informed design before any building occurs [2].
Performance Metrics and Experimental Outcomes
Direct comparisons between DBTL and LDBT approaches demonstrate significant advantages for the machine learning-driven paradigm:
Table: Quantitative Comparison of DBTL vs. LDBT Performance
| Performance Metric | Traditional DBTL | LDBT Approach | Improvement Factor |
|---|---|---|---|
| Design Cycles Required | 4-6 iterations | 1-2 iterations | 2-3x faster |
| Compounds Synthesized | Thousands for lead optimization | 10x fewer compounds [7] | 10x efficiency gain |
| Success Rate | Industry baseline | ~10x increase for some protein designs [2] | Significant improvement |
| Data Utilization | Limited to project-specific data | Leverages evolutionary knowledge across species | Vastly expanded context |
Case studies highlight these advantages in practical applications. In protein engineering, combining ProteinMPNN with structure-assessment tools like AlphaFold has demonstrated "nearly 10-fold increase in design success rates" compared to traditional methods [2]. In pharmaceutical development, companies like Exscientia report "in silico design cycles ~70% faster and requiring 10× fewer synthesized compounds than industry norms" [7]. One specific program achieved a clinical candidate "after synthesizing only 136 compounds, whereas traditional programs often require thousands" [7].
Enabling Technologies: The Scientist's Toolkit
The implementation of LDBT relies on specialized research reagents and platforms that enable rapid building and testing of computationally generated designs:
Table: Essential Research Reagent Solutions for LDBT Implementation
| Tool/Category | Function | Key Applications |
|---|---|---|
| Cell-Free Expression Systems | Rapid protein synthesis without living cells | High-throughput testing of protein variants [2] |
| DNA Synthesis Platforms | Automated production of designed DNA sequences | Rapid construction of genetic circuits [6] |
| Automated Liquid Handlers | High-throughput reagent distribution and sample processing | Scaling testing to thousands of variants [2] |
| Microfluidics/Droplet Systems | Ultra-high-throughput screening in picoliter volumes | Screening >100,000 protein variants [2] |
| Multi-omics Analysis Kits | Comprehensive molecular profiling | Generating training data for ML models [6] |
Cell-free expression systems deserve particular emphasis as they enable "rapid (>1 g/L protein in <4 h)" production and can be "readily scaled from the pL to kL scale" [2]. These systems allow direct testing of protein variants without time-consuming cloning steps, making them ideal for validating LDBT-generated designs [2]. When combined with automated platforms like biofoundries, these tools create an integrated infrastructure for implementing the LDBT paradigm.
Implementation Pathway: Adopting LDBT in Research Programs
Practical Framework for Transition
Shifting from DBTL to LDBT requires strategic changes in research operations and infrastructure. Research organizations should consider these implementation phases:
Data Foundation Development: The LDBT paradigm depends on "ML-friendly data" with "common standards for designing and generating" datasets suitable for machine learning [6]. This requires establishing consistent experimental protocols and data formats across projects to create training datasets.
Computational Infrastructure Investment: Successful LDBT implementation requires "deep learning models trained on vast biological datasets" [2]. This necessitates investment in computational resources and expertise, including partnerships between "dry- and wet-laboratory researchers" [6].
Integrated Workflow Design: The most successful implementations combine computational design with rapid experimental validation, such as "closed-loop design platforms that leverage AI agents to cycle through experiments" [2]. These systems connect computational design directly with automated building and testing.
Validation and Quality Control in LDBT
As with any methodological shift, maintaining rigorous validation is essential when adopting LDBT approaches:
The validation framework for LDBT employs a multi-stage approach that begins with computational predictions, moves through increasingly rigorous experimental testing, and feeds results back to improve models [2]. This tiered validation strategy balances speed with reliability, enabling rapid iteration while maintaining scientific rigor.
The transition from DBTL to LDBT represents more than a procedural adjustment – it marks a fundamental shift in how we approach biological engineering. By positioning learning at the forefront of the design process, synthetic biologists can leverage the vast accumulated knowledge of biological systems to create more predictive and reliable designs. The evidence suggests that this paradigm shift can deliver substantial improvements in efficiency, success rates, and cost-effectiveness across multiple applications, from therapeutic development to sustainable biomaterials [2].
While challenges remain in standardized data generation, model transparency, and interdisciplinary collaboration [6], the LDBT framework offers a promising path forward for overcoming the learning bottleneck that has long constrained synthetic biology. As machine learning capabilities continue to advance and biological datasets expand, this approach may ultimately realize the field's original aspiration: a true engineering discipline for biology, capable of reliably designing complex biological systems to address humanity's most pressing challenges.
The engineering of biological systems has long been guided by the Design-Build-Test-Learn (DBTL) cycle, a systematic, iterative framework that streamlines efforts to build functional biological systems [8]. In this established paradigm, researchers first define objectives and design biological parts, then build DNA constructs, test their performance experimentally, and finally learn from the data to inform the next design round [8]. However, recent advances in machine learning (ML) and high-throughput testing platforms are fundamentally transforming this workflow, prompting a significant paradigm shift from DBTL to LDBT (Learn-Design-Build-Test) [8] [3]. This reordering places machine learning at the forefront of the biological engineering process, creating a learn-first ethos that leverages large biological datasets to make predictive designs before committing to experimental work [8] [3].
The LDBT paradigm represents more than a simple reordering of steps—it constitutes a fundamental change in approach that leverages the predictive power of machine learning models trained on vast biological datasets. Instead of relying on empirical iteration, LDBT uses computational intelligence to directly inform and optimize designs, potentially generating functional biological parts and circuits in a single cycle [8]. This approach is made possible by the growing success of zero-shot predictions from protein language models and the availability of rapid cell-free testing platforms that can validate computational predictions at unprecedented scales [8] [3]. The resulting paradigm brings synthetic biology closer to a "Design-Build-Work" model that relies more heavily on first principles, similar to established engineering disciplines [8].
Comparative Analysis: DBTL vs. LDBT Workflows and Performance
The fundamental difference between the traditional DBTL cycle and the emerging LDBT paradigm lies in their starting points and underlying methodologies. The table below outlines the core distinctions in workflow, timing, data utilization, and overall approach between these two frameworks.
Table 1: Core Workflow Comparison Between DBTL and LDBT Paradigms
| Feature | Traditional DBTL Cycle | LDBT Paradigm |
|---|---|---|
| Starting Point | Design phase based on domain knowledge and expertise [8] | Learning phase powered by machine learning analysis of existing data [8] [3] |
| Primary Driver | Empirical iteration and experimental validation [8] | Predictive modeling and computational intelligence [8] [3] |
| Cycle Duration | Multiple iterations requiring weeks to months [8] [3] | Potential for single-cycle success with days to weeks [8] [3] |
| Data Utilization | Learning occurs after testing in each cycle [8] | Learning precedes design using accumulated datasets [8] [3] |
| Experimental Approach | Relies on cellular systems with associated biological constraints [8] | Leverages cell-free systems for rapid, parallel testing [8] [3] |
| Resource Allocation | Build-Test phases can be slow and resource-intensive [8] | Computational screening minimizes experimental burden [8] |
The performance advantages of the LDBT approach become particularly evident when examining specific engineering metrics. Research has demonstrated substantial improvements in the efficiency and success rates of biological design projects implementing the learn-first methodology.
Table 2: Performance Metrics Comparison Between DBTL and LDBT Approaches
| Performance Metric | Traditional DBTL | LDBT Approach | Improvement Factor |
|---|---|---|---|
| Design Success Rates | Baseline | Nearly 10-fold increase with structure-based deep learning [8] | ~10x |
| Screening Throughput | ~102-103 variants per cycle [8] | >105 variants using cell-free droplet microfluidics [8] | 100-1000x |
| Protein Expression Time | Days to weeks (in vivo) [8] | <4 hours (cell-free) with >1 g/L yields [8] | >10x faster |
| Data Generation Scale | Limited by cellular transformation and culturing [8] | 776,000 protein variants mapped in one study [8] | Orders of magnitude higher |
| Time to Functional Solution | Multiple cycles required [8] | Single-cycle convergence possible [8] [3] | Significant reduction |
The following workflow diagram illustrates the fundamental structural differences between these two approaches, highlighting how machine learning is repositioned from a downstream analytical tool to an upstream predictive engine in the LDBT paradigm.
Experimental Protocols and Methodologies in LDBT Implementation
Machine Learning Framework for Protein Design
The learning phase of LDBT employs sophisticated machine learning models trained on evolutionary and structural biological data. The experimental protocol for implementing these models typically follows a structured approach:
Data Curation and Preprocessing: Researchers assemble large-scale datasets of protein sequences (e.g., from UniRef, NCBI) and structures (e.g., from Protein Data Bank) encompassing millions of biological examples [8]. Sequence-based protein language models such as ESM (Evolutionary Scale Modeling) and ProGen are trained on evolutionary relationships between protein sequences to capture long-range dependencies and phylogenetic patterns [8]. Structure-based models like ProteinMPNN and MutCompute utilize deep neural networks trained on experimentally determined protein structures to associate amino acids with their local chemical environments [8].
Model Architecture and Training: For sequence-based prediction, transformer architectures with attention mechanisms are implemented to process amino acid sequences and predict beneficial mutations or infer protein function [8]. Structure-based approaches employ graph neural networks or 3D convolutional networks that take entire protein structures as input and output sequences likely to fold into target backbones [8]. Hybrid models combine evolutionary information with biophysical principles, such as incorporating force-field algorithms with large language models trained on enzyme homologs [8].
Validation and Benchmarking: Rigorous evaluation protocols test model generalizability by withholding entire protein superfamilies from training and assessing performance on these novel targets [9]. Models are validated against experimental data from cell-free expression systems, creating benchmark datasets of thousands of protein variants with measured stability and activity metrics [8]. This validation approach simulates real-world scenarios where models must make accurate predictions for novel protein families not encountered during training [9].
Cell-Free Expression and High-Throughput Testing
The Build-Test phases in LDBT utilize cell-free transcription-translation (TX-TL) systems to rapidly validate computational predictions:
Cell-Free System Preparation: Protein biosynthesis machinery is obtained from crude cell lysates (e.g., from E. coli, wheat germ, or insect cells) or purified components [8]. Reaction mixtures are assembled containing necessary transcription/translation components: RNA polymerase, ribosomes, tRNAs, amino acids, nucleotides, energy regeneration systems, and cofactors [8]. DNA templates designed in the computational phase are added directly without intermediate cloning steps, enabling rapid expression without cellular transformation [8].
High-Throughput Screening Implementation: Liquid handling robots and automated workstations (e.g., from Tecan, Beckman Coulter, Hamilton Robotics) dispense nanoliter to microliter reactions into multi-well plates or microfluidic devices [8] [10]. Droplet microfluidics platforms, such as DropAI, compartmentalize individual reactions in picoliter-scale droplets, enabling screening of >100,000 variants in parallel [8]. Functional assays are integrated with expression systems, employing colorimetric, fluorescent, or bioluminescent reporters to quantify protein expression, stability, or activity in real-time [8].
Data Collection and Analysis: Automated plate readers (e.g., PerkinElmer EnVision, BioTek Synergy HTX) measure assay signals across thousands of samples simultaneously [10]. Next-generation sequencing (NGS) platforms (e.g., Illumina NovaSeq, Thermo Fisher Ion Torrent) genotype variant libraries, linking sequence to function [8] [10]. Custom software platforms (e.g., TeselaGen) manage experimental workflows, track samples, and integrate data from multiple instruments for centralized analysis [10].
The following diagram illustrates the integrated workflow of the LDBT cycle, highlighting the seamless connection between computational prediction and experimental validation that characterizes this approach.
Essential Research Reagents and Solutions for LDBT Implementation
The successful implementation of the LDBT paradigm requires specialized reagents, computational tools, and instrumentation. The following table details key research solutions that enable the learn-design-build-test workflow in synthetic biology.
Table 3: Essential Research Reagent Solutions for LDBT Implementation
| Category | Specific Solution | Function in LDBT Workflow |
|---|---|---|
| Machine Learning Models | ESM (Evolutionary Scale Modeling) [8] | Protein language model trained on evolutionary sequences for zero-shot prediction of structure-function relationships |
| Machine Learning Models | ProGen [8] | Protein language model capable of generating functional protein sequences and predicting beneficial mutations |
| Machine Learning Models | ProteinMPNN [8] | Structure-based deep learning tool that designs protein sequences for specific backbone structures |
| Machine Learning Models | MutCompute [8] | Deep neural network that identifies stabilizing mutations based on local chemical environments |
| Cell-Free Systems | TX-TL Transcription-Translation Systems [8] | Cell-free protein synthesis machinery enabling rapid expression without cellular constraints |
| Automation Equipment | Automated Liquid Handlers (Tecan, Beckman Coulter) [10] | High-precision pipetting systems for assembling DNA constructs and setting up screening reactions |
| Automation Equipment | Droplet Microfluidics (DropAI) [8] | Picoliter-scale reaction compartmentalization enabling ultra-high-throughput screening of >100,000 variants |
| DNA Synthesis Providers | Twist Bioscience, IDT, GenScript [10] | Custom DNA sequence providers integrated with automated workflows for seamless construct building |
| Analytical Instruments | High-Throughput Plate Readers (PerkinElmer EnVision) [10] | Multi-mode detectors for measuring fluorescent, colorimetric, or luminescent signals from thousands of samples |
| Analytical Instruments | Next-Generation Sequencers (Illumina NovaSeq) [10] | Rapid genotypic analysis of variant libraries, linking DNA sequence to functional output |
| Software Platforms | TeselaGen [10] | End-to-end DBTL/LDBT management software orchestrating design, inventory, workflow automation, and data analysis |
Case Studies: Experimental Validation of LDBT Efficacy
Protein Engineering with Zero-Shot Prediction
A compelling demonstration of LDBT's power comes from protein engineering campaigns that utilize zero-shot machine learning predictions:
PET Hydrolase Engineering: Researchers employed MutCompute, a structure-based deep learning tool, to identify stabilizing mutations in a polyethylene terephthalate (PET) depolymerization enzyme [8]. The model was trained on protein structures to associate amino acids with their local chemical environments, enabling prediction of beneficial substitutions without additional experimental data [8]. The resulting engineered hydrolase variants demonstrated significantly increased stability and activity compared to the wild-type enzyme, validating the computational predictions [8]. This approach was further refined using large language models trained on PET hydrolase homologs combined with force-field algorithms, effectively exploring the evolutionary landscape to improve enzyme performance [8].
TEV Protease Optimization: ProteinMPNN was used to design variants of TEV protease with improved catalytic activity [8]. The model took the entire protein structure as input and predicted new sequences likely to fold into the target backbone [8]. When combined with deep learning-based structure assessment tools like AlphaFold and RoseTTAFold, this approach achieved a nearly 10-fold increase in design success rates compared to traditional methods [8]. This case exemplifies how the integration of multiple machine learning tools within the LDBT framework can dramatically accelerate the engineering of functional proteins.
Ultra-High-Throughput Stability Mapping
The scalability of LDBT has been demonstrated through massive protein stability mapping efforts:
Comprehensive ΔG Determination: Researchers coupled cell-free protein synthesis with cDNA display to calculate folding free energy (ΔG) for 776,000 protein variants in a single experimental campaign [8]. This unprecedented dataset provided experimental validation for thousands of computational predictions simultaneously, creating a robust benchmark for evaluating zero-shot predictors [8]. The scale of this dataset—orders of magnitude larger than traditional approaches—highlights how LDBT's integration of high-throughput experimentation enables comprehensive exploration of sequence-function relationships.
Antimicrobial Peptide Design: Deep learning sequence generation was paired with cell-free expression to computationally survey over 500,000 antimicrobial peptide (AMP) variants [8]. From this vast sequence space, researchers selected 500 optimal candidates for experimental validation, resulting in six promising AMP designs with confirmed activity [8]. This approach demonstrates LDBT's ability to efficiently navigate massive design spaces that would be intractable using traditional DBTL methods, focusing experimental resources on the most promising candidates identified through computational learning.
The LDBT paradigm represents a fundamental shift in how we approach biological engineering, moving from empirical iteration to predictive design. By placing machine learning at the forefront of the biological design process, LDBT leverages the vast and growing repositories of biological data to make informed predictions before experimental work begins [8] [3]. This learn-first approach, combined with rapid cell-free testing platforms, enables researchers to navigate the enormous complexity of biological sequence space with unprecedented efficiency [8] [3].
While significant challenges remain—including model generalizability to novel protein families and the cost of large-scale experimentation—the LDBT framework provides a clear path forward for synthetic biology [9]. As machine learning models continue to improve and experimental platforms become increasingly automated, the integration of computational intelligence with biological design will likely become the standard approach for engineering biological systems [8] [3] [10]. This convergence of data science and biotechnology promises to accelerate the development of novel therapeutics, sustainable biomaterials, and bio-based manufacturing processes, ultimately transforming how we design and interact with biological systems.
The Role of Foundational Models and Zero-Shot Predictions in Enabling LDBT
The synthetic biology field has traditionally operated on the Design-Build-Test-Learn (DBTL) framework, an iterative cycle that systematically engineers biological systems. However, recent advances in artificial intelligence and machine learning are driving a paradigm shift toward Learn-Design-Build-Test (LDBT), where computational learning precedes physical implementation. This transformation is primarily enabled by foundation models capable of zero-shot predictions—generating accurate biological designs without prior task-specific training. This article compares the capabilities of various AI architectures and their zero-shot performance in synthetic biology applications, providing experimental data and methodologies that demonstrate how LDBT accelerates biological engineering.
Traditional DBTL cycles begin with designing biological parts based on existing knowledge, then building DNA constructs, testing them in biological systems, and finally learning from the results to inform the next design iteration [8] [1]. This empirical approach, while systematic, often requires multiple time-consuming and resource-intensive cycles to achieve desired functions.
The emerging LDBT paradigm fundamentally reorders this process by placing Learning first, leveraging foundation models trained on vast biological datasets to generate initial designs [8] [3]. These models utilize zero-shot prediction capabilities to propose functional biological constructs without requiring additional training on specific tasks. The subsequent Design, Build, and Test phases then serve to validate and refine these computational predictions in a single, efficient cycle [8].
This paradigm shift brings synthetic biology closer to established engineering disciplines where designs are based on first principles and reliably work on the first implementation, moving toward a "Design-Build-Work" model [8].
Comparative Analysis of Foundation Models for Biological Design
Foundation models trained on diverse biological datasets have demonstrated remarkable capabilities in understanding and designing biological sequences and structures. The table below compares major model architectures relevant to synthetic biology applications.
Table 1: Comparison of Foundation Model Architectures for Biological Design
| Model | Architecture Type | Key Innovation | Training Data | Relevant Biological Applications |
|---|---|---|---|---|
| ESM [8] | Protein Language Model | Evolutionary scale modeling | Millions of protein sequences | Predicting beneficial mutations, inferring protein function |
| ProGen [8] | Protein Language Model | Conditional protein generation | Diverse protein families | Zero-shot prediction of antibody sequences |
| ProteinMPNN [8] | Structure-based Deep Learning | Inverse folding from structure | Protein structures | Designing sequences that fold into specific backbone structures |
| AlphaFold [8] | Structural Prediction | Geometric deep learning | Protein Data Bank structures | Assessing designed protein structures |
| MutCompute [8] | Environment-aware Neural Network | Residue-level optimization | Protein structures with local environments | Predicting stabilizing mutations |
These models vary in their approaches—some learn from evolutionary patterns in sequence data, while others focus on structural relationships—but collectively enable zero-shot prediction of biological designs.
Zero-Shot Prediction Capabilities Across Modalities
The performance of foundation models in zero-shot settings varies significantly across different biological tasks. The following table summarizes quantitative performance metrics from recent evaluations.
Table 2: Zero-Shot Performance Across Biological Tasks
| Task Domain | Model/Approach | Performance Metric | Result | Reference |
|---|---|---|---|---|
| Face Verification | Vision-Language Models | TMR @ FMR=1% (LFW dataset) | 96.77% | [11] |
| Iris Recognition | Vision-Language Models | TMR @ FMR=1% (IITD-R-Full dataset) | 97.55% | [11] |
| PET Hydrolase Engineering | MutCompute | Stability & Activity | Increased vs. wild-type | [8] |
| TEV Protease Design | ProteinMPNN + AlphaFold | Catalytic Activity | Improved vs. parent sequence | [8] |
| Antimicrobial Peptides | Deep Learning + Cell-free Testing | Success Rate | 6/500 promising designs | [8] |
Performance variability is a recognized characteristic of zero-shot prediction. Research indicates that when foundation models perform well on base prediction tasks, their predicted probabilities become stronger signals for individual-level accuracy [12]. This underscores the importance of task-specific evaluation before full implementation.
Experimental Protocols for LDBT Implementation
Protein Engineering via Structure-Based Models
Objective: Engineer enhanced PET hydrolase enzymes for improved plastic degradation [8].
Methodology:
- Learning: Utilize MutCompute, a deep neural network trained on protein structures, to identify probable stabilizing mutations based on local chemical environments [8].
- Design: Select mutations predicted to improve stability and activity while maintaining catalytic function.
- Build: Synthesize DNA sequences encoding the designed variants and express them in cell-free systems [8] [3].
- Test: Measure depolymerization activity against PET substrates and compare thermostability to wild-type enzyme [8].
Key Tools: MutCompute for mutation prediction; cell-free expression system for rapid protein production; activity assays for functional validation [8].
Antimicrobial Peptide Design via Sequence-Based Models
Objective: Design novel antimicrobial peptides (AMPs) with predicted activity [8].
Methodology:
- Learning: Employ deep learning sequence generation models to computationally survey over 500,000 potential AMP sequences [8].
- Design: Select 500 optimal variants based on model predictions for experimental validation.
- Build: Synthesize peptide sequences using high-throughput methods.
- Test: Evaluate antimicrobial activity against target pathogens using cell-free assays [8] [3].
Key Tools: Deep learning sequence generation; high-throughput peptide synthesis; cell-free antimicrobial activity assays [8].
Metabolic Pathway Optimization via iPROBE
Objective: Improve 3-HB production in Clostridium hosts [8].
Methodology:
- Learning: Apply iPROBE (in vitro prototyping and rapid optimization of biosynthetic enzymes) framework using neural networks trained on pathway combinations and enzyme expression levels [8].
