LDBT vs. DBTL: How Machine Learning and Cell-Free Systems Are Reshaping Synthetic Biology
This article provides a comparative analysis for researchers and drug development professionals on the evolving engineering cycles in synthetic biology.
LDBT vs. DBTL: How Machine Learning and Cell-Free Systems Are Reshaping Synthetic Biology
Abstract
This article provides a comparative analysis for researchers and drug development professionals on the evolving engineering cycles in synthetic biology. It explores the foundational principles of the traditional Design-Build-Test-Learn (DBTL) cycle and the emerging, data-driven Learn-Design-Build-Test (LDBT) paradigm. The scope covers the methodological shift driven by machine learning and rapid cell-free testing, addresses practical challenges and optimization strategies, and offers a critical validation of both approaches through performance metrics and application case studies, ultimately outlining the future implications for biomedical research and therapeutic development.
Core Concepts: Deconstructing the DBTL Cycle and the New LDBT Paradigm
The Design-Build-Test-Learn (DBTL) cycle represents a foundational framework in synthetic biology and engineering biology, applying rigorous engineering principles to the design and assembly of biological components [1] [2]. This iterative process provides a systematic methodology for developing biological systems with predicted functions, enabling researchers to engineer organisms for specific applications such as producing biofuels, pharmaceuticals, and other valuable compounds [1]. A fundamental challenge in biological engineering lies in the inherent complexity of living systems, where even rationally designed biological components can behave unpredictably when introduced into cellular environments [1] [2]. Unlike classical engineering disciplines that utilize well-characterized, man-made building blocks, synthetic biology often relies on partially characterized biological parts implemented within dynamic living systems that remain poorly understood [2].
The DBTL framework addresses this challenge through continuous iteration, where each cycle generates new data and insights to inform subsequent designs [3]. This approach has become synonymous with advanced synthetic biology workflows, particularly with the rise of automated biofoundries that streamline each phase of the cycle [4]. The structured nature of DBTL allows researchers to navigate the vast design space of biological systems methodically, gradually converging on optimal solutions through empirical testing and data-driven learning [3]. As the field evolves, new variations such as the LDBT (Learn-Design-Build-Test) cycle are emerging, proposing a reordering of the traditional sequence to leverage machine learning predictions before experimental building and testing [5] [6]. Nevertheless, the established DBTL cycle remains the predominant framework for engineering biological systems across academic and industrial contexts.
The Four Phases of the DBTL Cycle
Design Phase
The Design phase initiates the DBTL cycle by defining objectives for the desired biological function and specifying the genetic components needed to achieve it [5]. This stage encompasses both biological design and operational design [2]. Biological design involves specifying desired cellular functions, such as producing a target compound or generating detectable signals in response to analytes [2]. Operational design focuses on creating experimental procedures and protocols that will efficiently generate the required data [2].
During Design, researchers identify appropriate biological parts—including enzymes, reporters, and regulatory sequences—and determine how to assemble them to implement the desired function [2]. This process draws upon domain knowledge, expertise, and computational modeling approaches [5]. With the growing universe of characterized biological parts, standardized registries that catalog these components under various biological contexts become increasingly valuable [2]. The end product of the Design phase is one or more DNA sequences comprising multiple genetic parts that are predicted to generate the target functions in a specific biological context [2].
Table 1: Key Activities and Outputs in the Design Phase
| Activity Category | Specific Activities | Primary Outputs |
|---|---|---|
| Biological Design | Define target functions; Identify biological parts; Computational modeling | Functional specifications; DNA sequence designs |
| Operational Design | Design experimental protocols; Define performance specifications; Plan data capture | Experimental plans; Measurement protocols |
| Computational Support | Design-of-experiment (DoE) approaches; Optimization algorithms; DNA assembly planning | Optimized design libraries; Assembly strategies |
Build Phase
The Build phase translates designed DNA sequences into physical biological constructs [2]. This process primarily consists of DNA assembly, incorporation of the assembled DNA into a host organism, and verification of the correctly assembled sequence [2]. DNA assembly uses molecular biology techniques—often enhanced by robotic automation—to combine multiple DNA fragments according to the specifications from the Design phase [1] [2]. Complex constructs frequently require multiple hierarchical assembly rounds, where initial rounds assemble individual transcriptional units or large genes, and subsequent rounds combine these into complete pathways or circuits [2].
A key innovation in modern Build processes is the emphasis on modular design of DNA parts, which enables assembly of greater variety by interchanging individual components [1]. Automation has dramatically reduced the time, labor, and cost of generating multiple constructs, allowing for increased throughput with shortened development cycles [1]. After assembly, constructs are typically cloned into expression vectors and verified using colony qPCR, Next-Generation Sequencing (NGS), or other analytical methods [1]. The final output is a physical DNA molecule or library of DNA molecules comprising the specified sequence(s), ready for functional testing [2].
Test Phase
In the Test phase, researchers experimentally assess whether the built biological constructs perform their intended functions [5]. This involves introducing the constructs into characterization systems—which may include in vivo chassis like bacteria, yeast, or mammalian cells, or in vitro cell-free systems—and measuring their performance against objectives defined during the Design phase [5]. For metabolic engineering applications, testing typically involves growing engineered organisms and assaying for desired functions, such as quantifying product formation or measuring metabolic activity [2].
Comprehensive testing may require sophisticated analytical techniques including proteomics, liquid chromatography-mass spectrometry, gas chromatography-mass spectrometry, and next-generation DNA/RNA sequencing [2]. The Test phase generates critical performance data—such as product titer, yield, rate, enzyme activities, and dynamic response ranges—that enable assessment of the current design's efficacy [2]. Advances in high-throughput screening methodologies, including liquid handling robots and microfluidics, have significantly accelerated this phase by enabling parallel testing of thousands of variants [1] [5]. Cell-free transcription-translation systems have emerged as particularly valuable testing platforms because they circumvent complexities of living cells, allowing rapid assessment of genetic circuit performance within hours rather than days [5] [6].
Learn Phase
The Learn phase closes the DBTL cycle by analyzing data collected during testing to extract actionable insights for the next iteration [5]. This stage involves comparing experimental results with design objectives to identify successful elements and limitations of the current design [1] [5]. Learning mechanisms range from traditional statistical evaluations to advanced machine learning techniques that identify complex patterns within high-dimensional data [7] [3].
In the Learn phase, researchers develop or refine mathematical models—both statistical and mechanistic—of the engineered biological system [2]. The integration of multi-omics data with metabolic models, for instance, has proven valuable for identifying genetic interventions that improve titer, rate, and yield of engineered pathways [2]. These insights directly inform the Design phase of the subsequent DBTL cycle, creating a continuous improvement loop [1]. The learning generated also contributes to broader biological knowledge, helping to address fundamental challenges in predicting how foreign DNA will affect cellular function [1].
DBTL in Action: Experimental Protocol for Metabolic Engineering
To illustrate the practical implementation of a DBTL cycle, consider the optimization of dopamine production in Escherichia coli as documented in a 2025 study [7]. This example demonstrates a knowledge-driven DBTL approach that incorporates upstream in vitro investigation before full cycling.
Design and Build Protocol
The dopamine pathway was designed using two key enzymes: 4-hydroxyphenylacetate 3-monooxygenase (HpaBC) from native E. coli metabolism to convert L-tyrosine to L-DOPA, and L-DOPA decarboxylase (Ddc) from Pseudomonas putida to catalyze dopamine formation [7]. The host strain (E. coli FUS4.T2) was engineered for high L-tyrosine production through genomic modifications, including depletion of the transcriptional dual regulator TyrR and mutation of the feedback inhibition in chorismate mutase/prephenate dehydrogenase (TyrA) [7].
DNA Assembly and Verification:
- Genes were cloned into pET and pJNTN plasmid systems using standard molecular biology techniques [7].
- Constructs were verified through colony qPCR and sequencing before transformation into the production strain [7].
- Ribosome Binding Site (RBS) engineering was employed to fine-tune the relative expression levels of HpaBC and Ddc, using the UTR Designer tool to modulate RBS sequences [7].
Test and Learn Protocol
Cultivation Conditions:
- Minimal medium containing 20 g/L glucose, 10% 2xTY medium, MOPS buffer, vitamin B6, phenylalanine, and trace elements was used [7].
- Cultures were supplemented with appropriate antibiotics and induced with 1 mM IPTG [7].
Dopamine Quantification:
- Dopamine production was measured using high-performance liquid chromatography (HPLC) with electrochemical detection [7].
- Results were normalized to biomass (mg product/g biomass) and volume (mg/L) [7].
Learning and Iteration:
- Initial testing revealed the impact of GC content in the Shine-Dalgarno sequence on RBS strength and translation efficiency [7].
- This learning informed subsequent RBS redesign, culminating in a dopamine production strain achieving 69.03 ± 1.2 mg/L (34.34 ± 0.59 mg/g biomass)—a 2.6 to 6.6-fold improvement over previous state-of-the-art production [7].
Workflow Visualization: DBTL Cycle
The Scientist's Toolkit: Essential Research Reagents and Platforms
Successful implementation of DBTL cycles relies on specialized reagents, tools, and platforms that streamline each phase of the workflow.
Table 2: Essential Research Reagents and Platforms for DBTL Implementation
| Category | Specific Tools/Reagents | Function in DBTL Cycle |
|---|---|---|
| DNA Assembly & Parts | JBEI-ICE Registry; SynBioHub; Type IIS restriction enzymes (Golden Gate) | Catalog biological parts; Standardize assembly; Manage design metadata [8] |
| Host Engineering | E. coli production strains (e.g., FUS4.T2); Chromosomal integration systems; CRISPR-Cas9 tools | Provide metabolic background; Enable stable genetic modifications [7] |
| Analytical Methods | HPLC with electrochemical detection; LC-MS/MS; NGS; Colony qPCR | Quantify metabolites; Verify sequences; Validate constructs [7] [1] |
| Cell-Free Systems | TX-TL transcription-translation kits; Crude cell lysates; Non-canonical amino acids | Rapid testing of protein function; Bypass cellular complexity [5] [6] |
| Automation & Software | Liquid handling robots; Microfluidics; SBOLDesigner; UTR Designer | Increase throughput; Enable high-throughput screening; Facilitate design [1] [5] [8] |
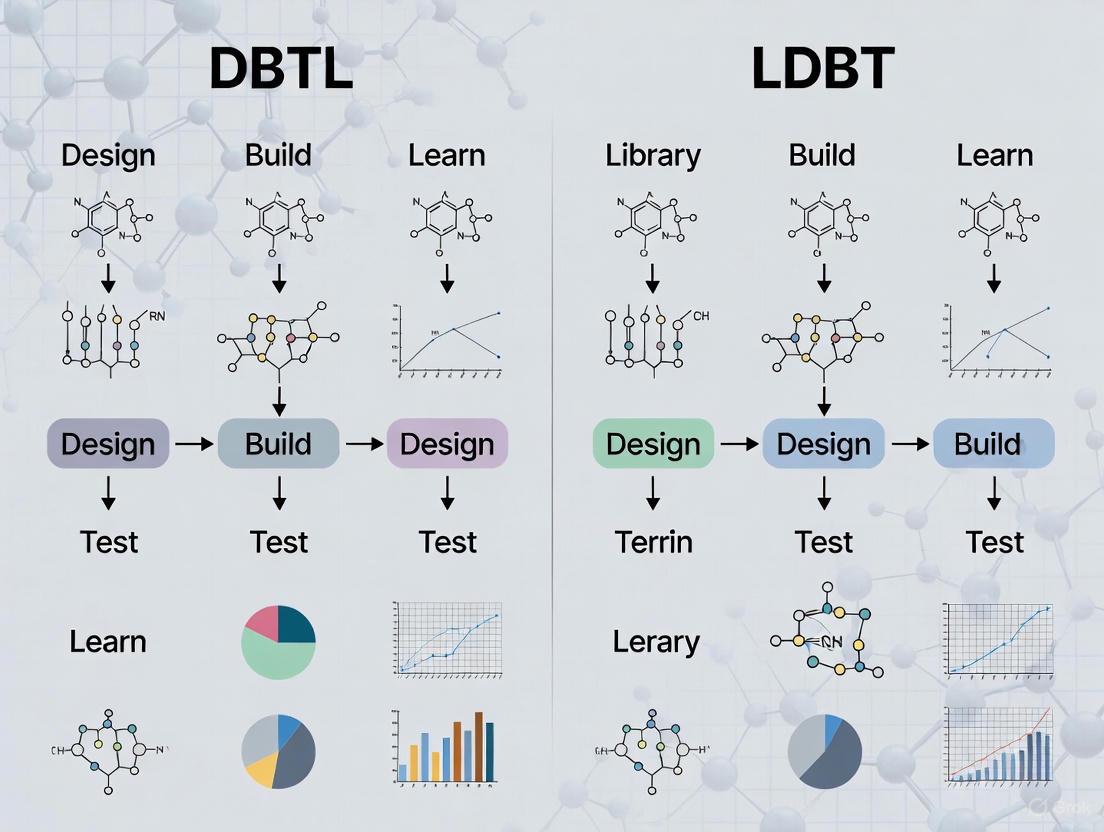
Emerging Evolution: From DBTL to LDBT
The established DBTL cycle faces ongoing refinement, most notably through the proposed LDBT (Learn-Design-Build-Test) paradigm that reorders the cycle to prioritize learning [5] [6]. This approach leverages machine learning models trained on large biological datasets to make zero-shot predictions about protein structure and function before experimental design and building [5]. Advances in protein language models (ESM, ProGen), structure-based design tools (ProteinMPNN, MutCompute), and functional predictors (Prethermut, DeepSol) enable increasingly accurate computational predictions that can guide biological design [5].
When combined with rapid cell-free testing platforms, LDBT promises to accelerate biological engineering by reducing dependency on costly and time-consuming trial-and-error experimentation [5] [6]. This evolution toward learning-first approaches represents a potential paradigm shift from iterative empirical optimization toward predictive engineering, potentially moving synthetic biology closer to the Design-Build-Work model used in established engineering disciplines like civil engineering [5]. Nevertheless, the core principles and workflow of the traditional DBTL cycle remain fundamental to synthetic biology, providing the foundational framework upon which these next-generation methodologies are being built.
The DBTL cycle's established framework continues to enable systematic engineering of biological systems, with ongoing enhancements through automation, data management platforms, and machine learning integration further increasing its power and efficiency for biotechnology applications.
Synthetic biology has long been governed by the Design-Build-Test-Learn (DBTL) cycle, a systematic framework for engineering biological systems. This iterative process involves designing genetic constructs, building them in the laboratory, testing their performance, and learning from the results to inform the next design iteration [1]. While this approach has enabled significant advances, it is inherently constrained by its reactive nature; learning occurs only after resource-intensive build and test phases, leading to multiple time-consuming and costly cycles [5]. However, a transformative paradigm shift is now underway, fueled by advances in machine learning (ML) and high-throughput experimental platforms. The emerging Learn-Design-Build-Test (LDBT) framework reorders this sequence, placing Learning at the forefront through data-driven prediction and zero-shot design [5]. This repositioning transforms synthetic biology from an empirical, trial-and-error discipline toward a more predictive engineering science, potentially collapsing multiple DBTL cycles into a single, efficient LDBT cycle that brings the field closer to a "Design-Build-Work" model [5] [6]. For researchers and drug development professionals, this shift promises to dramatically accelerate the development of therapeutic proteins, optimized biosynthetic pathways, and other bio-based products.
The Limitation of the Traditional DBTL Cycle
The traditional DBTL cycle, while systematic, faces significant challenges in predictability and efficiency. The core limitation lies in biological complexity: the impact of introducing foreign DNA into a cell is often difficult to predict due to non-linear, high-dimensional interactions between genetic parts and host cell machinery [9]. This complexity forces the engineering process away from rational design and into a regime of ad hoc tinkering [9].
- The Build-Test Bottleneck: The physical construction of genetic designs and their experimental testing are often the slowest and most resource-intensive phases. In vivo testing, in particular, is hampered by cellular processes like transformation and culturing, which are time-consuming and can be complicated by factors such as metabolic burden and toxicity to the host cell [5] [1].
- Iterative Learning Delay: In the DBTL cycle, learning is retrospective. Insights that could guide design are only generated after builds are tested, creating a lag in knowledge acquisition. This often necessitates several rounds of iteration to converge on a functional system, making the overall process costly and slow [5] [3].
Table 1: Core Challenges in the Traditional DBTL Cycle
| Challenge | Impact on Engineering Workflow |
|---|---|
| Unpredictable Biological Interactions | Limits the power of purely rational design, requiring extensive experimental iteration [9]. |
| Slow In Vivo Build/Test Phases | Creates a bottleneck, extending project timelines from weeks to months or years [5]. |
| Retrospective Learning | Delays the incorporation of critical insights into the design process, increasing the number of required cycles [3]. |
| Combinatorial Explosion | Vast design spaces make it experimentally infeasible to test all promising variants [3]. |
The LDBT Paradigm: A Machine-Learning First Approach
The LDBT paradigm addresses the core limitations of DBTL by leveraging modern machine learning to pre-emptively generate knowledge. In this new framework, the cycle begins with the Learn phase, where ML models trained on vast biological datasets are used to make zero-shot predictions about sequence-structure-function relationships before any physical design is initiated [5] [6].
The "Learn-First" Rationale
The rationale for this reordering is the growing success of ML models in making accurate functional predictions from sequence or structural data alone. These models have been trained on the entirety of available protein sequences and structures, effectively internalizing evolutionary and biophysical constraints [5]. This allows researchers to "interrogate" the model to generate designs with a high probability of success, effectively compressing the learning from many potential DBTL cycles into a single, upfront computational step [5] [9]. This approach is particularly powerful for navigating the vast combinatorial space of biological sequences, where testing all variants is impossible [3].
Key Machine Learning Technologies
The LDBT approach is enabled by several classes of machine learning models:
- Protein Language Models (e.g., ESM, ProGen): Trained on evolutionary relationships across millions of protein sequences, these models can predict beneficial mutations, infer function, and design novel, functional protein sequences in a zero-shot manner [5].
- Structure-Based Models (e.g., ProteinMPNN, MutCompute): These tools take protein structures as input and design sequences that fold into that backbone or optimize residues for stability and function. When combined with structure-prediction tools like AlphaFold, they significantly increase design success rates [5].
- Functional Prediction Models (e.g., Prethermut, Stability Oracle, DeepSol): These supervised learning models are trained on specific functional properties like thermodynamic stability or solubility, allowing for the in silico screening of designs for desired characteristics [5].
The LDBT Workflow in Practice
The practical implementation of the LDBT cycle integrates computational learning with rapid experimental validation. The workflow is illustrated in the following diagram, which highlights the streamlined, single-pass nature of the process driven by initial learning.
Phase 1: Learn
The cycle begins by harnessing machine learning models that have been pre-trained on massive biological datasets. These models encapsulate complex patterns of sequence evolution, structural stability, and functional fitness [5] [9]. Researchers can query these models to predict the properties of hypothetical sequences or to generate entirely new sequences with desired functions, a capability known as zero-shot design [5]. For example, a protein language model can be prompted to generate novel antimicrobial peptide sequences, which are then filtered for predicted activity and low toxicity before any DNA is synthesized [5].
Phase 2: Design
In this phase, the insights and pre-validated designs from the Learn phase are translated into specific, buildable DNA sequences. The design process is now guided and constrained by the ML predictions, ensuring a higher probability of success. This may involve selecting optimal codons for expression, assembling genetic circuits from parts with predicted compatibility, or designing libraries focused on the most promising regions of sequence space as identified by the models [6].
Phase 3: Build
The designed DNA constructs are synthesized and prepared for testing. To maintain the speed of the LDBT cycle, this phase often leverages automated DNA synthesis and assembly workflows [1]. The use of cell-free systems is particularly advantageous here, as they eliminate the need for time-consuming steps like cloning and transformation into living cells. Synthesized DNA can be directly added to cell-free reactions for expression, bypassing a major bottleneck of in vivo methods [5].
Phase 4: Test
The final phase involves the high-throughput experimental validation of the built designs. Cell-free transcription-translation (TX-TL) systems are a cornerstone of the LDBT approach for this purpose [5] [6]. These systems:
- Are highly rapid, producing proteins in less than 4 hours [5].
- Are scalable, enabling parallel testing of thousands of reactions from picoliter to kilo-liter scales [5].
- Offer fine control over the reaction environment, improving reproducibility [6].
- Can be coupled with robotics and microfluidics for ultra-high-throughput screening, as demonstrated by platforms like DropAI that screen over 100,000 reactions [5].
The data generated from this testing phase can be fed back to further refine and retrain the ML models, creating a virtuous cycle of improving predictive power for future LDBT campaigns.
Quantitative Comparison: DBTL vs. LDBT
The theoretical advantages of the LDBT cycle are borne out in practical metrics and experimental benchmarks. The following table summarizes key performance differentiators.
Table 2: Performance Comparison of DBTL vs. LDBT Approaches
| Metric | Traditional DBTL | LDBT Approach |
|---|---|---|
| Cycle Time | Weeks to months per cycle [1] | Days to weeks, with cell-free testing in hours [5] |
| Primary Learning Mode | Retrospective (post-testing) analysis [3] | Prospective, zero-shot prediction from pre-trained models [5] |
| Typical Cycles to Success | Multiple iterative cycles required [3] | Potential for single-cycle success [5] |
| Throughput of Test Phase | Limited by in vivo transformation and growth [1] | Ultra-high-throughput; >100,000 variants using microfluidics [5] |
| Handling of Design Space | Limited exploration due to low throughput [3] | Capable of navigating vast combinatorial spaces computationally [5] [3] |
| Resource Intensity | High (repeated cycles, labor, materials) [1] | Lower per project (faster convergence), but requires computational investment [6] |
Experimental Evidence and Case Studies
Simulation-based studies and real-world experiments demonstrate the efficacy of the LDBT approach:
- Enzyme Engineering: A pre-trained protein language model was used to design libraries for engineering a biocatalyst, resulting in successful enantioselective bond formation. This showcases the zero-shot design capability without additional model training [5].
- Antimicrobial Peptide (AMP) Discovery: Researchers combined deep learning-based sequence generation with cell-free expression. They computationally surveyed over 500,000 AMP sequences, selected 500 optimal variants for experimental testing, and identified six promising designs, demonstrating efficient navigation of a massive sequence space [5].
- Pathway Optimization: The iPROBE (in vitro prototyping and rapid optimization of biosynthetic enzymes) method uses a neural network trained on pathway combinations to predict optimal enzyme sets and expression levels, leading to a 20-fold improvement in product yield in a host organism [5].
- In Silico DBTL Benchmarking: A kinetic model-based framework demonstrated that ML methods like gradient boosting and random forest are highly effective in low-data regimes for guiding combinatorial pathway optimization, underscoring the power of learning in iterative strain improvement [3].
Essential Tools and Protocols for Implementing LDBT
Adopting the LDBT framework requires a suite of computational and experimental tools. The following table details key resources that constitute a modern LDBT toolkit.
Table 3: Research Reagent Solutions for the LDBT Workflow
| Tool Category | Example Solutions | Function in LDBT Workflow |
|---|---|---|
| Machine Learning Models | Protein Language Models (ESM, ProGen), Structure-Based Design Tools (ProteinMPNN, MutCompute), Stability Predictors (Stability Oracle) [5] | Enables the "Learn" phase by generating and pre-validating designs with desired properties. |
| Cell-Free Expression Systems | TX-TL systems from E. coli, wheat germ, or mammalian cell lysates; purified component systems [5] | Accelerates the "Build" and "Test" phases by enabling rapid, high-throughput protein expression without living cells. |
| High-Throughput Screening Platforms | Droplet microfluidics (e.g., DropAI), automated liquid handlers, microplate readers [5] | Allows parallel testing of thousands of designs, generating large datasets for model validation or retraining. |
| DNA Synthesis & Assembly | Automated gene synthesis, high-throughput molecular cloning workflows [1] | Facilitates the rapid physical construction of computationally designed DNA sequences. |
Detailed Experimental Protocol: Coupling ML Design with Cell-Free Testing
This protocol outlines a standard workflow for validating machine-learning-generated protein variants.
A. Learn & Design Phase:
- Objective Definition: Clearly define the target protein property (e.g., improved thermostability, higher enzymatic activity, novel binding).
- Model Selection & Interrogation:
- For zero-shot design, use a protein language model (e.g., ESM) or a structure-based tool (e.g., ProteinMPNN) to generate a library of candidate sequences.
- For optimization, use a predictive model (e.g., Stability Oracle) to score and filter a large mutational library, selecting the top-ranked variants for testing.
- DNA Sequence Design: Convert the selected protein sequences into DNA sequences with optimal codons for the chosen cell-free expression system.
B. Build Phase:
- DNA Synthesis: Order the designed sequences as linear DNA fragments or as cloned genes from a commercial synthesis provider.
- Template Preparation: If using linear fragments, amplify them via PCR to ensure sufficient quantity and purity. If using plasmids, purify them using standard miniprep or maxiprep kits.
C. Test Phase:
- Cell-Free Reaction Setup:
- Use a commercial cell-free kit or a homemade E. coli lysate system [5].
- In a 96-well or 384-well microplate, mix the cell-free reaction master mix with each DNA template (~10-20 ng/µL final concentration).
- Include positive and negative controls (e.g., a known well-expressing protein and a no-DNA control).
- Incubation and Measurement:
- Incubate the reaction plate at 30°C for 4-16 hours with shaking.
- Monitor expression if using a fluorescent protein. For enzymes, stop the reaction and assay for activity using a colorimetric or fluorescent substrate in a high-throughput plate reader.
- Data Analysis: Compare the activity or yield of the ML-designed variants to the wild-type or parent control. The top performers identified in this single round of testing are strong candidates for further development.
The transition from DBTL to LDBT represents a fundamental maturation of synthetic biology. By placing machine learning at the forefront of the design process, the LDBT framework leverages the vast and growing body of biological data to make predictive, zero-shot engineering a reality. When combined with the experimental acceleration provided by cell-free systems and high-throughput screening, this paradigm significantly shortens the path from concept to functional biological system. For the field of drug development, this shift is particularly impactful, promising to streamline the discovery and optimization of therapeutic proteins, vaccines, and biosynthetic pathways for small-molecule drugs. As machine learning models become more sophisticated and cell-free platforms more robust, the LDBT approach is poised to become the standard for a new era of predictable, efficient, and scalable biological design.
Synthetic biology has established itself as a premier engineering discipline by adopting and adapting core principles from traditional engineering fields. The foundational framework for this biological engineering endeavor has been the Design-Build-Test-Learn (DBTL) cycle, a systematic, iterative process that has streamlined efforts to build biological systems [10]. This cyclic methodology closely resembles approaches used in established engineering disciplines such as mechanical engineering, where iteration involves gathering information, processing it, identifying design revisions, and implementing those changes [10]. However, the field is now undergoing a paradigm shift driven by computational advances. The emergence of machine learning (ML) is prompting a fundamental rethinking of this established workflow, potentially reorganizing it into a Learn-Design-Build-Test (LDBT) cycle where learning precedes design [10] [6]. This transition represents a significant evolution in how engineers approach biological design, moving from a build-then-learn to a learn-then-build philosophy.
The DBTL cycle begins with the Design phase, where researchers define objectives and design biological parts or systems using domain knowledge and computational modeling [10]. In the Build phase, DNA constructs are synthesized and introduced into characterization systems, which can include in vivo chassis or in vitro cell-free systems [10]. The Test phase experimentally measures the performance of the engineered constructs, while the Learning phase analyzes this data to inform the next design iteration [10]. This cyclic process has formed the backbone of synthetic biology's progress, but its reliance on empirical iteration has limitations in efficiency and predictability.
The Established Paradigm: Design-Build-Test-Learn (DBTL)
Theoretical and Practical Foundations
The DBTL framework finds its theoretical roots in broader engineering design theory. The process is not unique to synthetic biology but closely mirrors approaches in mechanical engineering, where physical laws model parameters like damping and stiffness [10]. More fundamentally, all design processes, including DBTL, can be viewed as evolutionary in nature [11]. They follow a cyclic iterative process where concepts are modified or recombined, prototyped, tested for utility, and the best candidates are selected for further iteration—directly analogous to biological evolution through natural selection [11].
