Integrating Multi-Omics Data to Decipher CRISPRi Responses: From Foundational Concepts to Clinical Translation
This article explores the powerful synergy between multi-omics technologies and CRISPR interference (CRISPRi) screens, a combination that is revolutionizing functional genomics and therapeutic discovery.
Integrating Multi-Omics Data to Decipher CRISPRi Responses: From Foundational Concepts to Clinical Translation
Abstract
This article explores the powerful synergy between multi-omics technologies and CRISPR interference (CRISPRi) screens, a combination that is revolutionizing functional genomics and therapeutic discovery. We cover foundational principles, detailing how genomics, transcriptomics, proteomics, and epigenomics data provide a systems-level context for interpreting CRISPRi phenotypes. The article delves into advanced methodologies for data integration, including network analysis and AI-driven approaches, and addresses key challenges in data harmonization and computational infrastructure. Through comparative analyses across cell types and states, we highlight how integrated multi-omics data validates findings and reveals cell-context-specific dependencies, offering a comprehensive guide for researchers and drug development professionals aiming to leverage these tools for precision medicine.
Laying the Groundwork: How Multi-Omics Data Provides Context for CRISPRi Responses
The integration of multi-omics data demands genetic tools that are both precise and reversible to accurately map genotype-phenotype relationships. CRISPR interference, or CRISPRi, has emerged as a foundational technology in this domain. It is a genetic perturbation technique that allows for sequence-specific repression of gene expression without introducing double-strand breaks (DSBs) in DNA, thereby avoiding the associated genomic instability and permanent knockout effects [1] [2]. By providing a highly specific and tunable means to perform gene knockdown, CRISPRi enables the functional characterization of genes within a physiological context, making it an indispensable tool for modern functional genomics and drug target validation [3] [4].
This technical guide details the core mechanism of CRISPRi, provides standardized experimental protocols, and situates its application within the framework of integrated omics research.
Core Mechanism and Design Principles
The Core Components of the CRISPRi System
The CRISPRi system is engineered from the Type II CRISPR-Cas9 system but is functionally distinct due to a key modification: the use of a catalytically dead Cas9 (dCas9). This variant contains point mutations (D10A and H840A in the case of S. pyogenes Cas9) that inactivate the RuvC and HNH nuclease domains, rendering the protein incapable of cutting DNA [1] [2]. The system functions as a DNA-binding complex guided by a single-guide RNA (sgRNA), which directs dCas9 to a specific genomic locus through Watson-Crick base pairing [3] [1].
The primary mechanism of transcriptional repression is steric hindrance. Once bound to its target DNA sequence, which must be adjacent to a short Protospacer Adjacent Motif (PAM, e.g., 5'-NGG-3' for SpCas9), the dCas9-sgRNA complex physically blocks the progression of RNA polymerase (RNAP), thereby halting transcription [1] [4]. The repression is highly efficient, achieving up to 99.9% silencing in prokaryotes and over 90% in human cells [1].
To enhance repression efficiency in eukaryotic cells, dCas9 is often fused to a transcriptional repressor domain. The most commonly used is the Krüppel-associated box (KRAB) domain. When recruited to a gene's promoter, dCas9-KRAB induces heterochromatin formation, leading to a more potent and stable gene silencing, with repression levels reaching up to 99% in human cells [3] [1] [5].
gRNA Design for Optimal CRISPRi Efficiency
The design of the sgRNA is a critical determinant of CRISPRi success. Unlike CRISPR knockout, which targets early exons, CRISPRi gRNAs are most effective when targeting specific windows near the Transcription Start Site (TSS). The table below summarizes the key design parameters for CRISPRi gRNAs.
Table 1: Guidelines for CRISPRi gRNA Design Targeting the TSS
| Design Parameter | Optimal Targeting Window | Rationale | Key Considerations |
|---|---|---|---|
| Target Region | -50 to +300 bp relative to the TSS [6] [2] | This region is critical for transcription initiation and early elongation. | Targeting within the first 100 bp downstream of the TSS is often most effective [2]. |
| DNA Strand | Non-template strand for strongest repression (for dCas9 without KRAB) [1] | The RNAP helicase activity may weaken repression when sgRNA binds the template strand. | When using dCas9-KRAB, targeting either strand can be effective [6]. |
| gRNA Specificity | 20 nt base-pairing sequence | Ensures on-target binding. | Use design tools (e.g., CHOP-CHOP, E-CRISP) to minimize off-targets with similar sequences [6]. |
| Chromatin State | Accessible, nucleosome-free regions | Local chromatin accessibility impacts dCas9 binding efficiency [1]. | Consider integrating with ATAC-seq or other epigenomic data to inform target site selection. |
Experimental Workflow and Protocol
A typical CRISPRi experiment involves the generation of a stable cell line, delivery of sgRNAs, and phenotypic analysis. The workflow below outlines the key steps.