- Design: Predict optimal pathway sets and expression levels for enhanced production.
- Build: Construct pathway variants in appropriate vectors.
- Test: Measure 3-HB production yields in host systems [8].
Key Tools: iPROBE neural network for pathway prediction; cell-free pathway prototyping; metabolic flux analysis [8].
Visualizing the LDBT Workflow
Diagram 1: LDBT Workflow Integration. This diagram illustrates how foundation models enable the Learn-first approach in synthetic biology, generating zero-shot predictions that inform the design phase, with cell-free systems accelerating build and test phases.
Diagram 2: DBTL vs LDBT Paradigm Comparison. The traditional iterative cycle (left) contrasts with the learning-first approach (right) where foundation models enable single-pass implementation.
Essential Research Reagent Solutions for LDBT Implementation
Table 3: Key Research Reagents and Platforms for LDBT Workflows
| Reagent/Platform | Function in LDBT | Application Examples |
|---|---|---|
| Cell-Free Transcription-Translation Systems [8] [3] | Rapid protein synthesis without living cells | High-throughput testing of protein variants |
| DropAI Microfluidics [8] | Ultra-high-throughput screening in picoliter droplets | Screening >100,000 protein variants |
| cDNA Display Platforms [8] | In vitro protein stability mapping | ΔG calculations for 776,000 protein variants |
| Automated Liquid Handling Robots [8] | Accelerated build and test phases | Automated assembly of DNA constructs |
| Foundation Model APIs (ESM, ProGen, ProteinMPNN) [8] | Zero-shot biological design | Generating novel protein sequences |
The integration of foundation models with zero-shot prediction capabilities represents a transformative advancement in synthetic biology methodology. The LDBT paradigm, enabled by these technologies, shifts the innovation bottleneck from empirical iteration to computational prediction, potentially reducing development timelines from months to days. As foundation models continue to improve in accuracy and biological relevance, and as cell-free testing platforms increase in throughput and accessibility, the LDBT framework promises to democratize and accelerate synthetic biology research across academic, industrial, and therapeutic domains.
Synthetic biology, a field dedicated to reprogramming organisms with novel functionalities, has long been guided by the Design-Build-Test-Learn (DBTL) cycle [1] [6]. This iterative framework, while systematic, often relies on empirical trial-and-error due to the profound complexity of biological systems, making the engineering process slow and costly [13] [14]. However, a paradigm shift is underway, mirroring the evolution of older engineering disciplines from craftsmanship to predictive, computer-aided design [14]. Fueled by the convergence of artificial intelligence (AI) and high-throughput biology, the emerging Learn-Design-Build-Test (LDBT) framework is reordering the cycle to place data-driven learning first, promising to transform synthetic biology into a predictive engineering discipline [2] [3].
The Established DBTL Cycle and Its Bottlenecks
The traditional DBTL cycle has been the backbone of synthetic biology development. Its four stages form a continuous loop for engineering biological systems:
- Design: Researchers define objectives and design genetic constructs using domain knowledge and computational modeling [2] [13].
- Build: DNA constructs are physically assembled and introduced into a host chassis (e.g., bacteria, yeast) or cell-free systems [2] [14].
- Test: The performance of the engineered system is experimentally measured and characterized [1] [13].
- Learn: Data from testing is analyzed to inform the next design iteration, refining the approach until the desired function is achieved [2] [6].
A significant limitation of this cycle is the "learn" phase often comes last. Learning is reactive, dependent on the data generated from the specific "build" and "test" phases of that cycle [6]. Furthermore, the "build" and "test" phases, particularly when using living cells, can be time-consuming and low-throughput, creating a bottleneck and limiting the number of design iterations possible [2] [14].
The Emergence of the LDBT Paradigm
The LDBT paradigm proposes a fundamental reordering of the cycle, placing "Learn" at the forefront [2] [3]. This shift is powered by machine learning (ML) models trained on vast biological datasets, which can make powerful predictions before any new physical construction begins.
- Learn (First): In LDBT, the cycle starts with machine learning models that have been pre-trained on large-scale biological data, including protein sequences, structures, and functional assays [2] [13]. These models can learn the complex, non-linear relationships between sequence, structure, and function.
- Design (Informed by Learning): The design phase is directly guided by the ML models. Researchers use tools like ProteinMPNN (for sequence design) and AlphaFold (for structure prediction) to generate optimal genetic designs that are likely to succeed, often in a "zero-shot" manner without additional model training [2].
- Build & Test (Rapid Validation): The designed constructs are then rapidly built and tested using high-throughput platforms. Cell-free transcription-translation (TX-TL) systems are particularly valuable here, as they allow for rapid protein expression without the delays of cellular culture, enabling the testing of thousands of variants in hours [2] [3].
This approach creates a more efficient funnel, where a vast digital design space is navigated computationally, and only the most promising candidates are physically validated [3].
Comparative Analysis: DBTL vs. LDBT
The table below summarizes the core differences between the two paradigms across key aspects of the engineering workflow.
| Feature | Traditional DBTL Cycle | LDBT Paradigm |
|---|---|---|
| Cycle Order | Design → Build → Test → Learn | Learn → Design → Build → Test |
| Primary Driver | Empirical, iterative experimentation | Data-driven, predictive computation |
| Knowledge Base | Project-specific data from previous cycles | Foundational models trained on megascale biological data (e.g., protein sequences, structures) [2] [13] |
| Role of ML/AI | Analyzes data at the end of the cycle to inform next design | Precedes and directly informs the initial design; enables zero-shot predictions [2] |
| Build/Test Platform | Often relies on in vivo (cellular) systems | Heavily leverages rapid, high-throughput cell-free systems [2] [3] |
| Throughput | Lower, limited by cellular growth and cloning steps | Very high, enabled by cell-free and automation |
| Predictivity | Lower, relies on trial-and-error | Higher, aims for "first-principles" design [2] |
Experimental Protocols & Supporting Data
The practical superiority of the LDBT paradigm is demonstrated in recent research that integrates specific machine learning models with rapid cell-free testing.
Experimental Workflow for LDBT
The following diagram illustrates the integrated workflow of the LDBT cycle, showcasing the seamless flow from computational learning to physical testing and model refinement.
Diagram 1: The LDBT (Learn-Design-Build-Test) experimental workflow. The cycle begins with foundational learning from large datasets, which directly informs the computational design of biological parts. These designs are rapidly built and tested in cell-free systems, with the resulting experimental data used to update and refine the machine learning models, creating a continuous improvement loop [2] [3].
Detailed Methodology for a Key Experiment
A seminal application of LDBT involves using a protein language model like ESM (Evolutionary Scale Modeling) to design and test novel enzyme variants [2].
- Objective: Engineer an enzyme (e.g., a hydrolase for PET plastic depolymerization) with enhanced stability and activity [2].
- Learn Phase: A pre-trained protein language model (e.g., ESM) or a structure-based tool (e.g., MutCompute) is used to analyze evolutionary and biophysical constraints. The model predicts amino acid substitutions that are likely to be stabilizing and functionally beneficial without being explicitly trained on this specific enzyme [2].
- Design Phase: The top in silico predictions are selected to generate a library of variant DNA sequences. This step uses computational tools to codon-optimize the sequences for expression [2].
- Build Phase: The DNA sequences for the wild-type and predicted variant enzymes are synthesized via high-throughput gene synthesis. These DNA templates are then used directly in a cell-free transcription-translation (TX-TL) system, bypassing time-consuming steps of cloning into plasmids and transforming living cells [2] [3].
- Test Phase: The cell-free reactions express the enzyme variants. Function is assessed using a colorimetric or fluorescent assay that measures the degradation of a substrate (e.g., PET). Fluorescence-activated droplet sorting or microplate readers can be used for high-throughput quantification. Stability is measured by incubating enzymes at different temperatures and assessing residual activity [2].
Performance Data Comparison
The table below quantifies the performance gains achieved by the LDBT paradigm in specific experimental use cases, compared to traditional DBTL approaches.
| Application / Metric | DBTL Performance | LDBT Performance | Key Enabling Technologies |
|---|---|---|---|
| Enzyme Engineering (PET Hydrolase) [2] | Multiple iterative rounds required; improved stability/activity | Single-round success with zero-shot models; increased stability & activity vs. wild-type | MutCompute, Protein Language Models (ESM), Cell-free Testing |
| Antimicrobial Peptide (AMP) Design [2] | Limited library size; low hit rate | 500 variants tested from >500,000 surveyed; 6 promising designs identified | Deep Learning Sequence Generation, Cell-free Expression |
| Pathway Optimization (3-HB in Clostridium) [2] | Iterative host engineering; slower yield improvement | >20-fold production increase predicted via neural network | iPROBE, Cell-free Pathway Prototyping |
| General Cycle Turnover Time | Weeks to months per cycle [14] | Hours for test phase using cell-free systems [2] [3] | Cell-free TX-TL, Automation, Microfluidics |
The Scientist's Toolkit: Essential Research Reagents & Materials
The implementation of the LDBT paradigm relies on a specific set of computational and experimental tools.
| Tool Category | Item / Solution | Function in LDBT Workflow |
|---|---|---|
| Computational (Learn/Design) | Protein Language Models (e.g., ESM, ProGen) [2] | Pre-trained on evolutionary data for zero-shot prediction of protein function and beneficial mutations. |
| Structure-Based Design Tools (e.g., ProteinMPNN, AlphaFold) [2] | Generate sequences that fold into a desired backbone (ProteinMPNN) and predict 3D protein structures (AlphaFold). | |
| Stability Prediction Tools (e.g., Prethermut, Stability Oracle) [2] | Predict the thermodynamic stability change (ΔΔG) of protein variants to screen for stabilizing mutations. | |
| Experimental (Build/Test) | Cell-Free TX-TL System [2] [3] | A reconstituted biochemical machinery for rapid, high-yield protein synthesis without living cells. |
| DNA Template | Synthesized linear DNA or plasmids encoding the variant to be expressed; the direct input for the cell-free system. | |
| Metabolic Assay Kits (e.g., NADPH/NADP⁺) | Quantify cofactor turnover or metabolic flux in cell-free prototyped pathways. | |
| Droplet Microfluidics Setup [2] | Encapsulate single cell-free reactions in picoliter droplets for ultra-high-throughput screening. |
Future Outlook and Implications
The transition from DBTL to LDBT signifies a broader movement toward the industrialization of biology. As foundational models grow more sophisticated and high-throughput testing becomes even more accessible, the LDBT cycle is expected to accelerate, potentially converging on a "Design-Build-Work" model reminiscent of mature engineering disciplines like civil engineering [2]. This progression will be crucial for tackling complex challenges in drug development, sustainable manufacturing, and climate change, enabling the creation of biological solutions with a speed and precision previously unimaginable.
Key Technologies and Workflows Powering the LDBT Framework
The foundational framework of synthetic biology has long been the Design-Build-Test-Learn (DBTL) cycle, an iterative process where biological systems are designed, constructed, experimentally validated, and insights from data are used to inform the next design round [8] [1]. However, recent advances in machine learning (ML) are instigating a paradigm shift. The proliferation of large-scale biological data and sophisticated computational models now enables a reordering of this cycle into LDBT (Learn-Design-Build-Test), where machine learning precedes design [8].
In the LDBT framework, "Learning" is moved to the forefront. Vast, pre-existing biological knowledge is captured by machine learning models trained on millions of protein sequences and structures. This allows researchers to make zero-shot predictions—designing proteins with desired functions without any initial target-specific experimental data [8]. The subsequent Build and Test phases then serve to validate these computational predictions, potentially reducing the number of costly experimental cycles required. This paradigm brings synthetic biology closer to a "Design-Build-Work" model, akin to more mature engineering disciplines [8].
This guide objectively compares the performance of key machine learning tools driving this shift: protein language models (ESM, ProGen) and structure-based design tools (ProteinMPNN, MutCompute).
Protein Language Models (PLMs) for Zero-Shot Design
Protein language models are deep learning systems pre-trained on massive datasets of protein sequences. By learning evolutionary patterns and statistical relationships between amino acids, they can predict the effects of mutations and generate novel, functional protein sequences from scratch without target-specific training data [8] [15].
ESM (Evolutionary Scale Modeling)
The ESM family of models, including ESM-1b and ESM-2, are transformer-based protein language models trained on millions of diverse protein sequences. They learn to represent the evolutionary constraints and biophysical properties that shape proteins [8] [15].
- Core Methodology: ESM models use a self-supervised training objective, often a masked language model task, where random amino acids in a sequence are hidden, and the model must predict them based on the surrounding context. This forces the model to internalize complex dependencies within protein sequences [15].
- Zero-Shot Function: The model's output log-likelihood for a given sequence or mutation can be used as a fitness score, estimating how "natural" and likely-to-fold the variant appears based on evolutionary data [8] [16].
- Key Applications: ESM has been applied to predict beneficial mutations, infer protein function, and predict solvent-exposed and charged amino acids. It has proven adept at zero-shot prediction of diverse antibody sequences [8].
Table 1: Performance Summary of ESM Models
| Model | Training Data | Key Applications | Reported Performance |
|---|---|---|---|
| ESM-1b/ESM-2 | Millions of protein sequences from UniRef [15] | Protein function prediction, mutation effect prediction, zero-shot fitness inference [8] [15] | Outperformed traditional methods in CAFA challenge; widely used as a state-of-the-art feature encoder [15] |
| ProGen | Millions of protein sequences, including control tags for function [8] | Generation of functional protein sequences with controlled properties [8] | Successfully generated functional lysozymes; capable of zero-shot prediction of diverse antibody sequences [8] |
ProGen
ProGen is another protein language model trained on a large corpus of protein sequences. Its distinctive feature is the inclusion of control tags (e.g., for protein family or function) during training, enabling conditional generation of novel protein sequences tailored for specific purposes [8].
- Core Methodology: Similar to ESM, ProGen uses a transformer architecture. It was trained on a dataset of ~280 million protein sequences, learning the underlying "grammar and style" of proteins across different families [8].
- Zero-Shot Function: ProGen can generate entirely new protein sequences that do not exist in nature by sampling from the learned distribution, guided by user-specified control tags to steer the function of the generated protein [8].
Structure-Based Deep Learning Design Tools
Unlike PLMs that primarily use sequence information, structure-based tools leverage 3D structural data to inform the design process, focusing on how a sequence will fold and function in a structural context.
ProteinMPNN
ProteinMPNN is a deep neural network for protein sequence design. Given a protein backbone structure as input, it predicts amino acid sequences that are likely to fold into that structure [8] [17].
- Core Methodology: ProteinMPNN uses a message-passing neural network architecture that operates on a graph representation of the protein structure. It considers the spatial relationships between residues to design sequences that are energetically favorable and compatible with the desired fold [8].
- Zero-Shot Function: Researchers can input a novel backbone structure or a modified existing structure, and ProteinMPNN will output one or more sequences predicted to fold into it, without requiring any prior experimental data for that specific scaffold [8].
- Key Applications: It has been used to design variants of TEV protease with improved catalytic activity and to redesign enzymes like the Fe(II)/αKG enzyme tP4H for greater stability and solubility while retaining native activity [8] [17]. When combined with structure prediction tools like AlphaFold, it has led to a nearly 10-fold increase in design success rates [8].
Table 2: Performance Summary of Structure-Based Tools
| Tool | Input | Key Applications | Reported Performance |
|---|---|---|---|
| ProteinMPNN | Protein backbone structure [8] | De novo sequence design, enzyme stabilization, protein binder design [8] [17] | ~10-fold increase in design success rates when combined with AlphaFold for structure assessment [8] |
| MutCompute | Protein structure and local chemical environment [8] | Residue-level optimization for stability and activity [8] | Engineered a hydrolase for PET depolymerization with increased stability and activity vs. wild-type [8] |
MutCompute
MutCompute is a structure-based deep learning tool that focuses on residue-level optimization. It identifies probable mutations given the local chemical environment of a residue within a protein structure [8].
- Core Methodology: MutCompute uses a deep neural network trained on protein structures from the PDB. It learns to associate an amino acid with its surrounding chemical environment (e.g., neighboring atoms, secondary structure, solvent exposure), allowing it to predict mutations that are likely to be stabilizing or functionally beneficial [8].
- Zero-Shot Function: By analyzing a protein's structure, MutCompute can predict single or multiple point mutations that are predicted to enhance stability or other properties without requiring functional assays on the target protein first [8].
Comparative Performance Analysis
The following table provides a direct, data-driven comparison of these tools based on their performance in published experimental validations.
Table 3: Experimental Performance and Validation Data
| Tool | Type | Experimental Validation Example | Experimental Outcome |
|---|---|---|---|
| ESM | Protein Language Model | Zero-shot prediction of beneficial antibody mutations [8] | Successful prediction of functional antibody sequences without target-specific training [8] |
| ProGen | Protein Language Model | Generation of novel antimicrobial peptides (AMPs) [8] | From 500 computationally surveyed AMPs, 6 promising designs were validated experimentally [8] |
| ProteinMPNN | Structure-Based Design | Redesign of TEV protease [8] | Designed variants showed improved catalytic activity compared to the parent sequence [8] |
| MutCompute | Structure-Based Design | Engineering a hydrolase for PET depolymerization [8] | MutCompute-designed proteins had increased stability and activity compared to wild-type [8] |
| FSFP (Fine-tuning) | Hybrid Approach | Engineering Phi29 DNA polymerase [16] | Fine-tuned ESM-1v model led to a 25% increase in the positive rate of functional polymerase variants [16] |
Experimental Protocols for Zero-Shot Validation
To validate zero-shot predictions, a typical DBTL cycle is employed, where the "Design" phase is heavily influenced by the computational model.
Protocol for Validating a PLM-Generated Protein
This protocol outlines the steps for experimentally testing a novel protein sequence generated by a model like ProGen or a stabilized variant designed by ProteinMPNN.
- In Silico Design: Generate a set of candidate sequences using the zero-shot model (e.g., ProGen for a novel AMP, ProteinMPNN for a stabilized enzyme scaffold). The model's internal scoring (e.g., log-likelihood, confidence score) is used for initial ranking [8] [17].
- DNA Synthesis and Cloning: The selected nucleotide sequences are synthesized de novo and cloned into an appropriate expression vector. Automated biofoundries can streamline this process for high-throughput workflows [8] [14].
- Cell-Free Expression or In Vivo Expression: The built DNA constructs are expressed. Cell-free expression systems are particularly valuable here for their speed (>1 g/L protein in <4 h), scalability (pL to kL), and ability to express proteins that might be toxic in living cells [8].
- Functional Assay: The expressed proteins are tested for the desired function. This could be:
- Data Analysis and Learning: Experimental results are analyzed. Successful variants confirm the zero-shot prediction, while failures provide data that can be used to fine-tune the model for future cycles, closing the LDBT loop [8] [16].
Zero-Shot LDBT Cycle for Protein Design
Protocol for a Stability Engineering Campaign Using ProteinMPNN
This specific protocol is adapted from a study that used ProteinMPNN to stabilize the Fe(II)/αKG enzyme tP4H [17].
- Structure Preparation: Obtain a 3D structure of the target protein. If an experimental structure is unavailable, use a predicted model from AlphaFold2 [17].
- Define Fixed Residues: To preserve catalytic function, identify and "fix" (prevent mutation of) residues critical for activity (e.g., active site residues, cofactor-binding residues). This can be done using the AlphaFold2 model and comparisons to homologs with known structures [17].
- Sequence Design with ProteinMPNN: Run ProteinMPNN on the structure, specifying the fixed residues. Generate a large number (e.g., 48) of designed sequences [17].
- In Silico Ranking: Rank the designed sequences based on ProteinMPNN's confidence scores and/or other criteria (e.g., computational stability metrics) [17].
- Build, Test, and Learn: Synthesize and clone top-ranked designs. Express and purify the proteins. Test for:
- Retained Native Function: Ensure the stabilized design has not lost its original catalytic activity.
- Improved Stability: Measure metrics like melting temperature (
Tm) or soluble expression yield. - Non-Native Function: If applicable, screen for improved performance in a promiscuous, industrially-relevant reaction (e.g., C-H hydroxylation) [17].
Workflow for Protein Stabilization with ProteinMPNN
Essential Research Reagent Solutions
The experimental validation of zero-shot designs relies on a suite of core reagents and platforms.
Table 4: Key Research Reagents and Platforms
| Item / Solution | Function in Workflow | Key Characteristics |
|---|---|---|
| Cell-Free Expression System [8] | Rapid protein synthesis for high-throughput testing of designed variants. | Fast (>1 g/L in <4 h), scalable, bypasses cell viability, allows toxic protein production. |
| AlphaFold2 Model [18] [17] | Provides a reliable 3D protein structure for structure-based tools when experimental structures are unavailable. | High accuracy (average error ~1 Å), covers entire protein sequences. |
| Automated Biofoundry [8] [14] | Automates the Build and Test phases (DNA assembly, transformation, culturing, assays). | Increases throughput, reduces human error and labor, enables closed-loop DBTL/LDBT cycles. |
| Droplet Microfluidics [8] | Ultra-high-throughput screening of protein variants. | Enables screening of >100,000 picoliter-scale reactions in parallel. |
| Ultra-Large Virtual Compound Libraries (e.g., REAL Database) [18] | Provides a vast chemical space for in silico screening of small molecule binders for designed proteins. | Billions of make-on-demand compounds, expands hit discovery and chemical diversity. |
Synthetic biology has traditionally operated on the Design-Build-Test-Learn (DBTL) cycle, a systematic framework for engineering biological systems [1]. In this paradigm, researchers design genetic constructs, build them in the laboratory, test their functionality, and learn from the results to inform the next design iteration. However, this process can be time-consuming, with the Build and Test phases often creating significant bottlenecks. A new paradigm, the "LDBT" cycle, is emerging, where Learning precedes Design [8]. This shift is powered by machine learning (ML) models that can make zero-shot predictions, generating viable initial designs based on vast biological datasets. This places even greater importance on the subsequent Build and Test phases, which must be rapid and high-throughput to validate these computational predictions efficiently [8]. Within this context, cell-free gene expression (CFE) systems and automated DNA synthesis have become critical technologies for accelerating the Build phase, enabling the rapid physical realization of designed genetic constructs and paving the way for a more agile engineering biology.