This evolutionary perspective reveals that all design methods exist on a spectrum characterized by population size (throughput) and generation count (number of iterations) [11]. The exploratory power of any design approach is the product of these two factors, yet this power always pales compared to the vastness of biological design space [11]. Successful navigation of this space relies on two forms of learning: exploration (searching the fitness landscape) and exploitation (using prior knowledge to constrain and guide the search) [11].
Implementation and Challenges
In practical implementation, the DBTL cycle has been propelled by massive improvements in DNA sequencing and synthesis technologies. The cost of sequencing a human genome dropped from approximately $10 million in 2007 to around $600, enabling the accumulation of vast genomic databases that form the basis for redesigning biological systems [12]. Similarly, advances in DNA assembly methodologies like Gibson assembly have overcome limitations of conventional cloning, enabling seamless assembly of combinatorial genetic parts and even entire synthetic chromosomes [12].
Despite these technical advances, significant challenges have persisted in the DBTL cycle. The "learning" stage has proven particularly difficult due to the complexity and heterogeneity of biological systems, interactions between components, and variations in experimental setups [12]. While synthetic biologists can decipher data to create draft blueprints, many still resort to top-down approaches based on likelihoods and trial-and-error rather than genuine rational design [12]. This limitation has motivated the integration of more sophisticated computational approaches, particularly machine learning, to overcome these bottlenecks.
Table 1: Key Stages of the Traditional DBTL Cycle
| Stage | Core Activities | Primary Tools & Technologies | Major Challenges |
|---|---|---|---|
| Design | Define objectives; Design parts/system using domain knowledge | Computational modeling; Domain expertise; Biophysical principles | Limited predictive power of models; Complexity of biological systems |
| Build | Synthesize DNA; Assemble constructs; Introduce into chassis | DNA synthesis; Cloning; Genome editing; Cell-free systems | Time-consuming cloning; Cellular toxicity; Genetic instability |
| Test | Measure performance experimentally | Omics technologies; Fluorescence assays; Analytics | Low throughput; Cellular context effects; Difficulty of measurement |
| Learn | Analyze data; Compare to objectives; Inform next design | Statistical analysis; Data interpretation | Complexity & heterogeneity; Black box nature of biology; Incomplete knowledge |
The Emerging Paradigm: Learn-Design-Build-Test (LDBT)
The Machine Learning Revolution
The proposed LDBT cycle represents a fundamental reordering of the synthetic biology workflow, placing "Learning" at the forefront through machine learning [10]. This shift is made possible by the development of sophisticated protein language models and structural prediction tools that can leverage vast biological datasets to detect patterns in high-dimensional spaces, enabling more efficient and scalable design [10]. These models are trained on millions of protein sequences or hundreds of thousands of structures, allowing researchers to make increasingly accurate zero-shot predictions that improve the functionality of protein parts without additional training [10].
Several classes of machine learning models are driving this transition. Sequence-based protein language models such as ESM and ProGen are trained on evolutionary relationships between protein sequences embedded across phylogeny [10]. These models excel at predicting beneficial mutations and inferring protein functions, having proven adept at zero-shot prediction of diverse antibody sequences [10]. Structure-based models like MutCompute and ProteinMPNN use deep neural networks trained on protein structures to associate amino acids with their chemical environments, enabling prediction of stabilizing and functionally beneficial substitutions [10]. When combined with structure assessment tools like AlphaFold, these approaches have demonstrated nearly 10-fold increases in design success rates [10].
Enabling Technologies: Cell-Free Systems and Automation
The practical implementation of the LDBT paradigm is facilitated by the parallel development of high-throughput cell-free transcription-translation (TX-TL) systems [10] [6]. These systems circumvent complexities associated with living host cells—such as metabolic burden and genetic instability—enabling rapid assessment of genetic circuit performance within hours rather than days or weeks [6]. Cell-free expression leverages protein biosynthesis machinery from crude cell lysates or purified components to activate in vitro transcription and translation, producing more than 1 g/L of protein in under 4 hours [10].
The integration of cell-free systems with liquid handling robots and microfluidics has dramatically scaled testing capabilities. For example, DropAI leveraged droplet microfluidics and multi-channel fluorescent imaging to screen over 100,000 picoliter-scale reactions [10]. Biofoundries worldwide have institutionalized these high-throughput automated workflows, with facilities collaborating through the Global Biofoundry Alliance established in 2019 [12]. This infrastructure provides the massive, high-quality datasets required to train effective machine learning models for biological design.
Table 2: Machine Learning Approaches in the LDBT Paradigm
| ML Approach | Representative Tools | Primary Application | Key Strengths |
|---|---|---|---|
| Sequence-Based Language Models | ESM [10], ProGen [10] | Predicting beneficial mutations; Inferring protein function | Captures long-range evolutionary dependencies; Zero-shot prediction capability |
| Structure-Based Models | MutCompute [10], ProteinMPNN [10] | Residue-level optimization; Sequence design for target structures | Associates amino acids with local chemical environment; High success rates when combined with structure assessment |
| Stability Prediction | Prethermut [10], Stability Oracle [10] | Predicting thermodynamic stability changes of mutants | Predicts ΔΔG of proteins; Identifies stabilizing/destabilizing mutations |
| Solubility Prediction | DeepSol [10] | Predicting protein solubility from primary sequence | Maps sequence features (k-mers) to solubility; Helps screen expressible variants |
| Hybrid Approaches | Physics-informed ML [10] | Combining statistical power with physical principles | Leverages both data patterns and biophysical principles; Enhanced explanatory capability |
Comparative Analysis: DBTL vs. LDBT
Workflow and Philosophical Differences
The fundamental distinction between DBTL and LDBT lies in their starting points and underlying philosophies. The traditional DBTL cycle begins with design based on existing domain knowledge and hypotheses, representing a hypothesis-driven approach [10]. In contrast, the LDBT cycle starts with learning from vast datasets, employing a data-driven approach that uses machine learning to uncover hidden patterns and relationships before any design occurs [6]. This learn-first approach enables researchers to refine design hypotheses before constructing biological parts, potentially circumventing costly trial-and-error [6].
This philosophical shift also changes the role of iteration in the engineering process. While DBTL requires multiple cycles to gain knowledge, with Build-Test phases being particularly slow, LDBT aims to leverage pre-existing knowledge embedded in machine learning models to reduce iteration needs [10]. Given the increasing success of zero-shot predictions, it may be possible to reorganize the cycle such that Learn-Design allows an initial set of answers to be quickly built and tested, potentially generating functional parts and circuits in a single cycle [10]. This brings synthetic biology closer to a Design-Build-Work model that relies on first principles, similar to disciplines like civil engineering [10].
Practical Implementation and Efficiency Gains
The efficiency advantages of LDBT manifest most clearly in its handling of vast biological design spaces. The combinatorial nature of potential DNA sequence variations generates a landscape of possibilities too extensive for exhaustive exploration [6]. LDBT's machine learning component navigates this space intelligently through active learning techniques, strategically selecting the most informative sequence variants to test experimentally [6]. This approach maximizes information gain per experiment, reducing redundancy and focusing efforts on promising design regions [6].
Case studies demonstrate LDBT's practical efficacy. Researchers have paired deep-learning sequence generation with cell-free expression to computationally survey over 500,000 antimicrobial peptides, selecting 500 optimal variants for experimental validation, resulting in 6 promising designs [10]. In pathway engineering, in vitro prototyping and rapid optimization of biosynthetic enzymes (iPROBE) uses neural networks with training sets of pathway combinations to predict optimal pathway sets, improving 3-HB production in Clostridium by over 20-fold [10]. These examples showcase LDBT's ability to achieve rapid convergence on high-performance constructs with fewer iterations than conventional methods.
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Core Experimental Platforms
The implementation of both DBTL and LDBT cycles relies on a sophisticated toolkit of experimental platforms and reagents. Cell-free gene expression systems form a cornerstone of the emerging LDBT paradigm, enabling rapid testing without the constraints of living cells [10]. These systems leverage protein biosynthesis machinery from various organisms and can be customized through modular reagent exchanges [10]. Their flexibility allows incorporation of non-canonical amino acids and post-translational modifications, positioning them as versatile platforms for high-throughput synthesis and testing [10].
Automation and microfluidics constitute another critical component. Robotic liquid handling systems enable the scale-up of assembly and testing protocols, while droplet microfluidics allows massive parallelization of reactions [10] [6]. These technologies interface closely with advanced analytical methods, including next-generation sequencing and mass spectrometry, to collect multi-omics data at single-cell resolution [12]. The integration of these platforms in biofoundries provides the industrial-scale infrastructure needed for modern biological engineering.
Mathematical modeling remains an essential tool for studying gene regulatory circuits, whether in traditional DBTL or ML-enhanced LDBT approaches [13]. Models serve as logical machines to derive the implications of biological hypotheses, with mathematical language providing a powerful reasoning system for building arguments too intricate to hold in our heads [13]. The definition of a circuit—representing interactions between entities and the computing logic of such interactions—provides a map for building mathematical models where nodes represent molecular species and edges denote interactions or biochemical reactions [13].
For machine learning implementation, specific architectures have proven particularly valuable. Neural networks alongside classic ensemble methods capture nonlinear relationships between sequence features and functional outputs [6]. These models are trained on biological features encompassing promoter strengths, ribosome binding site sequences, codon usage biases, and secondary structure propensities [6]. The continuous improvement of these models through iterative experimental validation creates a virtuous cycle of enhanced predictive capability.
Table 3: Essential Research Reagent Solutions for Synthetic Biology Workflows
| Reagent/Platform | Primary Function | Application in DBTL/LDBT | Key Advantages |
|---|---|---|---|
| Cell-Free TX-TL Systems | In vitro transcription-translation | Rapid testing in Build-Test phases; Dataset generation for ML | Bypasses living cells; High throughput; Tunable environment |
| CRISPR-Based Editing | Targeted genome modification | Building constructs in host chassis; Creating mutator strains | Precision; Versatility across organisms; Multiplexing capability |
| DNA Synthesis & Assembly | De novo DNA construction | Building genetic designs; Library construction | Scalability; Speed; Independence from template availability |
| Droplet Microfluidics | Miniaturized reaction compartments | Ultra-high-throughput screening; Single-cell analysis | Massive parallelization; Reduced reagent costs |
| Protein Language Models | Protein sequence-function prediction | Learning phase; Zero-shot design | Evolutionary insight; No required structural data |
| Structure Prediction Tools | Protein structure/function prediction | Learning and Design phases | Environmental context; Stabilizing mutation identification |
| Multi-Omics Analytics | Comprehensive molecular profiling | Testing and Learning phases | Systems-level insight; Data richness for ML training |
Future Perspectives and Implications
Technological Convergence and Democratization
The convergence of machine learning with synthetic biology promises to fundamentally reshape biotechnology development timelines. The ability to quickly iterate designs based on predictive learning could dramatically shorten development windows for bio-based products, from pharmaceuticals to sustainable chemicals [6]. Furthermore, the reduced dependency on labor-intensive cloning and cellular culturing steps may democratize synthetic biology research, opening avenues for smaller labs and startups to participate in cutting-edge bioengineering without extensive infrastructure [6].
This technological convergence also enables more nuanced understanding of genotype-to-phenotype relationships. Traditional methods often struggle with the stochasticity and context-dependence inherent to biological systems, but the iterative learning and validation offered by the LDBT cycle helps disentangle these complexities through continual refinement of predictive models [6]. Each loop through the cycle yields improved biological insight and enhanced design rationales, fostering a virtuous circle of discovery and engineering [6].
Meta-Synthetic Biology and Evolutionary Engineering
Looking forward, the field appears to be moving toward what might be termed "meta-synthetic biology"—controlling not just biological function but the evolutionary processes themselves [14]. Research has demonstrated that mutation rates and spectra can be manipulated both globally across genomes and locally at specific genes or genomic regions [14]. Under specific conditions, mutational space can be considerably reduced—for instance, predominantly to G->T mutations on transcribed strands—constraining evolutionary paths and making outcomes more predictable [14].
This control over evolutionary processes enables new engineering paradigms. Rather than solely designing static biological systems, engineers can now design systems with specified evolutionary trajectories [14]. This might include creating microbes that are genetically hyper-stable for robust performance in bioreactors, or microbiome therapies that evolve in the gut to become personalized to host genetics [14]. Such approaches represent a ultimate synthesis of engineering and evolution, potentially resolving the apparent paradox between rational design and evolutionary tinkering [15].
The evolution of engineering paradigms in synthetic biology from DBTL to LDBT represents more than just a reordering of workflow stages—it signifies a fundamental shift in how biological engineers approach design. The traditional DBTL cycle, with its roots in classical engineering disciplines, has provided a systematic framework for biological innovation [10]. However, the integration of machine learning and high-throughput testing platforms is now enabling a more data-driven, predictive approach that places learning at the forefront of biological design [10] [6].
This paradigm shift promises to accelerate synthetic biology toward its ultimate goal: high-precision biological design with predictable outcomes [12]. By leveraging the growing power of machine learning models trained on expanding biological datasets, and combining these with rapid experimental validation through cell-free systems and automation, the field appears poised to overcome the limitations of iterative trial-and-error that have constrained its progress [10] [6] [12]. The continued convergence of biological engineering with computational intelligence and experimental ingenuity sets the stage for transforming how biological systems are understood, designed, and deployed for human benefit [6].
The Design-Build-Test-Learn (DBTL) cycle has long been the foundational framework of synthetic biology, representing an iterative process where researchers design biological systems, build them, test their functionality, and learn from the outcomes to inform the next design round [5]. However, recent advancements in machine learning (ML) and data generation technologies are catalyzing a fundamental restructuring of this paradigm. The emerging Learn-Design-Build-Test (LDBT) cycle represents a transformative approach where machine learning precedes design, leveraging vast biological datasets to make predictive, zero-shot designs that dramatically accelerate biological engineering [5] [6]. This shift from a build-test-learn cycle to a learn-first methodology is poised to reshape synthetic biology, moving the field closer to a "Design-Build-Work" model reminiscent of more established engineering disciplines [5]. This technical guide examines the core drivers enabling this transition, with particular focus on the integration of machine learning and megascale data generation through cell-free testing platforms.
The LDBT Framework: Core Components and Workflow
The LDBT framework fundamentally reorders the synthetic biology workflow, placing learning at the forefront of the engineering process. This reorientation leverages pre-existing knowledge encoded in machine learning models to generate more intelligent initial designs, potentially reducing or eliminating the need for multiple iterative cycles.
The LDBT Workflow Diagram
The following diagram illustrates the core structure and information flow of the LDBT paradigm:
Comparative Analysis: DBTL vs. LDBT Cycles
Table 1: Fundamental differences between traditional DBTL and the emerging LDBT paradigm
| Aspect | Traditional DBTL Cycle | LDBT Cycle |
|---|---|---|
| Starting Point | Design phase based on limited knowledge and hypotheses | Learning phase leveraging pre-trained ML models on vast datasets [5] |
| Primary Driver | Empirical experimentation and iterative testing | Predictive computational modeling and zero-shot design [5] |
| Data Utilization | Data generated from previous cycles informs subsequent designs | Pre-existing megascale datasets and foundational models enable intelligent first-pass designs [5] [6] |
| Cycle Duration | Multiple lengthy iterations often required | Potential for single-cycle success through accurate prediction [6] |
| Resource Intensity | High resource consumption across multiple build-test phases | Resource concentration on validated, high-probability designs [6] |
| Knowledge Foundation | Domain expertise and incremental learning from own experiments | Collective biological knowledge encoded in ML models [5] |
| Experimental Approach | Heavy reliance on in vivo systems and cellular constraints | Cell-free testing platforms for rapid, parallel validation [5] [6] |
Machine Learning as the Foundation of the Learn Phase
The initial "Learn" phase in LDBT is powered by sophisticated machine learning models trained on massive biological datasets. These models capture complex relationships between biological sequences, structures, and functions that would be impossible to discern through traditional analysis.
Key Machine Learning Approaches in LDBT
Table 2: Machine learning model types and their applications in the LDBT Learn phase
| Model Type | Examples | Training Data | Key Applications | Capabilities |
|---|---|---|---|---|
| Protein Language Models | ESM [5], ProGen [5] | Evolutionary relationships in protein sequences [5] | Predicting beneficial mutations [5], inferring protein function [5], designing antibody sequences [5] | Zero-shot prediction of diverse sequences [5], capturing long-range evolutionary dependencies [5] |
| Structure-Based Models | MutCompute [5], ProteinMPNN [5] | Experimentally determined protein structures [5] | Residue-level optimization [5], designing sequences for specific backbones [5], enzyme engineering [5] | Predicting stabilizing mutations [5], associating amino acids with local chemical environment [5] |
| Functional Prediction Models | Prethermut [5], Stability Oracle [5], DeepSol [5] | Experimental measurements of protein properties [5] | Predicting thermodynamic stability changes [5], predicting protein solubility [5], multi-property optimization [5] | Predicting ΔΔG of mutations [5], mapping sequence-fitness landscapes [5] |
| Hybrid & Augmented Models | Physics-informed ML [5], evolutionary-augmented models [5] | Combined datasets (sequences, structures, biophysics) [5] | Exploring evolutionary landscapes [5], engineering specialized enzymes [5], simultaneous multi-parameter optimization [5] | Combining predictive power of statistical models with explanatory strength of physical principles [5] |
Technical Implementation of ML Models in LDBT
The machine learning infrastructure supporting LDBT requires specialized implementation approaches:
Data Preparation and Feature Engineering
- Biological Feature Extraction: ML models leverage a broad spectrum of biological features including promoter strengths, ribosome binding site sequences, codon usage biases, and secondary structure propensities [6]. These features are encoded in high-dimensional vectors that capture both local and global sequence properties.
- Training Data Curation: Models are trained on diverse datasets ranging from millions of protein sequences [5] to hundreds of thousands of structures [5]. Data quality assurance involves removing redundant sequences, balancing representation across protein families, and integrating experimental measurements from standardized assays.
Model Architectures and Training
- Neural Network Architectures: State-of-the-art neural network architectures alongside classic ensemble methods capture nonlinear relationships between sequence features and functional outputs [6]. Graph-transformer architectures learn pairwise representations of residues for predicting protein stability [5].
- Active Learning Integration: ML components employ active learning techniques to intelligently navigate vast genetic design spaces by strategically selecting the most informative sequence variants to test experimentally [6]. This approach maximizes information gain per experiment, reducing redundancy and focusing efforts on promising design regions.
Zero-Shot Prediction Capabilities The most significant advancement enabling LDBT is the development of models capable of zero-shot prediction—generating functional designs without additional training on specific targets [5]. For example:
- Protein language models trained on evolutionary relationships can predict beneficial mutations and infer protein function without target-specific fine-tuning [5].
- Structure-based models like ProteinMPNN can design sequences that fold into specified backbones with nearly 10-fold increased success rates when combined with structure assessment tools like AlphaFold [5].
Megascale Data Generation Through Cell-Free Testing Platforms
The "Build" and "Test" phases in LDBT are accelerated through cell-free transcription-translation (TX-TL) systems, which enable rapid, high-throughput experimental validation of computationally designed biological parts.
Cell-Free Testing Workflow
The experimental pipeline for cell-free testing in LDBT involves the following automated workflow:
Technical Specifications of Cell-Free Testing Platforms
Cell-free systems provide a biochemical environment containing the essential components for transcription and translation without the constraints of living cells.
System Components and Configuration
- Cellular Machinery Source: Protein biosynthesis machinery obtained from crude cell lysates or purified components [5]. Systems can be derived from organisms across the tree of life, including E. coli, yeast, and mammalian cells [5].
- Reaction Customization: Modular reaction environments allow facile customization through addition of specific energy sources, nucleotide triphosphates, amino acids, and cofactors [5]. This enables optimization for different protein types and experimental goals.
- Non-Canonical Incorporation: Capable of incorporating non-canonical amino acids and performing post-translational modifications like glycosylation and phosphorylation [5], expanding the chemical diversity of testable designs.
Performance Characteristics and Capabilities
- Speed and Yield: Rapid protein production achieving >1 g/L protein in <4 hours [5], dramatically faster than in vivo expression systems.
- Throughput Capacity: Systems like DropAI leverage droplet microfluidics to screen upwards of 100,000 picoliter-scale reactions [5], enabling truly megascale data generation.
- Tolerance and Flexibility: Capable of expressing products toxic to living cells and testing under diverse conditions that would be lethal to whole organisms.
Quantitative Advantages of Cell-Free Testing
Table 3: Performance comparison between traditional in vivo testing and cell-free testing platforms
| Parameter | Traditional In Vivo Testing | Cell-Free Testing Platforms | Improvement Factor |
|---|---|---|---|
| Testing Cycle Time | Days to weeks (including cloning, transformation, and cellular growth) [6] | Hours (direct template addition to reactions) [5] [6] | 10-100x faster [6] |
| Throughput Capacity | ~10^3-10^4 variants per campaign | ~10^5-10^6 variants using microfluidics [5] | 100-1000x higher throughput [5] |
| Resource Consumption | High (media, antibiotics, cellular growth requirements) | Minimal (nanoliter-scale reactions) [5] | 1000x reduction in reagent use [5] |
| Environmental Control | Limited by cellular homeostasis and metabolic constraints | Precise control over redox potential, energy charge, and molecular composition [6] | Unprecedented parameter control |
| Data Generation Rate | Limited by cellular growth rates and assay scalability | Ultra-high-throughput mapping (e.g., 776,000 variants for stability mapping) [5] | Megascale data acquisition |
| Assay Flexibility | Constrained by cellular viability and genetic stability | Compatible with diverse conditions, including toxic compounds [5] | Expanded experimental design space |
Integrated LDBT Implementation: Case Studies and Performance Metrics
The integration of machine learning and cell-free testing creates a synergistic framework where each component enhances the other's capabilities in a virtuous cycle of improvement.
Experimental Protocol for Integrated LDBT Implementation
Phase 1: Learn-First Design Initiation
- Objective Definition: Clearly specify functional requirements for the biological system, including performance metrics, environmental constraints, and optimization priorities.
- Model Selection: Choose appropriate pre-trained ML models based on the design challenge (e.g., protein language models for sequence optimization, structure-based models for stability engineering).
- Zero-Shot Design Generation: Utilize selected models to generate initial design candidates without additional training, leveraging the evolutionary and structural knowledge embedded in the models [5].
Phase 2: High-Throughput Experimental Validation
- DNA Template Preparation: Synthesize DNA templates for top candidate designs using high-throughput oligo synthesis and assembly methods.
- Cell-Free Reaction Assembly: Utilize liquid handling robots to assemble cell-free reactions in 96, 384, or 1536-well plates, or employ microfluidic devices for picoliter-scale reactions [5].
- Expression and Assaying: Incubate reactions to enable protein expression, followed by functional assaying using colorimetric, fluorescent, or other high-throughput detection methods [5].
- Data Collection: Implement automated data acquisition systems to quantitatively measure performance metrics for each variant.
Phase 3: Model Refinement and Iteration
- Data Processing and Quality Control: Apply automated processing pipelines to normalize data, remove outliers, and compile results into structured datasets.
- Model Retraining (Optional): For particularly challenging design problems, use the newly generated data to fine-tune models for improved prediction on similar systems.
- Design Prioritization: Apply updated models to identify the most promising candidates for further development or scale-up.
Case Study: Enzyme Engineering via LDBT
A demonstrated application of LDBT involves engineering a hydrolase for polyethylene terephthalate (PET) depolymerization [5]:
- Learning Phase: MutCompute, a deep neural network trained on protein structures, identified probable mutations given the local chemical environment [5].
- Design Phase: The model predicted specific amino acid substitutions predicted to increase stability and activity [5].
- Build Phase: DNA templates for selected variants were synthesized and expressed in cell-free systems [5].
- Test Phase: Expressed variants were rapidly assayed for PET depolymerization activity, confirming that proteins with MutCompute-designed mutations showed increased stability and activity compared to wild-type [5].
Further optimization was achieved by augmenting the approach with large language models trained on PET hydrolase homologs and force-field-based algorithms, enabling more comprehensive exploration of the evolutionary landscape [5].
Performance Metrics and Validation
Table 4: Quantitative outcomes from LDBT implementation in protein engineering campaigns
| Engineering Campaign | Traditional DBTL Results | LDBT Approach Results | Improvement Factor |
|---|---|---|---|
| PET Hydrolase Engineering | Not specified in literature | MutCompute-designed variants showed increased stability and activity vs wild-type [5] | Significant improvement in key parameters [5] |
| TEV Protease Engineering | Not specified in literature | ProteinMPNN-designed variants improved catalytic activity vs parent sequence [5] | Design success rates increased nearly 10-fold with structure assessment [5] |
| Antimicrobial Peptide Design | Traditional screening of limited libraries | 500 optimal variants selected from computational survey of >500,000; 6 promising designs validated [5] | Highly efficient design-to-validation pipeline [5] |
| Biosynthetic Pathway Optimization | Multi-round iterative strain engineering | iPROBE used neural network on pathway combinations to improve 3-HB titer in Clostridium by >20-fold [5] | Dramatic reduction in optimization time [5] |
| General Protein Engineering | Multiple rounds of site-saturation mutagenesis | Linear supervised models trained on >10,000 reactions accelerated identification of favorable variants [5] | Data-driven acceleration of engineering campaigns [5] |
Essential Research Reagents and Tools for LDBT Implementation
Successful implementation of the LDBT framework requires specific research reagents and computational tools that enable the seamless integration of machine learning and rapid experimental validation.
Research Reagent Solutions for LDBT
Table 5: Essential reagents, tools, and platforms for implementing LDBT workflows
| Category | Specific Tools/Reagents | Function in LDBT Pipeline | Key Features |
|---|---|---|---|
| Machine Learning Models | ESM [5], ProGen [5], ProteinMPNN [5], MutCompute [5] | Learn-phase: Generating intelligent designs based on patterns in biological data | Zero-shot prediction capabilities [5], attention mechanisms for long-range dependencies [5], structure-based design [5] |
| Cell-Free Systems | TX-TL systems [5], PURE system [5], species-specific lysates [5] | Build/Test-phase: Rapid expression and testing of designed variants | Bypass cellular growth constraints [5], high-throughput compatibility [5], customizable reaction conditions [5] |
| Automation Platforms | Liquid handling robots [5], microfluidic devices [5] | Build/Test-phase: Enabling megascale parallel experimentation | Nanoliter-to-microliter reaction assembly [5], integration with screening assays [5], walk-away operation [5] |
| Assay Technologies | cDNA display [5], fluorescence-based assays [5], colorimetric readouts [5] | Test-phase: Quantitative measurement of function and properties | Ultra-high-throughput compatibility [5], quantitative output for ML training [5], minimal cross-reactivity [5] |
| Data Integration Tools | Automated data processing pipelines [6], active learning algorithms [6] | Learn-phase: Continuous model improvement from experimental data | Strategic selection of informative variants [6], maximized information gain per experiment [6], closed-loop optimization [6] |
The LDBT framework represents a fundamental shift in synthetic biology methodology, moving the field from empirical iteration toward predictive engineering. By placing learning at the forefront through machine learning models trained on megascale biological data, and accelerating validation through cell-free testing platforms, LDBT dramatically accelerates the design process for biological systems. The integration of these technologies creates a virtuous cycle where each experiment enhances predictive models, which in turn generate more intelligent designs for subsequent testing. As this paradigm gains broader adoption, it promises to transform synthetic biology from a trial-and-error discipline to a truly predictive engineering science, enabling more rapid development of novel therapeutics, sustainable materials, and bio-based solutions to global challenges. Future advancements will likely focus on fully automated closed-loop systems combining AI-driven design with robotic experimentation, further reducing development timelines and expanding the complexity of addressable biological engineering challenges.
Execution and Impact: Machine Learning and Cell-Free Systems in Action
The engineering of biological systems has traditionally been guided by the Design-Build-Test-Learn (DBTL) cycle, an iterative framework where insights from testing one design inform the next round of design hypotheses [10]. While systematic, this process can be time-consuming and resource-intensive, often requiring multiple rounds of iteration to converge on a functional protein or genetic circuit. The recent and rapid integration of advanced machine learning (ML) is fundamentally reshaping this paradigm. A new framework, termed LDBT (Learn-Design-Build-Test), places learning at the forefront [10] [6]. In this model, ML algorithms are used to learn directly from vast biological datasets—including evolutionary sequences, structural information, and experimental measurements—to generate informed designs before any physical building or testing occurs [10]. This "learn-first" approach enables zero-shot design, where models can generate novel, functional protein sequences without additional training or iterative experimental feedback for a specific task [10] [16]. This whitepaper provides an in-depth technical guide to the key machine learning tools—protein language models and structure-based design tools—that are making this paradigm shift possible, empowering researchers to accelerate the development of novel therapeutics and enzymes.
Protein Language Models for Zero-Shot Design
Protein language models (pLMs) treat amino acid sequences as texts written in a 20-letter alphabet. By training on millions of natural protein sequences, they learn the underlying "grammar" and "syntax" of proteins, capturing evolutionary constraints and functional patterns. This allows them to generate novel, functional sequences from scratch or predict the effects of mutations without explicit structural or functional data.
ESM (Evolutionary Scale Modeling) Models
The ESM family of models, based on the Transformer architecture, is trained on a masked language modeling objective, learning to predict randomly omitted amino acids in a sequence based on their context [16]. This process forces the model to internalize complex biophysical and evolutionary relationships.
- Core Architecture and Training: ESM models are bidirectional encoders. During pre-training, they process sequences with a portion of residues masked, learning to generate information-rich, context-aware embeddings for every position in a sequence. The largest ESM-2 model has 15 billion parameters, while the newer ESM-3 has scaled to 98 billion parameters [16].
- Zero-Shot Applications: A key application is the zero-shot prediction of mutational effects. Models like ESM-1v are designed to score the likelihood of sequence variants, effectively predicting fitness landscapes without experimental data [16] [17]. Furthermore, the embeddings generated by ESM models can be used directly as features for downstream prediction tasks (e.g., predicting stability or function) without task-specific fine-tuning, a process known as transfer learning [16].
- Performance and Scaling: Studies show that model performance in transfer learning scales with size but is also influenced by data availability. While larger models like ESM-2 15B perform best with large datasets, medium-sized models like ESM-2 650M and ESM C 600M offer a practical balance of performance and computational cost, especially when data is limited [16]. For embedding-based tasks, mean pooling (averaging embeddings across all sequence positions) has been found to be a surprisingly effective and superior compression strategy compared to more complex methods [16].
ProGen Models
In contrast to ESM's masked modeling, the ProGen family employs an autoregressive, generative approach, similar to GPT models in natural language processing. It is trained to predict the next amino acid in a sequence, making it inherently powerful for generating entire novel protein sequences from scratch.
- Core Architecture and Training: ProGen3 is a suite of models using a sparse mixture-of-experts architecture. It was trained on the Profluent Protein Atlas v1 (PPA-1), a curated dataset of 3.4 billion protein sequences totaling 1.1 trillion amino acid tokens [18] [19]. Model sizes range from 112 million to 46 billion parameters [18].
- Zero-Shot Applications: ProGen3 can perform de novo protein generation and critical tasks like infilling (designing sequences for a given protein backbone or functional motif) [18] [19]. Its training enables it to generate diverse, functional proteins across numerous families, including complex proteins like antibodies and gene editors, in a zero-shot manner.
- Performance and Scaling: Profluent's research demonstrates clear scaling laws for biological design. Larger ProGen3 models produce more diverse, valid, and functional sequences. For instance, the ProGen3-46B model generated 59% more diverse sequences (measured by unique sequences at 30% identity) than a 3B parameter model [18]. Furthermore, these large models benefit most from alignment with limited laboratory data, allowing their outputs to be fine-tuned for specific properties like stability or binding affinity, with one study showing a correlation with protein fitness improving from 33.1% to 67.3% after alignment [18].
Table 1: Comparison of Major Protein Language Model Families
| Feature | ESM (e.g., ESM-2, ESM-3) | ProGen (e.g., ProGen3) |
|---|---|---|
| Primary Architecture | Bidirectional Encoder (BERT-like) | Autoregressive Decoder (GPT-like) |
| Training Objective | Masked Language Modeling | Next Token Prediction |
| Core Strength | Context-aware embeddings, variant effect prediction | De novo sequence generation, infilling |
| Typical Zero-Shot Use | Predicting fitness of mutants, transfer learning via embeddings | Generating novel, full-length functional proteins |
| Model Scale | Up to 98B parameters (ESM3) | Up to 46B parameters |
| Key Differentiator | Excels at understanding and scoring existing sequences | Excels at creating entirely new sequences |
Structure-Based Tools for Zero-Shot Design
While pLMs operate primarily on sequence information, another class of tools uses protein structural data to design sequences that fold into specific three-dimensional shapes or interact with target molecules.
ProteinMPNN
ProteinMPNN is a deep learning-based protein sequence design tool that takes a protein backbone structure as input and outputs sequences that are predicted to fold into that structure.
- Core Methodology: It uses a graph neural network architecture where each residue is a node, and edges are defined by spatial proximity [20]. The network performs message passing to incorporate information from the local structural environment, then decodes the optimal amino acid identity for each position. A key innovation is its use of random autoregressive decoding, which allows for the efficient design of symmetric proteins and the generation of diverse sequence solutions for a single backbone [20].
- Zero-Shot Application: Given a backbone structure (from de novo design or a natural protein), ProteinMPNN can generate a high-fidelity sequence in a single pass, without requiring iterative optimization. It has been successfully used to design novel proteins, including variants of TEV protease with improved activity [10].
LigandMPNN
LigandMPNN is a generalization of ProteinMPNN that explicitly incorporates the atomic context of non-protein molecules, making it indispensable for designing enzymes, binders, and sensors.
- Core Methodology: The architecture extends ProteinMPNN by adding two additional graphs: a protein-ligand graph and a fully connected ligand graph [20]. This allows the model to perform message passing between ligand atoms and from ligand atoms to protein residues, explicitly modeling interactions like hydrogen bonding and metal coordination. It was trained on protein structures with associated small molecules, nucleotides, and metals from the PDB [20].
- Zero-Shot Application and Performance: LigandMPNN enables the zero-shot design of protein sequences that bind specific small molecules, DNA, or metals. It significantly outperforms both Rosetta and the original ProteinMPNN on native sequence recovery for residues interacting with these molecules [20]. For example, its sequence recovery for residues near metals is 77.5%, compared to 40.6% for ProteinMPNN, and it has been used to design over 100 experimentally validated binders [20].
MutCompute
MutCompute is a deep learning tool focused on residue-level optimization within a structural context. It identifies probable mutations that stabilize a protein or enhance its function based on the local chemical environment.
- Core Methodology: MutCompute uses a deep neural network trained on protein structures to associate an amino acid with its surrounding chemical environment, learning the statistical preferences for specific residues in specific structural contexts [10].
- Zero-Shot Application: It predicts stabilizing or functionally beneficial substitutions in a zero-shot manner. Its success is demonstrated in projects like engineering a hydrolase for PET depolymerization, where designs from MutCompute showed increased stability and activity compared to the wild-type enzyme [10].
Table 2: Comparison of Structure-Based Protein Design Tools
| Tool | Primary Input | Core Methodology | Key Strength | Demonstrated Zero-Shot Application |
|---|---|---|---|---|
| ProteinMPNN | Protein Backbone Structure | Graph Neural Network with message passing | Fast, robust sequence design for a given backbone | Designing stable de novo proteins, enzyme variants with improved activity [10] [20] |
| LigandMPNN | Backbone + Ligand Atoms | Extension of ProteinMPNN with ligand-to-protein graphs | Designing proteins that interact with small molecules, DNA, metals | Creating high-affinity small-molecule binders and sensors [20] |
| MutCompute | Protein Structure | Deep neural network trained on local structural environments | Identifying stabilizing mutations and local functional enhancements | Engineering a PET-depolymerizing hydrolase with improved stability and activity [10] |
Integrated Experimental Protocols for Validation
The transition from in silico zero-shot designs to physically validated proteins requires robust experimental workflows. The following protocols are commonly used to test and validate the outputs of ML models.
Protocol for High-Throughput Protein Characterization using Cell-Free Systems
This protocol leverages cell-free transcription-translation (TX-TL) systems to rapidly express and test designed protein sequences, perfectly aligning with the accelerated LDBT cycle [10] [6].
- DNA Template Preparation: Designed protein sequences are codon-optimized for the expression system. DNA templates are generated via high-throughput gene synthesis or PCR assembly.
- Cell-Free Reaction Setup: Reactions are assembled in microtiter plates using commercial or homemade cell-free extracts (e.g., from E. coli). Each reaction contains the DNA template, nucleotides, amino acids, energy sources, and an appropriate buffer.
- Protein Expression: Reactions are incubated for several hours (typically 4-6 hours) at a constant temperature (e.g., 30-37°C) to allow for protein synthesis.
- Functional Assay:
- For enzymes: Reactions are supplemented with a fluorogenic or chromogenic substrate. Activity is measured by monitoring fluorescence or absorbance over time using a plate reader.
- For binding proteins (e.g., antibodies): An immunoassay (e.g., ELISA) can be configured directly in the plate. For example, following expression, the plate is transferred to a refrigerator to stop the reaction, then a target antigen is added to bind to the synthesized antibody, which is detected via a labeled secondary reagent.
- Data Analysis: Fluorescence/absorbance reads are normalized to controls. Expression levels and functional outputs are correlated with design parameters to validate model predictions and provide data for future learning cycles.
Protocol for Validating Enzyme Variants with Improved Activity
This protocol details the validation of designed enzyme variants, such as those generated by ProteinMPNN or MutCompute [10] [17].
- Cloning and Expression: Designed sequences are cloned into an appropriate expression vector (e.g., a pET vector for bacterial expression) and transformed into an expression host like E. coli BL21(DE3).
- Protein Production: Cultures are grown to mid-log phase, and protein expression is induced with IPTG. Cells are grown further before harvesting by centrifugation.
- Purification: Cell pellets are lysed, and the recombinant protein is purified using affinity chromatography (e.g., Ni-NTA resin for His-tagged proteins). The purified protein is dialyzed into an appropriate assay buffer.
- Kinetic Assay: Purified enzyme is mixed with varying concentrations of substrate in a buffer. The reaction progress is monitored spectrophotometrically or fluorometrically to measure the initial velocity (v₀) at each substrate concentration [S].
- Data Analysis: The Michaelis-Menten equation is fitted to the data (v₀ vs. [S]) to determine the catalytic efficiency (kcat/KM). A significant improvement in kcat/KM over the wild-type or parent enzyme confirms a successful design.
Essential Research Reagent Solutions
The following table details key reagents and materials essential for the build and test phases of protein design workflows.
Table 3: Key Research Reagents for Experimental Validation
| Reagent / Material | Function in Workflow | Example Application |
|---|---|---|
| Cell-Free TX-TL System | Provides the biochemical machinery for rapid, cell-free protein synthesis from DNA templates [10] [6]. | High-throughput screening of protein expression and function [10]. |
| High-Throughput Gene Synthesis | Enables the physical construction of designed DNA sequences without traditional cloning. | Generating DNA templates for hundreds of ML-designed protein variants in parallel. |
| Affinity Chromatography Resin | Rapid purification of recombinant proteins based on an affinity tag (e.g., His-tag). | Isolating designed enzyme variants for kinetic characterization [17]. |
| Fluorogenic/Chromogenic Substrate | A molecule that produces a measurable signal (fluorescence/color) upon enzyme activity. | Quantifying the catalytic activity of designed enzymes in high-throughput assays [10]. |
| Microtiter Plates & Plate Reader | The platform and detector for parallelized, high-throughput assays. | Measuring fluorescence/absorbance from hundreds of cell-free or enzymatic reactions simultaneously. |
Workflow Visualization
The following diagrams illustrate the core concepts and experimental workflows described in this whitepaper.
The DBTL vs. LDBT Cycle in Synthetic Biology
Integrated LDBT Workflow with Cell-Free Testing
The advent of powerful machine learning tools like ESM, ProGen, ProteinMPNN, and LigandMPNN is fundamentally transforming protein engineering. By enabling effective zero-shot design, these tools are catalyzing a critical shift from the iterative DBTL cycle to the predictive LDBT paradigm. This allows researchers to start with a rich foundation of knowledge learned from data, dramatically accelerating the design process for antibodies, enzymes, and gene editors. As these models continue to scale and improve—guided by real-world experimental feedback in integrated workflows—they promise to unlock a new era of precision and speed in biological design, with profound implications for drug development and biotechnology.
Synthetic biology is traditionally defined by the Design-Build-Test-Learn (DBTL) cycle, an iterative process for engineering biological systems. In this framework, the "Build and Test" phases often create a significant bottleneck, requiring time-consuming cloning, transformation, and culturing in living cells. High-throughput cell-free transcription–translation (TX-TL) systems have emerged as a disruptive technology that dramatically accelerates these phases. By using crude cellular extracts or purified components to activate in vitro transcription and translation, these systems bypass the constraints of living cells, enabling rapid protein synthesis and circuit characterization directly from DNA templates.
A transformative shift in this paradigm is the move from DBTL to LDBT (Learn-Design-Build-Test), where machine learning (ML) precedes design. In the LDBT cycle, the role of high-throughput cell-free testing becomes even more critical. It serves as the physical engine for megascale data generation required to train foundational ML models and provides the rapid validation step for zero-shot computational predictions. This synergy creates a virtuous cycle: ML models generate better designs, while high-throughput cell-free testing provides the large, high-quality datasets needed to improve those very models, effectively closing the loop between prediction and experimentation [5] [6].
The Technology of High-Throughput Cell-Free Systems
Cell-free gene expression (CFE) systems leverage the protein biosynthesis machinery from crude cell lysates or purified components to execute transcription and translation in vitro. The fundamental advantage lies in their openness and controllability. Researchers can directly manipulate the molecular environment by adding DNA templates, substrates, or inhibitors without concerns for cell viability, toxicity, or transport across cell walls [5] [21].
Recent advances have focused on enhancing the throughput, speed, and scalability of these systems. Key technological innovations include:
- Liquid Handling Robots and Biofoundries: Automated systems, such as the ExFAB, can rapidly assemble thousands of cell-free reactions in microtiter plates, standardizing the "Build" phase and enabling the testing of vast combinatorial spaces [5].
- Microfluidics: The development of picoliter-scale droplet microfluidics represents a breakthrough. Platforms like DropAI can generate and screen over 1,000,000 unique reactions per hour, reducing reagent consumption and increasing experimental density by orders of magnitude [21].
- Fluorescent Color-Coding (FluoreCode): To manage the immense combinatorial complexity, strategies like FluoreCode use distinct fluorescent colors and intensities to tag and trace the composition of each microreactor. This allows for the deconvolution of thousands of simultaneous experiments through multi-channel imaging [21].
Quantitative Performance of Modern TX-TL Systems
The capabilities of modern cell-free systems have expanded significantly, supporting a wide range of applications from basic research to biomanufacturing. The table below summarizes the key performance metrics of advanced platforms.
Table 1: Performance Metrics of Advanced Cell-Free TX-TL Systems
| TX-TL System / Platform | Key Features | Reported Protein Yield (Batch Mode) | Throughput and Scale | Primary Applications Cited |
|---|---|---|---|---|
| All-E. coli Toolbox 3.0 [22] | Incorporates endogenous E. coli transcription machinery; improved ATP regeneration | ~4 mg/mL eGFP | Standard reactions: 2-20 µL | Gene circuit prototyping, synthetic cells, bacteriophage T7 production (1013 PFU/mL) |
| DropAI Platform [21] | AI-driven, droplet microfluidics, FluoreCode tagging | 2.1-fold decrease in unit cost, 1.9-fold yield increase for sfGFP | ~1,000,000 combinations/hour; 250 pL droplets | High-throughput optimization of CFE system composition |
| Coupled TX-TL Systems [23] | Combined transcription & translation in a single reaction; market-dominant method | Not specified (valued for speed and labor reduction) | Adaptable to high-throughput well-plate formats | Enzyme engineering, pathway prototyping, therapeutic development |
Experimental Protocols for High-Throughput Screening
This section details a specific methodology for ultra-high-throughput screening using microfluidics, as exemplified by the DropAI platform.
Protocol: DropAI for Combinatorial Optimization of CFE Systems
Objective: To rapidly screen massive combinatorial libraries of CFE system components (e.g., energy sources, additives, cofactors) to identify optimal formulations for high-yield protein synthesis [21].
Materials:
- Microfluidic Device: Designed for droplet generation and merging.
- CFE Reaction Mix: E. coli or B. subtilis cell lysate, buffer, nucleotides, amino acids, and a DNA template encoding a reporter protein (e.g., sfGFP).
- Component Libraries: Satellite solutions containing the CFE additives to be screened (e.g., different energy sources, coenzyme A, NAD, tRNA).
- Fluorescent Dyes: For color-coding satellite droplets (FluoreCode system).
- Fluorinated Oil & Surfactant: For emulsion stabilization (e.g., PEG-PFPE).
- Stabilizers: Poloxamer 188 (P-188) and Polyethylene glycol 6000 (PEG-6000) to enhance emulsion stability.
- Imaging System: Fluorescence microscope with multi-channel capabilities.
Methodology:
- Droplet Library Generation:
- A microfluidic device generates a stream of "carrier" droplets (~70 µm diameter), each containing the core CFE mix with the sfGFP DNA template.
- Simultaneously, the device generates pools of "satellite" droplets (~36 µm diameter), each containing a unique set of components from the libraries to be tested. Each satellite droplet type is labeled with a unique fluorescent color and intensity (FluoreCode).
- The microfluidics system sequentially merges one carrier droplet with multiple satellite droplets (e.g., four) to create a final merged droplet that is a unique combinatorial combination.
- The FluoreCode of the merged droplet precisely identifies its biochemical composition [21].
Incubation and Expression:
- The emulsion, containing hundreds of thousands of picoliter-scale reactors, is incubated at a defined temperature (e.g., 30°C) to allow for in-droplet gene expression.
- The addition of P-188 and PEG-6000 is critical to maintain emulsion stability throughout the incubation period, preventing droplet coalescence [21].
High-Throughput Imaging and Analysis:
- After incubation, the droplets are imaged using a multi-channel fluorescence microscope.
- The FluoreCode channels are read to decode the composition of each droplet.
- The sfGFP fluorescence intensity in each droplet is measured to quantify the protein synthesis yield corresponding to each specific combination of components.
Machine Learning and In Silico Optimization:
- The experimental data (composition and yield) is used to train a machine learning model (e.g., a linear supervised model or neural network).
- The established model predicts the contribution of each component and identifies novel, high-yield combinations that were not tested experimentally.
- The top-predicted formulations are then validated in vitro, confirming the model's accuracy and finalizing the optimized CFE system [21].
Diagram 1: High-Throughput Screening with DropAI.
Successful implementation of high-throughput cell-free experiments relies on a core set of reagents and tools. The following table details essential components and their functions.
Table 2: Essential Research Reagents for High-Throughput Cell-Free TX-TL
| Reagent / Resource | Function / Description | Example Use-Cases |
|---|---|---|
| Cell Lysate | Crude extract containing the core TX-TL machinery (RNA polymerase, ribosomes, translation factors). | E. coli lysate (e.g., myTXTL [22]), B. subtilis lysate, or reconstituted systems (PURE). |
| Energy Source | Regenerates ATP to fuel transcription and translation. | Maltodextrin/d-ribose [22], phosphoenolpyruvate (PEP), creatine phosphate. |
| DNA Template | Genetic program encoding the protein or circuit to be expressed. | Plasmid DNA or linear PCR product. Can include sigma factor-specific promoters (e.g., P70a [24]) or T7 promoters. |
| Fluorescent Reporters | Proteins whose fluorescence indicates successful expression and quantifies yield. | deGFP, eGFP, sfGFP, mCherry [24] [21] [22]. |
| Stabilizers & Crowding Agents | Mimic intracellular crowding, stabilize emulsions, and improve protein folding. | PEG-6000, PEG-8000 [22], Poloxamer 188 (P-188) [21], Ficoll. |
| Microfluidic Setup | Generates, merges, and incubates picoliter-scale droplet reactors. | DropAI platform for ultra-high-throughput combinatorial screening [21]. |
High-throughput cell-free TX-TL platforms are fundamentally reshaping the synthetic biology workflow. By drastically accelerating the "Build" and "Test" phases, they are not only streamlining the traditional DBTL cycle but also serving as an essential enabler for the emerging LDBT paradigm. The integration of microfluidics, automated imaging, and machine learning creates a powerful, closed-loop system for biological design. This convergence allows researchers to move from empirical, iterative tuning toward a more predictive engineering discipline, where the massive data generated from rapid cell-free testing directly fuels learning algorithms. As these platforms become more accessible and robust, they promise to accelerate the development of novel therapeutics, biosensors, and sustainable bio-manufacturing processes, ultimately bridging the gap between digital design and physical biological systems [5] [21] [6].
Protein engineering is a cornerstone of modern biotechnology, essential for developing novel therapeutics, industrial biocatalysts, and diagnostic tools. The traditional framework for these efforts has been the Design-Build-Test-Learn (DBTL) cycle, a systematic, iterative process where researchers design a protein, build the DNA construct, test its properties, and learn from the results to inform the next design iteration [1]. However, the increasing integration of computational power is driving a paradigm shift. A new framework, Learn-Design-Build-Test (LDBT), is emerging, where machine learning (ML) models pre-trained on vast biological datasets are used to generate optimal designs from the outset, fundamentally accelerating the engineering workflow [5] [6]. This in-depth technical guide explores the application of these cycles in protein engineering, with a focused examination of strategies for enhancing key protein properties: stability, solubility, and enzymatic activity. We provide a detailed analysis of current methodologies, supported by structured data and experimental protocols, to serve researchers and drug development professionals navigating this rapidly advancing field.
DBTL vs. LDBT: A Comparative Workflow Analysis
The core distinction between the traditional DBTL cycle and the modern LDBT approach lies in the initial phase and the flow of information.
- The Traditional DBTL Cycle: This cycle begins with a Design phase based on existing domain knowledge and biophysical principles. Researchers design DNA sequences for protein variants, which are then synthesized and cloned in the Build phase. The Test phase involves expressing and characterizing these variants to measure properties like stability and activity. Finally, the Learn phase analyzes the experimental data to generate hypotheses for the next design round, creating a potentially slow, iterative loop [1].
- The Emerging LDBT Cycle: The LDBT cycle reframes this process by starting with Learning. Here, machine learning models—such as protein language models (e.g., ESM, ProGen) and structure-based tools (e.g., ProteinMPNN, Stability Oracle)—leverage massive datasets to predict sequence-structure-function relationships [5]. This learned knowledge directly informs the Design of protein variants with a high probability of success. These designs are then Built and Tested, often using rapid, high-throughput systems like cell-free protein expression [5] [6]. This "learn-first" approach can reduce the need for multiple iterative cycles, moving synthetic biology closer to a "Design-Build-Work" model [5].
The following diagram illustrates the logical sequence and key components of these two contrasting engineering cycles.
Core Strategies for Engineering Key Protein Properties
Enhancing Protein Stability
Thermal stability is a critical parameter for industrial and therapeutic enzymes, as it directly influences shelf-life, reaction rate, and operational tolerance to harsh conditions [25]. Both computational and experimental strategies are employed.
Computational & ML-Guided Tools:
- Stability Oracle: A graph-transformer model trained on stability data and protein structures that predicts the change in folding free energy (ΔΔG) upon mutation, helping to identify stabilizing mutations [5].
- Prethermut: A machine learning tool trained on thermodynamic stability changes that predicts the effects of single- or multi-site mutations on stability [5].
- B-Factor Analysis: A traditional strategy that identifies flexible regions in a protein structure (often via molecular dynamics simulations) for rigidification through mutations to reduce "wobble" and enhance stability [25].
Experimental Strategy: Short-Loop Engineering This recent strategy targets rigid "sensitive residues" within short loops, which are often overlooked by B-factor analysis that focuses on highly flexible regions [25]. The methodology involves identifying cavities in short loops and filling them with hydrophobic residues that have large side chains (e.g., Tyr, Phe, Trp, Met). This enhances stability through strengthened hydrophobic interactions and structural constraints without necessarily forming new hydrogen bonds or salt bridges [25].
Table 1: Quantitative Improvements in Enzyme Stability via Short-Loop Engineering
| Enzyme | Source Organism | Mutation | Half-Life Improvement (Fold vs. Wild-Type) | Primary Stabilizing Mechanism |
|---|---|---|---|---|
| Lactate Dehydrogenase | Pediococcus pentosaceus | A99Y | 9.5 | Cavity filling & enhanced hydrophobic interactions [25] |
| Urate Oxidase | Aspergillus flavus | Not Specified | 3.11 | Cavity filling [25] |
| D-Lactate Dehydrogenase | Klebsiella pneumoniae | Not Specified | 1.43 | Cavity filling [25] |
Detailed Protocol: Short-Loop Engineering for Stability
- Identify Short Loops: Analyze the target protein structure to identify loop regions consisting of only a few amino acid residues.
- Virtual Saturation Screening: Use a computational tool like FoldX to calculate the unfolding free energy (ΔΔG) for virtual saturation mutagenesis at every residue in the short loop. Residues where multiple mutations yield ΔΔG < 0 (stabilizing) are identified as "critical sites" [25].
- Saturation Mutagenesis & Expression: Construct a saturation mutagenesis library for the identified critical site(s) and express the variant proteins.
- Stability Assay: Measure the half-life of the variant enzymes at elevated temperatures compared to the wild type. The half-life can be determined by incubating the enzyme at a specific temperature and periodically measuring residual activity.
- Validation via Molecular Dynamics (MD): For top-performing variants, run MD simulations to confirm reduced cavity volume and increased rigidity in surrounding structural domains [25].
Improving Protein Solubility
Poor solubility can lead to protein aggregation and loss of function, making it a major challenge in recombinant protein production.
Computational & ML-Guided Tools:
- DeepSol: A deep learning-based tool that predicts protein solubility directly from the primary sequence by mapping k-mers (short sequence fragments) to solubility outcomes [5]. This allows for the in silico screening of designs before moving to the Build phase.
Optimizing Enzymatic Activity
Enhancing catalytic efficiency, substrate specificity, and enantioselectivity is central to developing effective biocatalysts.
Computational & ML-Guided Tools:
- Protein Language Models (ESM, ProGen): These models, trained on millions of evolutionary-related sequences, can predict beneficial mutations and infer function, proving adept at zero-shot design of diverse functional sequences like antibodies [5].
- MutCompute: A deep neural network that uses local structural environments to predict functionally beneficial substitutions. It has been successfully used to engineer a hydrolase for improved polyethylene terephthalate (PET) depolymerization [5].
- ProteinMPNN: A structure-based sequence design tool that, when combined with structure prediction tools like AlphaFold, has led to a nearly 10-fold increase in the success rate of designing functional proteins, such as improved TEV protease variants [5].
- De Novo Enzyme Design: Tools like Rosetta and RFdiffusion allow for the design of novel enzyme active sites from scratch, enabling the creation of catalysts for new-to-nature reactions [26].
Experimental Strategy: Ultra-High-Throughput Screening with Cell-Free Systems
- Cell-Free Protein Synthesis: This system uses transcription-translation machinery from cell lysates to express proteins directly from DNA templates, bypassing time-consuming cloning and transformation steps [5]. It is rapid (>1 g/L protein in <4 hours), scalable, and ideal for producing toxic proteins [5].
- Coupling with Microfluidics: Platforms like DropAI leverage droplet microfluidics to screen hundreds of thousands of picoliter-scale cell-free reactions, generating massive datasets for activity mapping [5].
- Application Example: Researchers have coupled cell-free synthesis with cDNA display to map the stability (ΔG) of 776,000 protein variants, creating a vast benchmark dataset for ML predictors [5].