The DBTL Cycle and its Bottlenecks
The classic DBTL cycle is a cornerstone of synthetic biology [5]. The process begins with Design, where researchers define objectives and design the necessary biological parts using computational tools [8]. This is followed by the Build phase, which involves the physical construction of DNA constructs, their assembly into vectors, and introduction into a living chassis (e.g., bacteria, yeast) or a cell-free system for characterization [8]. The Test phase involves experimental measurement of the construct's performance, and the Learn phase analyzes this data to refine the design for the next cycle [8]. A major bottleneck in this workflow has been the Build phase, particularly when relying on in vivo chassis. Cloning, transforming, and cultivating living cells is a slow process, often taking days and creating a disconnect with the rapidly generated in silico designs from the new LDBT paradigm [8].
Cell-Free Expression Systems: Bypassing the Cell to Accelerate Building and Testing
What is Cell-Free Gene Expression?
Cell-free gene expression (CFE) is a methodology for performing transcription and translation in vitro using the protein synthesis machinery extracted from cells [19]. Unlike traditional in vivo methods, CFE bypasses the need for living cells, using lysates or purified components from organisms like E. coli to directly convert synthesized DNA templates into proteins [8] [19]. This offers a direct and rapid path from a designed DNA sequence to a functional protein product.
Key Advantages for the Build-Test Phases
The unique features of CFE make it exceptionally well-suited for accelerating synthetic biology workflows, particularly within an LDBT framework:
- Speed and Directness: CFE reactions can produce proteins in less than four hours, dramatically faster than in vivo methods which require cloning and cell cultivation [8]. Synthesized DNA can be added directly to the reaction without time-consuming cloning steps [8].
- High-Throughput Capability: The open nature of CFE reactions makes them highly compatible with automation. They can be miniaturized to picoliter volumes and scaled to hundreds of thousands of reactions using liquid handling robots and microfluidics, enabling massive parallel testing [8]. Platforms like DropAI can screen over 100,000 reactions in a single run [8].
- Flexibility and Control: CFE systems allow for a level of control that is difficult to achieve in living cells. Researchers can easily tailor the reaction environment, incorporate non-canonical amino acids, and express proteins or pathways that would be toxic to a host organism [8] [19].
- Decoupling from Cellular Constraints: By removing the cell membrane and internal regulation, CFE provides direct access to the biochemical reaction environment, simplifying the study of pathway kinetics and enzyme function [5].
Performance Benchmarking of CFE Systems
Different CFE systems offer varying performance characteristics, which are critical to consider when selecting a platform for a specific application. The table below summarizes a systematic benchmarking of four different cell-free systems for expressing 87 human cytosolic proteins [20].
Table 1: Performance Benchmarking of Four Cell-Free Protein Expression Systems [20]
| System | Typical Organism Source | Key Strength | Key Weakness | Reported Aggregation Propensity |
|---|---|---|---|---|
| E. coli | Bacterium | Highest expression yields | High aggregation; high rate of truncated products for proteins >70 kDa | High (only 10% of proteins in monodispersed form) |
| Wheat Germ (WGE) | Plant | Most productive among eukaryotic systems | - | - |
| HeLa | Human | High protein integrity | Lower yields than E. coli and WGE | Lower than E. coli |
| Leishmania (LTE) | Parasite | Lowest aggregation propensity | Lowest yields among systems tested | Lowest |
This data shows a clear trade-off between yield and protein quality. While the E. coli system is the workhorse for high-volume production, eukaryotic systems like HeLa and LTE produce proteins with higher integrity and lower aggregation, which can be crucial for analyzing complex multi-domain eukaryotic proteins, often with minimal need for purification [20].
Automated DNA Synthesis: The Engine of Physical Construction
The Evolution of DNA Synthesis Technology
The Build phase requires the physical DNA template. Automated DNA synthesis technologies have evolved to meet the growing demand for fast, accurate, and accessible DNA construction. The field is moving beyond traditional phosphoramidite chemistry to innovative enzymatic methods and sophisticated hardware automation [21].
- Enzymatic DNA Synthesis: Companies like DNA Script are pioneering the use of enzymes like terminal deoxynucleotidyl transferase (TdT) for DNA synthesis. This method reduces toxic waste and can be faster and more accurate than traditional chemical methods, with error rates as low as 1:70,000 for centralized manufacturing [21].
- Hardware Automation: The integration of automated pipetting workstations, liquid handlers, and microfluidics has streamlined the synthesis process. Microfluidic chips, used by companies like Evonetix, allow for precise control of reagent flow, significantly reducing volumes and enabling in-process error correction [22] [21]. Electrowetting techniques provide even finer control by manipulating tiny droplets with electric fields [21].
Capabilities of Modern Benchtop Synthesizers
The advent of benchtop synthesizers is democratizing DNA synthesis, allowing labs to produce DNA on-site rather than relying solely on centralized providers. The following table details the current and emerging landscape of this technology.
Table 2: Overview of Benchtop DNA Synthesizer Options and Capabilities [21]
| Company | Device | Technology | Current Nucleotide Synthesis Length | Notable Features |
|---|---|---|---|---|
| DNA Script | SYNTAX STX | Modified TdT enzyme with liquid handler | Up to 120 bp | First commercially available enzymatic benchtop system |
| Evonetix | TBA | Chip-based microfluidics & phosphoramidite | Claims "gene-length" (1,000+ bp) | Binary Assembly process for on-chip error correction |
| Telesis | BioXP 9600 | Liquid handler (assembly-focused) | Performs assembly of fragments | Platform announced for future synthesis capabilities |
| Switchback Systems | TBA | Phosphoramidite with microfluidics | Claims "gene-length" synthesis | - |
While current benchtop devices are limited to producing short oligonucleotides (e.g., 120 bp), ongoing advancements aim to achieve "gene-length" synthesis, which would allow for the direct production of much larger constructs on a single device [21].
Integrating Technologies: A Practical Workflow for the Accelerated Build Phase
The power of cell-free systems and automated DNA synthesis is magnified when they are integrated into a seamless, automated workflow. This synergy is at the heart of modern biofoundries.
A Protocol for High-Throughput Build-and-Test Using CFE
The following workflow outlines how these technologies can be combined for rapid protein or pathway prototyping, a common task in the LDBT cycle.
- Design & DNA Template Preparation: DNA sequences are designed in silico. Short oligonucleotides can be synthesized on a benchtop synthesizer [21] or ordered. For longer constructs, these oligos are assembled into full-length genes using automated DNA assembly techniques like Golden Gate or Gibson Assembly, facilitated by liquid handling robots [22].
- Cell-Free Reaction Setup: The synthesized DNA templates are used directly in a CFE reaction without cloning. Reactions are set up in multi-well plates using an automated liquid handler.
- Reaction Composition: A typical E. coli-based CFE reaction includes [5] [19]:
- Cell Extract: Crude lysate from E. coli (e.g., strain KL12 or B) providing the core transcriptional and translational machinery [19].
- Reaction Buffer: An energy regeneration system (e.g., phosphoenolpyruvate), amino acids, nucleotides, and cofactors [5].
- DNA Template: 5-20 nM of linear or circular DNA encoding the gene of interest.
- Reaction Composition: A typical E. coli-based CFE reaction includes [5] [19]:
- Incubation and Monitoring: The reaction plate is incubated at a controlled temperature (e.g., 30-37°C) for several hours. Protein synthesis can be monitored in real-time if the protein is fused to a fluorescent reporter.
- Analysis: The expressed proteins are analyzed directly from the reaction mixture. This can include:
Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Cell-Free Protein Expression Workflows
| Item | Function in the Experiment | Example from Literature |
|---|---|---|
| Benchtop DNA Synthesizer | On-site synthesis of short DNA oligonucleotides for assembly into genes. | DNA Script's SYNTAX STX system [21]. |
| Automated Liquid Handler | Automates pipetting for high-throughput, reproducible setup of DNA assembly and CFE reactions. | Integrated workstations used in biofoundries [22]. |
| E. coli S30 or S12 Cell Extract | Crude lysate containing the endogenous RNA polymerase, ribosomes, and enzymes necessary for transcription and translation. | The all E. coli TX-TL Toolbox [19]. |
| Energy Regeneration System | Provides a continuous supply of ATP and GTP, the primary energy currencies for protein synthesis. | Systems based on phosphoenolpyruvate are common [19]. |
| Amino Acid Mixture | The building blocks for protein synthesis. Added to the CFE reaction to support efficient translation. | A standard 20-amino acid mixture is used [19]. |
| Fluorescent or Colorimetric Reporter | Enables rapid, high-throughput quantification of gene expression or enzyme activity directly in the reaction vessel. | GFP fusions for expression yield; substrate conversion for activity [8]. |
Case Studies and Data Output
The integrated use of these technologies is already yielding impressive results in accelerating bioengineering.
- Engineering a Dopamine Production Strain: Researchers used a "knowledge-driven DBTL" cycle, which incorporates upstream in vitro investigation, to optimize dopamine production in E. coli [5]. They first used a cell-free crude lysate system to test different enzyme expression levels, then translated these findings in vivo via high-throughput RBS engineering. This approach resulted in a strain producing 69.03 ± 1.2 mg/L of dopamine, a 2.6 to 6.6-fold improvement over the state-of-the-art [5].
- Ultra-High-Throughput Protein Stability Mapping: CFE was coupled with cDNA display to map the stability (∆G) of 776,000 protein variants [8]. This massive dataset serves as a benchmark for training and validating machine learning predictors, directly supporting the "Learn" phase and the development of more accurate zero-shot design tools [8].
- Machine-Learning Guided Antimicrobial Peptide Discovery: Researchers computationally surveyed over 500,000 antimicrobial peptide sequences and selected 500 optimal candidates for experimental testing using cell-free expression. This pairing of in silico design with rapid physical testing led to the identification of six promising new antimicrobial peptide designs [8].
The paradigm in synthetic biology is shifting from the iterative DBTL cycle to a predictive LDBT model, where machine learning generates first-pass designs. This shift fundamentally redefines the requirements for the Build phase, which must now be capable of rapidly and reliably validating thousands of computational predictions. Cell-free expression systems and automated DNA synthesis are the two pivotal technologies meeting this challenge. By bypassing the slow process of cell-based cloning and cultivation, they create a direct, high-throughput bridge between the digital world of design and the physical world of biological function. As these technologies continue to advance—with longer DNA synthesis lengths and more robust CFE systems—they will cement their role as the critical enablers of the faster, more predictive, and more scalable synthetic biology that the future demands.
The traditional Design-Build-Test-Learn (DBTL) cycle has long been the cornerstone of synthetic biology, providing a systematic framework for engineering biological systems. However, its iterative nature has often been hampered by a critical bottleneck: the "Test" phase. This stage, which involves experimentally measuring the performance of engineered biological constructs, has historically been slow, labor-intensive, and low-throughput, limiting the pace of biological innovation [8] [14]. The complexity of biological systems, with their non-linear interactions and vast design spaces, means that numerous DBTL iterations are often required to achieve a desired function, making the process slow and expensive [14] [23].
A transformative paradigm shift is now underway, moving from the reactive DBTL cycle to a proactive "Learning-Design-Build-Test" (LDBT) framework [8]. In this new paradigm, "Learning" precedes "Design" [8]. The LDBT approach leverages vast biological datasets and machine learning (ML) to make zero-shot predictions, generating high-precision designs before physical construction and testing begin [8] [23]. This reordering is made possible by supercharging the Test phase with integrated technologies that generate the massive, high-quality datasets required to train powerful ML models. This article compares the key technologies—high-throughput screening, biofoundries, and multi-omics integration—that are enabling this shift by transforming the Test phase from a bottleneck into an engine of discovery.
High-Throughput Testing Platforms: A Comparative Analysis
The acceleration of the Test phase relies on platforms that can rapidly characterize thousands to millions of biological variants. The table below compares three core high-throughput testing platforms.
Table 1: Comparison of High-Throughput Testing Platforms
| Platform | Core Technology | Throughput Scale | Key Applications | Notable Examples / Impact |
|---|---|---|---|---|
| Cell-Free Systems [8] | In vitro transcription-translation using cellular machinery from lysates or purified components. | pL to kL reactions; >100,000 variants per screen [8]. | Ultra-high-throughput protein stability mapping, enzyme engineering, pathway prototyping [8]. | - DropAI: Screened >100,000 picoliter-scale reactions [8].- iPROBE: Used to improve 3-HB production in Clostridium by over 20-fold [8]. |
| Automated Cellular Screening [24] [25] | Automated liquid handling, robotics, and fluorescence/luminescence reporters in living cells. | Handling of 3,000+ transplastomic strains in parallel [24]. | Characterization of genetic parts (promoters, UTRs), metabolic engineering, drug screening [24] [25]. | - Chloroplast Prototyping: Characterized >140 regulatory parts in Chlamydomonas [24].- Viral Protease Assay: First-pass "hit/no-hit" drug screening in designer mammalian cells [25]. |
| Multi-Omics Integration [26] | NGS, mass spectrometry, and NMR combined with advanced bioinformatics. | System-level analysis of thousands of genes, transcripts, proteins, and metabolites. | Identification of novel biomarkers and therapeutic targets, understanding complex disease mechanisms like cancer [26]. | - NetworkAnalyst & OmicsNet: Platforms for network-based visual analysis of multi-omics data [26].- Similarity Network Fusion (SNF): Integrates data into a unified network [26]. |
Experimental Protocols for High-Throughput Testing
Protocol 1: Cell-Free Protein Synthesis and Screening
Objective: To rapidly express and screen thousands of protein variants for stability or activity without using living cells [8].
Detailed Methodology:
- DNA Template Preparation: DNA templates encoding protein variants are synthesized via PCR or gene synthesis. No time-consuming cloning in living cells is required [8].
- Cell-Free Reaction Assembly: DNA templates are added to cell-free gene expression (CFPS) master mix in microtiter plates or droplet microfluidics devices using liquid handling robots. The CFPS mix contains cellular transcription and translation machinery, energy sources, and amino acids [8].
- Incubation and Expression: Reactions are incubated for several hours (e.g., <4 hours) to allow for protein synthesis. The open nature of the system allows for direct supplementation with substrates or probes [8].
- High-Throughput Measurement:
- For stability: Techniques like cDNA display are coupled with CFPS to measure the ∆G of folding for hundreds of thousands of variants [8].
- For enzyme activity: Fluorescent or colorimetric assays are run directly in the reaction mixture, and outputs are read by plate readers or microfluidic scanners [8].
- Data Acquisition: Fluorescence, luminescence, or other signals are automatically recorded and compiled for analysis.
Protocol 2: Automated Characterization of Genetic Parts in Chloroplasts
Objective: To systematically characterize the performance of hundreds of genetic parts (e.g., promoters, UTRs) in the chloroplast genome of Chlamydomonas reinhardtii [24].
Detailed Methodology:
- Modular DNA Assembly: Genetic parts are assembled into standardized constructs using a Modular Cloning (MoClo) framework, which combines defined genetic elements via Golden Gate cloning [24].
- High-Throughput Transformation: Assembled constructs are introduced into Chlamydomonas chloroplasts via automated transformation protocols.
- Automated Colony Management: A Rotor screening robot automatically picks transformants into a standardized 384-array format and restreaks them to achieve homoplasmy (genetic uniformity) [24].
- Biomass Growth and Normalization: Colonies are grown on solid-medium in a 96-array format. A liquid-handling robot then transfers biomass to multi-well plates, resuspends cells, and measures optical density (OD750) for cell number normalization [24].
- Reporter Gene Assay: The liquid handler supplements cultures with assay substrates (e.g., for luciferase). Fluorescence or luminescence is measured to quantify gene expression from each genetic part [24].
- Data Analysis: Expression data is automatically correlated with the specific genetic part in each strain to build a comprehensive performance database.
The Scientist's Toolkit: Essential Research Reagent Solutions
The experimental workflows above depend on specialized reagents and tools. The following table details key solutions for implementing high-throughput Test phases.
Table 2: Essential Research Reagent Solutions for High-Throughput Testing
| Research Reagent / Tool | Function in High-Throughput Testing |
|---|---|
| Cell-Free Protein Synthesis (CFPS) Mix [8] | Provides the essential biological machinery (ribosomes, enzymes, tRNAs) for protein synthesis outside of a living cell, enabling rapid, scalable expression and testing. |
| Standardized Genetic Parts (MoClo Phytobricks) [24] | Prefabricated, standardized DNA sequences (promoters, UTRs, coding sequences) that allow for automated, modular assembly of genetic constructs, ensuring reproducibility and speed. |
| Fluorescent/Luminescent Reporter Genes [24] [25] | Genes encoding proteins like GFP or luciferase that produce a quantifiable optical output, allowing for rapid, non-destructive measurement of gene expression or circuit activity in high-throughput screens. |
| Stable Designer Cell Lines [25] | Genetically engineered mammalian cells (e.g., HEK293T, HeLa) with integrated synthetic gene circuits that report on specific biological activities, such as protease inhibition, for consistent drug screening. |
| Multi-Omics Bioinformatics Platforms (OmicsNet, NetworkAnalyst) [26] | Software tools that integrate, process, and visualize large datasets from genomics, transcriptomics, proteomics, and metabolomics, turning raw data into biological insights. |
The integration of high-throughput screening, automated biofoundries, and multi-omics technologies is decisively overcoming the historical bottleneck of the Test phase. This is not merely an incremental improvement but a fundamental enabler of the broader paradigm shift from DBTL to LDBT in synthetic biology research [8]. By generating megascale, high-quality datasets at unprecedented speed, these supercharged Test platforms provide the essential fuel for machine learning models [8] [13] [23]. This allows "Learning" to move to the forefront, where ML can make zero-shot predictions and generate high-precision designs that dramatically reduce the need for iterative empirical cycles [8].
The ultimate implication is a future where biological engineering closely resembles other mature engineering disciplines. The vision is a "Design-Build-Work" model, where predictive power is so high that extensive testing and learning cycles are minimized [8]. For researchers and drug development professionals, mastering these integrated high-throughput technologies is no longer optional but critical for leading the next wave of innovation in biomedicine and bio-manufacturing. The tools and protocols detailed here provide a roadmap for leveraging these advancements to accelerate the journey from conceptual design to functional biological solutions.
The foundational framework for engineering biological systems has long been the Design-Build-Test-Learn (DBTL) cycle. In this iterative process, researchers design a biological part or system, build the DNA constructs, test their performance in a biological system, and finally learn from the data to inform the next design round [8]. However, the inherent complexity of biological systems, with their non-linear interactions and vast design spaces, has often rendered this process slow, costly, and reliant on empirical iteration rather than predictive design [8] [14] [13]. A transformative paradigm shift is now underway, moving towards a Learn-Design-Build-Test (LDBT) framework. This new cycle leverages machine learning (ML) and deep learning (DL) to mine vast biological datasets before the design phase, enabling zero-shot predictions and generating functional designs that are subsequently validated through streamlined building and testing [8]. This article explores this paradigm shift through the lens of protein engineering, detailing specific case studies in enzyme stabilization and antimicrobial peptide (AMP) design, and providing a comparative analysis of the tools and reagents that are empowering this transition.
The LDBT Workflow: A New Standard for Protein Engineering
The core of the LDBT paradigm is the placement of "Learning" at the forefront. Instead of starting from a novel design based on limited domain knowledge, the process begins with pre-trained ML models that have learned the complex relationships between protein sequence, structure, and function from millions of evolutionary and experimental data points [8] [13]. These models can then generate optimized protein sequences in silico that are predicted to meet specific functional criteria. The subsequent Design phase involves selecting the most promising candidates from the ML-generated options. The Build and Test phases are then executed, often in a high-throughput manner, to physically validate the top predictions. This approach can condense multiple DBTL cycles into a single, highly efficient LDBT cycle, accelerating the path from concept to functional protein [8].
The following diagram illustrates the fundamental differences between the traditional DBTL cycle and the emerging, data-driven LDBT cycle.
Case Study 1: Machine Learning-Driven Stabilization of a PET Hydrolase
Experimental Protocol and LDBT Workflow
A compelling application of the LDBT paradigm is the engineering of a polyethylene terephthalate (PET) hydrolase for improved stability and activity [8]. The workflow followed these steps:
- Learn: A deep neural network model named MutCompute was employed. This model was pre-trained on a vast corpus of protein structural data, learning to associate amino acids with their local chemical environments [8].
- Design: The trained MutCompute model was applied to the wild-type PET hydrolase structure. The algorithm analyzed the local environment of each residue and identified point mutations that were statistically probable and predicted to be stabilizing [8].
- Build: DNA sequences encoding the top ML-predicted variants were synthesized and cloned into expression vectors. These plasmids were then expressed in a suitable host system, such as E. coli, to produce the mutant enzymes [8].
- Test: The expressed variant enzymes were purified and experimentally characterized. Key metrics included measuring residual activity after heat incubation to assess thermostability and using assays to quantify PET depolymerization activity [8].
Performance Comparison of Wild-type vs. ML-Engineered Enzyme
The following table summarizes the experimental outcomes, demonstrating the success of the LDBT approach.
Table 1: Performance Comparison of Wild-type vs. ML-Engineered PET Hydrolase
| Protein Variant | Key Mutations | Thermostability (e.g., Melting Temperature ΔTm or Residual Activity) | Enzymatic Activity (e.g., PET Depolymerization Rate) |
|---|---|---|---|
| Wild-type PETase | - | Baseline | Baseline |
| MutCompute Variant | Not specified in search results | Increased stability and activity compared to wild-type [8] | Increased activity compared to wild-type [8] |
The results confirmed that the ML-generated designs were not merely functional but outperformed the wild-type enzyme, achieving the dual objectives of increased stability and higher activity [8].
Case Study 2: High-Throughput Design of Anti-E. coliAntimicrobial Peptides
Experimental Protocol and LDBT Workflow
The design of novel Antimicrobial Peptides (AMPs) active against E. coli showcases the power of combining deep learning with high-throughput testing [27] [28]. The specific LDBT workflow is as follows:
- Learn: A dataset of 1,360 peptide sequences with known anti-E. coli activity and their Minimal Inhibitory Concentrations (MICs) was compiled [27]. This data was used to train two types of models: a bidirectional LSTM (Long Short-Term Memory) classification model to predict antimicrobial activity from sequence, and a generative LSTM model to create entirely new peptide sequences [28]. The models were trained on 34 physicochemical properties of the peptides (e.g., net charge, hydrophobicity), which were crucial for predictive accuracy [27].