Integrated Experimental Workflow: Combining LDBT with Cell-Free Testing
The following diagram and protocol detail an integrated workflow that merges the LDBT paradigm with high-throughput experimental validation, a powerful approach for modern protein engineering campaigns.
Detailed Protocol: ML-Guided Engineering with Cell-Free Testing
- Learn (ML-Driven Design): Input the wild-type protein sequence and structural information (experimental or from AlphaFold) into a pre-trained model like ProteinMPNN or a protein language model. Generate a library of sequence variants predicted to have improved target properties (e.g., stability, activity) [5].
- Build (Cell-Free Synthesis):
- DNA Template Prep: Synthesize the DNA sequences encoding the top ML-predicted variants. This can be done via pooled gene synthesis. For cell-free systems, these templates often only require a promoter and gene of interest, without the need for full plasmid vectors [5].
- Cell-Free Reaction: Combine the DNA templates with a cell-free transcription-translation system (e.g., derived from E. coli lysate) and incubate for several hours to express the protein variants [5].
- Test (High-Throughput Assay):
- Automation: Use liquid handling robots to set up thousands of cell-free reactions in microtiter plates [6].
- Functional Screening: For enzymatic activity, assays can be coupled to NADH depletion (measured by absorbance) or product formation (measured by HPLC or fluorescence). For stability, thermal shift assays or limited proteolysis coupled to mass spectrometry can be used in a high-throughput format [5] [26].
- Ultra-High-Throughput Option: For libraries of >100,000 variants, employ droplet microfluidics to encapsulate single DNA templates and cell-free reagents into picoliter droplets for parallelized screening and sorting [5].
- Learn (Data Integration & Model Refinement): Collect the functional data from the test phase and use it to retrain or fine-tune the initial machine learning model. This improves the model's predictive power for subsequent design rounds, creating a virtuous cycle of improvement [5] [6].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2: Key Reagents and Materials for Advanced Protein Engineering Workflows
| Item | Function/Application | Example Use-Case |
|---|---|---|
| Cell-Free Protein Synthesis System | Rapid in vitro expression of proteins from DNA templates without living cells. | High-throughput screening of ML-designed variants; expression of toxic proteins [5]. |
| Droplet Microfluidics Platform | Encapsulates single reactions in picoliter droplets for ultra-high-throughput screening. | Screening >100,000 protein variants in a single experiment (e.g., DropAI) [5]. |
| Machine Learning Software (e.g., ESM, ProteinMPNN) | Predicts protein structure, function, and optimal sequences for design. | Zero-shot design of stabilized enzymes or novel protein scaffolds [5]. |
| Automated Biofoundry | Robotic platforms that automate molecular biology steps (e.g., pipetting, cloning). | Enables fully automated DBTL/LDBT cycles; reduces human error and increases throughput [26]. |
| FoldX Software | Calculates protein stability and the effect of mutations (ΔΔG). | Virtual screening of mutation libraries to identify stabilizing substitutions (e.g., in short-loop engineering) [25]. |
The field of protein engineering is undergoing a profound transformation, driven by the convergence of machine learning and high-throughput experimental biology. The shift from a reactive DBTL cycle to a proactive LDBT framework places predictive power at the forefront of the design process. As detailed in this guide, strategies like short-loop engineering and ML-guided design are providing precise, rational methods to enhance stability, solubility, and activity. The integration of these computational approaches with rapid construction and testing platforms, such as cell-free systems and microfluidics, creates a powerful, closed-loop engineering environment. This paradigm not only accelerates the development of biocatalysts and biotherapeutics but also expands the scope of what is engineerable, paving the way for novel enzymes and functions that can address pressing challenges in medicine and industry.
The traditional Design-Build-Test-Learn (DBTL) cycle has long been the cornerstone of synthetic biology and therapeutic development. This iterative process involves designing biological parts, building genetic constructs, testing their functionality, and learning from the results to inform the next design cycle [5]. However, the Build and Test phases often create significant bottlenecks, particularly in therapeutic applications where working with living cells is time-consuming, low-throughput, and constrained by cellular viability [27]. These limitations become particularly problematic in two key areas: metabolic pathway prototyping for therapeutic compound production and the design of novel antimicrobial peptides (AMPs) to address antibiotic resistance.
A fundamental paradigm shift is emerging: the LDBT cycle (Learn-Design-Build-Test). This approach leverages advanced machine learning (ML) on existing biological datasets to generate optimized designs before any physical assembly occurs [5] [6]. When this learning-first strategy is combined with rapid cell-free testing platforms, it creates a powerful, accelerated workflow for developing biotherapeutics. This whitepaper details the technical implementation of the LDBT framework, showcasing its transformative potential in accelerating pathway prototyping and AMP design for therapeutic applications.
Technical Foundations: Core Technologies of the LDBT Framework
The Machine Learning (Learn) Component
The initial "Learn" phase utilizes sophisticated ML models trained on vast biological datasets to predict functional sequences and optimize designs. For protein and peptide engineering, key computational approaches include:
- Protein Language Models (PLMs): Models like ESM (Evolutionary Scale Modeling) and ProGen are trained on millions of natural protein sequences, allowing them to capture evolutionary constraints and generate novel, functional sequences. These can perform "zero-shot" prediction of beneficial mutations for stability and activity without additional training [5].
- Structure-Based Design Tools: Tools such as ProteinMPNN and MutCompute use deep neural networks trained on protein structures to design sequences that fold into desired backbone structures or optimize residues based on their local chemical environment [5].
- Feature-Based Prediction: For complex multifunctional molecules like AMPs, models can be fine-tuned for specific activities. The DLFea4AMPGen strategy, for instance, uses fine-tuned BERT models to predict antibacterial, antifungal, and antioxidant activities simultaneously, subsequently extracting key feature fragments that contribute to these activities [28].
These models effectively compress the "Learn" phase of multiple traditional DBTL cycles, providing a highly informed starting point for the "Design" phase.
The Build & Test Component: Cell-Free Systems
Cell-free gene expression (CFE) systems provide the rapid, high-throughput experimental platform essential for the LDBT workflow. These systems utilize the transcriptional and translational machinery from cell lysates, activated in vitro by adding energy sources and nucleotide precursors [5] [27].
Table 1: Advantages of Cell-Free Systems for Therapeutic Prototyping
| Advantage | Impact on Therapeutic Development |
|---|---|
| Speed | Protein expression and testing in hours, not days (e.g., >1 g/L protein in <4 hours) [5]. |
| High-Throughput | Scalability from picoliter droplets to manufacturing scales, enabling screening of >100,000 variants [5]. |
| Freedom from Cell Viability | Expression of toxic proteins or pathways (e.g., certain AMPs) that would kill living host cells [5]. |
| Direct Environmental Control | Precisely controlled reaction conditions for improved reproducibility and testing of compound effects [27]. |
| Open Access | Direct manipulation of the reaction environment, including incorporation of non-canonical amino acids [5]. |
The synergy is clear: ML models generate intelligent design libraries, and cell-free systems enable their ultra-rapid empirical validation. This creates a tight, fast loop where testing data can further refine the ML models, leading to continuous improvement.
Accelerating Metabolic Pathway Prototyping for Therapeutics
The production of complex therapeutic molecules often requires introducing multi-enzyme biosynthetic pathways into host organisms. The LDBT framework dramatically accelerates the debugging and optimization of these pathways.
Methodology for Cell-Free Pathway Prototyping
The following diagram illustrates the integrated LDBT workflow for prototyping a biosynthetic pathway, such as for a novel antibiotic or therapeutic compound.
Figure 1: The LDBT workflow for rapid metabolic pathway prototyping.
A key technical implementation is the "Mix-and-Match" approach for combinatorial pathway assembly [27]. This involves:
- Preparation of Specialized Lysates: Individual enzyme components are overexpressed in separate host cultures (e.g., E. coli). Cells are lysed, and the crude lysates, each enriched with a specific overexpressed enzyme, are prepared.
- Combinatorial Assembly: These lysates are mixed in various combinations to construct different pathway variants directly in the cell-free reaction.
- Rapid Testing: The assembled pathways are activated with substrates and cofactors. Product formation is quantified using high-throughput analytics like liquid chromatography-mass spectrometry (LC-MS) or coupled fluorescent assays.
This method bypasses the need to engineer a single living organism to express the entire pathway, a process that can take weeks. The entire Build-Test cycle for dozens of pathway combinations can be completed in a single day [27].
Case Study: iPROBE for Pathway Optimization
The in vitro Prototyping and Rapid Optimization of Biosynthetic Enzymes (iPROBE) methodology exemplifies the LDBT paradigm. In one application, a neural network was trained on a dataset of pathway enzyme combinations and their expression levels. The model then predicted the optimal sets to maximize the production of 3-hydroxybutyrate (3-HB), a potential platform chemical. This ML-guided design, rapidly built and tested in cell-free systems, led to a more than 20-fold improvement in titer when the optimal pathway was implemented in a Clostridium host [5].
Revolutionizing Antimicrobial Peptide (AMP) Design
The antimicrobial resistance crisis demands new antibiotics. AMPs are promising candidates, but their rational design is challenging due to the complex relationship between their sequence, structure, and activity. The LDBT framework is proving highly effective in tackling this challenge.
Methodology for Deep Learning-Driven AMP Discovery
The DLFea4AMPGen strategy provides a robust protocol for de novo AMP design, perfectly aligning with the LDBT paradigm [28]. The workflow is illustrated below.
Figure 2: The LDBT workflow for de novo design of antimicrobial peptides.
Step 1: Learn (Model Training & Feature Extraction)
- Model Fine-tuning: A pre-trained protein language model (e.g., Mindspore ProteinBERT/MP-BERT) is fine-tuned on specialized datasets for antibacterial (ABP-MPB), antifungal (AFP-MPB), and antioxidant (AOP-MPB) activities [28].
- Multifunctional Prediction: The fine-tuned models screen large peptide libraries (e.g., >23,000 peptides) to identify sequences predicted to possess multiple bioactivities.
- Interpretable AI with SHAP: The SHapley Additive exPlanations (SHAP) method is used to interpret the model. It quantifies the contribution of each amino acid position to the predicted activity, identifying Key Feature Fragments (KFFs) — typically 13-amino-acid-long fragments with the highest impact [28].
Step 2: Design (Sequence Generation & Library Construction)
- KFFs are classified into subfamilies based on phylogenetic analysis and amino acid frequency patterns.
- For each subfamily, a "plausible sequence subspace" is constructed by systematically arranging the most frequent amino acids at each position. This strategy intelligently reduces the virtual library size from an intractable 20^13 sequences to a manageable number of high-likelihood candidates [28].
Step 3: Build & Test (Rapid Synthesis and Validation)
- Selected candidate peptides are rapidly synthesized, often using cell-free systems to produce functional peptides quickly and potentially bypassing solubility issues [5].
- Peptides are tested in high-throughput assays for antimicrobial activity against a panel of drug-resistant pathogens, antifungal activity, and cytotoxicity (e.g., hemolysis) [28].
Quantitative Outcomes and Clinical Relevance
This LDBT approach yields dramatically higher success rates than traditional methods. The DLFea4AMPGen platform achieved a 75% experimental success rate, with 12 out of 16 designed peptides exhibiting at least two types of predicted activity. One designed peptide, D1, showed potent broad-spectrum activity against multidrug-resistant clinical isolates, both in vitro and in vivo in sepsis model mice [28].
Table 2: Key Design Considerations and Strategies for Clinically Viable AMPs
| Challenge | Structural-Based Design Strategy | Impact |
|---|---|---|
| Proteolytic Degradation | Incorporation of D-amino acids; Terminal acetylation/amidation; Peptide cyclization [29]. | Increased metabolic stability and extended half-life in vivo. |
| Hemolytic Toxicity | Modulation of hydrophobicity and charge; Selective amino acid substitution (e.g., reducing positive charge) [29]. | Improved therapeutic index and safety profile. |
| High Production Cost | Design of shorter peptides (<12 amino acids) retaining activity; Use of cost-effective recombinant production [30] [29]. | Economically viable manufacturing for therapeutics. |
| Bacterial Resistance | Combination therapy; Targeting intracellular targets in addition to membrane disruption [29]. | Reduced propensity for resistance development. |
Table 3: Key Research Reagent Solutions for LDBT in Therapeutics
| Reagent / Resource | Function / Purpose | Example / Specification |
|---|---|---|
| S30 or S12 T7 E. coli Extract | Core component of cell-free transcription-translation (TX-TL) systems; provides ribosomal and enzymatic machinery [27]. | Prepared from high-density cultures of engineered E. coli strains (e.g., BL21 Star); optimized for protein yield. |
| Energy Solution | Fuels the cell-free reaction by providing ATP, GTP, and energy regeneration (e.g., via phosphoenolpyruvate). | Includes amino acids, nucleotides, cofactors, and an energy source like phosphoenolpyruvate (PEP) [27]. |
| DNA Template | Encodes the gene or pathway to be expressed. | PCR product or linear DNA fragment; plasmid DNA; no need for cloning for rapid testing [5]. |
| Antimicrobial Peptide Databases | Provides curated datasets for training and benchmarking machine learning models. | APD6 (Antimicrobial Peptide Database) houses over 5,000 natural and synthetic AMP records with activity data [31]. |
| Microfluidic/Droplet System | Enables ultra-high-throughput testing by compartmentalizing reactions into picoliter volumes. | Platforms like DropAI for screening >100,000 cell-free reactions in parallel [5]. |
The integration of a learning-first LDBT paradigm with rapid cell-free testing platforms represents a transformative advancement for therapeutic development. In the critical areas of pathway prototyping for complex therapeutics and the design of novel antimicrobial peptides, this approach directly addresses the core bottlenecks of traditional DBTL cycles. By leveraging machine learning to distill knowledge from existing data and guide design, and employing cell-free systems to decouple testing from the constraints of cell growth, researchers can now iterate with unprecedented speed and scale. This accelerated workflow promises to significantly shorten the development timeline for urgently needed therapeutics, from new antibiotics to complex biopharmaceuticals, marking a new era in synthetic biology-driven medicine.
Synthetic biology is fundamentally engineered through iterative cycles of Design, Build, Test, and Learn (DBTL). This framework enables researchers to systematically design biological systems, build DNA constructs, test their performance, and learn from the data to inform the next design iteration [32]. Biofoundries have emerged as specialized facilities that automate this DBTL cycle, integrating robotic liquid handling systems, computational design, and high-throughput analytics to accelerate biological engineering [32] [33]. However, a paradigm shift is now underway, moving from the traditional DBTL cycle to a new LDBT (Learn-Design-Build-Test) framework where machine learning and prior knowledge precede initial design, potentially culminating in a single, efficient cycle that generates functional biological systems [5]. This transformation is critically enabled by closed-loop systems that integrate automation with intelligent control algorithms, creating self-optimizing experimental platforms that dramatically accelerate the pace of biological innovation [5].
Biofoundry Infrastructure and the DBTL Cycle
Core Components of a Biofoundry
Biofoundries provide integrated, automated infrastructure for high-throughput synthetic biology. Their operational mantra centers on the DBTL cycle, with each phase leveraging specific technologies [32] [33]:
- Design: Utilizes computer-aided biological design software (e.g., Cello for genetic circuits, j5 for DNA assembly)
- Build: Employs robotic liquid handling systems for automated DNA construction and strain engineering
- Test: Implements high-throughput screening and multi-omics characterization
- Learn: Applies computational modeling and bioinformatic analysis to refine designs
This integrated approach allows biofoundries to serve as nucleating hubs for industrial translation, providing accessible infrastructure for both academic researchers and commercial entities [33]. The Global Biofoundry Alliance (GBA), established in 2019 with over 30 member organizations worldwide, coordinates international efforts to standardize and advance biofoundry capabilities [32].
Quantitative Capabilities of Biofoundry Platforms
Table 1: Representative Biofoundry Output Metrics and Capabilities
| Metric Category | Exemplar Performance | Context and Application |
|---|---|---|
| Strain Engineering | 215 strains across 5 species in 90 days | DARPA challenge for 10 small molecule production [32] |
| DNA Construction | 1.2 Mb DNA built | DARPA timed pressure test [32] |
| Screening Throughput | >100,000 picoliter-scale reactions | DropAI droplet microfluidics platform [5] |
| Assay Development | 690 custom assays | Target molecule quantification in DARPA challenge [32] |
| Pathway Prototyping | 20-fold product improvement | iPROBE for 3-HB production in Clostridium [5] |
The LDBT Paradigm Shift: Learning Before Design
Theoretical Foundation
The emerging LDBT framework represents a fundamental reordering of the synthetic biology workflow, positioning "Learn" before "Design" [5]. This approach leverages machine learning models trained on vast biological datasets to make informed initial designs, potentially reducing or eliminating the need for multiple DBTL cycles:
- Zero-shot predictions: Protein language models (ESM, ProGen) enable functional protein design without additional experimental training data [5]
- Structure-based design: Tools like ProteinMPNN and MutCompute use deep neural networks to predict sequences that fold into desired structures or optimize local chemical environments [5]
- Hybrid approaches: Physics-informed machine learning combines statistical pattern recognition with biophysical principles for enhanced predictive power [5]
This paradigm shift brings synthetic biology closer to established engineering disciplines where designs are based on first principles and proven models, potentially achieving a "Design-Build-Work" outcome in a single cycle [5].
Implementation Workflow
LDBT Workflow: Machine learning precedes biological design
Closed-Loop Systems in Biofoundries
Principles of Closed-Loop Control
Closed-loop systems in biofoundries implement continuous feedback control between testing and design phases. These systems automatically adjust experimental parameters based on real-time measurements, creating self-optimizing platforms [5]. The fundamental control principle involves:
- Measurement: High-throughput sensors quantify biological performance (e.g., metabolite concentrations, fluorescence)
- Interpretation: Algorithms compare results against target specifications
- Actuation: Parameters are automatically adjusted (e.g., DNA sequence designs, pathway compositions, expression levels)
- Iteration: The cycle repeats until performance targets are met
This approach mirrors physiological closed-loop controlled (PCLC) medical devices that automatically adjust physiological variables through feedback from physiological sensors [34].
Implementation Architectures
Table 2: Closed-Loop Control Modalities in Biofoundries
| Control Type | Mechanism | Biofoundry Application |
|---|---|---|
| AI-Directed | Machine learning agents use predictive models to select next experiments | Fully automated DBTL cycles with minimal human intervention [32] |
| Optimization-Based | Algorithms like PID controllers adjust parameters to minimize performance error | Titration of enzyme expression levels in metabolic pathways [5] |
| Model-Predictive | Physics-informed ML models forecast outcomes to guide design | iPROBE pathway optimization using neural networks [5] |
| Adaptive Sampling | Active learning selects informative experiments for model training | Protein engineering campaigns using iterative saturation mutagenesis [5] |
Enabling Technologies for Automated Biofoundries
Cell-Free Expression Systems
Cell-free platforms have become critical biofoundry components that enable rapid building and testing phases [5]. These systems leverage transcription-translation machinery from cell lysates or purified components to express proteins without living cells, offering distinct advantages for automation:
- Speed: Protein production (>1 g/L) in under 4 hours [5]
- Scalability: Operation from picoliter to kiloliter scales [5]
- Tolerance: Production of toxic proteins impossible to make in vivo [5]
- Flexibility: Direct manipulation of reaction conditions and addition of non-canonical components [5]
When combined with liquid handling robots and microfluidics, cell-free systems enable ultra-high-throughput testing of thousands of protein variants or pathway configurations [5].
Machine Learning and AI Integration
Machine learning has become the driving force behind modern biofoundries, transforming their operational capabilities [5] [32]:
- Protein language models: Capture evolutionary dependencies in amino acid sequences for structure-function prediction
- Fitness landscape mapping: Models sequence-activity relationships across chemical space to engineer specialized enzymes
- Pathway optimization: Neural networks predict optimal enzyme combinations and expression levels for metabolic engineering
- Closed-loop AI agents: Autonomous systems that cycle through design-build-test iterations with minimal human intervention [5]
The integration of ML creates a virtuous cycle where high-throughput biofoundry data trains better models, which in turn design more effective experiments.
Research Reagent Solutions for Automated Biofoundries
Table 3: Essential Research Reagents for Biofoundry Operations
| Reagent / Material | Function | Application Example |
|---|---|---|
| Cell-Free Expression Kits | In vitro transcription and translation | Rapid protein synthesis without cloning [5] |
| DNA Assembly Master Mixes | Automated DNA construction | High-throughput plasmid assembly (e.g., Golden Gate, Gibson) [32] |
| Biosensors | Real-time metabolite monitoring | Closed-loop control of pathway flux [5] |
| Protein Stability Reagents | High-throughput stability mapping | ∆G calculations for 776,000 protein variants [5] |
| Non-Canonical Amino Acids | Expanded genetic code | Incorporation of novel functionalities into proteins [5] |
| Automated DNA Synthesis Kits | Oligo pool generation | Library construction for directed evolution [5] |
Experimental Protocols for Automated Biofoundries
Ultra-High-Throughput Protein Engineering Protocol
This protocol combines cell-free expression with cDNA display for protein stability mapping, enabling characterization of hundreds of thousands of variants [5]:
- Design: Generate variant library using zero-shot predictions from protein language models (ESM, ProGen) or structure-based tools (ProteinMPNN)
- Build:
- Synthesize DNA templates via automated oligo pool synthesis
- Express protein variants in picoliter-scale cell-free reactions (≥1 million reactions per day)
- Test:
- Couple expressed proteins to cDNA via puromycin linkage
- Apply stability challenge (e.g., thermal denaturation, proteolysis)
- Separate folded/unfolded populations via affinity selection
- Sequence surviving variants via high-throughput sequencing
- Learn:
- Calculate ∆G values for each variant
- Train stability prediction models on dataset
- Update design rules for next iteration
This protocol generated stability data for 776,000 protein variants, creating benchmark datasets for zero-shot predictor evaluation [5].
Closed-Loop Metabolic Pathway Optimization
The iPROBE (in vitro prototyping and rapid optimization of biosynthetic enzymes) protocol enables rapid pathway optimization [5]:
- Learn Phase:
- Curate training set of pathway enzyme combinations and expression levels
- Train neural network on substrate-product relationships
- Design Phase:
- Use trained model to predict optimal pathway sets
- Design DNA constructs with varying enzyme ratios
- Build Phase:
- Assemble pathway variants via automated DNA assembly (e.g., using j5 software)
- Express in cell-free system or rapid prototyping chassis
- Test Phase:
- Measure product titers via high-throughput analytics (LC-MS, fluorescence)
- Feed data back to update model parameters
- Automatically select next design round based on performance
This approach achieved over 20-fold improvement in 3-HB production in Clostridium hosts [5].
Case Studies and Performance Metrics
DARPA Biofoundry Challenge
The Defense Advanced Research Projects Agency (DARPA) administered a timed pressure test requiring a biofoundry to research, design, and develop strains to produce 10 small molecules in 90 days [32]. The challenge demonstrated biofoundry capabilities:
- Molecule diversity: Targets ranged from simple chemicals (1-hexadecanol) to complex natural products (epicolactone) and compounds with no known biological synthesis (tetrahydrofuran)
- Output metrics: Construction of 1.2 Mb DNA, 215 strains across five species, 690 custom assays
- Success rate: Target molecule or close analog produced for 6 of 10 molecules
- Approach flexibility: Employed diverse strategies including native pathway engineering, heterologous expression, and cell-free systems
This case illustrates how biofoundries can tackle complex, multifaceted challenges under demanding constraints [32].
Automated Antimicrobial Peptide Discovery
A closed-loop biofoundry workflow integrated deep learning with cell-free expression for antimicrobial peptide (AMP) discovery [5]:
- Learn: Computationally survey over 500,000 AMP sequences using deep learning models
- Design: Select 500 optimal variants balancing multiple properties (activity, toxicity, stability)
- Build: Synthesize and express selected variants in cell-free system
- Test: Evaluate antimicrobial activity and cytotoxicity
- Output: Identify 6 promising AMP designs with validated activity
This approach demonstrates the LDBT paradigm, where machine learning precedes physical experimentation to efficiently navigate vast design spaces.
Implementation Diagram for Closed-Loop Biofoundry
Closed-loop automation in biofoundries
Future Perspectives and Challenges
The integration of automation through biofoundries and closed-loop systems faces several development frontiers [32] [33]:
- Data standardization: Historical biological data often lacks standardized measurement and annotation required for optimal machine learning
- Analytical bottlenecks: High-throughput analytics remain a limitation for full DBTL automation
- Model generality: Developing ML models that transfer across biological systems and contexts
- Infrastructure access: Expanding biofoundry access to academic and industrial researchers
- Regulatory frameworks: Establishing standards for biologically manufactured products
The Global Biofoundry Alliance is actively addressing these challenges through working groups on metrology, reproducibility, and data quality [32] [33]. As these infrastructures mature, biofoundries are poised to dramatically accelerate the engineering biology innovation pipeline, supporting the growing bioeconomy through more sustainable and efficient biomanufacturing processes.
Navigating Challenges: Practical Hurdles and Optimization Strategies for LDBT
The paradigm for engineering biological systems is undergoing a fundamental shift. The traditional Design-Build-Test-Learn (DBTL) cycle, while systematic, relies heavily on empirical iteration, making it time-consuming and resource-intensive [5]. Emerging from synthetic biology research is a proposed reordering to the Learn-Design-Build-Test (LDBT) cycle, where machine learning (ML) and foundational models precede physical construction [5]. This paradigm shift places unprecedented importance on data strategy. The success of LDBT hinges on the creation of high-quality, megascale datasets that can train models capable of accurate zero-shot predictions, ultimately aiming for a "Design-Build-Work" model akin to more established engineering disciplines [5]. This technical guide details the strategies for assembling the data foundation required to power this transition, addressing the critical balance between data quality and quantity for researchers and drug development professionals.
Data Quality vs. Quantity: Finding the Goldilocks Zone
The construction of effective foundational models requires a strategic balance between the volume and the caliber of data. The relationship between these two factors is not linear, and understanding their interaction is crucial for efficient resource allocation.
The Role of Data Quality
Data quality encompasses attributes such as accuracy, reliability, consistency, and completeness [35]. High-quality data provides the foundation for accurate predictions and reliable models. The adage "garbage in, garbage out" is particularly salient in machine learning; noise, outliers, and irrelevant attributes within a dataset can lead to inaccurate results and misleading biological insights [35]. Furthermore, biased or poor-quality data can have severe consequences, as demonstrated by cases where ML models perpetuated societal biases in hiring, facial recognition, and healthcare risk algorithms, leading to discriminatory outcomes and significant financial costs [36].
The Role of Data Quantity
The amount of data required depends on the complexity of the biological problem, the algorithm employed, and the number of features in the dataset [35]. In general, more data can increase model accuracy, as it allows the algorithm to learn more robust patterns and generalize better to unseen data. This is especially true for building foundational models that aim to capture the complex relationships in biological sequences, structures, and functions [5]. The drive for megascale data generation is a key motivation behind adopting high-throughput cell-free platforms, which can generate data for hundreds of thousands of protein variants [5].
Strategic Balance and the "Goldilocks Zone"
Striking the right balance is paramount. An excessive volume of poor-quality data can overwhelm resources and complicate models without improving performance, while too little data will fail to capture the underlying complexity [35]. The "Goldilocks Zone" represents the optimal balance where the dataset is sufficiently large and diverse to be representative, yet of high enough quality to be reliable. The concept of a data flywheel is critical here: starting with a well-structured, high-quality dataset improves model performance, which in turn can be used to generate more high-quality data more efficiently, creating a virtuous cycle of improvement [36]. In the context of DBTL cycles, research suggests that when the number of strains to be built is limited, starting with a larger initial cycle is favorable over distributing the same number of strains evenly across multiple cycles, as it provides a richer initial dataset for the learning phase [3].