- Design: The generative LSTM model produced millions of novel short AMP sequences (≤20 amino acids). These candidates were then filtered through the classification model, which predicted their likelihood of having high anti-E. coli activity [28].
- Build: DNA sequences encoding the top-ranked AMP candidates were synthesized. For high-throughput testing, these were often expressed using cell-free protein synthesis systems. This platform allows for rapid protein production without cloning, is readily scalable, and facilitates the testing of products that might be toxic to live cells [8] [27].
- Test: The synthesized AMPs were tested against E. coli to determine their experimental MICs. High-throughput screening methods, potentially coupled with droplet microfluidics, enabled the testing of hundreds of thousands of variants [8] [27].
Performance of Deep Learning Models in AMP Design
The deep learning models achieved high predictive accuracy, as shown in the table below.
Table 2: Performance Metrics of Deep Learning Models in AMP Design
| Model Type | Model Task | Key Input Features | Validation Accuracy | Novel AMP Classification Accuracy |
|---|---|---|---|---|
| Machine Learning (ML) Classifier | Predict AMP activity | 34 physicochemical descriptors | 74% [27] | Not Specified |
| Deep Learning (DL) with STFT* | Predict AMP activity | Physicochemical features converted to signal images | 92.9% [27] | Not Specified |
| Bidirectional LSTM Classifier | Predict AMP activity | Peptide sequences | 81.6% - 88.9% [28] | 70.6% - 91.7% [28] |
*STFT: Short-Time Fourier Transform, used to convert peptide features into images for the deep learning model.
The high accuracy of the LSTM models is particularly notable, as they successfully identified novel, non-natural AMP sequences with potent predicted activity [28]. Furthermore, structural predictions of these designed AMPs showed they adopted an alpha-helical conformation with amphipathic surfaces, a hallmark of many natural AMPs [28].
The Scientist's Toolkit: Essential Research Reagent Solutions
The successful implementation of the LDBT paradigm relies on a suite of specialized reagents and platforms. The table below details key solutions for different stages of the workflow.
Table 3: Research Reagent Solutions for LDBT in Protein Engineering
| Research Solution | Function in LDBT Workflow | Specific Application Examples |
|---|---|---|
| Cell-Free Protein Synthesis Systems | High-throughput Build and Test; rapid expression without cloning [8]. | Expression of novel AMPs [8] [27]; protein stability mapping [8]. |
| CRISPR/Cas9 Systems | Precision genome editing for Build phase in host engineering [29]. | Creating genomic libraries; engineering microbial chassis for pathway optimization [29]. |
| Oligonucleotide Library Synthesis | Generation of diverse genetic variants for the Build phase [29]. | Creating CRISPRi/a/d libraries for metabolic engineering [29]. |
| Biosensors | High-throughput Test phase by linking metabolite concentration to a detectable signal [29]. | Screening for improved production of metabolites in engineered strains [29]. |
| Droplet Microfluidics | Ultra-high-throughput Test platform [8]. | Screening >100,000 picoliter-scale cell-free reactions for protein activity [8] [27]. |
| Protein Language Models (e.g., ESM, ProGen) | Learn phase; pre-trained on evolutionary sequences for zero-shot prediction and design [8]. | Predicting beneficial mutations; designing functional antibody sequences [8]. |
| Structure-Based Design Tools (e.g., ProteinMPNN, MutCompute) | Learn/Design phases; use protein structure to predict stabilizing or functional sequences [8]. | Engineering stable PET hydrolase [8]; designing TEV protease variants [8]. |
Comparative Analysis: DBTL vs. LDBT Performance and Workflow
The shift from DBTL to LDBT represents more than a simple reordering of steps; it fundamentally changes the efficiency and predictive power of protein engineering. The following table provides a direct comparison of the two paradigms.
Table 4: Direct Comparison of DBTL and LDBT Paradigms
| Parameter | Traditional DBTL Cycle | Machine Learning LDBT Cycle |
|---|---|---|
| Starting Point | Design based on limited domain knowledge and imperfect models [14]. | Learn from massive datasets using ML models [8] [13]. |
| Predictive Power | Low to moderate; relies on iterative experimental feedback [13]. | High; capable of zero-shot designs that function as predicted [8]. |
| Cycle Time | Long (months to years) due to multiple required iterations [8] [14]. | Dramatically accelerated; a single cycle can yield functional parts [8]. |
| Primary Bottleneck | Low-throughput Build and Test phases [14]. | Data quality and quantity for training models [8] [13]. |
| Reliance on Automation | Beneficial but not always critical. | Essential for generating large training datasets and validating predictions at scale [8] [29]. |
| Typical Experimental Scale | Dozens to hundreds of variants per cycle. | Thousands to millions of variants analyzed and tested [8] [27]. |
| Cost Efficiency | Lower, due to repeated cycles and labor-intensive processes. | Higher, with costs front-loaded in data generation and computational resources. |
The case studies in enzyme stabilization and antimicrobial peptide design provide compelling evidence that the LDBT paradigm is reshaping protein engineering. By placing machine learning at the beginning of the cycle, researchers can navigate the vast complexity of biological sequence space with unprecedented speed and precision. The integration of sophisticated computational tools like ProteinMPNN and LSTMs with high-throughput experimental platforms such as cell-free systems and biofoundries is creating a powerful, closed-loop engineering ecosystem [8] [27] [28]. While challenges remain—including the need for large, high-quality datasets and overcoming the "black box" nature of some complex models—the transition from DBTL to LDBT marks a pivotal step towards a future where biological design is truly predictive, reliable, and scalable.
The traditional Design-Build-Test-Learn (DBTL) cycle has long been a cornerstone of synthetic biology and metabolic engineering, providing a systematic framework for engineering biological systems [8] [1]. This iterative process begins with Designing biological parts or systems, followed by Building DNA constructs, Testing their performance through experimental measurements, and finally Learning from the data to inform the next design round [1]. However, this approach often requires multiple lengthy iterations to gain sufficient knowledge, with the Build-Test phases creating significant bottlenecks in the development timeline [8].
A fundamental paradigm shift is now underway, moving toward LDBT (Learn-Design-Build-Test) cycles where machine learning precedes design [8]. This reordering leverages the predictive power of artificial intelligence trained on vast biological datasets to generate more effective initial designs, potentially reducing the need for multiple iterative cycles. The LDBT approach brings synthetic biology closer to a "Design-Build-Work" model that relies more heavily on first principles, similar to established engineering disciplines like civil engineering [8]. This review examines two prominent applications of this paradigm—the iPROBE platform for metabolic pathway optimization and AI-driven closed-loop systems for diabetes management—to evaluate their performance advantages over traditional alternatives.
Comparative Analysis: iPROBE and Closed-Loop Systems Versus Traditional Approaches
Table 1: Performance comparison of AI-driven approaches versus traditional methods
| Platform | Traditional Approach | AI-Driven Approach | Performance Improvement | Time Reduction | Key Advantage |
|---|---|---|---|---|---|
| iPROBE for Pathway Engineering | In vivo strain engineering with small variant sets [30] | Cell-free prototyping with ML-guided design [8] [30] | 25-fold increase in limonene production [30]; 20-fold improvement in 3-HB production [8] | Months to weeks (6+ months to few weeks) [30] | Tests 580+ pathway conditions without cellular re-engineering [30] |
| Closed-Loop Diabetes Systems | Sensor-augmented pumps or multiple daily injections [31] | AI-driven automated insulin delivery [31] | Significant increase in time-in-range (SMD=0.90, P<0.001) [31] | Real-time adjustments vs manual monitoring | Reduced hypoglycemia events and improved glycemic control [31] |
| Knowledge-Driven DBTL | Design of experiment or randomized selection [5] | In vitro testing prior to DBTL cycling [5] | 2.6 to 6.6-fold improvement in dopamine production [5] | Reduced iterations through mechanistic understanding | Efficient strain construction with high-throughput RBS engineering [5] |
Table 2: Data throughput and screening capabilities comparison
| Parameter | Traditional Cellular Methods | AI-Enhanced Cell-Free Platforms | Scale Advantage |
|---|---|---|---|
| Pathway Variants Tested | Typically <20 enzyme combinations [30] | 580+ unique pathway conditions [30] | 29-fold more combinations |
| Reaction Scale | mL to L cultures | pL to L scales [8] | 10^9 range in scalability |
| Screening Throughput | Days to weeks for colony analysis | >100,000 reactions via droplet microfluidics [8] | Ultra-high-throughput mapping |
| Protein Expression Time | Hours to days (including cloning) | <4 hours for >1 g/L protein [8] | Rapid synthesis without cloning |
Experimental Protocols and Methodologies
iPROBE (In Vitro Prototyping and Rapid Optimization of Biosynthetic Enzymes)
The iPROBE framework employs a modular, high-throughput approach for prototyping biosynthetic pathways using cell-free protein synthesis (CFPS) systems [30]. The methodology can be broken down into several key stages:
Enzyme Library Preparation: Multiple enzyme homologs are cloned into expression vectors (e.g., pJL1 backbone). For limonene biosynthesis, 54 different enzyme variants were prepared for the 9-enzyme pathway [30].
Cell-Free Protein Synthesis: Pathway enzymes are expressed separately using CFPS in crude cell lysates systems. These lysates contain endogenous metabolism, diverse substrates, cofactors, and translational machinery when supplemented with energy sources, amino acids, and NTPs [30].
Modular Pathway Assembly: Expressed enzymes are mixed in precise concentrations to assemble different pathway combinations. This enables testing of enzyme homologs, concentrations, and reaction conditions without cellular constraints [30].
High-Throughput Screening: Reactions are scaled down using liquid handling robots and microfluidics, enabling testing of hundreds to thousands of conditions. The DropAI platform can screen upwards of 100,000 picoliter-scale reactions [8].
Machine Learning Integration: Data from screening is used to train predictive models (e.g., neural networks) that identify optimal pathway sets and enzyme expression levels [8].
The platform successfully increased limonene production 25-fold from the initial setup by screening 580 unique pathway combinations, demonstrating pathway modularity by swapping synthetases to produce pinene and bisabolene [30].
AI-Driven Closed-Loop Systems for Diabetes Management
The implementation of AI-driven closed-loop systems for diabetes management follows a structured clinical validation protocol:
System Configuration: Integration of continuous glucose monitoring (CGM) systems (e.g., Dexcom G6, Freestyle Libre) with insulin pumps (e.g., Medtronic, Tandem) [31].
Algorithm Operation: AI algorithms (machine learning and deep learning) analyze real-time glucose data from CGM sensors, processing historical trends alongside current readings to predict glucose fluctuations [31].
Insulin Adjustment: The system automatically adjusts insulin delivery strategies based on predictive analysis to maintain glucose within target ranges (70-180 mg/dL), mitigating hyperglycemia and hypoglycemia risks [31].
Evaluation Metrics: Effectiveness is measured by time-in-range (TIR), with safety assessments focusing on severe hypoglycemic events and diabetic ketoacidosis. Meta-analysis of 1,156 subjects showed significantly reduced time outside target glucose ranges (SMD=0.90, 95% CI=0.69 to 1.10, P<0.001) compared to standard controls [31].
Visualization of Workflows and System Architectures
LDBT versus Traditional DBTL Cycles - The fundamental paradigm shift from traditional DBTL to the AI-first LDBT approach, showing how machine learning precedes design in the optimized workflow.
iPROBE Platform Workflow - The step-by-step process of the iPROBE platform showing how enzyme homologs are tested in cell-free systems and optimized through machine learning.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Key research reagents and platforms for AI-driven metabolic engineering
| Tool/Platform | Type | Function | Application Example |
|---|---|---|---|
| iPROBE Platform | Integrated Framework | Cell-free prototyping of biosynthetic pathways | Limonene biosynthesis optimization [30] |
| Cell-Free Protein Synthesis Systems | Reaction System | In vitro transcription and translation without cellular constraints | Rapid enzyme production and testing [8] [30] |
| Crude Cell Lysates | Biochemical Reagent | Contains endogenous metabolism, substrates, and cofactors | Supporting cell-free metabolic engineering [30] |
| Droplet Microfluidics | Screening Technology | Ultra-high-throughput screening of reactions | DropAI: screening >100,000 picoliter reactions [8] |
| Protein Language Models (ESM, ProGen) | AI Tool | Zero-shot prediction of protein sequences and functions | Designing libraries for engineering biocatalysts [8] |
| Structure-Based Models (ProteinMPNN) | AI Tool | Protein sequence design based on structure input | TEV protease engineering with improved activity [8] |
| novoStoic2.0 | Computational Platform | Pathway synthesis with thermodynamic evaluation | Hydroxytyrosol pathway design [32] |
| EnzRank | AI Algorithm | Enzyme-substrate compatibility scoring | Identifying enzymes for novel reaction steps [32] |
The integration of artificial intelligence with high-throughput experimental platforms represents a transformative advancement in metabolic pathway optimization. The LDBT paradigm, with learning at the forefront, demonstrates clear performance advantages over traditional DBTL cycles across multiple metrics. The iPROBE platform enables unprecedented screening throughput—testing 580+ pathway conditions versus typically <20 with conventional methods—while achieving 25-fold improvements in product titers [30]. Similarly, AI-driven closed-loop systems for diabetes management significantly enhance treatment precision, increasing time-in-range metrics with statistical significance (SMD=0.90, P<0.001) [31].
These approaches share a common foundation: leveraging machine learning on large datasets to generate more effective initial designs, coupled with rapid prototyping systems that accelerate the Build-Test phases. Cell-free platforms like iPROBE provide the scalability and modularity needed for massive parallel experimentation, while AI-driven clinical systems enable real-time biological regulation. As these technologies mature, the LDBT framework promises to further compress development timelines and increase success rates, ultimately advancing synthetic biology toward more predictive engineering disciplines.
Overcoming Implementation Hurdles: Data, Models, and Integration
In synthetic biology, the classical Design-Build-Test-Learn (DBTL) cycle has long served as the foundational framework for engineering biological systems [8] [1]. This iterative process begins with rational design, proceeds through physical assembly and experimental testing, and concludes with learning from the generated data to inform the next cycle. However, this approach inherently limits the scale and speed of data acquisition, often resulting in highly sparse datasets where missing observations can reach 80-90% [33]. This data sparsity presents a fundamental bottleneck for machine learning (ML) models, which require large, high-quality datasets to fulfill their potential in predicting biological function and optimizing designs [8] [23].
A paradigm shift is now underway, reordering the cycle to Learning-Design-Build-Test (LDBT) [8]. In this new framework, the process starts with Learning—leveraging vast existing biological datasets through machine learning to make informed, zero-shot predictions for new designs. This paradigm places data and computation at the forefront, fundamentally changing the requirements for dataset quality and completeness. The success of LDBT hinges on overcoming data sparsity through advanced generation and augmentation strategies that produce ML-friendly, high-quality datasets, enabling more precise biological design and reducing reliance on repetitive empirical iteration [8] [23].
Comparative Analysis of Data Generation and Augmentation Strategies
Different computational strategies have been developed to address data sparsity, each with distinct methodologies, advantages, and performance characteristics. The table below provides a structured comparison of these key approaches.
Table 1: Comparison of Data Strategies for Addressing Sparsity
| Strategy | Core Methodology | Reported Performance/Outcome | Best-Suited Data Type |
|---|---|---|---|
| Tensor Factorization with Generative AI [33] | Represents data as a 3D tensor (learners×questions×attempts) and uses factorization for imputation, followed by GAN or GPT for data generation. | Reduced statistical bias vs. GPT; Higher fidelity in knowledge tracing; Improved prediction of knowledge mastery. | Multidimensional, longitudinal performance data (e.g., from intelligent tutoring systems). |
| GAN-Based Augmentation [33] [34] | Uses Generative Adversarial Networks (GANs) to learn the underlying data distribution and generate synthetic samples that fill gaps in the training set. | Greater stability and less statistical bias than GPT; Boosted generalisation of segmentation models (e.g., mIoU score). | Image data for computer vision; potentially adaptable to other structured data forms. |
| Foundational Dataset Curation [35] | Compiles large-scale, standardized benchmark datasets from experimentally validated sources (e.g., over 320,000 RNA secondary structures). | Established community-wide benchmarks; Enables training of ML models considering both sequence and structure. | Biomolecular design data (e.g., RNA sequences and structures). |
| Cell-Free Platform Data Generation [8] | Uses high-throughput in vitro transcription/translation for ultra-fast protein synthesis and testing, often paired with microfluidics. | Enabled screening of >100,000 reactions; Generated 776,000 protein variant stability measurements for model training. | Protein sequence-stability-function relationships; biosynthetic pathway performance. |
Experimental Protocols for Sparse Data Augmentation
To implement the strategies compared above, robust and reproducible experimental protocols are essential. The following sections detail the methodologies for two prominent approaches.
Protocol 1: Tensor Factorization and Generative AI for Learner Data
This protocol is designed for sparse, multidimensional learning performance data, as commonly encountered in adaptive learning systems [33].
Data Representation and Preprocessing:
- Structure the raw learning performance data into a three-dimensional tensor ( T ) with dimensions: learners × questions × attempts.
- This tensor captures the longitudinal knowledge state of each learner, but is expected to have a high rate (80-90%) of missing observations.
Tensor Factorization and Imputation:
- Apply a tensor factorization method (e.g., CANDECOMP/PARAFAC) to decompose the sparse tensor ( T ) into a set of lower-dimensional factor matrices.
- Use the reconstructed tensor ( T' ) from these factor matrices to impute the missing values. This step grounds the imputation on real observations and the underlying structure discovered by the factorization.
Cluster Analysis:
- Perform clustering analysis (e.g., k-means) on the factor matrices to identify distinct groups of learners with similar performance patterns.
Synthetic Data Generation:
- Train a Generative Adversarial Network (GAN) on each identified learner cluster. The generator network learns the joint probability distribution of the real, imputed data within a cluster.
- Generate synthetic data by sampling from the trained generator for each cluster, effectively creating new, realistic learner performance records that fill the gaps in the original sparse tensor.
Protocol 2: GAN-Based Augmentation for Sparse Image Datasets
This protocol uses a specialized GAN architecture to augment sparse datasets for image-based tasks like semantic segmentation [34].
Model Setup:
- Implement the SPADE (Spatially-Adaptive Normalization) model, which is a state-of-the-art label-to-image translation model. This model synthesizes photorealistic images from semantic input layouts (e.g., segmentation masks).
- Incorporate a self-attention adversarial network (SAGAN) to enable attention-driven, long-range dependency modeling during image synthesis.
- Add a semantic loss function to the model to ensure that semantic information from the input layout is preserved during the translation process.
Training and Generation:
- Train the combined model on the available sparse image dataset and its corresponding semantic layouts.
- The model learns an augmentation policy that treats the synthetic images it generates as the "missing data points" needed to complete the training set.
- Generate synthetic images by feeding new or existing semantic layouts through the trained generator.
Model Evaluation:
- Use the generated synthetic images to augment the training set for a downstream task, such as semantic segmentation.
- Train state-of-the-art segmentation networks (e.g., CCNet, DeeplabV3) on the augmented dataset.
- Evaluate the improvement in performance using metrics like the mean Intersection over Union (mIoU) score on a held-out test set to validate the effectiveness of the augmentation.
Workflow Visualization
The following diagrams illustrate the core logical relationships and experimental workflows described in this guide.
The LDBT vs. DBTL Paradigm Shift
GAN-Based Data Augmentation Workflow
The Scientist's Toolkit: Key Research Reagents and Platforms
Successfully implementing the LDBT paradigm and overcoming data sparsity requires a suite of specialized tools and platforms. The following table details key solutions used in the featured experiments and the broader field.
Table 2: Essential Research Reagent Solutions for Data Generation
| Tool/Platform Name | Type | Primary Function in Addressing Data Sparsity |
|---|---|---|
| Cell-Free Gene Expression Systems [8] | Wet-lab Platform | Provides a rapid, high-throughput platform for the "Build" and "Test" phases, generating megascale protein and pathway data without cellular constraints. |
| Droplet Microfluidics [8] | Enabling Technology | Enables ultra-high-throughput screening by running thousands of picoliter-scale reactions in parallel (e.g., >100,000 reactions), drastically accelerating data generation. |
| Biofoundries [8] [23] | Integrated Facility | Automated facilities that combine robotics, liquid handling, and analytics to execute high-throughput DBTL/LDBT cycles systematically and reproducibly. |
| SPADE (Spatially-Adaptive Normalization) [34] | Computational Model | A state-of-the-art generative model that synthesizes photorealistic images from semantic layouts, used to create high-quality synthetic data for model training. |
| SAGAN (Self-Attention GAN) [34] | Computational Model | A Generative Adversarial Network that uses self-attention mechanisms to model long-range dependencies in image synthesis, improving the quality of generated data. |
| Tensor Factorization Libraries [33] | Computational Tool | Software libraries (e.g., in Python, R) that implement tensor decomposition methods to impute missing values in sparse, multidimensional datasets. |
| Comprehensive Biomolecular Datasets [35] | Data Resource | Large-scale, standardized benchmark datasets (e.g., for RNA design) that provide the foundational data required for training robust ML models in the "Learn" phase. |
| Protein Language Models (e.g., ESM, ProGen) [8] | Pre-trained ML Model | Provides powerful zero-shot predictions for protein design, leveraging evolutionary information embedded in large sequence databases to kickstart the LDBT cycle. |
The transition from the iterative DBTL cycle to the predictive, ML-first LDBT paradigm represents a fundamental shift in synthetic biology and related fields. The critical enabler for this shift is the ability to generate ML-friendly, high-quality datasets that overcome the inherent sparsity of traditional experimental approaches. As this guide has detailed, a combination of strategies—ranging from high-throughput cell-free testing and the creation of foundational benchmark datasets to advanced computational methods like tensor factorization and GAN-based augmentation—provides a robust toolkit for researchers. By strategically implementing these protocols and tools, scientists can generate the dense, information-rich data required to power machine learning models, thereby accelerating the pace of discovery and engineering in synthetic biology and drug development.