Table 1: Comparison of Data Quality versus Data Quantity
| Aspect | Data Quality | Data Quantity |
|---|---|---|
| Primary Focus | Accuracy, consistency, completeness, and relevance of data points [35]. | Volume and scale of the collected data. |
| Key Risk | Biased, inaccurate, or noisy data leading to flawed models and erroneous conclusions [36]. | Insufficient data failing to capture the complexity of the biological system, leading to overfitting. |
| Impact on Model | Directly affects the reliability, fairness, and real-world applicability of predictions [36]. | Influences the model's ability to generalize and identify complex, non-intuitive patterns [3]. |
| Acquisition Focus | Rigorous curation, validation, cleaning, and normalization processes. | High-throughput technologies, automated data generation, and scalable experimental platforms. |
Megascale Data Generation: Experimental Methodologies and Protocols
Accelerating the Build-Test phases is critical for megascale data generation. Here, cell-free systems and biofoundries have emerged as transformative technologies.
Cell-Free Protein Synthesis for Ultra-High-Throughput Testing
Cell-free gene expression (CFE) platforms leverage the protein biosynthesis machinery from cell lysates or purified components to activate in vitro transcription and translation [5]. This methodology offers several distinct advantages for data generation:
- Speed and Scalability: CFE is rapid, producing over 1 g/L of protein in less than 4 hours, and is readily scalable from picoliter to kiloliter scales [5]. This enables the testing of a massive number of genetic designs orders of magnitude faster than in vivo methods.
- Modularity and Control: Synthesized DNA templates can be directly added to the system without time-consuming cloning steps. The reaction environment is highly modular, allowing for facile customization and the incorporation of non-canonical amino acids [5].
- Tolerance to Toxicity: CFE enables the production of proteins or pathways that would be toxic to a live cell, expanding the functional space that can be experimentally probed [5].
Protocol: Ultra-High-Throughput Protein Stability Mapping
This protocol details a method for generating stability data (ΔG) for hundreds of thousands of protein variants, creating a vast dataset for training or benchmarking machine learning models [5].
- Library Design: Design a DNA library encoding the protein variants of interest (e.g., via site-saturation mutagenesis).
- Cell-Free Expression: Express the protein variants in a picoliter-scale cell-free reaction system.
- cDNA Display: Couple the in vitro synthesized proteins to their encoding mRNA via a puromycin linker, creating a stable protein-cDNA fusion.
- Denaturant Titration: Subject the protein-cDNA fusions to a gradient of a chemical denaturant (e.g., guanidinium chloride).
- Stability Measurement: Use a functional assay or a binding assay to determine the fraction of unfolded protein at each denaturant concentration. The cDNA tag allows for the amplification and sequencing of variants that remain folded at a given condition.
- Data Analysis: Fit the denaturation data to a thermodynamic model to calculate the folding free energy (ΔG) for each variant.
The dataset generated from this protocol—such as the 776,000 protein variants cited—is quintessential for building foundational models of protein stability [5].
Biofoundries and Automated Workflows
Biofoundries integrate automation, robotics, and data management to execute DBTL cycles at a massive scale. They are increasingly leveraging cell-free platforms alongside high-throughput in vivo workflows [5]. A key methodology is the use of closed-loop systems where AI agents design experiments, robots build and test the constructs, and the resulting data is automatically fed back to the AI to inform the next round of designs [5]. This automation is critical for achieving the megascale required for robust model training.
Table 2: Essential Research Reagent Solutions for Megascale Data Generation
| Reagent / Solution | Function in Experimental Protocol |
|---|---|
| Cell-Free Extract | The core catalytic machinery for in vitro transcription and translation, typically derived from E. coli, wheat germ, or insect cells [5]. |
| Energy Regeneration System | A mix of compounds (e.g., phosphoenolpyruvate, creatine phosphate) to sustain ATP levels during prolonged cell-free reactions. |
| Non-Canonical Amino Acids | Enables the incorporation of novel chemical functionalities into proteins, expanding the diversity of sequences and functions that can be explored [5]. |
| cDNA Puromycin Linker | A critical reagent for cDNA display protocols, creating a physical link between a synthesized protein and its genetic code for high-throughput screening [5]. |
| Droplet Microfluidics Chips | Enables the partitioning of reactions into millions of picoliter-scale droplets, allowing for ultra-high-throughput screening of enzymatic activities or binding events [5]. |
A Framework for Data Management and Model Training
With megascale data generated, a structured framework for management and learning is essential to translate data into predictive power.
Simulated DBTL Cycles for Model Benchmarking
A significant challenge in evaluating ML methods for synthetic biology is the lack of public multi-cycle datasets. A proposed solution is a mechanistic kinetic model-based framework [3]. This involves:
- Model Construction: Developing a detailed kinetic model of a metabolic pathway embedded within a physiologically relevant cell model (e.g., an E. coli core kinetic model).
- In Silico Library Generation: Simulating the effect of combinatorial perturbations (e.g., changes in enzyme concentrations via promoter/RBS libraries) on the product flux.
- Cycle Simulation: Using the kinetic model as a "ground truth" simulator to test different ML algorithms and DBTL strategies over multiple virtual cycles. This allows for the systematic comparison of methods like gradient boosting and random forests, and has shown these methods to be robust to training set biases and experimental noise in the low-data regime [3].
Machine Learning Integration and Recommendation Algorithms
Machine learning transforms the "Learn" phase from a retrospective analysis to a predictive and generative engine.
- Zero-Shot Prediction: Pre-trained protein language models (e.g., ESM, ProGen) and structure-based models (e.g., ProteinMPNN, AlphaFold) can generate functional designs without additional experimental data from the target system, effectively enabling the LDBT cycle [5].
- Active Learning: This ML technique reduces the required data volume by allowing the model to strategically query the most informative data points for experimental testing. This creates a more efficient DBTL cycle by focusing resources on designs that will maximize learning [35].
- Automated Recommendation: Algorithms can be designed to propose new strain designs for the next DBTL cycle. These typically use an ensemble of ML models to create a predictive distribution of performance and sample new designs based on a balance of exploration (testing uncertain regions of design space) and exploitation (improving high-performing designs) [3].
The diagram below illustrates the workflow of this integrated, data-centric framework.
The transition from DBTL to LDBT cycles represents a fundamental evolution in synthetic biology and metabolic engineering, positioning machine learning and data as the primary drivers of biological design. Success in this new paradigm is contingent on a strategic and integrated approach to data. This requires not only the generation of megascale datasets through advanced experimental platforms like cell-free systems and biofoundries but also an unwavering commitment to data quality and a structured framework for managing the learning process. By finding the "Goldilocks Zone" between data quantity and quality, leveraging simulated environments for benchmarking, and implementing intelligent recommendation systems, researchers can build the powerful foundational models needed to realize the promise of predictive biological engineering.
The central aspiration of synthetic biology—to rationally reprogram organisms with predictable outcomes—is fundamentally challenged by the inherent complexity of biological systems. This complexity manifests as context-dependence, where genetic parts function differently across cellular environments, and unforeseen interactions between synthetic constructs and host machinery [37] [38]. Traditionally, the field has relied on the Design-Build-Test-Learn (DBTL) cycle, an iterative engineering workflow. However, the "Learn" phase often constitutes a bottleneck, as extracting definitive design principles from complex, heterogeneous biological data has proven difficult [37]. This limitation has sustained a reliance on empirical iteration rather than predictive design.
A paradigm shift is emerging to address this core challenge. Recent advances propose reordering the cycle to LDBT (Learn-Design-Build-Test), where machine learning (ML) leverages vast biological datasets before the design phase [5] [6]. This learning-first approach aims to embed predictive power at the outset of the design process, enabling in silico models to better navigate biological complexity. This guide details the technical strategies and methodologies for implementing this approach, providing researchers with a framework to manage context-dependence and unforeseen interactions computationally, thereby accelerating the path to high-precision biological design.
The Paradigm Shift: From DBTL to LDBT
The traditional DBTL cycle begins with a design hypothesis based on existing knowledge. Researchers then build the DNA construct, test it in a biological system (in vivo or in vitro), and finally learn from the results to inform the next design iteration [5] [37]. This process, while systematic, can be slow, and its success is often constrained by the designer's initial assumptions and the limited scope of testable designs.
The LDBT cycle inverts this process. It starts with a comprehensive Learning phase, where machine learning models are trained on large-scale biological data—from public databases, proprietary datasets, or high-throughput experiments—to learn the complex relationships between DNA sequence, biological context, and functional output [5] [6]. This learned model then directly informs the Design of new genetic parts or systems. The subsequent Build and Test phases serve to validate the computational predictions and, crucially, to generate new high-quality data that can be fed back to further refine the ML models.
This shift is transformative because it uses computational power to pre-navigate the vast biological design space. By learning from data first, the LDBT cycle mitigates the trial-and-error nature of traditional DBTL, reducing the number of iterative cycles needed to achieve a functional system and helping to manage complexity from the outset [5].
In Silico Strategies for Managing Biological Complexity
Machine Learning for Predictive Design
Machine learning models are uniquely suited to disentangle the high-dimensional, non-linear relationships that characterize biological systems. They can be trained on diverse data types to predict functional outcomes, thereby managing complexity in silico.
- Protein Language Models (e.g., ESM, ProGen): These models, trained on millions of evolutionary-related protein sequences, learn the "grammar" of proteins. They enable zero-shot prediction of protein function, stability, and mutations without additional experimental data, effectively capturing long-range dependencies within sequences that are difficult to model with traditional biophysics [5].
- Structure-Based Models (e.g., ProteinMPNN, MutCompute): These tools use deep neural networks trained on protein structures to design sequences that fold into desired backbone structures (ProteinMPNN) or to identify stabilizing mutations given a local chemical environment (MutCompute). When coupled with structure prediction tools like AlphaFold, they significantly increase the success rate of protein design [5].
- Function-Specific Predictors: Specialized ML tools target specific protein properties critical for application success. For instance, Prethermut and Stability Oracle predict the thermodynamic stability changes (ΔΔG) upon mutation, while DeepSol predicts protein solubility from sequence. These predictors help screen out non-functional variants before moving to the experimental stage [5].
Accounting for Isoform Diversity in Drug Targeting
Biological complexity is exemplified by alternative splicing, which can generate multiple protein isoforms from a single gene with distinct, sometimes opposing, functions. A purely gene-centric view can miss critical interactions. An in silico analysis of cancer drug-target interactions revealed that 76% of drugs either miss a potential target isoform or target other isoforms with varied expression in normal tissues, potentially explaining off-target effects or lack of efficacy [39].
Methodology for In Silico Isoform Analysis:
- Data Curation: Integrate drug-target interaction data from sources like the Drug Gene Interaction Database (DGIdb) with isoform information from Ensembl and protein structure data from the PDB.
- Binding Site Mapping: Extract sequence-level ligand binding sites from structural data. Perform multiple sequence alignments of all protein isoforms from the same gene against the canonical binding site sequence.
- Expression Analysis: Analyze transcript-specific expression (e.g., using RNA-Seq TPM data from TCGA and GTEx) across relevant normal and diseased tissues to understand isoform context.
- Structural Modeling: Construct 3D homology models of different isoforms and analyze differences in ligand binding pocket architectures (size, shape, electrostatics) that could affect drug binding [39].
Table 1: Impact of Alternative Splicing on Drug-Target Interactions (Case Study on Cancer Drugs)
| Metric | Finding | Implication for Drug Discovery |
|---|---|---|
| Genes with ≥5 Protein Isoforms | 618 out of 1,434 drug-target genes | Highlights the prevalence of proteome diversity overlooked in a one-gene-one-target model. |
| Drugs with Potential Off-target Isoform Effects | 76% of analyzed drugs | Suggests a major contributor to unexpected toxicities or lack of clinical efficacy. |
| Key Example: VEGFA | Isoform switching from anti-angiogenic (VEGFA165b) to pro-angiogenic (VEGFA165) in cancer. | Targeting the canonical isoform without context may be ineffective or counterproductive. |
| Key Example: BCL2L1 | Switching from pro-apoptotic (Bcl-xs) to anti-apoptotic (Bcl-xl) enables cancer cell survival. | Drugs designed against one isoform may not work if a functional switch occurs. |
De Novo Design of Context-Responsive Genetic Parts
Promoters are a classic example of context-dependent parts. A billboard model of promoter regulation, where transcription factor regulatory elements (TFREs) act as independent, additive modules, enables rational design. Research has shown that by profiling host cell transcription factor expression and identifying non-cooperative, modular TFREs, researchers can design synthetic promoters in silico with predictable activities in specific contexts, such as CHO cells for biopharmaceutical production [38].
Experimental Protocol for Billboard Promoter Design:
- Host Context Profiling: Perform RNA-seq on the host cell line (e.g., CHO-K1) across different growth phases and conditions. Identify stably expressed transcription factors (TFs) [38].
- TFRE Selection: Obtain cognate binding sites (TFREs) for the stably expressed TFs from databases (e.g., TFcheckpoint). Test these TFREs in varying homotypic and heterotypic promoter architectures to identify sets that display position-insensitive, additive function [38].
- Model Building: Construct a library of heterotypic promoters. Measure promoter activity (e.g., using a SEAP reporter gene) and use multiple linear regression to model activity as a function of TFRE copy numbers. The regression coefficients (β) represent the relative transcriptional activity of a single copy of each TFRE [38].
- In Silico Design & Validation: Use the model to design new promoter sequences in silico by combining TFREs to achieve a target activity level. Build and test the designed promoters to validate predictive accuracy [38].
The Experimental Engine: Cell-Free Systems for Rapid Model Validation and Training
To close the LDBT loop, the Build-Test phases must be rapid and high-throughput. Cell-free transcription-translation (TX-TL) systems are ideal for this role. These systems use the protein biosynthesis machinery from cell lysates or purified components to express proteins without living cells [5] [6].
Advantages for Managing Complexity:
- Speed: Protein production and testing can be completed in hours, not days [5].
- Decoupling: Removes the confounding effects of cell viability, metabolic burden, and genetic instability, simplifying the system and yielding more interpretable data [6].
- High-Throughput: Can be integrated with liquid handling robots and microfluidics to screen thousands of variants (e.g., >100,000 reactions in DropAI) [5].
- Control: The reaction environment is highly controllable, allowing for the precise manipulation of conditions to study context-dependence systematically [5].
These systems are not just for validation; they are powerful data generators. By enabling ultra-high-throughput testing of protein variants or genetic circuits, they produce the large, high-quality datasets needed to train and refine the machine learning models at the start of the LDBT cycle [5].
A Practical Toolkit for Implementation
Table 2: Research Reagent Solutions for an LDBT Workflow
| Reagent / Tool Category | Specific Examples | Function in Workflow |
|---|---|---|
| Machine Learning Models | ESM, ProGen, ProteinMPNN, MutCompute, Prethermut, Stability Oracle, DeepSol | Perform zero-shot or data-informed design of proteins and genetic systems, predicting function and stability to navigate complexity in silico. |
| Cell-Free Protein Synthesis System | E. coli lysate, CHO lysate, PURExpress | Provide a rapid, high-throughput platform for building and testing designed genetic constructs without the noise of a living cell. |
| High-Throughput Screening Platform | Droplet microfluidics, Automated liquid handling robots (in biofoundries) | Enable the testing of thousands of design variants in parallel, generating the megascale data required for training robust ML models. |
| Data Resources | Drug Gene Interaction Database (DGIdb), Ensembl, BioLiP, TCGA, GTEx | Provide foundational data on drug-target interactions, protein isoforms, structures, and tissue-specific expression for model training and context-analysis. |
Overcoming biological complexity is the defining challenge of predictive biological design. The framework outlined here—centered on the LDBT cycle—provides a robust roadmap. By leveraging machine learning to learn first from large datasets, designing with isoform and context-specificity in mind, and validating with rapid cell-free systems, researchers can systematically manage context-dependence and unforeseen interactions. This integrated, in silico-driven approach is critical for accelerating the development of robust synthetic biology applications, from engineered therapeutics to sustainable bioproduction.
Synthetic biology has long been governed by the Design-Build-Test-Learn (DBTL) cycle, an iterative framework for engineering biological systems. However, recent technological advancements are prompting a fundamental rethinking of this paradigm. The convergence of artificial intelligence (AI) and cell-free protein synthesis (CFPS) platforms is enabling a new, more efficient approach: the Learn-Design-Build-Test (LDBT) cycle [5] [6] [40]. In this reordered framework, the process begins with machine learning (ML) models that leverage vast biological datasets to inform and optimize designs before any physical building occurs [5]. This learning-first approach is particularly powerful when combined with the speed and flexibility of cell-free systems for the subsequent Build and Test phases.
Cell-free systems have emerged as a transformative technology by decoupling gene expression from the constraints of living cells [41]. These platforms utilize the transcriptional and translational machinery from cell lysates or purified components to synthesize proteins in vitro, offering unprecedented control over the reaction environment [41]. This technical guide explores the cutting-edge methodologies for optimizing these systems along three critical dimensions: scalability, cost-efficiency, and reaction fidelity, positioning them as the engine for next-generation synthetic biology workflows within the emerging LDBT paradigm.
Core Concepts: DBTL vs. LDBT in Synthetic Biology
The traditional DBTL cycle begins with researchers designing biological parts based on domain knowledge and objectives [5]. These designs are then built (e.g., through DNA synthesis and assembly) and introduced into living cells for testing. The resulting data is analyzed during the Learn phase to inform the next design iteration [5]. While effective, this approach often requires multiple, time-consuming cycles to achieve desired functions, with the Build-Test phases acting as a particular bottleneck [5].
The LDBT cycle represents a paradigm shift by placing Learning at the forefront [5] [6]. Powered by ML, this initial phase utilizes pre-trained models on megascale biological datasets—including millions of protein sequences and structures—to generate high-quality, zero-shot predictions for optimal designs [5]. This computational "Learning" precedes the physical "Design" of genetic constructs. The subsequent "Build" and "Test" phases are dramatically accelerated using CFPS platforms, which enable rapid in vitro expression and validation without the need for cellular cloning and cultivation [5] [41]. This reordering, from DBTL to LDBT, aims to transform synthetic biology into a more predictive engineering discipline, reducing reliance on empirical iteration and moving closer to a "Design-Build-Work" model [5].
Table 1: Comparison of DBTL and LDBT Cycles in Synthetic Biology
| Cycle Phase | Traditional DBTL Cycle | LDBT Cycle |
|---|---|---|
| Entry Point | Design based on domain knowledge and objectives [5] | Learn from large datasets using machine learning models [5] [6] |
| Key Technologies | Computational modeling, DNA synthesis, in vivo chassis [5] | Protein language models (e.g., ESM, ProGen), structure-based tools (e.g., ProteinMPNN) [5] |
| Build Phase | DNA assembly and introduction into living cells (bacteria, yeast, etc.) [5] | Rapid in vitro expression using cell-free transcription-translation (TX-TL) systems [5] [41] |
| Test Phase | Measurement in living systems, which can be slow and constrained by cellular viability [5] | High-throughput testing in cell-free systems, enabling direct control of reaction conditions [5] [6] |
| Primary Advantage | Systematic, iterative framework | Potential for single-cycle success via predictive design and rapid testing [5] |
| Primary Challenge | Build-Test phases can be slow, requiring multiple iterations [5] | Dependence on quality and scale of training data for machine learning models [5] |
Optimization Strategies for Cell-Free Systems
Enhancing Scalability and Throughput
Achieving scalability in CFPS involves moving from microliter-scale reactions in academic labs to industrially relevant volumes and throughput. Key strategies include:
Integration with Automation: Liquid-handling robots and digital microfluidics enable the precise setup of thousands of parallel cell-free reactions [41]. This automation is crucial for screening large DNA libraries, such as those generated during the ML-guided "Design" phase of LDBT. Biofoundries are increasingly leveraging these automated CFPS workflows to accelerate the DBTL cycle [41].
Miniaturization and Microfluidics: Technologies like droplet microfluidics allow the encapsulation of individual cell-free reactions in picoliter-volume droplets [5]. This approach, as demonstrated by the DropAI platform, enables the screening of over 100,000 distinct reactions in a single experiment, generating the massive datasets required for training and refining ML models [5] [41].
Lyophilization for Stability and Distribution: Freeze-drying (lyophilization) of pre-assembled cell-free reactions creates stable, shelf-stable pellets that can be rehydrated on demand [41]. This not only simplifies workflow logistics but also facilitates the distribution of standardized cell-free platforms to diverse settings, supporting the democratization of synthetic biology [41].
Improving Cost-Efficiency
The perceived high cost of CFPS has been a barrier to its widespread adoption. Optimization focuses on reducing reagent costs and increasing protein yield:
Crude Lysate Optimization over Reconstituted Systems: While fully reconstituted systems like the PURE system offer high control, they are prohibitively expensive for large-scale applications [41]. Using optimized crude cell lysates from organisms like E. coli is a more cost-effective strategy. Research focuses on improving extract preparation protocols to maximize the activity and longevity of the transcriptional-translational machinery while minimizing costs [41].
Enhanced Energy Regeneration Systems: Cell-free reactions require a constant supply of energy (e.g., ATP). Moving from expensive energy sources like phosphoenolpyruvate (PEP) to more cost-effective alternatives like creatine phosphate or maltodextrin-based systems significantly reduces operational costs and extends reaction duration, thereby improving yield [41].
High-Yield Reaction Optimization: Increasing the protein yield per unit volume of reaction directly improves cost-effectiveness. This is achieved by optimizing the "master mix"—fine-tuning the concentrations of essential components like magnesium ions (Mg²⁺), potassium ions (K⁺), nucleoside triphosphates (NTPs), and amino acids [41]. Such optimization can yield more than 1 gram of protein per liter of reaction in under 4 hours [5].
Ensuring Reaction Fidelity and Control
Reaction fidelity refers to the accuracy of protein synthesis and the reliability of the system in reporting on the designed function. Key optimization areas include:
Source and Quality of Lysates: The choice of lysate source (e.g., E. coli, wheat germ, insect cells, mammalian systems) dictates the fidelity of protein production, particularly for complex eukaryotic proteins requiring specific post-translational modifications [41] [42]. Matching the lysate source to the protein of interest is critical.
Tunable Reaction Environment: The open nature of CFPS allows for direct manipulation of the reaction biochemistry. This includes the incorporation of:
Standardization and Reproducibility: The use of defined, purified components and standardized protocols minimizes batch-to-batch variability in lysate production and reaction assembly. This is essential for generating high-quality, reproducible data for ML model training and validation within the LDBT framework [6].
Table 2: Key Optimization Targets for Cell-Free Systems
| Optimization Dimension | Key Parameters | Impact on Performance |
|---|---|---|
| Scalability | Reaction volume (pL to kL), degree of automation, integration with microfluidics [5] [41] | Determines screening throughput and feasibility for industrial biomanufacturing |
| Cost-Efficiency | Lysate source and preparation, energy system efficiency, protein yield per unit cost [41] | Affects accessibility and economic viability for large-scale applications |
| Reaction Fidelity | Lysate source (E. coli, wheat germ, mammalian), ability to perform PTMs, control over redox environment [41] [42] | Determines the functional accuracy of synthesized proteins, especially complex biologics |
| Data Generation for ML | Reproducibility, quantitative output (e.g., fluorescence, enzymatic activity), compatibility with high-throughput readouts [5] [6] | Critical for creating high-quality datasets to train and validate machine learning models in the LDBT cycle |
Experimental Protocols for Optimization
High-Throughput Screening of Enzyme Variants Using CFPS
This protocol is designed for the rapid "Test" phase in an LDBT cycle, where thousands of ML-designed enzyme variants need to be functionally characterized.
- DNA Template Preparation: Use a cell-free compatible plasmid vector or linear PCR amplicon as the DNA template. The design, informed by ML models (e.g., ProteinMPNN, ESM), should incorporate a strong, cell-free compatible promoter (e.g., T7) [5] [41].
- CFPS Reaction Assembly in Microplates: On a liquid-handling robot, dispense a standardized CFPS master mix into a 384-well microplate. The master mix should consist of:
- Cell extract (e.g., E. coli S30 extract)
- Energy solution (creatine phosphate or maltodextrin-based)
- Amino acids mix (including any non-canonical amino acids if required)
- Cofactors (NAD+, CoA, etc.)
- Salts and buffer (Mg²⁺, K⁺, HEPES) [41]
- Template Addition and Reaction Initiation: Transfer the DNA library variants into the microplate wells. Seal the plate to prevent evaporation and initiate the protein synthesis reaction by incubating at a defined temperature (e.g., 30-37°C) for 4-6 hours [41].
- Functional Assay and Data Collection: After expression, directly assay the enzyme activity in the same well. This can be achieved by:
- Data Analysis and Model Feedback: The high-quality dataset of sequence-function relationships is fed back to the ML models to refine future design predictions, closing the LDBT loop [5] [6].
Protocol for Metabolic Pathway Prototyping and Optimization (iPROBE)
The in vitro Prototyping and Rapid Optimization of Biosynthetic Enzymes (iPROBE) methodology leverages CFPS to rapidly assemble and test metabolic pathways.
- Individual Enzyme Expression: Express each enzyme of the proposed biosynthetic pathway in separate, small-scale CFPS reactions. This confirms the activity of each component [5].
- Combinatorial Pathway Assembly: In a 96-well format, combine the crude cell-free expression lysates containing individual active enzymes in different ratios. This creates a combinatorial matrix of pathway combinations and enzyme stoichiometries without the need for purification [5] [41].
- Pathway Functionality Testing: Initiate the coupled metabolic reactions by adding the starting substrate to each well. Quantify the final product titer using analytical methods like HPLC or mass spectrometry after a defined incubation period [5] [41].
- Data-Driven Optimization: The resulting dataset of product yields for different enzyme combinations and ratios is used to train a neural network or other ML model. The model then predicts the optimal pathway configuration for in vivo implementation in a production host [5]. This approach has been used to improve product titers by over 20-fold in a microbial host [5].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Cell-Free Protein Synthesis
| Reagent/Material | Function | Examples & Key Characteristics |
|---|---|---|
| Cell Lysates | Provides the core transcriptional and translational machinery (ribosomes, tRNAs, RNA polymerase, translation factors). | E. coli S30 Extract [41], Wheat Germ Extract [41] [42], Rabbit Reticulocyte Lysate [41] [42], Insect Cell Lysate [41] [42]. Choice depends on protein origin and PTM requirements. |
| Energy Regeneration System | Maintains ATP and GTP levels to fuel protein synthesis over extended periods. | Creatine Phosphate/Creatine Kinase [41], Maltodextrin-based systems [41]. More cost-effective and stable than early systems like Phosphoenolpyruvate (PEP). |
| Amino Acid Mixture | Building blocks for protein synthesis. | 20 canonical amino acids. Can be supplemented with non-canonical amino acids (ncAAs) for incorporating novel chemical functionalities [5] [41]. |
| Cofactors & Salts | Essential for enzyme kinetics and maintaining proper reaction physiology. | Mg²⁺ (critical for ribosome function), K⁺, NAD+, CoA [41]. Concentrations are finely tuned for optimal yield. |
| DNA Template | Genetic blueprint for the protein to be expressed. | Plasmid DNA or linear PCR products with a cell-free compatible promoter (e.g., T7, SP6) [41]. |
| Automation-Compatible Vessels | Enable high-throughput, parallel experimentation. | 384-well microplates, nanoliter- to picoliter-scale droplet microfluidic chips [5] [41]. |
Case Study: Knowledge-Driven DBTL for Dopamine Production
A 2025 study exemplifies the power of integrating in vitro CFPS data into strain engineering, a hybrid approach that aligns with the LDBT philosophy [7]. The goal was to optimize an E. coli strain for dopamine production.
- In Vitro Learning (L): Researchers first used a crude E. coli cell lysate CFPS system to express the two key enzymes in the dopamine pathway (HpaBC and Ddc) and test their relative expression levels and activities in vitro. This upstream investigation provided mechanistic insights and identified optimal enzyme ratios before in vivo engineering [7].
- Design (D): Based on the in vitro data, a set of genetic constructs with varying Ribosome Binding Site (RBS) strengths was designed to fine-tune the expression levels of HpaBC and Ddc in the production strain [7].
- Build (B): The RBS library was built and introduced into the dopamine production host E. coli FUS4.T2 [7].
- Test (T): The resulting strain library was tested for dopamine production, leading to the identification of a top-performing strain [7].
Result: The knowledge-driven cycle, informed by initial cell-free testing, developed a strain producing 69.03 ± 1.2 mg/L of dopamine, a 2.6 to 6.6-fold improvement over previous state-of-the-art in vivo production methods [7]. This case demonstrates how CFPS can de-risk and accelerate the learning phase, even within a traditionally structured DBTL cycle.