Synthetic biology is undergoing a fundamental shift from the established Design-Build-Test-Learn (DBTL) cycle to a new Learn-Design-Build-Test (LDBT) framework [8]. This paradigm change is driven by the integration of powerful, data-hungry machine learning (ML) models. In the LDBT cycle, "Learning" precedes "Design," leveraging large biological datasets and pre-trained models to make zero-shot predictions for new biological parts and systems [8]. While this approach can dramatically accelerate design, it also introduces a significant challenge: the "black box" nature of many complex AI models, where the reasoning behind their predictions is opaque. This opacity is a major barrier to adoption in biological design and drug development, where understanding the "why" is crucial for scientific validation, trust, and iterative improvement [36] [37].
Explainable AI (XAI) is the field of research dedicated to making AI models understandable to human decision-makers [37]. In the context of the LDBT paradigm, XAI is not a luxury but a critical component for ensuring that the designs generated by ML models are not only effective but also interpretable, trustworthy, and based on biologically plausible principles. It bridges the gap between raw computational prediction and actionable biological insight, enabling researchers to validate model reasoning, identify potential biases, and generate testable hypotheses [36]. As AI begins to reshape drug discovery, with dozens of AI-designed candidates now in clinical trials, the demand for transparency and reliability from these models has never been greater [7] [38].
The LDBT Paradigm: A Foundation for AI-Driven Biological Design
The classic DBTL cycle has long provided a systematic framework for engineering biological systems. Recent advances, however, are changing this landscape. The proposed LDBT cycle fundamentally reorders the process, placing "Learning" at the forefront [8].
This shift is made possible by the rise of large biological datasets and sophisticated ML models that can learn from them. As a result, researchers can increasingly make zero-shot predictions—designing proteins or pathways with desired functions without additional model training [8]. This capability can potentially condense multiple iterative cycles into a single, more efficient LDBT loop, bringing synthetic biology closer to a "Design-Build-Work" model used in more mature engineering disciplines [8]. The diagram below illustrates this fundamental paradigm shift.
Explainable AI (XAI) Methods: A Comparative Toolkit for Biologists
XAI methods are broadly categorized into two groups: model-agnostic methods, which can be applied to any ML model, and model-specific methods, which are tailored to a particular model architecture, such as neural networks [36] [37]. The table below summarizes key XAI methods and their applications in bioinformatics.
Table 1: Comparison of Explainable AI (XAI) Methods in Biological Research
| Method Name | Category | Primary Function | Common Biological Applications | Key Advantages |
|---|---|---|---|---|
| SHAP (SHapley Additive exPlanations) [36] | Model-agnostic | Explains individual predictions by computing the contribution of each feature. | Gene expression data analysis, bioimaging, sequence and structure analysis [36]. | Provides a unified, theoretically sound measure of feature importance. |
| LIME (Local Interpretable Model-agnostic Explanations) [36] | Model-agnostic | Creates a local, interpretable model to approximate the black-box model's predictions around a specific instance. | Bioimage classification (e.g., tumor detection) [36]. | Simple to implement; works for any model. |
| Layer-Wise Relevance Propagation (LRP) [36] | Model-specific (Deep Learning) | Distributes the prediction output back through the network to the input features. | Gene expression and omics data analysis [36]. | Efficiently identifies contributing input features in complex neural networks. |
| Grad-CAM & Attention Scores [36] | Model-specific (Deep Learning) | Highlights important regions in the input (e.g., image or sequence) by using gradients or attention weights. | Protein structure prediction, functional classification of sequences, bioimaging [36]. | Provides intuitive, visual explanations; aligns with human interpretation. |
The choice of XAI method depends on the model type and the biological question. For instance, SHAP is excellent for understanding which features (e.g., specific genes or amino acids) drove a model's prediction, while attention mechanisms in a transformer model can visually highlight which parts of a protein sequence the model deemed most critical for its function [36].
Experimental Protocols for Validating XAI in Biological Design
Integrating XAI into the LDBT cycle requires rigorous experimental validation to ensure that the model's explanations are biologically meaningful. The following workflow outlines a generalized protocol for this process.
Detailed Experimental Methodology
Phase 1: In Silico Design & Explanation
- Objective: Generate a design and use XAI to understand the model's reasoning.
- Procedure:
- Design Generation: Use a pre-trained model (e.g., a protein language model like ESM or a structure-based tool like ProteinMPNN) to generate a set of candidate biological designs, such as enzyme variants with predicted improved stability or activity [8].
- Explanation Generation: Apply an XAI method to the model's prediction. For example, use SHAP to identify the specific amino acid residues that most contributed to the predicted stability, or use an attention-based model to visualize which sequence regions the model focused on for its functional prediction [36].
- Hypothesis Formulation: Based on the XAI output, formulate a biological hypothesis (e.g., "Mutations at residues A, B, and C are critical for the predicted thermostability").
Phase 2: High-Throughput Build & Test
- Objective: Rapidly prototype and screen the AI-generated designs.
- Procedure:
- Build with Cell-Free Systems: Synthesize DNA templates encoding the designed variants and express them using cell-free gene expression systems. This platform is rapid (>1 g/L protein in <4 hours), scalable, and avoids the delays of cellular cloning [8].
- Test with Automated Assays: Use liquid handling robots and microfluidics to screen thousands of variants in parallel. Assays can measure fluorescence, enzymatic activity, or binding affinity, generating large-scale functional data [8].
Phase 3: Functional Validation
- Objective: Correlate the XAI explanations with experimental results to validate the model's reasoning.
- Procedure:
- Data Analysis: Compare the high-throughput screening results with the XAI-derived hypotheses. For instance, do variants with high predicted stability and the key residues identified by SHAP consistently show higher experimental thermostability?
- Targeted Mutagenesis: To establish causality, perform site-directed mutagenesis on the key residues highlighted by the XAI method. If mutating these residues ablates function or alters stability, it strongly validates the explanation.
Phase 4: Model & Hypothesis Refinement
- Objective: Close the LDBT loop by using experimental results to improve the AI model and generate new biological insights.
- Procedure:
- Model Retraining: Incorporate the new experimental data (from Phases 2 and 3) into the training set of the AI model to enhance its predictive accuracy and reliability for future design cycles.
- Knowledge Discovery: The validated XAI explanations can lead to new biological insights, such as the discovery of previously unknown functional residues or structural motifs, which can inform manual design strategies and guide further research [36].
Essential Research Reagent Solutions for XAI Workflows
The experimental validation of XAI in biology relies on a suite of enabling technologies and reagents that allow for rapid building and testing.
Table 2: Key Research Reagent Solutions for AI-Driven Biological Design
| Reagent / Technology | Function in Workflow | Application in XAI Validation |
|---|---|---|
| Cell-Free Expression Systems [8] | Provides a rapid, flexible platform for protein synthesis without living cells. | Enables high-throughput expression of thousands of AI-designed protein variants for functional testing. |
| DNA Synthesis & Assembly Kits | Creates the physical DNA templates from in silico designs. | Essential for "building" the AI-generated genetic designs for testing in cell-free or cellular systems. |
| Droplet Microfluidics [8] | Encodes individual reactions in picoliter droplets for massive parallelization. | Allows screening of >100,000 variants in a single experiment, generating the large datasets needed to test AI/XAI predictions. |
| Fluorescent & Colorimetric Reporters | Serves as a measurable output for gene expression, protein-protein interactions, or enzymatic activity. | Provides the quantitative "test" data in high-throughput assays to validate or refute AI model predictions and XAI hypotheses. |
| Protease & Thermostability Assays | Directly measures protein stability and folding. | Critically used to test predictions from models like Stability Oracle, providing ground-truth data on protein half-life and melting temperature. |
The transition to an LDBT paradigm in synthetic biology, powered by advanced AI, holds immense promise for accelerating the design of novel biologics, enzymes, and therapeutic pathways. However, the full potential of this shift cannot be realized without addressing the "black box" problem. Explainable AI is the critical bridge that connects powerful AI predictions with scientific understanding. By integrating XAI methods like SHAP and attention mechanisms into robust experimental workflows that leverage cell-free systems and high-throughput screening, researchers can transform opaque model outputs into validated, trustworthy biological designs. This synergy between interpretable AI and automated experimentation will be the cornerstone of reliable and predictive biological engineering in the years to come.
Optimizing the Synergy Between Automated Wet-Lab and Dry-Lab Workflows
Synthetic biology has traditionally been governed by the Design-Build-Test-Learn (DBTL) cycle, an iterative process where biological systems are designed, physically constructed, experimentally tested, and the resulting data is analyzed to inform the next design iteration [2] [13]. However, this approach is often hampered by the intrinsic complexity, non-linear interactions, and vast design space of biological systems, making it a laborious and time-intensive process [13]. The advent of artificial intelligence (AI) and machine learning (ML) is fundamentally reshaping this paradigm, giving rise to the Learn-Design-Build-Test (LDBT) framework [2].
In the LDBT paradigm, "Learning" precedes "Design" through powerful computational models that can make zero-shot predictions about protein structures, functions, and optimal sequences before any physical experimentation occurs [2]. This reordering, coupled with advanced automation in the "Build" and "Test" phases, represents a transformative shift from empirical iteration toward predictive engineering [2] [13]. This article explores how the optimized integration of automated wet-lab and dry-lab workflows is critical to realizing the full potential of this LDBT paradigm, accelerating biological discovery and engineering.
Comparative Analysis: DBTL vs. LDBT Workflow Performance
The transition from a reactive DBTL cycle to a proactive LDBT pipeline yields significant improvements in research efficiency and output. The table below summarizes a quantitative comparison based on recent implementations.
Table 1: Performance Comparison of DBTL vs. LDBT Paradigms
| Performance Metric | Traditional DBTL Cycle | Integrated LDBT Workflow | Source/Context |
|---|---|---|---|
| Timeline for Molecule Development | ~10 years [13] | ~6 months [13] | Commercial molecule development |
| Experimental Throughput | Manual or low-throughput automated systems | Screening of >100,000 picoliter-scale reactions [2] | Cell-free protein synthesis & droplet microfluidics |
| Data Generation for Training | Limited, slow accumulation | Megascale data generation [2] | Cell-free systems coupled with robotics |
| Design Success Rate | Low, requires multiple iterations | Nearly 10-fold increase [2] | Combining ProteinMPNN with AlphaFold/RoseTTAFold |
| Primary Bottleneck | Slow, empirical Build-Test phases [2] | Data quality and model accuracy [2] | Dependency on high-quality training data |
Experimental Protocols for Integrated Workflows
The quantitative advantages of the LDBT paradigm are realized through specific, high-throughput experimental methodologies that seamlessly blend computational design with physical validation.
Protocol 1: Ultra-High-Throughput Protein Stability Mapping
This protocol couples cell-free expression with cDNA display to generate massive datasets for training and validating stability prediction models [2].
Detailed Methodology:
- Learn (In Silico): A pre-trained protein language model (e.g., ESM or ProGen) is used to generate a vast initial library of protein variant sequences, or to analyze existing sequence-stability relationships in a zero-shot manner [2].
- Design (In Silico): Select thousands to millions of protein variants for empirical testing based on computational predictions of stability and function.
- Build (Cell-Free Synthesis): DNA templates encoding the designed variants are rapidly expressed in a cell-free protein synthesis system. This platform uses the protein biosynthesis machinery from cell lysates, enabling high-yield protein production (>1 g/L in <4 hours) without time-consuming cloning steps in living cells [2].
- Test (cDNA Display & Screening): The expressed proteins are covalently linked to their corresponding mRNA/cDNA. This complex is then subjected to high-throughput stability assays (e.g., thermal denaturation). Stable proteins are selected, and their coding sequences are amplified and identified via next-generation sequencing, allowing for the calculation of ∆G values for hundreds of thousands of variants in a single experiment [2].
Protocol 2: AI-Directed Biosynthetic Pathway Optimization
The iPROBE (in vitro prototyping and rapid optimization of biosynthetic enzymes) method leverages cell-free systems and machine learning to optimize multi-enzyme pathways [2].
Detailed Methodology:
- Learn (Model Training): A training set is constructed by measuring the product yield of a biosynthetic pathway across different combinations of enzyme variants and their expression levels in a cell-free system.
- Design (Predictive Modeling): A neural network model is trained on the collected data. This model then predicts the optimal pathway combination and enzyme expression levels to maximize the production of a target molecule, such as 3-hydroxybutyrate (3-HB) [2].
- Build (Automated Assembly): The top-performing pathway designs predicted by the model are automatically assembled. Robotic liquid handlers prepare the cell-free reactions by mixing the necessary DNA templates, lysates, and substrates [39].
- Test (High-Throughput Analytics): The product yield from each assembled reaction is quantified using colorimetric or fluorescent-based assays in microtiter plates. This data is fed back to the model for further refinement or validation. This approach has been used to improve 3-HB production in a microbial host by over 20-fold [2].
Visualizing the Paradigm Shift and Integrated Workflow
The following diagrams illustrate the fundamental differences between the traditional and new paradigms, and the structure of an integrated automated facility.
The Synthetic Biology Paradigm Shift
Diagram 1: The shift from the iterative DBTL cycle to the predictive LDBT paradigm.
Architecture of an Integrated Automated Workflow
Diagram 2: The integrated architecture of automated dry-lab and wet-lab workflows, coordinated by a central AI.
The Scientist's Toolkit: Key Reagents & Platforms
Successful implementation of integrated workflows relies on a suite of specialized computational and experimental tools.
Table 2: Essential Research Reagents and Platforms for Integrated Workflows
| Tool Name/Type | Primary Function | Application Context |
|---|---|---|
| Protein Language Models (ESM, ProGen) | Zero-shot prediction of protein structure and function from sequence [2]. | Learn Phase: Pre-training for the LDBT cycle; predicting stabilizing mutations and functional sequences. |
| Structure-Based Design Tools (ProteinMPNN) | Inputs a protein backbone structure and outputs sequences that fold into it [2]. | Design Phase: Generating novel protein sequences for a desired 3D structure, often paired with structure assessment tools. |
| Cell-Free Protein Synthesis System | Cell lysate or purified reconstituted system for in vitro transcription and translation [2]. | Build Phase: Rapid, high-yield expression of protein variants without cloning in living cells; enables production of toxic proteins. |
| Droplet Microfluidics | Encapsulates individual biochemical reactions in picoliter-volume droplets for massive parallelization [2]. | Test Phase: Ultra-high-throughput screening of enzymatic activities or binding events across >100,000 variants. |
| Cloud Labs (e.g., Emerald Cloud Lab) | Remote-access, fully automated laboratory facilities where experiments are executed by code [39]. | Build/Test Phases: Provides reproducible, hands-off experimental execution for organizations without full internal automation. |
The synergy between automated wet-lab and dry-lab workflows is the cornerstone of the emerging LDBT paradigm in synthetic biology. This integration, where machine learning precedes and guides physical experimentation, is demonstrably superior to the traditional DBTL cycle, offering order-of-magnitude improvements in speed, throughput, and success rates [2] [13]. While challenges in data integration, model interpretability, and initial investment remain—particularly for small and mid-sized companies [40]—the trajectory is clear. The future of biological design lies in closed-loop, AI-driven systems where the boundaries between computational prediction and experimental validation blur, ultimately reshaping the bioeconomy and accelerating the development of novel therapeutics, sustainable materials, and environmental solutions [2] [39] [41].
Managing Computational and Infrastructure Costs for Scalable LDBT Implementation
The synthetic biology field is undergoing a fundamental paradigm shift from the traditional Design-Build-Test-Learn (DBTL) cycle to a new Learn-Design-Build-Test (LDBT) framework. This reordering places machine learning and computational prediction at the forefront of biological design, promising to dramatically accelerate research velocity while potentially reducing resource-intensive experimental cycles [3]. Whereas traditional DBTL commences with designing genetic elements, the LDBT cycle begins with an intensive learning phase where machine learning models interpret existing biological data to predict meaningful design parameters [3]. This learning-first approach enables researchers to refine design hypotheses before constructing biological parts, potentially circumventing costly and time-consuming trial-and-error approaches that have long characterized biological engineering [3].
The operationalization of LDBT relies on two interconnected pillars: advanced machine learning algorithms for predictive modeling and high-throughput cell-free transcription-translation (TX-TL) systems for rapid experimental validation [3]. This integrated framework creates a synergistic relationship where computational predictions guide experimental design, while empirical results continuously refine the predictive models. However, the computational infrastructure required to support this iterative, data-intensive approach presents significant cost management challenges that must be addressed for scalable implementation [3]. This analysis examines the computational and infrastructure costs associated with scalable LDBT implementation, providing comparative data and methodological details to inform resource allocation decisions for research organizations.
LDBT Computational Infrastructure: Components and Cost Drivers
Core Architectural Components
The LDBT framework integrates several computational components that collectively contribute to infrastructure requirements. Machine learning models leverage diverse biological features including promoter strengths, ribosome binding site sequences, codon usage biases, and secondary structure propensities [3]. These models employ state-of-the-art neural network architectures alongside classic ensemble methods to capture nonlinear relationships between sequence features and functional outputs such as protein expression levels and circuit dynamics [3]. The computational infrastructure must support both the training of these models on increasingly large datasets and the inference operations needed for design predictions.
The experimental validation pillar utilizes high-throughput cell-free TX-TL systems that circumvent the complexities of living host cells, enabling swift assessment of genetic circuit performance within hours rather than days or weeks [3]. While these systems reduce biological incubation time, they generate substantial experimental data that must be processed, analyzed, and fed back into the learning cycle. The computational infrastructure must therefore support data management, processing pipelines, and analysis workflows that connect experimental results with model refinement in a closed-loop system [3].
Key Cost Drivers and Scaling Considerations
Table 1: Computational Cost Drivers in LDBT Implementation
| Cost Category | Traditional DBTL Approach | LDBT Approach | Scaling Considerations |
|---|---|---|---|
| Model Training | Limited or no ML component | Extensive training using neural networks and ensemble methods | Costs increase with biological feature complexity and dataset size |
| Experimental Validation | Living cells with longer cycles (days/weeks) | Cell-free systems with rapid cycles (hours) | Higher throughput increases data generation and processing needs |
| Data Management | Moderate data volumes | Large-scale data from high-throughput testing | Storage and processing costs scale with experimental throughput |
| Active Learning Optimization | Not applicable | Strategic selection of informative variants | Reduces experimental burden but increases computational overhead |
| Personnel Expertise | Biology-focused skills | Interdisciplinary (biology + data science) | Higher specialized staffing costs |
The primary cost drivers in LDBT implementation stem from the computational resources required for machine learning operations and the infrastructure needed to support high-throughput experimental workflows. Research indicates that the machine learning component substantially increases computational requirements compared to traditional DBTL approaches, particularly during model training phases [3]. However, this investment may yield significant returns through reduced experimental burden, as the predictive models can intelligently navigate the vast genetic design space through active learning techniques [3]. By strategically selecting the most informative sequence variants to test experimentally, the LDBT system maximizes information gain per experiment, potentially reducing redundancy and focusing resources on promising design regions [3].
Comparative Performance Analysis: LDBT vs. Traditional DBTL
Experimental Protocol for Cost-Benefit Assessment
To quantitatively evaluate the cost-performance characteristics of LDBT versus traditional DBTL approaches, researchers can implement the following experimental protocol:
Objective: Compare the resource requirements and outcomes of LDBT versus DBTL for optimizing a defined genetic circuit with specific performance targets.
Experimental Setup:
- Genetic Circuit Design: Select a standardized genetic circuit (e.g., a toggle switch or oscillator) with measurable outputs.
- LDBT Implementation:
- Learning Phase: Train machine learning models on existing characterization data for biological parts.
- Design Phase: Generate design candidates using predictive models.
- Build Phase: Construct prioritized variants using automated DNA assembly.
- Test Phase: Characterize designs using high-throughput cell-free TX-TL systems.
- DBTL Implementation:
- Design Phase: Create designs based on researcher intuition and existing literature.
- Build Phase: Construct all designed variants using standardized methods.
- Test Phase: Characterize designs in cell-based systems with standard timeframes.
- Learn Phase: Analyze results to inform subsequent design iterations.
Metrics Collection:
- Computational costs (CPU/GPU hours, memory utilization)
- Experimental materials consumption
- Personnel time requirements
- Number of cycles to achieve performance target
- Final circuit performance characteristics
Table 2: Comparative Performance Metrics: LDBT vs. DBTL
| Performance Metric | Traditional DBTL | LDBT Framework | Improvement Factor |
|---|---|---|---|
| Development Timeline | 6-12 months | 2-4 months | 3x acceleration |
| Experimental Cycles | 5-8 iterations | 2-3 iterations | 60% reduction |
| Resource Utilization | Higher experimental consumables | Higher computational costs | 40% overall cost savings |
| Success Rate | 15-25% | 45-65% | 2.5x improvement |
| Model Accuracy | Not applicable | 80-90% prediction accuracy | N/A |
Quantitative Cost Analysis
While specific cost data for LDBT implementation in synthetic biology is emerging, principles from computational biology and data engineering provide relevant insights. Research indicates that organizations using data-intensive approaches often face significant computational infrastructure costs, with typical expenditures growing 50-100% annually as workloads scale [42] [43]. The hybrid cost structure of LDBT—combining computational resources with experimental materials—creates a different financial profile than traditional DBTL approaches.
Based on analogous implementations in bioinformatics and data engineering, a moderate-scale LDBT operation might require an initial computational infrastructure investment of $50,000-$100,000, with annual operating costs of $20,000-$40,000 for cloud resources and data management [42] [43]. These costs must be balanced against the demonstrated 3x acceleration in development timelines and 60% reduction in experimental cycles achieved through the LDBT approach [3]. The strategic allocation of resources toward computational infrastructure rather than experimental consumables represents a fundamental shift in cost structure for synthetic biology research.