The optimization of cell-free systems for enhanced scalability, cost-efficiency, and reaction fidelity is fundamentally reshaping the practice of synthetic biology. These advancements are not merely incremental improvements but are enabling a foundational shift from the iterative DBTL cycle to the predictive, data-driven LDBT cycle. In this new paradigm, machine learning leverages vast biological knowledge to design biological parts with high success rates, while advanced cell-free platforms serve as the rapid-validation engine. The synergy between in silico learning and in vitro testing creates a powerful feedback loop, accelerating the journey from design to functional biological systems. As these technologies continue to mature and converge, they promise to usher in an era of truly programmable biology, with profound implications for drug development, biomanufacturing, and diagnostic innovation.
The classic Design-Build-Test-Learn (DBTL) cycle has long served as the foundational framework for synthetic biology engineering. However, the integration of advanced machine learning and massive datasets is prompting a fundamental re-evaluation of this paradigm. Emerging approaches propose inverting the cycle to Learning-Design-Build-Test (LDBT), where machine learning models trained on expansive biological data precede and guide the design phase [10]. This shift is particularly impactful in the realm of multi-omics data integration, where combining genomic, transcriptomic, proteomic, and metabolomic data provides a more comprehensive view of biological systems but also introduces significant computational challenges. The refinement of model accuracy through active learning and sophisticated multi-omics integration is therefore critical for accelerating biological engineering, reducing experimental iterations, and achieving predictive design in synthetic biology.
This technical guide details practical methodologies for implementing active learning frameworks and integrating diverse omics datasets to enhance predictive modeling within modern DBTL and LDBT cycles.
Active Learning Frameworks for Biological Data
Active learning creates an iterative feedback loop between a model and an experimentation process, strategically selecting the most informative data points to improve the model efficiently. Several techniques are particularly suited to biological data, which is often high-dimensional and costly to acquire.
Core Active Learning Strategies
- Uncertainty Sampling: The model identifies instances in the unlabeled data pool where its prediction confidence is lowest (e.g., in a classification task, it might select samples where the predicted class probabilities are most similar). Labeling these ambiguous points provides the maximum information gain. This is highly effective for optimizing biological assays where measurements are expensive.
- Diversity Sampling: This strategy aims to select a batch of data points that represent the broad diversity of the input space. It ensures the model learns from a comprehensive set of scenarios, preventing overfitting to a specific region and improving generalizability. This is crucial for capturing the inherent heterogeneity in biological systems, such as metabolic variation across cell populations [43].
- Query-by-Committee: A "committee" of multiple models is trained on the current labeled data. Instances where the committee members disagree the most are selected for labeling. This approach reduces the bias of a single model and effectively explores complex fitness landscapes in protein or pathway engineering.
Quantitative Impact of Active Learning
The table below summarizes the performance of various active learning strategies in different biological applications, demonstrating their efficiency in reducing the required training data.
Table 1: Performance of Active Learning Strategies in Biological Applications
| Strategy | Application Context | Key Performance Metric | Reported Outcome |
|---|---|---|---|
| Uncertainty Sampling | Protein Stability Prediction | Model Mean Squared Error (MSE) | Training MSE: 0.0009546, Test MSE: 0.0009198 [43] |
| Heterogeneity-Powered Learning | Metabolic Engineering (Triglyceride Production) | Predictive Accuracy from Single-Cell Data | High accuracy model enabling minimal operational suggestions for high yield [43] |
| Query-by-Committee | Enzyme Engineering | Design Success Rate | Nearly 10-fold increase in design success rates when combining ProteinMPNN with AlphaFold assessment [10] |
Experimental Protocol: Implementing "The Muddiest Point" for Model Diagnostics
"The Muddiest Point" is a simple yet powerful reflective technique adapted from classroom pedagogy to identify the most challenging concepts for learners [44]. In a machine learning context, it is repurposed to identify the data points or feature types that most challenge a predictive model.
Detailed Methodology:
- Model Inference and Confidence Scoring: After training an initial model on a seed dataset, run inference on a large, unlabeled pool of data (e.g., a library of protein sequences or single-cell metabolomic profiles).
- Identify Low-Confidence Predictions: Calculate a confidence score for each prediction. For regression, this could be the variance of an ensemble's output. For classification, it is typically the entropy of the predicted class probabilities.
- Select the "Muddiest" Instances: Rank all instances in the unlabeled pool by their confidence scores (lowest to highest).
- Prioritized Experimental Validation: The top k instances (e.g., the 100 most uncertain protein variants) are prioritized for experimental testing ("labeling").
- Model Retraining and Iteration: The newly acquired experimental data is added to the training set, and the model is retrained. This cycle is repeated until a desired performance threshold is met.
This protocol directly implements uncertainty sampling, ensuring that wet-lab resources are allocated to experiments that will most efficiently improve the model.
Multi-Omics Data Integration Techniques
Multi-omics integration seeks to combine data from different molecular layers (genome, transcriptome, proteome, metabolome) to create a unified model of a biological system. The high-dimensionality, heterogeneity, and noise of these datasets make integration non-trivial.
Data Integration Methodologies
- Early Integration: Also known as concatenation, this method involves merging raw or pre-processed data from different omics layers into a single, combined feature matrix before model training. While simple, it is highly susceptible to technical noise and dominance of one data type due to scale differences.
- Intermediate Integration: This approach uses models that can learn joint representations from multiple data types simultaneously. Deep generative models like Variational Autoencoders (VAEs) are particularly powerful for this, learning a shared latent space that captures the underlying biological state common to all omics modalities [45].
- Late Integration: Models are trained separately on each omics data type, and their outputs (e.g., predictions or extracted features) are combined in a final meta-model. This is flexible but may fail to capture more subtle, cross-omics interactions.
Computational Workflow for Multi-Omics Integration
The following diagram illustrates a typical workflow for integrating multi-omics data using a deep learning approach, highlighting the key steps from data collection to biological insight.
Experimental Protocol: Single-Cell Multi-Omics with RespectM
The RespectM protocol provides a detailed methodology for acquiring microbial single-cell level metabolomics (MSCLM) data, which is a powerful source for multi-omics integration and heterogeneity-powered learning [43].
Detailed Methodology:
- Sample Preparation and Cell Handling:
- Dilute the original microbial cell culture to a density of approximately 100 cells/μL to minimize the co-sampling of multiple cells during mass spectrometry imaging (MSI).
- Use a laser etching guided droplet microarray (LEM) on an ITO-coated glass slide to create isolated compartments for individual cell droplets, preventing cross-contamination.
- Matrix Application and Data Acquisition:
- Apply a chemical matrix for MALDI-MSI using a sublimation method to ensure a homogeneous, fine-grained coating and reduce background noise.
- Perform discontinuous mass spectrometry imaging with a low laser raster co-sampling rate (manually calculated at ~9.1% at the chosen density) to target individual cells.
- Acquire mass spectra, collecting data from thousands of single cells (e.g., 4,321 cells as reported).
- Data Processing and Analysis:
- Process raw spectral data using software like SCiLS Lab and open-source R packages (
sclmpute,MetNormalizer,Stream) for standardization, normalization, and imputation. - Integrate quality control (QC) samples (e.g., 10 blank matrix points per spot sequence) to correct for batch effects.
- Use the resulting single-cell metabolomic data (e.g., 600+ metabolites across 4,321 cells) to train deep neural networks (DNNs). The resulting heterogeneity-powered learning (HPL) model can predict optimal metabolic engineering strategies, such as suggesting gene overexpression targets for high triglyceride production [43].
- Process raw spectral data using software like SCiLS Lab and open-source R packages (
The Scientist's Toolkit: Key Reagents and Computational Tools
Successfully implementing these advanced techniques requires a suite of specialized reagents and software tools.
Table 2: Essential Research Reagent Solutions and Computational Tools
| Category | Item | Function / Description |
|---|---|---|
| Experimental Reagents | ITO-coated Glass Slides | Conductive slides required for MALDI Mass Spectrometry Imaging. |
| Sublimation Matrix (e.g., DHB) | A homogeneous chemical matrix applied to samples to enable laser desorption/ionization. | |
| Cell Lysis & Metabolite Stabilization Reagents | Reagents like methanol or acetonitrile to quickly quench metabolism and extract metabolites. | |
| Computational Tools | VAE Frameworks (e.g., PyTorch, TensorFlow) | Enable building custom deep generative models for intermediate multi-omics integration [45]. |
| Protein Language Models (e.g., ESM, ProGen) | Pre-trained models for zero-shot prediction of protein structure and function, used in the LDBT "Learn" phase [10]. | |
| Structure Prediction & Design (e.g., AlphaFold, ProteinMPNN) | Tools for predicting protein 3D structure and designing novel sequences that fold into a desired structure [10]. | |
Single-Cell Analysis Suites (e.g., SCiLS Lab, R packages sclmpute, Stream) |
Specialized software for processing, normalizing, and analyzing single-cell MSI data and visualizing trajectories [43]. | |
| Data Integration Platforms | Multi-Omics Data Harmonization Tools | Computational methods to reconcile data with varying formats, scales, and biological contexts prior to integration [46]. |
| Network Integration Software | Tools that map multi-omics datasets onto shared biochemical networks (e.g., metabolic pathways) to improve mechanistic understanding [46]. |
Pathway Visualization: The LDBT Cycle in Synthetic Biology
The core paradigm shift from a DBTL to an LDBT cycle fundamentally changes how machine learning and data are utilized in the bioengineering workflow. The following diagram contrasts these two cycles, highlighting the role of pre-trained models and data-first approaches.
The convergence of active learning strategies and sophisticated multi-omics data integration is fundamentally refining model accuracy in synthetic biology. By strategically guiding experimentation and building comprehensive models from diverse molecular data, these techniques are accelerating the transition from the iterative, empirical DBTL cycle to the more predictive, knowledge-forward LDBT paradigm. This shift, powered by machine learning and high-throughput data generation, promises to transform biological engineering into a more precise and predictive discipline, ultimately enabling the rational design of novel biological systems for therapeutics, manufacturing, and environmental sustainability.
The transition of biological designs from controlled laboratory environments (in vitro) to complex living systems (in vivo) represents a critical bottleneck in synthetic biology and therapeutic development. This whitepaper examines the fundamental challenges in this translational process and evaluates two contrasting engineering frameworks: the traditional Design-Build-Test-Learn (DBTL) cycle and the emerging Learn-Design-Build-Test (LDBT) paradigm. By integrating advanced technologies including organ-on-a-chip systems, machine learning-guided design, pharmacokinetic/pharmacodynamic (PK/PD) modeling, and advanced formulation strategies, we present a comprehensive technical roadmap for enhancing the predictive accuracy of in vitro models. This analysis specifically addresses the needs of researchers, scientists, and drug development professionals working to accelerate the translation of synthetic biological systems into effective living chassis.
The "in vitro to in vivo gap" describes the fundamental disconnect between biological performance in artificial laboratory environments and function within complex living organisms. Traditional in vitro models, while offering control and scalability, often fail to recapitulate the physiological complexity of living systems, leading to promising designs that fail upon transition to in vivo testing [47]. This gap is particularly problematic in drug development, where only an estimated 0.1% of nanomedicine research output successfully reaches clinical application, creating significant economic and scientific inefficiencies [48].
The limitations of conventional approaches stem from multiple factors. Two-dimensional cell cultures lack the three-dimensional architecture, biomechanical forces, and heterogeneous cell populations found in living tissues [47]. Furthermore, animal models, while providing a whole-organism context, often demonstrate poor predictive value for human physiology due to interspecies differences [47]. This translational challenge has prompted a critical reevaluation of engineering frameworks in synthetic biology, shifting from traditional iterative approaches to data-driven predictive methodologies that can bridge this divide more effectively.
Framework Analysis: DBTL vs. LDBT Cycles
The Traditional DBTL Cycle
The Design-Build-Test-Learn (DBTL) cycle has served as the cornerstone engineering framework in synthetic biology. This iterative process begins with Design, where researchers define objectives and create genetic designs based on domain knowledge and computational modeling. The Build phase involves synthesizing DNA constructs and introducing them into biological chassis. Subsequently, the Test phase experimentally measures system performance, followed by the Learn phase, where data analysis informs subsequent design iterations [5]. While systematic, this approach often requires multiple costly and time-consuming cycles to achieve desired functionality, particularly when initial designs are based on incomplete biological understanding.
The Emerging LDBT Paradigm
A paradigm shift is emerging with the Learn-Design-Build-Test (LDBT) framework, which positions machine learning at the forefront of the design process [5] [6]. In this model, the cycle begins with Learn, where machine learning models trained on vast biological datasets make zero-shot predictions about sequence-function relationships before any physical construction occurs. This learning-first approach informs the subsequent Design phase, where optimized genetic constructs are computationally generated. The Build and Test phases then validate these predictions, with high-throughput cell-free systems enabling rapid experimental feedback [6]. This reordering creates a more efficient, predictive engineering workflow that reduces reliance on empirical iteration.
Table 1: Comparative Analysis of DBTL and LDBT Frameworks
| Aspect | Traditional DBTL Cycle | LDBT Paradigm |
|---|---|---|
| Starting Point | Design based on existing knowledge | Learning from comprehensive datasets |
| Primary Driver | Empirical iteration | Predictive modeling |
| Build Phase | In vivo chassis (bacteria, mammalian cells) | Cell-free systems for rapid prototyping |
| Test Throughput | Lower (days to weeks) | Higher (hours to days) |
| Data Utilization | Sequential learning from each cycle | Pre-emptive learning from existing data |
| Resource Intensity | Higher (multiple iterations) | Lower (reduced iteration needs) |
Quantitative Framework Comparison
Table 2: Performance Metrics for Engineering Frameworks
| Metric | Traditional DBTL | LDBT Approach | Improvement Factor |
|---|---|---|---|
| Cycle Duration | Weeks to months | Hours to days | 5-10x faster [6] |
| Design Success Rate | Low (requires multiple iterations) | Higher (zero-shot prediction) | Nearly 10x increase with ProteinMPNN+AlphaFold [5] |
| Experimental Throughput | 10-100 variants per cycle | 100,000+ reactions with microfluidics [5] | 1000x increase |
| Data Generation Scale | Limited by in vivo constraints | Megascale data from cell-free systems [5] | Orders of magnitude higher |
| Resource Requirements | High (cloning, cellular culturing) | Lower (cell-free, automated) | Significant reduction [6] |
Enabling Technologies for Predictive Translation
Organ-on-a-Chip Systems
Organ-on-a-chip (OOC) technology represents a revolutionary approach to bridging the in vitro to in vivo gap by replicating human organ microarchitecture, microenvironment, and function in vitro [49]. These microfluidic devices culture living cells in continuous perfusion to create tissue-level structures that mimic organ physiology more accurately than conventional 2D cultures. By incorporating primary human cells, biomechanical forces, and dynamic fluid flow, OOC systems enable the study of complex human pathophysiology and drug responses with unprecedented fidelity [49]. For instance, CN Bio's PhysioMimix platform recreates complex human biology to accurately predict human drug responses, demonstrating particular value for modeling complex diseases like non-alcoholic steatohepatitis (MASH) where animal models have proven inadequate [49].
Machine Learning-Guided Protein Design
Machine learning algorithms have dramatically enhanced our ability to predict protein structure and function from sequence data. Protein language models like ESM and ProGen, trained on millions of evolutionary relationships, enable zero-shot prediction of beneficial mutations and protein functions [5]. Structure-based tools such as MutCompute and ProteinMPNN leverage deep neural networks to optimize protein stability and activity, with demonstrated success in engineering improved hydrolases for polyethylene terephthalate (PET) depolymerization [5]. These computational approaches are particularly powerful when integrated with high-throughput experimental validation, creating a virtuous cycle of model improvement and design optimization.
Pharmacokinetic/Pharmacodynamic (PK/PD) Modeling
Quantitative PK/PD modeling establishes mathematical relationships between drug exposure, target engagement, and physiological effects, enabling prediction of in vivo efficacy from in vitro data [50]. Remarkably, researchers have demonstrated that PK/PD models trained almost exclusively on in vitro data can accurately predict in vivo tumor growth dynamics by linking in vitro PD models with in vivo PK profiles corrected for fraction unbound drug [50]. In one case study, only a single parameter adjustment—the intrinsic cell growth rate in the absence of drug—was required to scale the PD model from in vitro to in vivo settings, highlighting the potential for robust translational prediction [50].
Advanced Formulation Platforms
Advanced formulation strategies are critical for translating nanoparticle designs into functional therapeutic products. Lipid-based platforms, including liposomes and lipid nanoparticles (LNPs), dominate clinically approved nanomedicines, with proven success in products like Doxil and COVID-19 mRNA vaccines [48]. Polymer-based platforms using materials like PLGA offer controlled release profiles, while hybrid systems address specific delivery challenges. The clinical translation of these platforms requires careful attention to Chemistry, Manufacturing, and Controls (CMC) considerations, particularly regarding batch-to-batch consistency, stability, and scalability under Good Manufacturing Practice (GMP) standards [48].
Table 3: Technology Platforms for Bridging the Translational Gap
| Technology | Mechanism | Applications | Limitations |
|---|---|---|---|
| Organ-on-a-Chip | Recreates human organ microarchitecture and microenvironment | Disease modeling, drug efficacy testing, toxicity assessment | Limited multi-organ integration, specialized equipment needed |
| Machine Learning-Guided Design | Predicts sequence-structure-function relationships from training data | Protein engineering, genetic circuit design, pathway optimization | Requires large, high-quality datasets, black box limitations |
| PK/PD Modeling | Mathematical modeling of drug exposure-response relationships | Predicting in vivo efficacy from in vitro data, dosing regimen optimization | Dependent on quality of input data, may require species scaling factors |
| Cell-Free Expression Systems | In vitro transcription-translation without cellular constraints | Rapid prototyping, toxic protein production, high-throughput testing | Limited to acute effects, no cellular context |
| Advanced Formulations | Stabilizes and delivers therapeutic nanoparticles | Nanomedicine development, controlled release, targeted delivery | Manufacturing complexity, stability challenges, immunogenicity concerns |
Experimental Protocols for Predictive Translation
Protocol: LDBT Cycle for Protein Engineering
Objective: Engineer an enzyme with enhanced thermostability using the LDBT framework.
Materials:
- Pre-trained protein language models (ESM, ProGen)
- Structure prediction tools (AlphaFold, RoseTTAFold)
- Cell-free transcription-translation system (TX-TL)
- Liquid handling robot or microfluidics platform
- Functional assay reagents specific to target enzyme
Procedure:
- Learn Phase: Input wild-type protein sequence into machine learning models (e.g., MutCompute, Stability Oracle) to predict stabilizing mutations.
- Design Phase: Generate variant sequences incorporating predicted mutations, filtering using structure assessment tools (AlphaFold) to verify proper folding.
- Build Phase: Synthesize DNA templates encoding designed variants and express proteins using cell-free TX-TL system.
- Test Phase: Assess enzyme thermostability (e.g., thermal shift assays, residual activity after heating) and function in high-throughput format.
- Model Refinement: Incorporate experimental results to retrain machine learning models for subsequent design rounds.
Expected Outcomes: Identification of stabilized enzyme variants with improved thermostability while maintaining catalytic function, achieved with fewer design cycles than traditional approaches.
Protocol: In Vitro to In Vivo Efficacy Prediction
Objective: Predict in vivo antitumor efficacy from in vitro data using PK/PD modeling.
Materials:
- Cancer cell lines (e.g., NCI-H510A for SCLC)
- Test compound (e.g., ORY-1001 LSD1 inhibitor)
- In vitro cell culture systems (2D and 3D)
- Target engagement assay reagents
- Biomarker detection assays (e.g., GRP for LSD1 inhibition)
- PK modeling software
Procedure:
- In Vitro PD Model Development:
- Expose cells to compound across multiple doses and timepoints
- Measure target engagement, biomarker levels, and cell viability
- Conduct both pulsed and continuous dosing regimens
- Fit ordinary differential equations to capture relationships between exposure, PD response, and cell growth
In Vivo PK Characterization:
- Administer compound to animal models
- Collect plasma concentration time courses
- Develop PK model (e.g., two-compartment model with first-order absorption)
- Calculate unbound plasma concentration
Model Integration and Prediction:
- Link in vitro PD model with in vivo PK via unbound drug concentration
- Adjust single parameter for intrinsic tumor growth rate
- Validate model by comparing predicted vs. observed in vivo tumor growth inhibition
Expected Outcomes: Accurate prediction of in vivo efficacy across multiple dosing regimens, enabling clinical trial design with reduced animal testing [50].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for Translational Biology
| Reagent/Category | Function | Example Applications |
|---|---|---|
| Primary Human Cells | Provide human-relevant biology | Organ-on-a-chip models, patient-specific assays |
| Cell-Free TX-TL Systems | Enable rapid protein expression without living cells | High-throughput protein engineering, circuit prototyping |
| Ionizable Lipids | Formulate lipid nanoparticles for nucleic acid delivery | mRNA vaccine development, gene therapy |
| PEGylated Lipids | Enhance nanoparticle circulation time | Stealth drug delivery systems, reduced RES clearance |
| Targeting Ligands | Direct therapeutics to specific tissues or cells | Active targeting strategies, improved therapeutic index |
| Stimuli-Responsive Polymers | Enable triggered release in specific microenvironments | pH-sensitive delivery, enzyme-activated systems |
| PhysioMimix Platforms | Recreate human organ biology in vitro | Disease modeling, drug efficacy testing [49] |
| Machine Learning Models | Predict sequence-structure-function relationships | Zero-shot protein design, library optimization |
Integrated Workflow for Predictive Translation
The integration of LDBT frameworks with advanced translational technologies represents a paradigm shift in synthetic biology and therapeutic development. By positioning machine learning at the forefront of biological design and leveraging human-relevant test systems like organ-on-a-chip platforms, researchers can significantly enhance the predictive power of in vitro experiments. This approach, complemented by quantitative PK/PD modeling and advanced formulation strategies, creates a more efficient path from concept to clinical application. While challenges remain in data integration, model interpretability, and multi-organ system development, the coordinated implementation of these technologies promises to substantially bridge the in vitro to in vivo gap, accelerating the development of effective biological designs and therapeutics for human applications.
Head-to-Head: Performance Metrics and Comparative Analysis of DBTL vs. LDBT
The foundational paradigm of synthetic biology, the Design-Build-Test-Learn (DBTL) cycle, is undergoing a profound transformation. Propelled by advances in artificial intelligence (AI) and machine learning (ML), a reordered approach—the Learn-Design-Build-Test (LDBT) cycle—is emerging as a powerful alternative. This whitepaper provides a quantitative comparison of these two frameworks, analyzing their impact on development timelines, success rates, and resource allocation in bioengineering and drug development. The data indicate a significant shift: while traditional DBTL cycles rely on iterative empirical testing to accumulate knowledge, the LDBT cycle leverages pre-trained ML models to front-load the learning phase, enabling more predictive design and reducing the need for multiple costly build-test iterations [10]. This analysis is critical for researchers and development professionals seeking to optimize R&D strategies for maximum efficiency and output.
The standard DBTL cycle is a systematic, iterative framework for engineering biological systems. The process begins with Design, where researchers define objectives and design biological parts using domain knowledge and computational modeling. This is followed by Build, involving DNA synthesis, assembly, and introduction into a chassis organism. The Test phase experimentally measures the performance of the constructed system, and the Learn phase analyzes this data to inform the next design round [10]. This cycle is a cornerstone of synthetic biology but often requires multiple turns to gain sufficient knowledge, with the Build and Test phases being particularly slow and resource-intensive [10].
The LDBT cycle represents a paradigm shift. It proposes that the data traditionally "learned" through iterative Build-Test phases may already be inherent in sophisticated machine learning algorithms. In this reordered cycle, Learn comes first, leveraging large biological datasets and pre-trained models (e.g., protein language models like ESM and ProGen) to make zero-shot predictions of functional biological components. This is followed by Design based on these computational insights, and a single, efficient round of Build and Test to validate the predictions [10]. This approach moves synthetic biology closer to a "Design-Build-Work" model, akin to established engineering disciplines where designs are reliable from the first iteration [10].
Quantitative Benchmarks: A Comparative Analysis
The following tables synthesize quantitative data on the performance and resource utilization of the DBTL and LDBT paradigms.
Table 1: Comparative Development Timelines and Success Rates
| Metric | Traditional DBTL / Conventional Methods | AI/ML-Driven LDBT Approaches | Source |
|---|---|---|---|
| Typical Preclinical Timeline | 5-6 years [51] | 18 months [51] [52] | |
| Target to Preclinical Candidate | ~4 years (industry average) [51] | ~30 months [51] | |
| Clinical Trial Phase 1 Success Rate | 40-65% [52] | 80-90% [52] | |
| Cycle Time for Directed Evolution | 7-14 days (industry goal) [53] | Enabled by modern computational tools [53] | |
| Data Point: Insilico Medicine (IPF drug) | N/A | Target to Phase 1 in 30 months [51] |
Table 2: Resource Allocation and Economic Impact
| Aspect | Traditional DBTL / Conventional Methods | AI/ML-Driven LDBT Approaches | Source |
|---|---|---|---|
| Average Cost to Bring a Drug to Market | Over $2 billion [51] | Significant reduction in R&D OpEx [51] | |
| Attrition Rate (Entering Clinical Trials) | ~90% [51] | AI predicts efficacy/safety, reducing failure rate [52] | |
| Value of Generative AI to Pharma Industry | N/A | $60-$110 billion annually (McKinsey estimate) [51] | |
| Market Size for AI in Drug Discovery (2030) | N/A | $8-$20 billion (from ~$2.6B in 2025) [51] |
Experimental Protocols: DBTL in Action
Knowledge-Driven DBTL for Dopamine Production inE. coli
A 2025 study exemplifies a knowledge-driven DBTL cycle for optimizing dopamine production, demonstrating a rigorous methodology for the "Test" and "Learn" phases [7].
1. Experimental Objective: To develop and optimize an E. coli strain for high-yield dopamine production by fine-tuning the expression of a two-enzyme pathway (HpaBC and Ddc) [7].
2. Protocol Details:
- Design & Build: A library of production strains was built using Ribosome Binding Site (RBS) engineering to modulate the translation initiation rate of the hpaBC and ddc genes. This created a range of relative expression levels for the two enzymes [7].
- Test: High-throughput cultivation was performed in a minimal medium with controlled carbon sources. The key performance indicators measured were:
- Dopamine Titer: The concentration of dopamine in the culture supernatant (in mg/L), quantified via analytical methods like HPLC.
- Biomass-Specific Yield: The mass of dopamine produced per unit of cell biomass (in mg/g_biomass), indicating metabolic efficiency [7].
- Learn: Data from the "Test" phase was analyzed to determine the optimal RBS combination that maximized both titer and yield. The study found that the GC content in the Shine-Dalgarno sequence was a critical factor influencing RBS strength and, consequently, pathway efficiency. This learning directly informed subsequent genetic designs [7].
3. Outcome: The optimized strain achieved a dopamine production of 69.03 ± 1.2 mg/L, a 2.6 to 6.6-fold improvement over previous state-of-the-art in vivo production methods [7].
LDBT forDe NovoEnzyme Design
The LDBT cycle employs a distinct, computation-heavy methodology for creating novel biocatalysts.
1. Experimental Objective: To design a novel enzyme capable of catalyzing a specific, desired reaction de novo.
2. Protocol Details:
- Learn: A pre-trained protein language model (e.g., ZymCTRL) or a structure-based diffusion model (e.g., RFdiffusion) is used. These models have been trained on millions of protein sequences and structures, encapsulating fundamental principles of protein folding and function [10] [54].
- Design: The learned model is conditioned with a description of the target function. This can be:
- Build: The most promising in silico designs are selected, and their DNA sequences are synthesized de novo using commercial gene synthesis services. These are then cloned into expression vectors [10].
- Test: The expressed proteins are purified and subjected to functional assays to measure catalytic activity, specificity, and stability. Cell-free expression systems are often used here for rapid, high-throughput testing [10].