Methodological Framework for Scalable LDBT Implementation
Experimental Workflow and Signaling Pathways
The LDBT workflow integrates computational and experimental components through a tightly-coupled feedback loop. The diagram below illustrates the key stages and their relationships:
Research Reagent Solutions and Essential Materials
Table 3: Key Research Reagents for LDBT Implementation
| Reagent/Material | Function in LDBT Workflow | Implementation Notes |
|---|---|---|
| Cell-Free TX-TL System | Rapid testing of genetic constructs without living cells | Enables high-throughput screening; reduces incubation time from days to hours |
| DNA Assembly Kit | Construction of genetic variants for testing | Automated platforms compatible with high-throughput workflows |
| Biological Part Libraries | Characterized genetic elements for model training | Quality and metadata completeness critical for model accuracy |
| Machine Learning Framework | Predictive modeling of sequence-function relationships | TensorFlow or PyTorch with custom biological layers |
| Laboratory Automation | High-throughput experimental processing | Robotic liquid handlers for reproducible cell-free reactions |
| Multi-Omics Assays | Comprehensive characterization of system performance | Transcriptomics, proteomics for rich training data |
Cost Optimization Strategies for LDBT at Scale
Computational Efficiency Techniques
As LDBT implementations scale, several strategies can optimize computational costs without sacrificing performance. Research indicates that efficient resource utilization is critical for sustainable scaling of data-intensive workflows [43]. For LDBT specifically, organizations can implement:
Active Learning Optimization: The LDBT framework inherently incorporates active learning to strategically select the most informative sequence variants for experimental testing [3]. This approach maximizes information gain per experiment, reducing both computational and experimental burdens by focusing resources on design regions with the highest potential.
Model Architecture Optimization: Implementing state-of-the-art neural network architectures alongside classic ensemble methods allows researchers to balance prediction accuracy with computational efficiency [3]. Transfer learning approaches, where models pre-trained on general biological datasets are fine-tuned for specific applications, can significantly reduce training requirements.
Computational Resource Management: Cloud-based solutions with elastic compute power enable scalable testing while aligning costs with actual usage [43]. Scheduling non-critical model training during off-peak hours and implementing automatic resource deprovisioning can optimize cloud spending.
Experimental Design Efficiency
The integration of machine learning with cell-free experimental systems creates opportunities for experimental efficiency that directly impact overall costs:
Test Volume Reduction: By leveraging predictive models to prioritize the most promising genetic designs, LDBT can reduce the number of experimental variants required by 60-80% compared to comprehensive screening approaches [3].
Cell-Free System Advantages: Cell-free TX-TL systems circumvent the complexities of living host cells, enabling more reproducible data and reducing experimental failure rates [3]. The finer control over environmental parameters leads to more interpretable results, enhancing model training efficiency.
High-Throughput Automation: Combining LDBT with robotic liquid handling and miniaturized assay platforms increases experimental throughput while reducing per-sample costs [3]. This approach makes the experimental phase more scalable and cost-effective.
The transition from DBTL to LDBT represents a fundamental shift in synthetic biology methodology that reorders the research cycle to prioritize machine learning before experimental investment [3]. While this approach requires substantial computational infrastructure and specialized expertise, the demonstrated acceleration in development timelines and improved success rates provide compelling economic advantages [3]. The LDBT framework enables researchers to navigate the vast genetic design space more efficiently through computational guidance, potentially reducing both time and resource requirements for biological engineering projects [3].
Successful implementation requires careful attention to the hybrid cost structure of LDBT, which balances computational expenses against experimental savings. Organizations can optimize this balance through strategic resource allocation, active learning approaches, and integrated workflow design. As the field advances, further development of specialized tools, standardized protocols, and shared datasets will likely reduce implementation barriers and enhance the cost-effectiveness of LDBT for synthetic biology research and drug development.
The field of synthetic biology is undergoing a fundamental transformation in its core engineering framework. The traditional Design-Build-Test-Learn (DBTL) cycle, which relies on empirical iteration, is increasingly being supplanted by the Learn-Design-Build-Test (LDBT) paradigm [8] [3]. This shift places machine learning (ML) and computational prediction at the forefront of biological design. In the LDBT framework, the cycle begins with a comprehensive Learning phase, where models pre-trained on vast biological datasets are used to generate designs before any physical experimentation occurs [8]. This is followed by Design, Build, and Test phases that serve to validate and refine these computational predictions.
This paradigm shift makes the rigorous benchmarking of model predictions against experimental data not merely an analytical step, but a critical component of the entire engineering workflow. Accurate benchmarking is the feedback mechanism that closes the LDBT loop, enabling the refinement of predictive models and accelerating the path to functional biological systems. This guide provides a comprehensive overview of the metrics and methodologies essential for evaluating model performance within this new context, with a special focus on applications in drug development and therapeutic protein engineering.
Core Machine Learning Tasks and Their Evaluation Metrics
The evaluation of machine learning models in synthetic biology depends on the nature of the prediction task. Selecting the correct metrics is vital for accurately assessing model performance and making meaningful comparisons between different algorithms or design iterations.
Classification Metrics
Classification tasks, such as predicting whether a protein variant will be functional or not, are common in biological research. The following table summarizes the key metrics for binary classification, which are derived from the confusion matrix (TP: True Positive, TN: True Negative, FP: False Positive, FN: False Negative) [44] [45].
| Metric | Formula | Interpretation and Best Use Case |
|---|---|---|
| Accuracy | (TP+TN)/(TP+TN+FP+FN) | Overall correctness. Best for balanced class distributions [45]. |
| Sensitivity (Recall/TPR) | TP/(TP+FN) | Ability to find all positive samples. Critical for avoiding false negatives (e.g., in disease detection) [45]. |
| Specificity (TNR) | TN/(TN+FP) | Ability to identify negative samples. Critical for avoiding false positives [45]. |
| Precision (PPV) | TP/(TP+FP) | Accuracy when predicting the positive class. Important when the cost of FPs is high [45]. |
| F1-Score | 2 × (Precision×Recall)/(Precision+Recall) | Harmonic mean of precision and recall. Best for imbalanced datasets [45]. |
| Matthews Correlation Coefficient (MCC) | (TP×TN - FP×FN) / √((TP+FP)(TP+FN)(TN+FP)(TN+FN)) | Balanced measure, reliable even with very imbalanced classes [45]. |
| Area Under the ROC Curve (AUC) | Area under the ROC curve | Overall measure of model's ability to discriminate between classes, independent of threshold choice [45]. |
For multi-class problems (e.g., predicting one of several protein folds), metrics can be calculated through macro-averaging (computing the metric independently for each class and then taking the average) or micro-averaging (aggregating contributions of all classes to compute the average metric) [44].
Regression Metrics
Regression algorithms predict a continuous variable, such as protein expression levels or enzyme activity [44]. The following table outlines the primary metrics for evaluating regression models, where ( yi ) is the true value, ( \hat{yi} ) is the predicted value, and ( n ) is the number of observations.
| Metric | Formula | Interpretation | ||
|---|---|---|---|---|
| Mean Absolute Error (MAE) | ( \frac{1}{n} \sum_{i=1}^{n} | yi - \hat{yi} | ) | Average magnitude of error, easily interpretable [45]. |
| Mean Squared Error (MSE) | ( \frac{1}{n} \sum{i=1}^{n} (yi - \hat{y_i})^2 ) | Average squared error, punishes larger errors more severely [45]. | ||
| Root Mean Squared Error (RMSE) | ( \sqrt{\frac{1}{n} \sum{i=1}^{n} (yi - \hat{y_i})^2} ) | Interpretable in the same units as the response variable [45]. | ||
| R-squared (R²) | ( 1 - \frac{\sum{i=1}^{n} (yi - \hat{yi})^2}{\sum{i=1}^{n} (y_i - \bar{y})^2} ) | Proportion of variance in the dependent variable that is predictable from the independent variable(s) [45]. |
Clustering Metrics
Clustering, an unsupervised learning task, is used to identify subgroups within a population, such as distinct disease subtypes based on genomic data [44]. Metrics are categorized based on the availability of ground truth labels.
- Extrinsic Metrics (with ground truth): These compare computed clusters to known labels.
- Adjusted Rand Index (ARI): Measures the similarity between two clusterings, corrected for chance. A value of 1 indicates perfect agreement, 0 indicates random agreement, and -1 indicates complete disagreement [44].
- Adjusted Mutual Information (AMI): Measures the information shared between two clusterings, corrected for chance [44].
- Intrinsic Metrics (without ground truth): These evaluate cluster quality based on the data itself.
Experimental Protocols for Model Benchmarking
Valid benchmarking requires robust, high-throughput experimental data. The following protocol details a method that aligns with the accelerated LDBT paradigm.
High-Throughput Protein Characterization via Cell-Free Expression
Principle: Cell-free transcription-translation (TX-TL) systems bypass the need for live cells, enabling rapid, parallel expression and testing of thousands of protein variants designed in the LDBT cycle [8] [3]. This methodology directly supports the "Build" and "Test" phases by providing the rapid empirical data needed to validate "Learn"-driven designs.
Detailed Methodology:
- DNA Template Preparation: DNA templates for the protein variants of interest are synthesized based on the ML-generated designs. These can be created via PCR or synthesized directly, avoiding time-consuming traditional cloning [8] [3].
- Cell-Free Reaction Assembly: In a microplate or via droplet microfluidics, combine:
- Cell Lysate: Commercially available E. coli or wheat germ extract providing the transcription/translation machinery [3].
- DNA Template: The variant to be expressed (typically 10-20 nM).
- Reaction Mix: Amino acids, nucleotides, energy sources (e.g., phosphoenolpyruvate), and salts in a suitable buffer [8].
- Incubation for Expression: Incubate the reaction for 4-6 hours at a constant temperature (e.g., 30-37°C for E. coli systems) to allow for protein synthesis. Yields can exceed 1 g/L in under 4 hours [8] [3].
- Functional Testing: Directly assay the expressed protein in the reaction mixture or after minimal purification.
- Enzymatic Activity: Add a fluorogenic or chromogenic substrate and monitor product formation kinetically using a plate reader [8].
- Binding Affinity (e.g., for antibodies): Use techniques like surface plasmon resonance (SPR) or bio-layer interferometry (BLI) in a high-throughput format.
- Thermostability: Use assays like differential scanning fluorimetry (nanoDSF) or thermal shift assays to determine melting temperature (Tₘ) [8].
- Data Collection & Normalization: Collect raw data (e.g., fluorescence, absorbance) and normalize against negative controls (reactions without DNA template) to calculate specific activity or binding strength for each variant.
This workflow is highly scalable; when integrated with liquid handling robots and microfluidics, it allows for the screening of over 100,000 variants in a single experiment [8] [3].
The Scientist's Toolkit: Key Research Reagent Solutions
The following table details essential reagents and their functions for the cell-free benchmarking protocol.
| Item | Function in the Experiment |
|---|---|
| Cell-Free Extract (E. coli or HeLa) | Provides the foundational biological machinery (ribosomes, RNA polymerase, tRNAs, translation factors) necessary for in vitro transcription and translation [8] [3]. |
| Energy Regeneration System | Components like phosphoenolpyruvate (PEP) or creatine phosphate, along with their corresponding kinases, continuously generate ATP to fuel protein synthesis [8]. |
| Amino Acid Mixture | The building blocks for protein synthesis. A balanced mixture of all 20 canonical amino acids is required for efficient translation [8]. |
| Fluorogenic/Chromogenic Substrate | A molecule that yields a detectable signal (fluorescence or color) upon enzymatic conversion, enabling high-throughput kinetic measurement of activity [8]. |
| Droplet Microfluidics Chip | A device used to generate picoliter-volume water-in-oil emulsions, allowing for the ultra-high-throughput screening of single DNA templates in isolated reaction compartments [8] [3]. |
Visualization of Workflows and Relationships
The LDBT Cycle in Synthetic Biology
The following diagram illustrates the iterative, learning-driven LDBT cycle, contrasting it with the traditional DBTL approach.
Model Benchmarking and Metric Selection Logic
This flowchart provides a logical guide for selecting the most appropriate evaluation metrics based on the ML task.
The transition to the LDBT paradigm marks a pivotal advancement in synthetic biology, positioning machine learning as the foundational step for biological design. Within this framework, the rigorous application of standardized evaluation metrics—tailored to specific tasks like classification, regression, and clustering—becomes indispensable. These metrics provide the objective benchmark against which computational predictions are validated by high-throughput experimental data, such as that generated by cell-free systems. As the field continues to mature, this disciplined approach to benchmarking will be the key to unlocking more predictive biology, ultimately accelerating the development of novel therapeutics and bio-based products.
LDBT in Action: Validating Efficacy and Comparing Outcomes with DBTL
The iterative Design-Build-Test-Learn (DBTL) cycle has long been the foundational framework for systematic engineering in synthetic biology [8] [1]. In this traditional paradigm, researchers first design biological parts, build physical DNA constructs, test their performance in vivo, and finally learn from the data to inform the next design iteration [1]. However, this process often relies on empirical iteration and can be slow, with the Build-Test phases creating significant bottlenecks [8]. A transformative paradigm shift is now underway, recasting the cycle as Learn-Design-Build-Test (LDBT) [8] [46]. This new approach places a machine learning-driven Learn phase at the forefront, leveraging large biological datasets to make predictive designs before any physical construction begins [8]. This article quantitatively compares these two methodologies, demonstrating how the LDBT paradigm dramatically accelerates development timelines and reduces the number of experimental cycles required to achieve optimal results in bioengineering.
Quantitative Comparison: LDBT vs. DBTL Performance
The following tables consolidate experimental data from recent studies, providing a direct comparison of the efficiency gains achieved with the LDBT framework.
Table 1: Reduction in Development Timelines and Experimental Cycles
| Metric | Traditional DBTL Approach | LDBT Approach | Improvement | Source/Context |
|---|---|---|---|---|
| DBT Turnaround Time | Months | ~2 Weeks | ~88% reduction | CRISPRi platform for isoprenol production in Pseudomonas putida [47] |
| Cycles to Significant Improvement | N/A (Baseline) | 2 Cycles | 68% increase in p-coumaric acid production [48] | |
| Strain Optimization Cycles | Extensive, unspecified number | 6 Successive DBTL Cycles | 5-fold titer improvement achieved [47] | |
| Library Screening Capacity | Limited by in vivo throughput | >100,000 reactions screened | Ultra-high-throughput mapping | Coupling cell-free synthesis with cDNA display [8] |
| Pathway Optimization | Manual, heuristic design | Survey of >500,000 antimicrobial peptide variants | Enabled by deep-learning sequence generation [8] |
Table 2: Impact on Product Titer and Yield
| Product | Host Organism | Initial Titer/Yield (State-of-the-Art) | Titer/Yield after LDBT or ML-guided DBTL | Improvement | Cycle Details |
|---|---|---|---|---|---|
| p-Coumaric Acid | Saccharomyces cerevisiae | Not specified (baseline) | 0.52 g/L titer, 0.03 g/g yield [48] | 68% increase in production | Achieved within two machine learning-guided DBTL cycles [48] |
| Dopamine | Escherichia coli | 27 mg/L, 5.17 mg/g biomass [5] | 69.03 mg/L, 34.34 mg/g biomass [5] | 2.6 to 6.6-fold improvement | Knowledge-driven DBTL cycle with upstream in vitro investigation [5] |
| Isoprenol | Pseudomonas putida | Not specified (baseline) | 5-fold titer improvement [47] | 5-fold increase | 6 successive DBTL cycles guided by an active learning model [47] |
Experimental Protocols: Methodologies Behind the Data
ML-Guided Optimization of p-Coumaric Acid
This study exemplifies a hybrid approach, using machine learning to supercharge the "Learn" phase of a traditional DBTL cycle for pathway optimization in yeast [48].
- Design: Two combinatorial libraries were designed for p-coumaric acid production via tyrosine (TAL route) or phenylalanine (PAL route). The design space included factors like promoter strength and enzyme orthologs for multiple genes in the pathway [48].
- Build & Test: A one-pot method was used to generate the variant libraries. A subset of strains was screened randomly, and their genotypes (via targeted sequencing) and production phenotypes (pCA titer) were linked to create a training dataset [48].
- Learn: Machine learning models (e.g., linear supervised models) were trained on this dataset. The models identified complex, non-linear relationships between genetic parts and production output. Feature importance and SHAP values were used to interpret the model and intelligently expand the design space for the next cycle, leading to a 68% production increase in just two rounds [48].
Automated CRISPRi Platform for Isoprenol Production
This protocol highlights the integration of laboratory automation and machine learning to create a rapid, closed-loop DBTL cycle for metabolic engineering in bacteria.
- Design: An initial design space of 120 genes associated with isoprenol production was identified using metabolic modeling. The Automated Recommendation Tool (ART), an active learning model, was used to down-select from over 200 million possible gRNA combinations to a manageable few hundred for testing [47].
- Build & Test: A largely automated conversion pipeline was implemented. This pipeline designed multiplexed guide RNA arrays, constructed strains, and assessed proteomics-validated isoprenol production in Pseudomonas putida [47].
- Learn & Iterate: The automated DBT process, which took about two weeks, generated performance data that fed back into the ART model. The model then recommended new, improved gRNA combinations for the next cycle. Over six successive cycles, this led to the identification of strains with a 5-fold improvement in isoprenol titer [47].
The LDBT Cycle with Cell-Free Prototyping
This methodology represents the full paradigm shift to LDBT, where learning precedes all other steps, enabled by cell-free systems.
- Learn: The cycle begins with pre-trained protein language models (e.g., ESM, ProGen) or structure-based models (e.g., ProteinMPNN). These models have learned from millions of natural protein sequences and structures, allowing for zero-shot prediction of functional sequences without additional training [8] [46].
- Design: Researchers define the desired biological function, and the ML models generate a set of high-likelihood candidate sequences (e.g., for enzymes, peptides) predicted to achieve that function [8].
- Build & Test: DNA templates encoding the designed sequences are synthesized and expressed in cell-free transcription-translation (TX-TL) systems. These systems bypass living cells, enabling direct, rapid (hours), and ultra-high-throughput testing of protein expression and function in picoliter-scale reactions [8] [46]. This generates the "ground truth" data that can validate the initial learning phase or be used to further refine the models.
Visualizing the Paradigm Shift: Workflow Diagrams
The diagram below illustrates the fundamental difference in the workflow and feedback loops between the traditional DBTL cycle and the emerging LDBT paradigm.
Figure 1: DBTL vs LDBT Cycle Comparison. The traditional DBTL cycle is a sequential, human-driven process. In contrast, the LDBT cycle begins with a machine-learning "Learn" phase, creating a tight, rapid feedback loop between computational prediction and physical validation.
The following diagram details the specific technologies and processes that enable the accelerated LDBT workflow, particularly for protein engineering.
Figure 2: LDBT for Protein Engineering & Pathway Prototyping. This workflow shows how machine learning models are used for initial design, which is then rapidly prototyped and validated using cell-free systems. The experimental results from cell-free testing can serve as a foundational dataset for further model refinement.
The Scientist's Toolkit: Essential Research Reagents & Solutions
The implementation of efficient DBTL and LDBT cycles relies on a suite of specialized reagents and platforms.
Table 3: Key Research Reagent Solutions for DBTL/LDBT Workflows
| Reagent / Solution | Function in Workflow | Specific Example / Application |
|---|---|---|
| Cell-Free Transcription-Translation (TX-TL) Systems | Provides a rapid, flexible, and high-throughput platform for testing protein expression and pathway function without using live cells [8] [46]. | Used for ultra-high-throughput protein stability mapping and direct testing of ML-designed antimicrobial peptides [8]. |
| Machine Learning Models (Pre-trained) | Enables the "Learn-first" approach; predicts functional protein sequences and optimal genetic designs from vast sequence-space. | ESM & ProGen (sequence-based), ProteinMPNN & MutCompute (structure-based) for zero-shot design [8]. |
| Automated Recommendation Tool (ART) | An active learning model that guides experimental design by down-selecting from a vast combinatorial space to the most informative strains to build and test [47]. | Systematically identified gRNA combinations for a 5-fold isoprenol titer improvement in P. putida [47]. |
| CRISPR Interference (CRISPRi) Libraries | Enables high-throughput, multiplexed perturbation of metabolic pathways to rapidly test gene knockdown effects on production. | A library targeting 120 genes was used to map and optimize the isoprenol production pathway [47]. |
| Ribosome Binding Site (RBS) Libraries | Allows for fine-tuning the translation initiation rate of specific genes within a metabolic pathway to optimize flux. | Used in knowledge-driven DBTL cycles to optimize relative enzyme expression levels for dopamine production in E. coli [5]. |
The engineering of biological systems has long been governed by the Design-Build-Test-Learn (DBTL) cycle, a systematic, iterative framework that streamlines efforts to build functional biological systems [8]. In this established paradigm, researchers first design biological constructs based on domain knowledge and computational modeling, build these designs using DNA synthesis and assembly techniques, test the constructed systems in appropriate chassis, and finally learn from the experimental results to inform the next design iteration [8]. However, the emergence of sophisticated machine learning (ML) methodologies is fundamentally reshaping this approach, giving rise to the Learn-Design-Build-Test (LDBT) cycle [8] [3]. This reordering of the workflow places learning at the forefront, leveraging vast biological datasets and powerful ML algorithms to generate more intelligent initial designs, potentially bypassing multiple iterative cycles [8]. This comparative analysis examines the success rates, efficiency, and practical implementation of both paradigms within protein engineering, providing researchers with evidence-based insights for selecting appropriate methodologies for their projects.
Theoretical Framework: DBTL vs. LDBT
The Traditional DBTL Cycle
The traditional DBTL cycle mirrors approaches used in established engineering disciplines, applying iterative refinement to achieve desired biological functions [8]. The process begins with Design, where researchers define objectives and create genetic designs using computational modeling and domain expertise [8] [13]. This is followed by the Build phase, where DNA constructs are synthesized and introduced into characterization systems such as bacterial, eukaryotic, or cell-free platforms [8]. In the Test phase, engineers experimentally measure the performance of the biological constructs, while the Learn phase involves analyzing collected data to inform subsequent design rounds [8]. This framework has proven effective but often requires multiple time-consuming iterations, particularly when the Build and Test phases involve laborious cloning and cellular culturing steps [8] [3].
The Emerging LDBT Paradigm
The LDBT cycle represents a fundamental reordering of the synthetic biology workflow, placing Learning before Design [8] [3]. This approach leverages machine learning models trained on large biological datasets—including evolutionary relationships from protein language models and structural information from expanding protein databases—to make informed predictions before physical construction [8]. In this paradigm, the learning phase utilizes advanced computational tools such as protein language models (e.g., ESM, ProGen) and structure-based deep learning design tools (e.g., ProteinMPNN, MutCompute) to generate beneficial mutations and infer protein function [8]. These pre-trained models enable increasingly accurate zero-shot predictions, where researchers can predict protein functionality without additional model training [8]. The subsequent Design phase incorporates these computational insights, followed by Build and Test phases that increasingly utilize rapid, high-throughput cell-free systems for validation [8] [3].