3. Outcome: Successful applications of this paradigm have led to the creation of novel luciferases and stabilized hydrolases with significantly improved activity, often with a high success rate in initial testing, reducing the need for multiple DBTL rounds [10] [54].
Workflow Visualization: DBTL vs. LDBT
The fundamental difference between the two cycles is their starting point and iterative nature. The diagram below illustrates the logical flow of each paradigm.
The Scientist's Toolkit: Key Research Reagents and Solutions
The implementation of both DBTL and LDBT cycles relies on a suite of specialized reagents, software, and platforms. The following table details key resources for building and testing engineered biological systems.
Table 3: Essential Research Reagents and Solutions for DBTL/LDBT Cycles
| Category | Item / Solution | Function / Application | Source / Example |
|---|---|---|---|
| Computational Design | Protein Language Models (pLMs) | Generate novel protein sequences and predict function from sequence alone. | ESM, ProGen [10] |
| Structure Prediction & Design Tools | Predict protein 3D structure from sequence (AF2) or generate sequences for a desired backbone (ProteinMPNN). | AlphaFold2, ProteinMPNN [10] [54] | |
| RFdiffusion | Generate novel protein backbone structures de novo conditioned on specific motifs (e.g., active sites). | RFdiffusion, RFdiffusion2 [54] | |
| DNA Assembly & Build | DNA Synthesis & Assembly | De novo gene synthesis and modular assembly of genetic constructs. | Twist Bioscience platform [55] |
| RBS Library Kits | Pre-designed DNA parts for fine-tuning gene expression levels in metabolic pathways. | Used in RBS engineering [7] | |
| Chassis & Expression | Production Strains | Engineered host organisms (e.g., E. coli) optimized for production of target compounds. | E. coli FUS4.T2 (dopamine production) [7] |
| Cell-Free Expression Systems | Rapid, high-throughput protein synthesis without the constraints of living cells. | Crude cell lysate systems [10] | |
| Testing & Analytics | High-Throughput Screening | Automated platforms for analyzing thousands of variants for activity, stability, or production titer. | Biofoundries, Droplet Microfluidics (e.g., DropAI) [10] |
| Analytical Standards & Kits | For quantifying reaction products (e.g., dopamine, enzymes) via HPLC, MS, or fluorescence. | Implied in testing protocols [7] |
The quantitative benchmarks and experimental protocols presented herein clearly delineate the operational and economic distinctions between the DBTL and LDBT cycles. The traditional DBTL cycle, while systematic, is inherently iterative and empirical, leading to extended timelines and high costs as knowledge is accumulated gradually through experimentation [51] [7].
In contrast, the LDBT cycle leverages the predictive power of machine learning to front-load the knowledge phase, fundamentally compressing development timelines—from years to months in preclinical stages—and improving early-stage success rates [10] [51] [52]. The adoption of LDBT is not merely an incremental improvement but a paradigm shift towards a more predictive and engineering-driven discipline. For researchers and drug development professionals, integrating AI and ML into the core of the R&D workflow is transitioning from a competitive advantage to a strategic necessity for achieving efficiency, scalability, and success in synthetic biology applications.
Engineering a PET Hydrolase via DBTL vs. LDBT Approaches
The enzymatic degradation of poly(ethylene terephthalate) (PET) presents a promising route toward addressing global plastic pollution. Engineering efficient PET hydrolases has traditionally followed the Design-Build-Test-Learn (DBTL) cycle, a systematic framework for biological engineering. However, a paradigm shift is emerging with the Learn-Design-Build-Test (LDBT) approach, which leverages machine learning and advanced testing platforms to accelerate and enhance the protein engineering process [5] [6]. This technical analysis compares these two methodologies through the specific lens of engineering PET hydrolases, examining how the repositioning of the "Learn" phase fundamentally changes strategy, efficiency, and outcomes.
The DBTL Cycle: A Traditional Framework for Biocatalyst Engineering
Core Principles and Workflow
The Design-Build-Test-Learn (DBTL) cycle is a cornerstone of synthetic biology, providing a systematic, iterative framework for engineering biological systems [1]. In the context of protein engineering:
- Design relies on domain knowledge, expertise, and computational modeling to define objectives and design protein variants [5].
- Build involves synthesizing DNA constructs, assembling them into vectors, and introducing them into expression systems [5] [1].
- Test experimentally measures the performance of engineered proteins (e.g., activity, stability, expression) [5].
- Learn analyzes collected data to inform the next design round, iterating until desired function is achieved [5].
This cycle closely resembles approaches in established engineering disciplines, where iteration involves gathering information, processing it, identifying design revisions, and implementing changes [5].
Case Study: PET Hydrolase Engineering via DBTL
The engineering of a leaf-branch compost cutinase (LCC) variant illustrates a traditional DBTL approach applied to PET degradation. The goal was to achieve economically viable industrial PET depolymerization, with key parameters including high solids loading (>150 g kg⁻¹) and product yield (>90%) [56].
Experimental Protocol:
- Design: Based on structural knowledge of LCC, researchers introduced specific mutations (F243I/D238C/S283C/Y127G) to improve stability and activity [56].
- Build: The engineered LCC variant (LCCICCG) was expressed in a microbial host system, typically Escherichia coli [57].
- Test: Enzymatic performance was evaluated using pretreated PET waste at industrially relevant solids loading (200 g kg⁻¹). Depolymerization yields were quantified by measuring released products [56].
- Learn: Results showed 90% depolymerization of post-consumer PET, but a critical learning revealed that 10% nonbiodegradable PET remained with high crystallinity (~30%) due to physical aging, limiting immediate reuse [56].
Table 1: DBTL Cycle Outcomes for LCCICCG Engineering
| DBTL Phase | Key Activities | Outcomes for PET Hydrolase |
|---|---|---|
| Design | Rational mutation based on structure | Four targeted mutations (F243I/D238C/S283C/Y127G) |
| Build | Expression in E. coli | Successful production of LCCICCG variant |
| Test | Depolymerization at 200 g kg⁻¹ | 90% conversion of pretreated PET waste |
| Learn | Analysis of limitations | Identified 10% non-biodegradable residual PET with high crystallinity |
This DBTL achievement was significant, establishing a benchmark for enzymatic PET recycling. However, the remaining 10% nonbiodegradable PET posed both environmental and economic challenges, with an estimated 80 kilotons of residual waste annually if implemented at scale [56].
The LDBT Paradigm: A Machine Learning-Driven Shift
Fundamental Restructuring of the Engineering Cycle
The LDBT cycle represents a transformative reordering of the traditional synthetic biology workflow, placing Learning before Design through machine learning (ML) [5] [6]. This paradigm shift is enabled by:
- Protein language models (e.g., ESM, ProGen) trained on evolutionary relationships between millions of protein sequences [5].
- Structure-based deep learning tools (e.g., ProteinMPNN, MutCompute) that predict sequences folding into specific backbones or identify stabilizing mutations [5].
- Zero-shot prediction capabilities that generate functional protein designs without additional training [5].
In LDBT, the learning phase leverages these ML models to mine evolutionary and biophysical information from vast datasets, enabling predictive design before any physical construction occurs [5] [6].
Case Study: TurboPETase Engineering via LDBT
The computational redesign of a hydrolase from bacterium HR29 into TurboPETase exemplifies the LDBT approach, addressing the limitation of residual nonbiodegradable PET left by LCCICCG [56].
Experimental Protocol:
- Learn: Researchers employed a protein language model trained on ~26,000 homologous PET hydrolase sequences to predict beneficial amino acid substitutions. The model used a Transformer encoder with absolute position embedding to process input sequences, sorting residue positions by mutation probability scores [56].
- Design: From 18 initial candidate positions, 7 embedded in the PET-binding groove were selected for experimental validation. After identifying destabilizing mutations, the GRAPE strategy was applied, using four complementary algorithms (FoldX, Rosettacartesianddg, ABACUS, DDD) to design stabilizing compensatory mutations [56].
- Build: The designed TurboPETase variant (BhrPETaseH218S/F222I/A209R/D238K/A251C/A281C/W104L/F243T) was expressed and purified [56].
- Test: The variant was evaluated against multiple benchmark PET hydrolases across temperature ranges (50°C–65°C) and at high solids loading (200 g kg⁻¹) [56].
Table 2: LDBT Cycle Outcomes for TurboPETase Engineering
| LDBT Phase | Key Activities | Outcomes for PET Hydrolase |
|---|---|---|
| Learn | Protein language model on 26K homologs | 18 candidate positions identified; evolutionary fitness predictions |
| Design | GRAPE strategy with 4 algorithms | 8 mutations combined with stability-activity balance |
| Build | Expression of final variant | Successful production of TurboPETase |
| Test | Depolymerization at 200 g kg⁻¹ | Nearly complete depolymerization in 8h; 61.3 ghydrolyzed PET L⁻¹ h⁻¹ maximum rate |
TurboPETase outperformed all benchmark enzymes, achieving nearly complete PET depolymerization within 8 hours at industrially relevant conditions (200 g kg⁻¹ solids loading) [56]. Kinetic and structural analysis suggested that a more flexible PET-binding groove facilitated targeting of more specific attack sites [56].
Comparative Analysis: DBTL vs. LDBT for PET Hydrolase Engineering
Workflow and Strategic Differences
The fundamental distinction between these approaches lies in their starting point and information flow. The following diagram illustrates the contrasting workflows:
Workflow Comparison: Traditional DBTL vs. ML-Driven LDBT
Performance and Efficiency Metrics
Table 3: Direct Comparison of DBTL vs. LDBT PET Hydrolase Engineering
| Parameter | DBTL Approach (LCCICCG) | LDBT Approach (TurboPETase) |
|---|---|---|
| Engineering Strategy | Structure-informed rational design | Protein language model + force-field algorithms |
| Key Mutations | 4 targeted mutations | 8 combinatorially optimized mutations |
| Depolymerization Yield | 90% at 200 g kg⁻¹ | ~99% (nearly complete) at 200 g kg⁻¹ |
| Reaction Time | Not specified for 90% yield | 8 hours for near-complete depolymerization |
| Maximum Production Rate | Not reported | 61.3 ghydrolyzed PET L⁻¹ h⁻¹ |
| Thermostability (Tm) | Not specified for LCCICCG | 84°C |
| Residual PET Waste | 10% (high crystallinity) | Minimal |
| Data Utilization | Limited to experimental results | Evolutionary information from 26,000 homologs |
| Design Space Exploration | Limited by rational design capacity | Vast sequence space via computational prediction |
The Role of Enabling Technologies
The implementation of LDBT critically depends on synergistic technological platforms:
Cell-Free Expression Systems: These platforms accelerate the Build and Test phases by leveraging protein biosynthesis machinery from cell lysates or purified components [5]. They enable:
- Rapid protein production (>1 g/L in <4 hours) without time-intensive cloning [5]
- Direct testing of expressed proteins, even toxic variants [5]
- High-throughput testing when combined with liquid handling robots and microfluidics (e.g., screening >100,000 reactions) [5]
Machine Learning Integration: ML addresses the complexity of sequence-structure-function relationships in proteins [5] [37]. Key applications include:
- Zero-shot prediction of functional sequences without additional training [5]
- Fitness landscape mapping to optimize multiple enzyme properties simultaneously [5]
- Active learning to strategically select informative variants for testing, maximizing information gain per experiment [6]
Research Reagent Solutions Toolkit
Table 4: Essential Research Tools for PET Hydrolase Engineering
| Research Tool | Type/Classification | Function in PET Hydrolase Engineering |
|---|---|---|
| Protein Language Models (ESM, ProGen) | Computational | Predict beneficial mutations and infer function from evolutionary sequences [5] |
| Structure-Based Design Tools (ProteinMPNN, MutCompute) | Computational | Design sequences for specific backbones or optimize residues for local environment [5] |
| Stability Prediction Tools (Prethermut, Stability Oracle) | Computational | Predict thermodynamic stability changes from mutations (ΔΔG) [5] |
| Cell-Free Transcription-Translation | Experimental Platform | Rapid protein expression without cloning; enable high-throughput testing [5] |
| GRAPE Strategy | Computational Framework | Combines FoldX, Rosetta, ABACUS, DDD for stability compensation mutations [56] |
| DropAI Microfluidics | Experimental Platform | Screen >100,000 picoliter-scale reactions for ultra-high-throughput testing [5] |
| RetroPath & Selenzyme | Computational | Automated pathway and enzyme selection for metabolic engineering [58] |
The comparison between DBTL and LDBT approaches for PET hydrolase engineering reveals a fundamental transition in synthetic biology methodology. The traditional DBTL cycle successfully produced the LCCICCG variant with 90% PET depolymerization, representing a significant engineering achievement. However, the LDBT paradigm has demonstrated superior outcomes through TurboPETase, achieving nearly complete depolymerization while balancing stability and activity constraints.
This performance advantage stems from LDBT's ability to leverage evolutionary information from thousands of homologs before physical construction, enabling more informed design decisions. The integration of machine learning with rapid testing platforms creates a virtuous cycle where each experiment enhances predictive models, progressively reducing dependency on empirical iteration.
For researchers engineering biocatalysts, particularly for challenging substrates like PET, the LDBT framework offers a more efficient path to optimal solutions. However, this approach requires specialized computational resources and expertise. As ML models and experimental infrastructure continue advancing, LDBT is positioned to become the dominant paradigm for biological design, potentially realizing the aspiration of synthetic biology for truly predictive engineering from first principles [5] [37].
The foundational framework of synthetic biology has long been the Design-Build-Test-Learn (DBTL) cycle, an iterative process that guides the engineering of biological systems. However, the convergence of artificial intelligence (AI) and high-throughput experimental platforms is catalyzing a fundamental paradigm shift. This transformation repositions the traditional cycle into a Learn-Design-Build-Test (LDBT) sequence, placing a data-driven learning phase at the forefront of biological engineering. This whitepaper provides an in-depth technical assessment of the qualitative advantages offered by the LDBT paradigm, specifically evaluating its transformative impact on predictability, resource optimization, and accessibility for researchers, scientists, and drug development professionals. The core distinction lies in the starting point: while the DBTL cycle begins with a design hypothesis based on existing domain knowledge, the LDBT cycle initiates with a comprehensive machine learning-driven analysis of vast biological datasets to inform and predict optimal design parameters from the outset [5] [6]. This reordering is more than procedural; it represents a new engineering philosophy for biology, moving the field closer to a "Design-Build-Work" model akin to more mature engineering disciplines like civil engineering [5].
The impetus for this shift stems from recognized bottlenecks in the traditional DBTL cycle. Although the "Build" and "Test" stages have been accelerated by advances in DNA synthesis and automation, the "Learn" phase has remained a critical challenge due to the complexity, heterogeneity, and non-linear interactions within biological systems [37] [9]. The LDBT paradigm directly addresses this bottleneck by leveraging machine learning (ML) and deep learning (DL) to navigate the high-dimensional design space of biological sequences and systems. By learning first from existing or purpose-generated megascale data, the LDBT framework transforms synthetic biology from an iterative, empirical practice into a more predictive and efficient science [5] [9]. This paper will dissect this transition through a technical lens, providing detailed methodologies and a comparative analysis to illustrate the profound advantages of the LDBT approach.
Core Conceptual Framework: DBTL vs. LDBT
The Traditional DBTL Cycle
The Design-Build-Test-Learn (DBTL) cycle is a systematic, iterative framework for engineering biological systems. Its stages are defined as follows:
- Design: Researchers define objectives and design genetic constructs using domain knowledge and computational modeling, often selecting and arranging standardized biological parts [5] [1].
- Build: The designed DNA constructs are physically synthesized and assembled into vectors, which are then introduced into a living chassis (e.g., bacteria, yeast) or a cell-free system [5] [1].
- Test: The performance of the engineered biological system is experimentally measured using functional assays, sequencing, and omics technologies to collect data on how well it meets the design objectives [5] [9].
- Learn: Data from the Test phase is analyzed to understand the system's behavior and to inform revisions for the next Design round, thus continuing the cycle until the desired function is achieved [5] [1].
A primary limitation of this cycle is its reactive nature; learning occurs after building and testing, often requiring multiple costly and time-consuming iterations to converge on a functional design [6].
The Emergent LDBT Paradigm
The LDBT cycle fundamentally reorders the process, initiating with a machine learning phase:
- Learn: The cycle begins by leveraging pre-trained machine learning models on large biological datasets—including protein sequences, structures, and omics data—to extract hidden patterns and infer design rules. This phase utilizes powerful zero-shot predictors and foundational models to gain predictive insight before any physical design is committed [5] [6].
- Design: Informed by the predictive models from the Learn phase, researchers generate optimized genetic designs. This stage employs AI-driven tools that can propose sequences or systems with a high probability of success, significantly de-risking the design process [5] [40].
- Build: Similar to the traditional cycle, this involves the physical construction of genetic material. However, this phase is accelerated by its integration with predictive design and the use of rapid, automated platforms like cell-free gene expression systems [5] [6].
- Test: The built constructs are rigorously characterized. The data generated not only validates the design but also serves as high-quality, ground-truthed data to further refine and retrain the machine learning models, creating a virtuous, self-improving cycle [5] [9].
This workflow redefines the role of experimentation. Instead of being the primary engine for generating knowledge, it serves to validate computationally derived predictions and to generate targeted data for continuous model improvement [6].
Comparative Workflow Visualization
The following diagram illustrates the fundamental structural differences and information flows between the traditional DBTL cycle and the emerging LDBT paradigm.
Qualitative Advantage 1: Enhanced Predictability
The foremost advantage of the LDBT cycle is its dramatic enhancement of predictive capability in biological design. By commencing with a learning phase powered by machine learning, the LDBT framework directly addresses the core challenge of biological complexity that has long hindered predictable engineering.
Technical Mechanisms for Improved Prediction
The LDBT paradigm leverages several advanced computational techniques to achieve superior predictability:
- Zero-Shot Prediction with Protein Language Models: Tools like ESM (Evolutionary Scale Modeling) and ProGen are trained on millions of protein sequences, allowing them to capture deep evolutionary relationships. These models can predict beneficial mutations, infer function, and generate novel, functional protein sequences without requiring additional, target-specific training data. For instance, these models have successfully designed diverse antibody sequences and predicted solvent-accessible amino acids with high accuracy [5].
- Structure-Based Sequence Design: Models such as ProteinMPNN and MutCompute use deep neural networks trained on protein structures. ProteinMPNN takes a protein backbone as input and outputs sequences that fold into that structure, leading to a nearly 10-fold increase in design success rates when coupled with structure assessment tools like AlphaFold [5]. MutCompute focuses on residue-level optimization by identifying probable mutations given the local chemical environment, which has been used to engineer hydrolases with increased stability and activity for PET depolymerization [5].
- Hybrid and Physics-Informed Models: Emerging approaches combine the pattern recognition strength of statistical models with the explanatory power of physical principles. For example, some models integrate large language models trained on homologs with force-field-based algorithms to explore the evolutionary landscape of enzymes more comprehensively [5].
Experimental Protocol: Validating Predictive Models
The following protocol outlines a standard methodology for validating the predictive power of an LDBT-driven protein engineering campaign.
- Objective: To experimentally validate the stability and activity of protein variants designed by a zero-shot protein language model.
- Design (Informed by ML):
- Input: A wild-type protein sequence and a defined functional objective (e.g., improve thermostability at 60°C).
- Model Inference: Use a pre-trained model (e.g., ESM, Stability Oracle) to generate a ranked list of candidate mutant sequences predicted to have higher folding stability (lower ΔΔG) and retained active site geometry.
- Build (Cell-Free Expression):
- DNA Template Preparation: Synthesize DNA templates encoding the top 20-50 candidate sequences via high-throughput oligo synthesis. No cloning is required.
- Cell-Free Reaction: Express proteins using a commercial or homemade cell-free transcription-translation system (e.g., derived from E. coli lysate) in a 96- or 384-well plate format. Incubate for 4-6 hours at 30°C [5] [6].
- Test (High-Throughput Assays):
- Stability Measurement: Use a high-throughput thermal shift assay. Add a fluorescent dye (e.g., SYPRO Orange) to the cell-free reaction mixture and run a thermal melt curve in a real-time PCR machine. The melting temperature (Tm) serves as a proxy for protein stability [6].
- Activity Screening: For an enzyme, couple the cell-free expression directly with a colorimetric or fluorogenic activity assay in the same well. For the PET hydrolase example, a released product could be detected colorimetrically [5].
- Data Collection: Use plate readers and automated imaging systems to collect fluorescence and absorbance data for all wells in parallel.
- Learn (Model Refinement):
Qualitative Advantage 2: Superior Resource Optimization
The LDBT cycle introduces a step-change in efficiency, dramatically optimizing the use of time, financial resources, and laboratory materials. This is achieved by minimizing trial-and-error and strategically guiding experimental efforts toward the most promising regions of the biological design space.
Quantitative Comparison of Resource Utilization
The table below summarizes the key resource optimization metrics differentiating DBTL and LDBT approaches, based on reported data from research campaigns and platform companies.
Table 1: Resource Optimization - DBTL vs. LDBT
| Resource Metric | Traditional DBTL Cycle | LDBT Paradigm | Key LDBT Enabler |
|---|---|---|---|
| Development Timeline | Months to years for multiple iterative cycles [9] | Potential reduction to weeks or months for a single cycle [5] [6] | Zero-shot prediction; rapid cell-free testing |
| Experimental Throughput | Limited by in vivo cloning and culturing (dozens to hundreds of variants) [1] | Ultra-high-throughput with microfluidics (>100,000 picoliter-scale reactions) [5] | Droplet microfluidics; automated biofoundries |
| Primary Cost Driver | Repeated cloning, transformation, and cell culture [1] | Upfront computational cost and DNA synthesis | Decoupling from cellular growth |
| Data Efficiency | Low; learning is confined to a single project's data | High; leverages foundational models trained on global data [5] | Pre-trained protein language models (ESM, ProGen) |
| Success Rate | Low initial success, improves with iteration | Higher initial success due to pre-screened designs [5] | AI-guided intelligent design |
Strategic Resource Allocation via Active Learning
A key technical mechanism for resource optimization in LDBT is active learning. Instead of testing a random or exhaustively designed library, the machine learning model strategically selects the most informative sequence variants to test experimentally. This "query-by-committee" or "bayesian optimization" approach maximizes the information gain per experiment, effectively reducing the number of Build-Test iterations required to converge on an optimal design [6]. For example, in a pathway optimization campaign using iPROBE (in vitro prototyping and rapid optimization of biosynthetic enzymes), a neural network was trained on a subset of pathway combinations to predict the optimal sets, leading to a 20-fold improvement in product titer in a host organism with minimal experimental effort [5]. This represents a profound shift from brute-force screening to intelligent, guided exploration.
Qualitative Advantage 3: Increased Accessibility and Democratization
The LDBT paradigm lowers significant barriers to entry, making advanced biological engineering accessible to a broader range of researchers and organizations beyond large, well-funded institutions.
Key Factors Enhancing Accessibility
- Reduction in Specialized Infrastructure Dependency: The integration of cell-free systems circumvents the need for maintaining microbial culture collections, sterile fermentation facilities, and extensive molecular cloning labs. Cell-free reactions are modular, reproducible, and can be run with minimal lab equipment [5] [6].
- Democratization of Design Expertise: AI-powered design platforms (e.g., cloud-based ProteinMPNN or ESM interfaces) allow researchers without deep expertise in protein biophysics to generate high-quality designs. This abstracts away the need for intricate, low-level knowledge of sequence-structure-function relationships [40].
- Lowered Operational Costs: While computational resources have a cost, they are often lower and more accessible than the recurring expenses of maintaining large-scale wet-lab operations for iterative testing. The ability to achieve success in fewer cycles also presents a significant overall cost reduction, making projects financially viable for smaller entities [6].
The Scientist's Toolkit: Essential Reagents for LDBT Workflows
The following table details key reagents and materials that form the core of an LDBT workflow, emphasizing the shift toward computational and cell-free resources.
Table 2: Research Reagent Solutions for LDBT Implementation
| Item | Function in LDBT Workflow | Example Use Case |
|---|---|---|
| Pre-trained ML Models (e.g., ESM, ProteinMPNN) | Provides zero-shot predictive power for the "Learn" phase; generates functional protein and genetic sequences. | Designing a novel antimicrobial peptide from first principles [5]. |
| Cell-Free Protein Synthesis Kit | A key "Build" component; enables rapid, high-throughput protein expression without living cells. | Expressing and testing 1000s of enzyme variants in a 384-well plate format within hours [5] [6]. |
| Droplet Microfluidics System | Part of the "Test" phase; allows for ultra-high-throughput screening by compartmentalizing reactions into picoliter droplets. | Screening a library of >100,000 protein variants for binding affinity using fluorescence-activated droplet sorting [5]. |
| Synthetic DNA Oligo Pools | The physical "Build" material; used to synthesize the computationally designed DNA sequences. | Ordering a pool of 500 designed gene variants for parallel cloning and expression [55]. |
| Fluorescent Biosensors / Dyes | Part of the "Test" phase; enables real-time, high-throughput measurement of product formation or protein stability. | Using a fluorogenic substrate to measure enzyme kinetics directly in a cell-free reaction [6]. |
Integrated Experimental Workflow: An LDBT Case Study
To synthesize the concepts of predictability, optimization, and accessibility, the following diagram and description outline a complete, integrated LDBT workflow for engineering a therapeutic enzyme.
Workflow Description: This workflow integrates all phases of the LDBT cycle for a specific application.
- Learn & Design: The process begins by querying a pre-trained protein language model with the wild-type enzyme sequence and the engineering goal (e.g., "increase solubility and activity at pH 5.5"). The model generates and ranks hundreds of candidate variant sequences based on evolutionary fitness and predicted stability.
- Build: The top 200 predicted sequences are synthesized as oligonucleotide pools and expressed directly in a high-throughput cell-free transcription-translation system, bypassing traditional cloning in cells [5].
- Test: The expressed variants are screened in parallel using a fluorogenic substrate for activity and a thermal shift assay for stability—all performed robotically in a microtiter plate. This generates a rich dataset linking sequence to function.
- Iterative Learning: The experimental results are fed back into the model to fine-tune its predictions for this specific protein family. This closed loop validates the initial predictions and continuously improves the model, creating a powerful, project-specific design tool. A lead candidate with confirmed high activity and stability is identified for further development.
This end-to-end process, which can be completed in a matter of weeks, exemplifies the synergistic advantages of the LDBT paradigm: it is predictive (AI-driven design), optimized (high-throughput, minimal iterations), and accessible (relies on commercially available cell-free kits and cloud computing).
The qualitative advantages of the LDBT paradigm over the traditional DBTL cycle are profound and multifaceted. By placing machine learning at the forefront of the biological engineering workflow, LDBT fundamentally enhances predictability through zero-shot and structure-based design tools, enabling researchers to navigate biological complexity with unprecedented accuracy. It achieves superior resource optimization by strategically guiding experiments through active learning, dramatically reducing development timelines and costs associated with iterative trial-and-error. Finally, the reliance on computational power and cell-free systems significantly increases accessibility, democratizing advanced bioengineering capabilities for startups, academic labs, and researchers without extensive infrastructure. As the underlying AI models continue to improve and high-throughput experimental platforms become more widespread, the LDBT cycle is poised to become the standard framework for synthetic biology. This will not only accelerate the development of novel therapeutics, sustainable chemicals, and advanced materials but also reshape the very practice of biological engineering into a more predictable, efficient, and inclusive discipline.
The Design-Build-Test-Learn (DBTL) cycle represents a cornerstone framework in synthetic biology and engineering biology for the systematic development and optimization of biological systems [1]. This iterative process enables researchers to engineer organisms for specific functions, from producing biofuels to pharmaceuticals [1]. While recent advancements have introduced machine learning-guided approaches and cell-free systems to accelerate these cycles [59], traditional DBTL methodologies maintain critical relevance in specific research and development contexts.
This technical guide examines the limitations and fit-for-purpose applications of traditional DBTL cycles, providing researchers with a structured framework for selecting appropriate methodological approaches. We present quantitative comparisons, detailed experimental protocols, and strategic implementation guidelines to inform decision-making for synthetic biology applications, particularly within drug development pipelines where conventional methods continue to offer distinct advantages despite emerging alternatives.
The Traditional DBTL Cycle: Core Components and Workflow
The DBTL cycle operates as an iterative framework for biological engineering, comprising four interconnected phases [1]:
Design: Applying rational principles to design biological components and pathways, often with an emphasis on modular DNA parts for constructing varied genetic assemblies [1].
Build: Assembling designed genetic constructs into expression vectors, increasingly through automated processes to reduce time, labor, and cost while increasing throughput [1].
Test: Analyzing constructed biological systems in functional assays to evaluate performance against design specifications [1].
Learn: Extracting insights from experimental data to inform subsequent design iterations, progressively refining systems toward desired functions [1].
The following diagram illustrates the cyclical nature and key activities of the traditional DBTL workflow:
Limitations of Traditional DBTL Approaches
Throughput and Scalability Constraints
Traditional DBTL cycles face significant throughput limitations, particularly in the Build phase where DNA synthesis methods struggle to meet growing demands for high-quality, gene-length sequences [60]. Manual testing methods create bottlenecks that restrict the exploration of large design spaces, ultimately slowing overall development cycles [1] [61]. While automation technologies offer potential solutions, their implementation requires substantial infrastructure investment and process re-engineering.
Biological Predictive Validity Challenges
A critical limitation emerges in the Test phase, where biological models often demonstrate poor predictive validity for human outcomes. In drug development, conventional animal models show limited correlation with human toxicity and efficacy, contributing to high failure rates in clinical trials [62]. Approximately 90% of drugs entering clinical trials fail to reach the market, with lack of efficacy—often stemming from inadequate model systems—being a primary cause [63].
Resource Intensiveness and Timeline Constraints
Traditional DBTL implementations require substantial time and financial investments. The preclinical phase alone typically consumes 3-6 years, with costs ranging from $1-6 million [63]. Bringing a new drug to market averages $985 million to over $2.8 billion, influenced significantly by the resource-intensive nature of traditional DBTL workflows and high failure rates [63].
Table 1: Resource Requirements and Success Rates in Traditional Drug Discovery
| Parameter | Value | Context |
|---|---|---|
| Preclinical Phase Duration | 3-6 years | Major contributor to overall development timeline [63] |
| Preclinical Costs | $1-6 million | Significant portion of overall R&D budget [63] |
| Clinical Trial Success Rate | ~10% | Only 10% of drug candidates transition from preclinical to clinical trials [63] |
| Market Approval Cost | $985M-$2.8B | Average cost including failed candidates [63] |
Fit-for-Purpose Applications: Where Traditional DBTL Excels
Early-Stage Proof-of-Concept Development
Traditional DBTL approaches remain preferred for initial proof-of-concept work where established protocols and standardized parts provide reliability. When engineering novel biological systems with limited precedent, the methodical nature of traditional DBTL allows comprehensive characterization and troubleshooting. The modular design of DNA parts enables researchers to assemble diverse construct variations through well-established assembly techniques [1].
Regulated Environments and Qualification Requirements
In highly regulated industries like pharmaceutical development, traditional methods with established regulatory precedents often present lower compliance risks. The FDA's Fit-for-Purpose Initiative acknowledges that certain drug development tools may be accepted based on thorough evaluation without formal qualification [64]. This creates scenarios where validated traditional methods outweigh potentially superior but unproven alternatives.
Resource-Limited Settings and Specialized Applications
For research environments with constraints in specialized instrumentation or computational infrastructure, traditional DBTL offers accessibility advantages. The manual testing nature, while slower, requires less capital investment than fully automated systems [61]. Similarly, specialized applications with unique requirements may lack optimized high-throughput solutions, making adaptable traditional approaches more practical.
Comparative Analysis: Traditional vs. Advanced DBTL Methodologies
Table 2: DBTL Methodology Comparison for Synthetic Biology Applications
| Characteristic | Traditional DBTL | ML-Guided DBTL | Cell-Free DBTL |
|---|---|---|---|
| Cycle Speed | Months | Weeks [59] | Days [59] |
| Throughput | Low (manual processes) | High (computational prediction) | Very High (cell-free systems) [59] |
| Data Requirements | Minimal initial data | Large training datasets required [59] | Minimal initial data |
| Implementation Cost | Lower initial investment | High computational infrastructure | Moderate (specialized reagents) |
| Regulatory Precedent | Established | Emerging | Limited |
| Flexibility | High (adaptable protocols) | Medium (model-dependent) | High (multiple reactions) [59] |
| Predictive Validity | Variable (model-dependent) | Improved with sufficient data [59] | Context-dependent |
Experimental Protocols for Traditional DBTL Implementation
Protocol 1: Modular DNA Assembly for Genetic Constructs
This foundational Build-phase protocol enables researchers to create multiple genetic construct variations through interchangeable biological parts [1]:
Design Phase: Select standardized biological parts with compatible assembly sites. Prioritize modularity to enable future iterations.
DNA Assembly:
- Combine double-stranded DNA fragments with appropriate vector backbones
- Use restriction enzyme-based or Gibson assembly methods
- Incubate according to optimized parameters for the selected method
Transformation and Verification:
- Introduce assembled constructs into expression vectors
- Transform into suitable bacterial strains
- Verify successful assembly through colony qPCR or sequencing [1]
- Culture positive clones for plasmid isolation
Functional Testing:
- Express constructs in target host systems
- Assess performance against design specifications
- Collect quantitative data for Learn phase analysis
Protocol 2: Colony Screening and Selection Workflow
This Test-phase protocol addresses the bottleneck of identifying successful constructs from assembly reactions [1]:
Plate Transformed Colonies:
- Spread transformation reactions on selective media plates
- Incubate until distinct colonies form (typically 12-16 hours)
Colony Selection Methods:
- Manual picking using sterile tips or inoculation loops
- Array selected colonies in multi-well plates containing growth media
- Incubate with shaking for protein expression
High-Throughput Screening:
- Process samples using functional assays relevant to the system
- For enzyme engineering, measure substrate conversion rates
- Record quantitative metrics for each variant
Data Collection and Analysis:
- Compile screening results with sequence information
- Identify performance trends and correlations
- Select lead candidates for further iteration
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Traditional DBTL Workflows
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Expression Vectors | DNA construct maintenance and expression | Selection of appropriate promoters, ORIs, and resistance markers critical [1] |
| DNA Assembly Master Mixes | Enzymatic assembly of DNA fragments | Restriction enzyme or Gibson assembly systems for modular construct creation [1] |
| Competent Bacterial Cells | Transformation and plasmid propagation | High-efficiency strains for library construction and maintenance [1] |
| Cell-Free Expression Systems | In vitro protein synthesis without cells | Rapid testing of enzyme variants; 1217 variants tested in 10,953 reactions demonstrated [59] |
| qPCR Reagents | Verification of successful assembly | Colony qPCR provides rapid validation before sequencing [1] |
| Next-Generation Sequencing Kits | Comprehensive construct verification | Essential for validating designed mutations in engineered enzymes [1] |
| Specialized Substrates | Functional assay components | Enable testing of enzyme activity; 1100+ unique reactions demonstrated for substrate profiling [59] |
Workflow Integration: Traditional DBTL in Practice
The implementation of a traditional DBTL cycle for enzyme engineering follows a structured pathway, as illustrated in the following workflow diagram:
Strategic Implementation Guidelines
When to Select Traditional DBTL Approaches
Project Stage Considerations:
- Initial concept validation with limited precedent
- Small-scale prototyping requiring flexible, adaptable methods
- Regulatory submissions requiring established methodologies
Resource Assessment:
- Limited access to high-throughput automation infrastructure
- Budget constraints prohibiting specialized computational resources
- Expertise availability favoring molecular biology over data science
Biological System Factors:
- Poorly characterized systems with limited training data for ML approaches
- Complex biological contexts where cell-free systems lack predictive validity
- Specialized host organisms without established high-throughput tools
Integration Strategies with Advanced Methods
Traditional DBTL need not function as an exclusive approach. Strategic integration with emerging technologies can enhance efficiency while maintaining reliability:
Hybrid Implementation: Use traditional methods for initial cycle iterations to generate high-quality data, then transition to ML-guided approaches once sufficient training data exists [59].
Complementary Validation: Employ cell-free systems for rapid preliminary screening [59], followed by traditional in vivo validation for lead candidates.
Phased Adoption: Gradually introduce automation technologies into specific DBTL phases where they offer the greatest efficiency gains while maintaining traditional approaches in other phases.
Traditional DBTL cycles maintain significant relevance in synthetic biology and drug development despite the emergence of advanced methodologies. Their fit-for-purpose application spans early-stage development, regulated environments, resource-constrained settings, and specialized biological contexts. The limitations of traditional approaches—particularly in throughput, predictive validity, and resource requirements—must be balanced against their advantages in reliability, regulatory precedent, and implementation accessibility.
Researchers should select DBTL methodologies through systematic assessment of project requirements, available resources, and stage-specific needs rather than defaulting to either traditional or advanced approaches exclusively. Strategic integration of traditional and emerging methods often provides the optimal path forward, leveraging the strengths of each approach while mitigating their respective limitations. As the field advances, traditional DBTL will continue evolving rather than disappearing, maintaining its foundational role in biological engineering while incorporating targeted technological enhancements to address its core limitations.
In the rapidly evolving field of synthetic biology, the structured execution of the Design-Build-Test-Learn (DBTL) cycle is fundamental to engineering biological systems. However, the traditional DBTL cycle is being transformed by the integration of artificial intelligence (AI), giving rise to a next-generation, Learning-driven Design-Build-Test (LDBT) paradigm. This shift is critical for researchers, scientists, and drug development professionals aiming to accelerate the discovery and development of novel therapeutics, sustainable materials, and other bio-based products [65] [40].
The core distinction lies in the sequence and automation of key activities. The traditional DBTL cycle is often a sequential, human-intensive process. In contrast, the LDBT cycle embeds AI and machine learning at its core, creating a continuous, automated learning loop that dramatically accelerates the pace of innovation [40]. This guide provides a decisive framework for selecting the optimal development cycle for your specific research and development goals, synthesizing quantitative data, experimental protocols, and strategic visualizations to inform your decision.
Comparative Analysis: DBTL vs. LDBT Cycles
Understanding the structural and functional differences between these two cycles is the first step in the selection process. The table below summarizes the core distinctions.
Table 1: A comparative overview of the traditional DBTL and AI-driven LDBT cycles.
| Feature | Traditional DBTL Cycle | AI-Driven LDBT Cycle |
|---|---|---|
| Core Workflow | A sequential process of Design, Build, Test, and Learn [66]. | An iterative, integrated cycle where Learning informs every subsequent Design step, often automated [40]. |
| Primary Driver | Human intuition and prior knowledge, supplemented by experimental data. | Data-driven insights and predictions generated by AI and machine learning models [65] [40]. |
| Learning Phase | A distinct, often final phase in the cycle. | A continuous, foundational activity that occurs in parallel with all other phases [40]. |
| Automation Level | Typically low to moderate, with significant manual intervention. | High, with the potential for fully automated "self-driving" laboratories [40]. |
| Cycle Speed | Can take months or years, often due to the "Test" bottleneck [65]. | Radically accelerated; AI can run thousands of simulations in hours, compressing cycles to days or weeks [65]. |
| Key Enabling Technologies | Molecular biology techniques (PCR, cloning), sequencing. | AI/ML models (LLMs, transformers), robotic automation, advanced bio-design tools (BDTs) [40]. |
The following diagram illustrates the logical relationship and workflow differences between the two cycles.
Quantitative Data Synthesis
The strategic shift from DBTL to LDBT is supported by compelling market data and performance metrics. The synthetic biology market itself is experiencing explosive growth, projected to grow from US$9.5 billion in 2020 to US$38.7 billion by 2027, underscoring the field's economic and technological significance [66].
The most significant quantitative differences, however, are observed in R&D efficiency. The table below compiles key performance indicators that highlight the transformative impact of the LDBT cycle.
Table 2: Key quantitative metrics comparing traditional and AI-accelerated development processes.
| Metric | Traditional Workflow | AI-Accelerated (LDBT) Workflow | Source / Example |
|---|---|---|---|
| Drug Discovery Timeline | 4-5 years (early stage) | 12 months (early stage) | Exscientia (OCD treatment to trials) [65] |
| DNA Synthesis Turnaround | >14 days (via vendor) | <1 day (in-house) | On-demand synthesis technology [67] |
| Time Reduction for Molecule Testing | Weeks for lab tests | Hours for AI simulations | AI-powered in silico modeling [65] |
| Protein Structure Prediction | Years of laborious work | Structures for 200M+ proteins predicted | DeepMind's AlphaFold [65] |
Experimental Protocols in AI-Driven Synthetic Biology
The LDBT cycle is not a theoretical concept but is implemented through concrete, cutting-edge experimental methodologies. Below are detailed protocols for two key experiments that exemplify this approach.
Protocol 1: AI-Guided Metabolic Pathway Optimization
Objective: To engineer a microbial host for the high-yield production of a valuable compound (e.g., a therapeutic molecule or biofuel) [65] [67].
Detailed Methodology:
- Design (AI-Powered):
- Data Aggregation: Feed AI models with massive datasets, including genomic data, experimental outcomes from prior cycles, and biochemical knowledge bases [67].
- Predictive Modeling: The AI model analyzes the metabolic network and precisely predicts the impact of genetic modifications (e.g., gene knock-outs, promoter swaps, enzyme engineering) on the host's metabolic flux and final product yield [67].
- Generate Design List: The AI outputs a prioritized list of specific genetic edits predicted to optimize the pathway.
Build (Automated):
- In-house DNA Synthesis: Utilize rapid, automated DNA synthesizers (e.g., enzymatic 'digital to biological converters') to create the genetic constructs designed by the AI overnight, on-demand [67].
- Automated Strain Engineering: Use robotic automation to perform high-throughput genome editing (e.g., via CRISPR) in the microbial host to create a library of engineered strains based on the AI's design list.
Test (High-Throughput):
- Cultivation and Screening: Grow the library of engineered strains in parallel, automated bioreactors.
- Analytics: Employ high-throughput analytics, such as mass spectrometry, to rapidly quantify the titers of the target compound and key metabolic intermediates for each strain.
Learn (Continuous):
- Data Feedback Loop: The experimental results (production yields, growth data) are fed back into the AI model.
- Model Retraining: The AI model is retrained on this new, high-quality data, improving its predictive accuracy for the next cycle of designs and closing the loop [67].
Protocol 2: De Novo Protein Design using Generative AI
Objective: To create a novel, functional protein (e.g., a therapeutic enzyme or binding protein) that does not exist in nature [65].
Detailed Methodology:
- Learn (Foundational Model Training):
Design (Generative):
- Define Functional Constraints: Provide the AI with a set of desired properties (e.g., "bind to target X," "catalyze reaction Y," "maximize thermal stability").
- Generate Candidate Sequences: The generative AI model creates thousands of novel amino acid sequences predicted to fold into structures that meet the desired functional constraints [65].
Build (Physical Instantiation):
- Gene Synthesis: Convert the top-ranking AI-designed protein sequences into DNA sequences, which are then synthesized in vitro [67].
- Protein Expression: Clone these synthetic genes into expression systems (e.g., E. coli, yeast) to produce the physical proteins.
Test (Functional Validation):
- Structural Validation: Use techniques like X-ray crystallography or cryo-electron microscopy to confirm the protein's structure matches the AI's prediction.
- Functional Assays: Perform biochemical and cellular assays to measure the novel protein's activity, specificity, and stability against the design objectives.
Learn (Model Refinement):
- The experimental data on which designs were successful or unsuccessful is used to refine and improve the generative AI model, enhancing its performance for future protein design challenges [40].
The Scientist's Toolkit: Key Research Reagent Solutions
Implementing the DBTL and LDBT cycles requires a suite of essential materials and technologies. The following table details the key reagents and tools that form the backbone of modern synthetic biology workflows.
Table 3: Essential research reagents and tools for synthetic biology workflows.
| Item | Function / Description | Relevance to Cycle |
|---|---|---|
| Automated DNA/RNA Synthesizer | Enables rapid, in-house production of genetic constructs (genes, mRNA) on-demand, replacing slow vendor outsourcing [67]. | Critical for LDBT; eliminates the "Build" bottleneck. |
| AI/ML Biodesign Tools (BDTs) | Software that uses AI to predict protein structures (e.g., AlphaFold), design genetic constructs, and optimize metabolic pathways [65] [40]. | The core engine of the LDBT cycle. |
| Rapid DNA Sequencing Platform | Provides the high-throughput data on genetic sequences and modifications that is essential for training and validating AI models [40]. | Foundational for both cycles, especially "Test" and "Learn". |
| Enzymatic "Digital-to-Biological" Converter | A technology that directly translates digital DNA sequence information into physical, synthesized DNA molecules [67]. | Key for LDBT, enabling a seamless digital-to-physical pipeline. |
| Robotic Liquid Handling Systems | Automates repetitive laboratory tasks such as pipetting, plating, and assays, enabling high-throughput experimentation [40]. | Essential for scaling the "Test" phase in LDBT. |
| Machine Learning-guided Experimental Tools (e.g., METIS) | Modular software that uses active learning to interactively suggest the next best experiment, optimizing systems with limited data [66]. | Embodies the "Learn" phase, guiding efficient experimental design. |
Decision Framework for Cycle Selection
Choosing between the DBTL and LDBT paradigms depends on your project's specific constraints and ambitions. The following diagram outlines the key decision-making pathway.
Framework Guidance:
- Choose the LDBT cycle if: Your project is characterized by high complexity (e.g., novel pathway engineering, de novo protein design), access to large and well-curated datasets is available, and you have the computational resources and AI/ML expertise in-house. This path is optimal for achieving breakthrough innovations and maximizing speed [65] [40].
- Choose the traditional DBTL cycle if: You are working on a well-understood biological system, lack large training datasets, or have limited access to AI tools and expertise. This path remains a robust and effective method for incremental improvements and projects with lower resource flexibility [66].
- Adopt a Hybrid Approach if: Your project is novel but you are in the early stages of building AI capabilities. Begin with a DBTL cycle to generate high-quality data, then progressively integrate AI tools into the "Learn" and "Design" phases as data accumulates, effectively transitioning towards a full LDBT cycle over time [40].
The transition from the traditional DBTL cycle to the AI-powered LDBT cycle represents a paradigm shift in synthetic biology and drug development. The LDBT framework offers a decisive advantage in speed, precision, and the ability to tackle biological complexity, as evidenced by its groundbreaking applications in drug discovery and protein design [65]. However, this powerful approach demands significant investment in data, technology, and expertise.
The decision framework provided herein empowers researchers and organizations to make a strategic, evidence-based choice. By honestly assessing your project's goals, available data, and resource constraints against this framework, you can select the most efficient and effective path forward. As the field continues to evolve, the integration of AI through the LDBT cycle will undoubtedly become the standard for those aiming to lead in the next wave of biotechnological innovation.
Conclusion
The comparison between DBTL and LDBT reveals a fundamental shift from empirical iteration towards a predictive, first-principles approach in synthetic biology. The integration of machine learning at the outset of the cycle, combined with rapid cell-free testing, demonstrably accelerates the engineering of biological systems, reduces resource-intensive trial-and-error, and enhances design predictability. For biomedical and clinical research, this LDBT paradigm promises to drastically shorten development timelines for novel therapeutics, including engineered cells, enzymes, and biosynthetic pathways. Future directions will hinge on developing more robust foundational models, standardizing cell-free platforms, and fully automating the LDBT cycle, ultimately moving the field closer to a 'Design-Build-Work' model that can reliably reshape the bioeconomy and advance personalized medicine.
References
- 1. DBTL Approach in Synthetic Biology | Molecular Devices [https://www.moleculardevices.com/applications/synthetic-biology/dbtl-approach]
- 2. - Design - Build - Test (DBTL) | EBRC Research Roadmap Learn [https://roadmap.ebrc.org/dbtl/]
- 3. Simulated Design–Build–Test–Learn Cycles for Consistent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10510747/]
- 4. Abstraction hierarchy to define biofoundry workflows and ... [https://www.nature.com/articles/s41467-025-61263-6]
- 5. combining machine learning and rapid cell-free testing [https://www.nature.com/articles/s41467-025-65281-2]
- 6. LDBT: Machine Learning Meets Rapid Cell-Free Testing [https://bioengineer.org/ldbt-machine-learning-meets-rapid-cell-free-testing/]
- 7. The knowledge driven DBTL cycle provides mechanistic ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02729-6]
- 8. evaluation of free management platforms for synthetic biology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6690502/]
- 9. Digital to Biological Translation: How the Algorithmic Data ... [https://www.mdpi.com/2674-0583/3/4/17]
- 10. LDBT instead of DBTL: combining machine learning and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12589603/]
- 11. Engineering is evolution: a perspective on design ... [https://www.nature.com/articles/s41467-024-48000-1]
- 12. Synthetic biology: Learning the way toward high-precision ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3002116]
- 13. The art of modeling gene regulatory circuits [https://www.nature.com/articles/s41540-024-00380-2]
- 14. Meta synthetic biology: controlling the evolution of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6302734/]
- 15. Evolutionary tinkering vs. rational engineering in the times of ... [https://lsspjournal.biomedcentral.com/articles/10.1186/s40504-018-0086-x]
- 16. Medium-sized protein language models perform well at ... [https://www.nature.com/articles/s41598-025-05674-x]
- 17. Machine learning-guided evolution of pyrrolysyl-tRNA ... [https://www.nature.com/articles/s41467-025-61952-2]
- 18. Introducing ProGen3 [https://www.profluent.bio/showcase/progen3]
- 19. Biology has scaling laws too - Air Street Press [https://press.airstreet.com/p/biology-has-scaling-laws-profluent-progen3]
- 20. Atomic context-conditioned protein sequence design using ... [https://www.nature.com/articles/s41592-025-02626-1]
- 21. AI-driven high-throughput droplet screening of cell-free ... [https://www.nature.com/articles/s41467-025-58139-0]
- 22. The all-E. coliTXTL toolbox 3.0: new capabilities of a cell-free ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8546610/]
- 23. Cell-free Protein Synthesis Market worth $308.9 million by ... [https://www.marketsandmarkets.com/PressReleases/cell-free-protein-synthesis.asp]
- 24. Quantitative modeling of transcription and translation of an ... [https://www.nature.com/articles/s41598-019-48468-8]
- 25. Short-loop engineering strategy for enhancing enzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11982487/]
- 26. Automated in vivo enzyme engineering accelerates biocatalyst... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11043082/]
- 27. Cell-Free Synthetic Biology for Pathway Prototyping [https://www.sciencedirect.com/science/article/pii/S0076687918301800]
- 28. DLFea4AMPGen de novo design of antimicrobial peptides ... [https://www.nature.com/articles/s41467-025-64378-y]
- 29. Peptide-based antibiotics: structure-driven strategies to ... [https://www.sciencedirect.com/science/article/pii/S0968089625004274]
- 30. Antimicrobial Peptides for Therapeutic Applications: A Review [https://pmc.ncbi.nlm.nih.gov/articles/PMC6268056/]
- 31. APD6: the antimicrobial peptide database is expanded to ... [https://pubmed.ncbi.nlm.nih.gov/40927993/]
- 32. Frontiers in biofoundry: opportunities and challenges [https://www.frontiersin.org/journals/synthetic-biology/articles/10.3389/fsybi.2025.1630026/full]
- 33. Biofoundries are a nucleating hub for industrial translation [https://pmc.ncbi.nlm.nih.gov/articles/PMC8546609/]
- 34. Closed-loop systems in medical devices [https://blog.johner-institute.com/regulatory-affairs/closed-loop-systems/]
- 35. Finding the balance between Data Quality and ... [https://dataloop.ai/blog/finding-the-balance-between-data-quality-and-data-quantity-for-an-accurate-ai-model/]
- 36. Why Data Quality Is More Important Than Ever in an AI-Driven ... [https://dataproducts.substack.com/p/why-data-quality-is-more-important]
- 37. Learning the way toward high-precision biological design [https://pmc.ncbi.nlm.nih.gov/articles/PMC10231942/]
- 38. In silico design of context-responsive mammalian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5737543/]
- 39. In silico analysis of alternative splicing on drug-target gene ... [https://www.nature.com/articles/s41598-019-56894-x]
- 40. The convergence of AI and synthetic biology: the looming ... [https://www.nature.com/articles/s44385-025-00021-1]
- 41. Automated and Programmable Cell-Free Systems for Scalable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12463565/]
- 42. Cell-Free Protein Expression System [https://www.datainsightsmarket.com/reports/cell-free-protein-expression-system-587327]
- 43. RespectM revealed metabolic heterogeneity powers deep ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10329182/]
- 44. Active Learning: How to Implement It In Classroom in 2025 [https://www.simplek12.com/blog/active-learning]
- 45. A technical review of multi-omics data integration methods [https://pubmed.ncbi.nlm.nih.gov/40748323/]
- 46. 2025 Trends: Multiomics [https://www.genengnews.com/topics/omics/2025-trends-multiomics/]
- 47. In Vitro vs In Vivo: A History of Modern Cell Culture [https://emulatebio.com/in-vitro-vs-in-vivo-cell-culture/]
- 48. Bridging the Gap: The Role of Advanced Formulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12579833/]
- 49. Organ-on-a-chip Bridging the in vitro to in vivo gap [https://cn-bio.com/resource/next-generation-drug-discovery-bridging-the-gap-with-organ-on-a-chip-technology/]
- 50. Predicting In Vivo Efficacy from In Vitro Data - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC7070804/]
- 51. How AI Is Irrevocably Altering the DNA of Drug Development [https://www.drugpatentwatch.com/blog/how-ai-is-already-changing-drug-development/?srsltid=AfmBOopT86zMN0bizRP2U0JxyJUaSnNfFdsN7ZaD0CuZ1QeexAqPFSgi]
- 52. The Impact of AI on Drug Development Strategies in 2025 [https://vasro.de/en/ai-drug-development-pipeline-2025/]
- 53. Reflections from Biotrans 2025: Charting the Future of… [https://www.biocatalysts.com/media-resources/biotrans-biocatalysis-future-trends]
- 54. The Emerging Role of Machine Learning in Biocatalysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550629]
- 55. Synthetic Biology Market Size and YoY Growth Rate, 2025 ... [https://www.coherentmarketinsights.com/market-insight/synthetic-biology-market-112]
- 56. Computational redesign of a hydrolase for nearly complete ... [https://www.nature.com/articles/s41467-024-45662-9]
- 57. Enzyme selection, optimization, and production toward ... [https://pubmed.ncbi.nlm.nih.gov/37594123/]
- 58. An automated Design-Build-Test-Learn pipeline for ... [https://www.nature.com/articles/s42003-018-0076-9]
- 59. Accelerated enzyme engineering by machine-learning ... [https://www.nature.com/articles/s41467-024-55399-0]
- 60. Optimizing the DBTL cycle to scale engineering biology [https://www.evonetix.com/white-papers/optimizing-the-design-build-test-learn-cycle-in-synthetic-biology]
- 61. DBTL bioengineering cycle for part characterization and ... [https://www.sciencedirect.com/science/article/pii/S2405896324017488]
- 62. Limitations of Animal Studies for Predicting Toxicity in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7185927/]
- 63. Dealing With The Challenges Of Drug Discovery [https://www.zeclinics.com/blog/dealing-with-the-challenges-of-drug-discovery]
- 64. Drug Development Tools: Fit-for-Purpose Initiative [https://www.fda.gov/drugs/development-approval-process-drugs/drug-development-tools-fit-purpose-initiative]
- 65. The Future of Drug Development: AI Meets Synthetic Biology [https://idr.co/blog/the-future-of-drug-development-ai-meets-synthetic-biology]
- 66. Global advances in synthetic biology [https://www.ddw-online.com/global-advances-in-synthetic-biology-20471-202211/]
- 67. Driving Synthetic Biology Forward Through Adoption of ... [https://medcitynews.com/2025/05/driving-synthetic-biology-forward-through-adoption-of-technology-enabling-rapid-dna-and-mrna-synthesis/]