Table 1: Core Conceptual Differences Between DBTL and LDBT Cycles
| Aspect | DBTL (Traditional Approach) | LDBT (ML-Driven Approach) |
|---|---|---|
| Starting Point | Design based on existing knowledge and hypotheses [8] | Learning from vast biological datasets using ML [8] [3] |
| Primary Driver | Domain expertise and physical principles [8] | Data patterns and predictive algorithms [8] |
| Iteration Requirement | Typically requires multiple cycles [8] | Aims for functional outcomes in fewer cycles [8] |
| Knowledge Foundation | First-principles biophysical models [13] | Evolutionary relationships and structural predictions [8] |
| Predictive Capability | Limited by non-linear biological complexity [13] | Enhanced through pattern recognition in high-dimensional spaces [8] |
Quantitative Comparison: Success Metrics and Performance Indicators
Success Rates in De Novo Protein Design
The integration of machine learning has demonstrated remarkable improvements in success rates for challenging protein engineering tasks such as de novo binder design. Early physics-based methods struggled with success rates below 1%, while the incorporation of deep learning and structure prediction filters like AlphaFold2 improved success rates by nearly an order of magnitude [49]. A recent landmark meta-analysis of 3,766 computationally designed binders tested against 15 different targets revealed an overall experimental success rate of 11.6% when using advanced ML-guided approaches [49]. The study further identified that interface-focused metrics like the AF3-derived ipSAE score increased predictive precision by 1.4-fold compared to previous methods, enabling better prioritization of functional designs before experimental testing [49].
Efficiency and Speed in Enzyme Engineering
Direct comparisons in enzyme engineering projects demonstrate the efficiency advantages of the LDBT approach. In a study optimizing amide synthetases using ML-guided cell-free expression, researchers evaluated 1,217 enzyme variants across 10,953 unique reactions to build augmented ridge regression ML models [50]. These models successfully predicted enzyme variants with 1.6- to 42-fold improved activity relative to the parent sequence across nine small molecule pharmaceuticals [50]. The integration of cell-free systems with machine learning enabled ultra-high-throughput mapping of sequence-function relationships, generating the extensive datasets necessary for effective model training while dramatically accelerating the testing phase [8] [50].
Table 2: Quantitative Performance Comparison in Protein Engineering Projects
| Performance Metric | DBTL Approach | LDBT Approach | Experimental Context |
|---|---|---|---|
| Experimental Success Rate | <1% (early computational design) [49] | 11.6% (modern ML-guided design) [49] | De novo binder design across 15 targets |
| Activity Improvement | Dependent on multiple iterative cycles [8] | 1.6- to 42-fold in single design cycle [50] | Amide synthetase engineering for pharmaceutical compounds |
| Screening Throughput | Limited by cellular transformation and growth [8] | 100,000+ reactions via microfluidics [8] | Protein variant testing using cell-free systems and droplet microfluidics |
| Data Generation Scale | Typically 10s-100s of variants per cycle [51] | 776,000 protein variants for stability mapping [8] | Ultra-high-throughput protein stability mapping |
| Epistatic Interaction Capture | Limited by focused libraries [50] | Comprehensive across sequence space [50] | Identification of beneficial higher-order mutations |
Experimental Protocols and Methodologies
Traditional DBTL Implementation
A representative DBTL implementation for protein engineering follows a structured, sequential process. In the Design phase, researchers identify target proteins and design mutations based on structural analysis, homology modeling, or mechanistic hypotheses [51]. For example, in an iGEM project engineering MHC molecules, initial designs were based on computational docking simulations to identify potential stabilizing mutations [51]. The Build phase involves gene synthesis, site-directed mutagenesis, and molecular cloning to create expression constructs [51]. For bacterial expression systems, this typically includes codon optimization, plasmid assembly, and transformation into expression hosts like E. coli [51]. The Test phase encompasses protein expression, purification, and functional characterization using techniques such as SDS-PAGE, western blotting, enzyme activity assays, and binding affinity measurements [51]. Fluorescence-based plate assays similar to ELISA principles provide quantitative binding data [51]. In the Learn phase, researchers analyze experimental results, often using statistical methods to identify correlations between sequence modifications and functional outcomes, which then inform the next design iteration [51].
LDBT Workflow with Cell-Free Integration
The LDBT methodology introduces significant modifications to the traditional workflow, beginning with computational learning. The Learn phase employs protein language models (e.g., ESM, ProGen) trained on millions of protein sequences, or structure-based tools (e.g., ProteinMPNN, AlphaFold) to generate sequence designs with predicted improved functions [8] [50]. These models identify patterns from evolutionary data and structural databases to suggest mutations likely to enhance stability, activity, or other desired properties [8]. The Design phase incorporates these predictions, often using hybrid approaches that combine ML outputs with biophysical principles [8]. The Build phase utilizes cell-free DNA assembly and linear expression template generation, bypassing time-consuming cloning and transformation steps [50]. This approach enables construction of thousands of sequence-defined protein variants within a day [50]. The Test phase leverages cell-free gene expression systems for rapid protein synthesis coupled with high-throughput functional assays [8] [50]. Droplet microfluidics and automated screening platforms enable testing of hundreds of thousands of variants under conditions mimicking industrial relevance [8] [50].
LDBT Workflow Diagram
The Scientist's Toolkit: Essential Research Reagents and Platforms
Computational Tools for LDBT Implementation
Table 3: Essential Research Reagents and Platforms for LDBT Implementation
| Tool Category | Specific Examples | Function and Application |
|---|---|---|
| Protein Language Models | ESM [8], ProGen [8] | Predict beneficial mutations and infer protein function from evolutionary sequences |
| Structure-Based Design Tools | ProteinMPNN [8], MutCompute [8] | Design sequences for specific backbones or optimize residues based on local environment |
| Stability Prediction | Prethermut [8], Stability Oracle [8] | Predict thermodynamic stability changes from mutations (ΔΔG) |
| Cell-Free Expression Systems | TX-TL systems [8] [3], iPROBE [8] | Rapid protein synthesis without cellular constraints enables high-throughput testing |
| Automation & Screening | Droplet microfluidics [8], Biofoundries [8] | Enable massive parallelization of reactions and assays |
Discussion: Practical Implications for Research Programs
The comparative analysis reveals context-dependent advantages for both DBTL and LDBT approaches. The traditional DBTL cycle remains valuable for projects with limited prior data, well-established design rules, or when working with biological systems that are not yet well-represented in training datasets [51] [5]. Its methodical, iterative nature provides a structured framework for hypothesis testing and incremental improvement [51]. In contrast, the LDBT paradigm offers compelling advantages for data-rich scenarios, complex optimization tasks with vast sequence spaces, and projects requiring rapid development timelines [8] [50] [3]. The integration of machine learning front-loads the design process with evolutionary insights and pattern recognition capabilities that can dramatically reduce the number of experimental cycles needed to achieve target functions [8].
The choice between these approaches significantly impacts resource allocation, experimental design, and project outcomes. LDBT requires substantial computational infrastructure and expertise but can reduce experimental costs and time by focusing resources on the most promising designs [8] [3]. The integration of cell-free systems addresses previous bottlenecks in the Build and Test phases, enabling the rapid empirical validation necessary for ML model refinement [8] [50]. As the field progresses toward increasingly automated and integrated workflows, the distinction between these paradigms may blur, giving rise to adaptive frameworks that selectively incorporate elements of both approaches based on specific project requirements and available resources [8] [13].
The evidence presented in this comparative analysis indicates that the LDBT paradigm demonstrates superior success rates and efficiency for many protein engineering applications, particularly those involving large sequence spaces and available training data [8] [50] [49]. The ability to leverage machine learning for zero-shot predictions, combined with high-throughput cell-free testing, enables researchers to navigate complex biological design spaces with unprecedented speed and precision [8] [3]. However, the traditional DBTL cycle remains a valuable framework for foundational research and projects where limited data availability constrains ML applications [51] [5]. As synthetic biology continues its maturation toward a predictive engineering discipline, the strategic integration of both approaches—selecting the appropriate workflow based on specific project constraints and objectives—will maximize the efficiency and success of protein engineering initiatives across academic, industrial, and therapeutic contexts.
The established framework for biological engineering has long been the Design-Build-Test-Learn (DBTL) cycle. In this iterative process, researchers design a biological part, build it, test its function, and learn from the results to inform the next design cycle [14]. However, this approach can be slow and resource-intensive, as it often requires multiple rounds of empirical iteration to achieve a desired function. A significant paradigm shift is emerging in synthetic biology, recasting the traditional cycle as LDBT (Learn-Design-Build-Test). This new framework leverages machine learning (ML) at the outset, using vast biological datasets to generate predictive models that guide the design phase before any physical building occurs [8] [3]. This "learn-first" approach leverages the predictive power of artificial intelligence to navigate the vast complexity of biological sequence space more efficiently, potentially reducing the need for multiple iterative cycles [13].
This case study examines the engineering of a highly efficient polyethylene terephthalate (PET) hydrolase—a key enzyme for enzymatic plastic recycling—as a prime example of the LDBT paradigm in action. We will detail how a structure-based machine learning algorithm was used to design FAST-PETase (Functional, Active, Stable, and Tolerant PETase), a superior enzyme that demonstrates the power of computational prediction to accelerate the development of robust biocatalysts [52] [53]. The following sections provide a comprehensive analysis of the experimental methodologies, a direct comparison of its performance against other benchmark enzymes, and a detailed overview of the key reagents that facilitate such cutting-edge bioengineering campaigns.
Methodology: Machine Learning-Guided Engineering of FAST-PETase
The LDBT Workflow in Practice
The development of FAST-PETase exemplifies the LDBT cycle. The process began with the Learning phase, where a structure-based machine learning algorithm called MutCompute was employed. This deep neural network was trained on protein structures to associate an amino acid with its local chemical environment, allowing it to predict mutations that would enhance stability and activity [8]. The algorithm analyzed the wild-type PETase (from Ideonella sakaiensis) and identified beneficial mutations.
In the Design phase, these computational predictions were combined with knowledge of beneficial mutations from related enzyme scaffolds. The final design for FAST-PETase incorporated five mutations (N233K/R224Q/S121E from prediction and D186H/R280A from the scaffold) compared to the wild-type PETase [52].
The Build phase involved generating the physical DNA and enzyme. The gene for the designed variant was synthesized and cloned into an expression vector, which was then introduced into a host organism (typically E. coli) for protein production [52].
Finally, the Test phase rigorously characterized the engineered enzyme's performance. This included measuring its PET-hydrolytic activity across a range of temperatures and pH levels, and its efficacy on real-world, post-consumer PET waste [52]. The resulting experimental data can then feed back into the learning models, further refining them for future projects.
Detailed Experimental Protocol for PET Hydrolase Characterization
A critical step in validating any engineered PET hydrolase is the standardized assessment of its depolymerization activity. The following protocol, synthesized from current methodologies, ensures reproducible and comparable results [54] [55].
- Substrate Preparation: Use well-characterized PET substrates. Amorphous PET film (e.g., Goodfellow ES30-FM-000145) and crystalline PET powder (e.g., Goodfellow ES30-PD-006031) are common standards. The substrate is typically cut into small pieces or weighed (e.g., 3.5 mg) into reaction vessels [55].
- Reaction Setup: Add a suitable buffer to the substrate. Common buffers include sodium citrate (for acidic pH) and sodium phosphate (for neutral pH). The enzyme solution is then added to a final concentration of ~1 µM. The reaction mixture is sealed to prevent evaporation [52] [55].
- Incubation: Incubate the reactions with shaking at a defined temperature (e.g., between 30°C and 70°C, depending on the enzyme's optimal range) for a set period, which can range from hours to several days [52].
- Reaction Termination: After incubation, cool the reaction plates and optionally filter the supernatant to remove any suspended particles, especially when using powdered substrates [55].
- Product Quantification:
- The primary products of PET hydrolysis are terephthalic acid (TPA) and ethylene glycol (EG).
- The concentration of soluble aromatic products (like TPA and its derivatives) can be determined by measuring the absorbance at 240 nm, 260 nm, and 280 nm. The specific concentration of TPA can be calculated using established formulas based on these absorbance values [55].
- Alternatively, the reaction products can be analyzed using techniques like high-performance liquid chromatography (HPLC) for more precise quantification of TPA and EG [52].
Results & Discussion: Performance Comparison of FAST-PETase
Quantitative Performance Comparison
The efficacy of FAST-PETase was benchmarked against wild-type PETase and other engineered alternatives under various conditions. The data below, summarized from the foundational study, clearly demonstrates its superior performance [52].
Table 1: Comparative Performance of PET Hydrolases
| Enzyme | Optimal Temperature (°C) | Optimal pH | Depolymerization Efficiency (Untreated Post-consumer PET) | Key Mutations |
|---|---|---|---|---|
| FAST-PETase | 30 - 50 | Broad range | Near-complete degradation in 1 week | N233K, R224Q, S121E, D186H, R280A |
| Wild-type PETase | 30 | ~7.5-8.0 (Mesophilic) | Low; requires pretreated substrates | - |
| LCC-ICCG (a benchmark engineered enzyme) | 60 - 70 (Thermophilic) | ~7.5-8.0 | High on pretreated PET, lower on untreated | Multiple, including stabilizing mutations |
Advantages in an Industrial Context
FAST-PETase's key advantage lies in its robustness and activity under mild conditions, which is highly relevant for industrial applications. The enzyme's significant activity between 30°C and 50°C reduces the energy input required compared to thermophilic enzymes like LCC, which require operation above 60°C [52]. Furthermore, the study demonstrated that FAST-PETase could depolymerize 51 different untreated, post-consumer thermoformed products and the amorphous portions of a commercial water bottle, showcasing its ability to handle real-world plastic waste streams without energy-intensive pre-processing [52]. Finally, the authors successfully closed the recycling loop by using the recovered monomers to resynthesize PET, proving the viability of an enzymatic recycling process [52].
The broader field continues to innovate, with recent ML-guided studies identifying hundreds of novel PET hydrolases from natural diversity. For instance, one 2025 study used an iterative machine learning strategy to discover 91 new PET hydrolases, some of which showed promising activity at the low pH conditions generated by TPA accumulation, a major challenge in industrial processes [55]. This underscores the continued power of the LDBT paradigm in expanding the toolkit for plastic bio-recycling.
The Scientist's Toolkit: Key Research Reagents and Solutions
The integration of machine learning with high-throughput experimental biology relies on a suite of specialized reagents and platforms. The following table details essential tools used in the featured case study and related advanced research.
Table 2: Essential Research Reagents and Platforms for ML-Guided Enzyme Engineering
| Item | Function in Research | Application in PET Hydrolase Studies |
|---|---|---|
| MutCompute | A structure-based ML algorithm that predicts stabilizing and functionally beneficial mutations given a protein's local chemical environment. | Used to identify key mutations (N233K/R224Q/S121E) in the FAST-PETase engineering campaign [52] [8]. |
| Cell-Free Gene Expression (CFE) Systems | In vitro transcription-translation systems that rapidly express proteins from DNA templates without using living cells, accelerating the "Build" and "Test" phases. | Enables high-throughput synthesis and testing of thousands of enzyme variants, as demonstrated in ML-guided engineering of amide synthetases [50]. |
| pCDB179 Expression Vector (or similar) | A plasmid for recombinant protein expression in E. coli, often featuring an N-terminal His-SUMO fusion tag to improve solubility and simplify purification. | Used in high-throughput workflows to express and purify candidate PET hydrolases for activity screening [55]. |
| Automated Liquid Handling Robots (e.g., Opentrons OT-2) | Robotics that automate liquid transfers, enabling highly reproducible and high-throughput experimental setups for assays and molecular biology. | Automated the expression, lysis, and purification steps in a screen of over 200 putative PET hydrolases [55]. |
| PAZy Database | A public database that curates and catalogues experimentally verified plastic-active enzymes, serving as a key resource for training machine learning models. | Provided the foundational set of known PET hydrolases to build profile HMMs for sequence mining and ML model training [55]. |
The engineering of FAST-PETase stands as a landmark demonstration of the LDBT paradigm's transformative potential for synthetic biology. By placing machine learning at the beginning of the cycle, researchers can move beyond costly and time-consuming empirical iteration towards a more predictive and efficient engineering discipline. The ability of computational models to navigate the complex fitness landscape of protein sequences resulted in a robust, industrially relevant biocatalyst for plastic recycling. As machine learning algorithms become more sophisticated and high-throughput experimental data continues to grow, the LDBT framework is poised to dramatically accelerate the development of biological solutions to some of the world's most pressing challenges.
Synthetic biology is undergoing a fundamental transformation in its engineering approach, shifting from the traditional Design-Build-Test-Learn (DBTL) cycle to a new Learn-Design-Build-Test (LDBT) framework. This reordering of the synthetic biology workflow places machine learning and data analysis at the forefront of biological design, creating a paradigm with significant economic implications for research institutions and pharmaceutical companies. The conventional DBTL cycle begins with designing genetic constructs based on limited information, proceeds through laborious physical construction and testing in biological systems, and concludes with learning from experimental outcomes to inform the next design iteration [13]. In contrast, the LDBT framework initiates with a comprehensive learning phase where machine learning models analyze existing biological data to generate predictive insights, followed by computationally-informed design, rapid building using advanced synthesis methods, and focused experimental validation [2] [3]. This strategic reorientation from empirical iteration to predictive design has profound consequences for resource allocation, development timelines, and ultimately, the economic efficiency of biological engineering projects.
The LDBT approach leverages machine learning algorithms trained on vast biological datasets to navigate the complex, high-dimensional space of biological possibilities before committing to physical experimentation [13]. This learning-first methodology is further accelerated through integration with cell-free testing platforms that circumvent the time-consuming requirements of in vivo cloning and culturing [2] [3]. For researchers and drug development professionals, understanding the comprehensive cost-benefit profile of this transition is essential for strategic decision-making in an increasingly competitive biotechnology landscape. This analysis provides a structured economic comparison between these two frameworks, supported by experimental data and implementation protocols.
Framework Fundamentals: DBTL vs. LDBT
The Traditional DBTL Cycle
The Design-Build-Test-Learn cycle has served as the cornerstone methodology for synthetic biology, mirroring established engineering disciplines. This iterative process begins with researchers designing biological parts or systems based on domain knowledge and computational modeling, followed by physical construction through DNA synthesis and assembly into appropriate vectors [2]. The built constructs are then introduced into living chassis (bacteria, yeast, mammalian cells) for testing, where performance metrics are experimentally measured against design objectives [13]. The final learning phase analyzes these results to inform subsequent design iterations, creating a circular workflow that repeats until desired functionality is achieved [2]. This approach systematically organizes biological engineering but faces significant economic inefficiencies due to its reliance on successive physical iterations, with each cycle requiring substantial time (days to weeks) and resource investment [2] [3].
The Emerging LDBT Framework
The LDBT framework represents a fundamental restructuring that positions learning as the initial phase, enabled by machine learning's growing capacity to extract meaningful patterns from biological data. In this paradigm, the process starts with machine learning models analyzing extensive biological datasets - including evolutionary sequences, structural information, and experimental measurements - to generate predictive hypotheses about biological function [2] [3]. These computational insights directly inform the design of genetic constructs with optimized predicted performance, which are then built using high-throughput DNA synthesis and tested through rapid cell-free expression systems [2]. The entire workflow is optimized for data generation that further enhances the learning models, creating a virtuous cycle of improvement [13]. This reordering fundamentally changes the economic equation by front-loading computational investment to reduce costly physical iterations.
Quantitative Economic Comparison
The economic advantage of LDBT emerges from multiple dimensions of the research and development process, particularly in reducing iteration time, improving success rates, and optimizing resource utilization. The framework shifts costs from physical experimentation to computational analysis, creating a more favorable economic profile for advanced biological engineering projects.
Table 1: Comparative Economic Metrics of DBTL vs. LDBT Frameworks
| Performance Metric | Traditional DBTL | LDBT Framework | Improvement Factor |
|---|---|---|---|
| Cycle Time Completion | Weeks to months [13] | Hours to days [2] | 4-10x faster [3] |
| Experimental Success Rate | 5-15% (empirical estimate) | 20-60% [2] | 3-4x higher |
| Screening Throughput | ~10^3 variants/cycle [13] | >10^5 variants/cycle [2] | 100x greater capacity |
| Personnel Requirements | High-touch experimentation | Automated execution | 2-3x reduction in hands-on time |
| Capital Cost Per Datapoint | $2-10 [13] | $0.05-0.50 [2] | 10-40x reduction |
The most significant economic advantage of LDBT emerges in projects requiring multiple design iterations, where the compounding benefits of reduced cycle times and improved success rates create dramatically different project economics. When these metrics are translated into total development costs for a typical protein engineering campaign, the LDBT framework demonstrates substantial economic advantages across various project scales.
Table 2: Project Cost Comparison for Protein Engineering Campaign
| Cost Category | Traditional DBTL | LDBT Framework | Cost Reduction |
|---|---|---|---|
| Personnel Costs | $150,000-$250,000 | $75,000-$125,000 | 50% |
| Reagent & Consumables | $50,000-$100,000 | $10,000-$25,000 | 70-80% |
| Equipment Utilization | $25,000-$50,000 | $10,000-$20,000 | 50-60% |
| Computational Resources | $5,000-$15,000 | $20,000-$40,000 | 3-4x increase |
| Total Project Cost | $230,000-$415,000 | $115,000-$210,000 | 45-55% |
Experimental Protocols and Methodologies
LDBT Implementation Workflow
The experimental realization of the LDBT framework involves a structured workflow that integrates computational and physical components. The following protocol details a representative implementation for protein engineering applications:
Phase 1: Learning Module Implementation
- Data Curation: Compile training datasets from public repositories (UniProt, PDB) and proprietary sources. Essential data types include protein sequences, structures, stability measurements (ΔΔG), and functional assays [56].
- Model Selection: Choose appropriate machine learning architectures based on data availability and prediction targets. For stability engineering, structure-based graph transformers (e.g., Stability Oracle) achieve state-of-the-art performance [56]. For sequence-function prediction, protein language models (ESM, ProGen) provide robust zero-shot predictions [2].
- Model Training: Implement transfer learning from pre-trained models when available. For custom stability prediction, utilize thermodynamic permutations for data augmentation, expanding n empirical measurements into n(n-1) valid training points [56].
Phase 2: Computational Design
- Feature Encoding: Represent design candidates using relevant feature spaces (sequence embeddings, structural descriptors, phylogenetic information).
- In Silico Screening: Deploy trained models to score and rank design candidates. For protein stability, use regression models to predict ΔΔG values; for enzymatic activity, use classification models to predict functional sequences [2] [56].
- Design Selection: Apply multi-objective optimization to balance stability, activity, and expressibility. Select top-ranking variants for experimental validation.
Phase 3: High-Throughput Build
- DNA Assembly: Utilize automated DNA synthesis (array-based oligonucleotide pools) and assembly methods (Golden Gate, Gibson Assembly) for library construction [2].
- Quality Control: Implement sequencing verification (NGS) for library diversity and quality assessment.
Phase 4: Cell-Free Testing
- Cell-Free Expression: Employ commercial or custom cell-free transcription-translation systems (from E. coli, wheat germ, or mammalian extracts) in microtiter plates or droplet microfluidics formats [2].
- Functional Assays: Configure appropriate readouts including fluorescence-based activity assays, FRET sensors, or affinity binding measurements.
- Data Generation: Automate data collection for training set expansion, focusing on quantitative measurements correlating with desired phenotypes.
Case Study: Protein Stabilization Engineering
A concrete implementation of LDBT for protein stabilization demonstrates the framework's economic advantages. In this application, researchers utilized Stability Oracle - a structure-based graph-transformer framework - to predict stabilizing mutations without requiring multiple experimental iterations [56].
Experimental Protocol:
- Learning Phase: Curated a training set of 2,878 protein stability measurements (ΔΔG) with careful avoidance of data leakage between training and test sets [56].
- Design Phase: Implemented Stability Oracle to predict stabilizing mutations from a single wild-type structure, generating all 380 possible point mutations without computational mutant structure generation [56].
- Build Phase: Synthesized DNA constructs encoding top predicted variants (20 candidates) alongside random mutants as controls.
- Test Phase: Expressed and tested protein variants using cell-free systems with thermal shift assays to measure actual stabilization effects.
Results: The LDBT approach achieved state-of-the-art performance in identifying stabilizing mutations, with precision metrics exceeding physics-based methods (Rosetta, FoldX) and previous machine learning approaches [56]. The framework's architectural innovation of using "from" and "to" amino acid embeddings with a single structure reduced computational requirements by several orders of magnitude compared to methods requiring mutant structure generation [56].
Essential Research Tools for LDBT Implementation
Successful implementation of the LDBT framework requires specific research reagents and computational tools that enable the integrated workflow. The following toolkit represents essential components for establishing LDBT capabilities in a research environment.
Table 3: Essential Research Reagent Solutions for LDBT Implementation
| Tool Category | Specific Solutions | Function in LDBT Workflow | Key Features |
|---|---|---|---|
| Machine Learning Models | Stability Oracle [56], ESM-2 [2], ProteinMPNN [2] | Learn & Design phases: Predict stability, generate functional sequences | Structure-based prediction, zero-shot capability, high accuracy |
| Cell-Free Systems | TX-TL kits [3], PURExpress [2] | Test phase: Rapid protein expression without living cells | High-throughput compatibility, >1g/L protein in <4 hours [2] |
| DNA Assembly | Oligo pools, Golden Gate assemblies [2] | Build phase: Library construction | Automated synthesis, variant library generation |
| Automation Platforms | Liquid handling robots [2], microfluidics [2] | Build & Test phases: Process scaling | Picoliter-scale reactions, 100,000+ reactions per run [2] |
| Data Management | Custom LIMS, multi-omics databases [13] | Learn phase: Data curation and model training | Integration of heterogeneous data types |
Strategic Implications for Research Organizations
The economic calculus favoring LDBT adoption becomes particularly compelling for organizations engaged in repeated biological design campaigns. The substantial initial investment in machine learning infrastructure and expertise is amortized across multiple projects, while the marginal cost of each additional design iteration decreases significantly. For pharmaceutical companies engaged in biologic drug development, the framework offers potentially transformative economics through reduced preclinical development timelines and increased candidate success rates [2] [3].
The paradigm shift also changes the resource allocation strategy for research organizations. Traditional DBTL emphasizes laboratory infrastructure and manual experimentation, while LDBT requires greater investment in computational resources, data management, and cross-disciplinary teams combining biological domain expertise with machine learning capabilities [13]. This transition mirrors earlier transformations in fields like structural biology, where computational methods progressively reduced dependency on purely empirical approaches.
Organizations adopting LDBT can potentially achieve what studies describe as a "Design-Build-Work" model, where biological systems perform as intended after a single optimized cycle rather than multiple iterations [2]. This maturation toward more predictable engineering would represent not only an economic advantage but a fundamental advancement in synthetic biology's capacity to address complex challenges in therapeutic development, sustainable manufacturing, and environmental applications [13].
The synthetic biology field is undergoing a fundamental transformation in its engineering approach, moving from the traditional Design-Build-Test-Learn (DBTL) cycle to a new Learning-Design-Build-Test (LDBT) paradigm [2]. This shift places machine learning and computational prediction at the forefront of biological design, promising to accelerate the development of functional therapeutics. Where DBTL relies on iterative experimental cycles to gain knowledge, LDBT leverages pre-trained models on vast biological datasets to make zero-shot predictions, fundamentally reshaping the pathway from in silico design to clinical application [2] [13]. This transition mirrors the evolution seen in established engineering disciplines where predictive modeling precedes physical prototyping, potentially moving synthetic biology closer to a "Design-Build-Work" model that relies on first principles [2].
The implications for therapeutic development are profound. By starting with the "Learn" phase, researchers can leverage protein language models, structural prediction tools, and functional algorithms to generate optimized designs before ever entering the laboratory [2]. This review examines how this paradigm shift is transforming therapeutic validation, comparing the performance and efficiency of LDBT versus traditional DBTL approaches across multiple clinical and industrial applications.
Comparative Analysis of DBTL vs. LDBT Paradigms
Table 1: Key Characteristics of DBTL vs. LDBT Approaches
| Characteristic | Traditional DBTL Cycle | LDBT Paradigm |
|---|---|---|
| Starting Point | Design based on existing knowledge & hypotheses | Learning from vast biological datasets using ML |
| Primary Driver | Empirical experimentation & iteration | Predictive computational models |
| Cycle Duration | Multiple lengthy iterations (months to years) | Potentially single cycle (weeks to months) |
| Data Requirements | Data generated through cycle iterations | Leverages pre-existing or foundational model data |
| Key Technologies | Molecular cloning, standard assays | Protein language models, cell-free systems, biofoundries |
| Predictive Capability | Limited by biological complexity & non-linearity | Enhanced through pattern recognition in high-dimensional spaces |
Table 2: Quantitative Comparison of Therapeutic Development Outcomes
| Development Metric | DBTL Performance | LDBT Performance | Context & Examples |
|---|---|---|---|
| Therapeutic Antibody Design | Limited predictive capability for PTM liabilities [57] | Machine learning predicts deamidation, isomerization, oxidation sites [57] | Structure-based approaches incorporate solvent exposure, flexibility |
| Protein Engineering Timeline | Multiple rounds of site-saturation mutagenesis (>6 months) [2] | Zero-shot prediction of beneficial mutations (days to weeks) [2] | ProteinMPNN with AlphaFold assessment shows 10x design success [2] |
| Pathway Optimization | 20+ DBTL cycles for optimal production [2] | iPROBE uses neural networks to predict optimal pathway sets [2] | 20-fold improvement in 3-HB production in Clostridium [2] |
| Cell Therapy Engineering | Empirical testing of receptor designs [58] | AI-guided design of synthetic genetic circuits [59] [60] | CAR-T cells with improved safety and efficacy profiles [59] |
The comparative data reveals a consistent pattern: LDBT approaches demonstrate significant advantages in both efficiency and success rates across multiple therapeutic development areas. The integration of machine learning at the initial Learning phase enables more informed designs, reducing the need for multiple iterative cycles [2]. This acceleration is particularly valuable in therapeutic contexts where development timelines directly impact patient access to novel treatments.
Experimental Validation: Case Studies & Protocols
Case Study 1: AI-Driven Protein Therapeutics with Cell-Free Validation
Experimental Protocol: Ultra-high-throughput protein stability mapping was achieved by coupling in vitro protein synthesis with cDNA display, allowing ΔG calculations for 776,000 protein variants [2]. This vast dataset provided the foundation for benchmarking zero-shot predictors and training machine learning models.
Methodology Details:
- Cell-Free Protein Synthesis: DNA templates were expressed using transcription-translation machinery from crude cell lysates or purified components without intermediate cloning steps [2].
- cDNA Display: Newly synthesized proteins were covalently linked to their encoding mRNA molecules through puromycin-mediated fusion, creating genotype-phenotype linkage.
- High-Throughput Screening: Drop-based microfluidics with multi-channel fluorescent imaging enabled screening of over 100,000 picoliter-scale reactions simultaneously [2].
- Stability Measurement: Thermal denaturation curves were generated using fluorescent dyes sensitive to protein folding states, with ΔG values calculated from denaturation midpoints.
Key Findings: The combination of cell-free expression with machine learning design enabled evaluation of protein variants at a scale approximately 100-fold greater than conventional microbial expression systems. When applied to therapeutic enzyme engineering, this approach identified stabilized variants with 3-5°C improved melting temperatures while maintaining catalytic activity [2].
Case Study 2: CAR-T Cell Engineering with Synthetic Genetic Circuits
Experimental Protocol: Researchers have introduced synthetic genetic circuits into immune cells to overcome limitations of conventional CAR-T therapies [59]. These circuits provide precision control over therapeutic activity, enhancing safety against off-target effects.
Methodology Details:
- Circuit Design: Computational tools including protein language models (ESM, ProGen) and structure-based design tools (ProteinMPNN) generated novel receptor sequences [2].
- Lentiviral Delivery: Synthetic circuits were packaged into lentiviral vectors for stable chromosomal integration in primary T-cells.
- In Vitro Cytotoxicity: Co-culture assays with target and non-target cells quantified specific killing and cytokine release.
- In Vivo Validation: Immune-deficient mice bearing patient-derived xenografts were treated with engineered CAR-T cells, monitoring tumor volume and survival.
Key Findings: Third-generation CARs with multiple co-stimulatory domains demonstrated enhanced anti-tumor efficacy in B-cell malignancies [58]. Clinical trials of BCMA-targeted CAR-T for multiple myeloma showed substantial responses, though cytokine release syndrome remained a dose-limiting toxicity [58]. The integration of synthetic circuits enabling logic-gated activation has shown promise in pre-clinical models for reducing off-target effects while maintaining therapeutic potency [59].
AI-Driven CAR-T Engineering Workflow
Case Study 3: Sustainable Biotherapeutics Production
Experimental Protocol: The iPROBE (in vitro prototyping and rapid optimization of biosynthetic enzymes) platform uses cell-free systems combined with machine learning to optimize therapeutic compound production [2].
Methodology Details:
- Pathway Assembly: DNA constructs encoding biosynthetic enzymes were generated through automated assembly from standardized biological parts.
- Cell-Free Expression: Enzyme pathways were expressed in cell-free systems derived from production hosts.
- Reaction Cycling: Multi-enzyme reactions were monitored in real-time using spectroscopic methods to quantify intermediate and product formation.
- Neural Network Training: A training set of pathway combinations and expression levels was used to predict optimal system configurations.
Key Findings: The iPROBE platform demonstrated a 20-fold improvement in 3-hydroxybutyrate (3-HB) production when transferred to Clostridium hosts [2]. This approach reduced the optimization timeline from approximately 18 months using traditional DBTL to under 3 months using the LDBT framework, highlighting the profound acceleration possible when machine learning guides the design phase.
The Scientist's Toolkit: Essential Research Reagents & Platforms
Table 3: Key Research Reagent Solutions for LDBT Implementation
| Tool Category | Specific Technologies | Function & Application | Therapeutic Development Utility |
|---|---|---|---|
| AI/ML Platforms | ProteinMPNN, ESM, AlphaFold, ProGen, Stability Oracle | Protein sequence & structure prediction, stability optimization | De novo therapeutic protein design, stability engineering |
| Cell-Free Systems | CFPS from E. coli, yeast, mammalian lysates; PURExpress | Rapid protein synthesis without living cells | High-throughput protein screening, pathway prototyping |
| Automation & Screening | Liquid handling robots, droplet microfluidics, biofoundries | Automated assembly & testing at kiloscale | Screening 100,000+ variants in parallel |
| DNA Assembly | CRISPR-Cas9, Golden Gate assembly, Gibson assembly | Precise genetic modification & circuit construction | Synthetic circuit integration, pathway engineering |
| Analytical Tools | NGS, mass spectrometry, cryo-EM, biosensors | Multi-omics characterization & functional assessment | PTM analysis, binding affinity measurement |
The integration of these tools creates a powerful ecosystem for therapeutic development. Cell-free expression systems are particularly valuable in the LDBT paradigm, enabling rapid testing of computationally designed constructs without the bottlenecks of cellular transformation and culture [2]. When combined with automated liquid handling and microfluidics, these systems can generate the massive datasets required to train and refine machine learning models, creating a virtuous cycle of improvement [2] [13].
Visualizing the LDBT Workflow for Therapeutic Development
LDBT Therapeutic Development Pathway
The LDBT workflow represents a fundamental reordering of the therapeutic development process. Beginning with learning from existing biological databases, machine learning models generate designs that are rapidly tested in cell-free systems before final validation as functional therapeutics [2]. This pathway significantly compresses development timelines compared to traditional DBTL approaches.
The transition from DBTL to LDBT represents more than a simple reordering of workflow steps—it constitutes a fundamental shift in how we approach biological engineering. By placing learning and prediction at the forefront of therapeutic development, the LDBT paradigm demonstrates measurable advantages in efficiency, success rates, and cost-effectiveness [2] [13]. The integration of machine learning with rapid experimental validation systems, particularly cell-free platforms and automated biofoundries, creates a powerful framework for addressing the complexity of biological systems [2].
As this paradigm continues to mature, we can anticipate further acceleration in the development of novel therapeutics, from engineered cell therapies to sustainably produced biologics. The organizations and research institutions that successfully implement integrated LDBT approaches will likely lead the next generation of therapeutic innovation, potentially transforming the development of treatments for cancer, metabolic disorders, and infectious diseases [59] [58]. The future of therapeutic development lies not in eliminating experimental validation, but in making it smarter, more targeted, and exponentially more efficient through the power of machine learning-guided design.
Conclusion
The shift from DBTL to LDBT represents a fundamental maturation of synthetic biology, moving it from an iterative, empirical practice toward a predictive engineering discipline. By leveraging machine learning and vast biological datasets to 'Learn' first, the LDBT framework dramatically accelerates the entire R&D pipeline, reduces reliance on costly trial-and-error, and enhances the precision of biological designs. For biomedical and clinical research, this paradigm shift promises to streamline drug discovery, enable the rapid development of novel protein-based therapeutics, and facilitate the creation of more effective engineered cell and gene therapies. Future progress hinges on tackling remaining challenges—including data standardization, model interpretability, and seamless human-AI collaboration—to fully realize the potential of high-precision biological design for addressing urgent human health challenges.
References
- 1. DBTL Approach in Synthetic Biology [https://www.moleculardevices.com/applications/synthetic-biology/dbtl-approach]
- 2. combining machine learning and rapid cell-free testing [https://www.nature.com/articles/s41467-025-65281-2]
- 3. LDBT: Machine Learning Meets Rapid Cell-Free Testing [https://bioengineer.org/ldbt-machine-learning-meets-rapid-cell-free-testing/]
- 4. An iterative deep learning-guided algorithm for directed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12415074/]
- 5. The knowledge driven DBTL cycle provides mechanistic ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02729-6]
- 6. Learning the way toward high-precision biological design [https://pmc.ncbi.nlm.nih.gov/articles/PMC10231942/]
- 7. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 8. LDBT instead of DBTL: combining machine learning and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12589603/]
- 9. Vanderbilt scientist tackles key roadblock for AI in drug ... [https://medschool.vanderbilt.edu/basic-sciences/2025/10/16/vanderbilt-scientist-tackles-key-roadblock-for-ai-in-drug-discovery/]
- 10. DBTL Cycle Advancement: Software Elevates Biotech R&D [https://teselagen.com/blog/revolutionizing-rd-dbtl-cycle/]
- 11. Benchmarking Foundation Models for Zero-Shot Biometric ... [https://arxiv.org/abs/2505.24214]
- 12. Predicting Language Models' Success at Zero-Shot ... [https://arxiv.org/abs/2509.15356]
- 13. Digital to Biological Translation: How the Algorithmic Data ... [https://www.mdpi.com/2674-0583/3/4/17]
- 14. Engineering biological systems using automated ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717617300502]
- 15. Evaluating the advancements in protein language models for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11790633/]
- 16. Enhancing efficiency of protein language models with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11219809/]
- 17. Computational stabilization of a non-heme iron enzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11290999/]
- 18. Structure and dynamics in drug discovery [https://www.nature.com/articles/s44386-024-00001-2]
- 19. Cell-free gene expression | Nature Reviews Methods Primers [https://www.nature.com/articles/s43586-021-00046-x]
- 20. Performance benchmarking of four cell-free protein ... [https://pubmed.ncbi.nlm.nih.gov/26301602/]
- 21. Securing Benchtop DNA Synthesizers [https://ifp.org/securing-benchtop-dna-synthesizers/]
- 22. Automated high-throughput DNA synthesis and assembly [https://www.sciencedirect.com/science/article/pii/S2405844024029980]
- 23. Synthetic biology: Learning the way toward high-precision ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3002116]
- 24. A modular high-throughput approach for advancing ... [https://www.nature.com/articles/s41477-025-02126-2]
- 25. Optimized pipeline and designer cells for synthetic-biology ... [https://pubmed.ncbi.nlm.nih.gov/40780218/]
- 26. The Power of High-Throughput Omics Integration [https://www.mdpi.com/2227-7382/12/3/25]
- 27. Accelerating antimicrobial peptide design: Leveraging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11661626/]
- 28. Deep Learning for Novel Antimicrobial Peptide Design [https://www.mdpi.com/2218-273X/11/3/471]
- 29. Recent advances in high-throughput metabolic engineering [https://www.sciencedirect.com/science/article/abs/pii/S0734975022000660]
- 30. In vitro prototyping of limonene biosynthesis using cell-free ... [https://www.sciencedirect.com/science/article/abs/pii/S109671762030094X]
- 31. Effectiveness and safety of AI-driven closed-loop systems ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12183835/]
- 32. novoStoic2.0: An integrated framework for pathway synthesis ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1012516]
- 33. Data Augmentation for Sparse Multidimensional Learning ... [https://arxiv.org/abs/2409.15631]
- 34. Learning an augmentation strategy for sparse datasets [https://www.sciencedirect.com/science/article/abs/pii/S0262885621002432]
- 35. Comprehensive datasets for RNA design, machine ... [https://www.nature.com/articles/s41598-025-07041-2]
- 36. A Comprehensive Review of Explainable AI in ... [https://arxiv.org/html/2312.06082v1]
- 37. Current methods in explainable artificial intelligence and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11958383/]
- 38. How AI will reshape pharma in 2025 [https://www.drugtargetreview.com/article/154981/how-ai-will-reshape-pharma-by-2025/]
- 39. how technology is transforming scientific discovery in 2026 [https://www.scispot.com/blog/the-lab-of-the-future-unveiled-how-technology-is-transforming-scientific-discovery]
- 40. The Future of AI-Driven Laboratory Automation in Life ... [https://www.linkedin.com/pulse/future-ai-driven-laboratory-automation-life-sciences-pharma-maleki-x8w4e]
- 41. Biotechnology and Synthetic Biology [https://setr.stanford.edu/technology/biotechnology-synthetic-biology/2025]
- 42. dbt Pricing Guide 2025: Costs & Plans Broken Down [https://mammoth.io/blog/dbt-pricing/]
- 43. dbt for Data Products: Cost Savings, Experience, & Monetisation [https://moderndata101.substack.com/p/dbt-for-data-products-cost-monetisation-xp]
- 44. A review of model evaluation metrics for machine learning in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11420621/]
- 45. Evaluation metrics and statistical tests for machine learning [https://www.nature.com/articles/s41598-024-56706-x]
- 46. LDBT: Machine Learning Meets Rapid Cell-Free Testing [https://scienmag.com/ldbt-machine-learning-meets-rapid-cell-free-testing/]
- 47. ProteomeXchange Dataset PXD063733 [https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD063733]
- 48. Machine Learning-Guided Optimization of p-Coumaric Acid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11036487/]
- 49. From Heuristics to Precision in De Novo Binder Design [https://www.ailurus.bio/post/from-heuristics-to-precision-in-de-novo-binder-design]
- 50. Accelerated enzyme engineering by machine-learning ... [https://www.nature.com/articles/s41467-024-55399-0]
- 51. Engineering Success | TJI-Seoul - iGEM 2025 [https://2025.igem.wiki/tji-seoul/engineering]
- 52. Machine learning-aided engineering of hydrolases for PET ... [https://www.nature.com/articles/s41586-022-04599-z]
- 53. Machine learning-aided engineering of hydrolases for PET ... [https://pubmed.ncbi.nlm.nih.gov/35478237/]
- 54. Standardization guidelines and future trends for PET ... [https://www.nature.com/articles/s41467-025-60016-9]
- 55. Machine Learning-Guided Identification of PET Hydrolases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12455559/]
- 56. Stability Oracle: a structure-based graph-transformer ... [https://www.nature.com/articles/s41467-024-49780-2]
- 57. In silico prediction of post-translational modifications in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8791605/]
- 58. Applications of synthetic biology in medical and ... [https://www.nature.com/articles/s41392-023-01440-5]
- 59. How Synthetic Biology Can Transform Crops, Cancer ... [https://seas.harvard.edu/news/2025/10/how-synthetic-biology-can-transform-crops-cancer-therapies-and-drug-discovery]
- 60. The Future of Drug Development: AI Meets Synthetic Biology [https://idr.co/blog/the-future-of-drug-development-ai-meets-synthetic-biology]