Diagram 1: CRISPRi Experimental Workflow
Key Research Reagent Solutions
The following table catalogues the essential materials required to establish a CRISPRi system.
Table 2: Essential Research Reagents for CRISPRi Experiments
| Reagent / Tool | Function / Description | Example Formats |
|---|---|---|
| dCas9 Repressor | The engineered, non-cutting core nuclease fused to a repressor domain (e.g., KRAB). | Lentiviral expression vector (e.g., pLV-dCas9-KRAB); stable cell line. |
| sgRNA Expression Vector | Delivers the targeting component; contains a promoter (e.g., U6) and scaffold sequence. | Lentiviral vector; all-in-one systems containing both dCas9 and sgRNA. |
| gRNA Design Tools | Bioinformatics software to design highly specific and efficient sgRNAs. | CHOP-CHOP, E-CRISP, CRISPR Direct [6]. |
| Lentiviral Packaging System | For producing viral particles to efficiently deliver constructs into a wide range of cell types. | Third-generation packaging plasmids (psPAX2, pMD2.G). |
| Induction System | Allows for temporal control over dCas9-KRAB expression. | Doxycycline-inducible TetO promoter system [3]. |
Detailed Protocol: Implementing a CRISPRi Knockdown
The following protocol is adapted from multiple established sources for use in mammalian cells [3] [2].
Step 1: Generate a Stable "Helper" Cell Line
- Introduce the dCas9-KRAB construct into your target cell line via lentiviral transduction.
- Use an inducible promoter (e.g., Tet-On) for controllable expression to prevent potential cytotoxicity from long-term dCas9 expression [3].
- Select and expand stable pools or clones using the appropriate antibiotic (e.g., puromycin). Validate dCas9-KRAB expression via immunoblotting or flow cytometry upon induction (e.g., with 1 µg/mL doxycycline for 48 hours).
Step 2: Design and Clone sgRNAs
- Identify the canonical TSS of your target gene from a trusted genome annotation database (e.g., RefSeq).
- Using a gRNA design tool, select 3-5 sgRNAs targeting the region from -50 to +300 bp relative to the TSS.
- Clone the top-ranked sgRNA sequences into a lentiviral sgRNA expression vector via golden gate or restriction cloning.
Step 3: Deliver sgRNA and Induce Knockdown
- Transduce the stable dCas9-KRAB helper cell line with the sgRNA-containing lentivirus at a low multiplicity of infection (MOI < 1) to ensure single copy integration.
- After 24-48 hours, select transduced cells with the appropriate antibiotic (e.g., blasticidin).
- To initiate knockdown, add doxycycline to the culture medium to induce dCas9-KRAB expression. A time course of 3-7 days is typical for observing maximal repression.
Step 4: Validate and Analyze Phenotype
- Knockdown Efficiency: Quantify mRNA levels using qRT-PCR 72-96 hours post-induction. Expect >80% repression with well-designed gRNAs [3].
- Protein Level Analysis: Confirm reduced protein expression via western blot or immunofluorescence 5-7 days post-induction.
- Phenotypic Screening: Perform functional assays relevant to your biological question (e.g., cell proliferation, migration, differentiation, or drug sensitivity).
Comparative Advantages in Functional Genomics
CRISPRi offers distinct advantages over other loss-of-function technologies, which are critical for interpreting omics data.
Table 3: CRISPRi vs. Alternative Gene Silencing Technologies
| Feature | CRISPRi | CRISPR Nuclease (KO) | RNAi (shRNA/siRNA) |
|---|---|---|---|
| Mechanism of Action | Transcriptional repression (DNA level) | DNA cleavage and mutagenesis (DNA level) | mRNA degradation/destabilization (cytoplasmic RNA level) |
| Reversibility | Yes (tunable and reversible) [2] [7] | No (permanent knockout) | Partially reversible (transient knockdown) |
| Specificity & Off-Targets | High specificity; minimal off-targets with careful design [3] [1] | High specificity, but off-target cleavage can occur [8] | High off-target effects due to competition with endogenous miRNA machinery [8] [9] |
| Tunability | Yes (via inducer dosage or sgRNA engineering) [4] | No (binary outcome) | Limited (depends on transfection efficiency) |
| Genetic Target Space | Can target non-coding RNAs, promoters, and introns [1] | Primarily coding exons | Primarily mRNA transcripts; inefficient for nuclear RNA [2] |
| Cytotoxicity / Genotoxicity | Low (no DNA damage) [2] [7] | High (DSBs cause genomic instability) [2] | Variable (can trigger immune responses) [8] |
Integration with Omics Data and Future Perspectives
CRISPRi is uniquely positioned for integration with multi-omics approaches. Its precision and reversibility make it ideal for perturb-seq-type experiments, where single-cell RNA sequencing is used to read out the transcriptional consequences of many individual genetic perturbations simultaneously [5]. This allows for the direct mapping of gene regulatory networks.
The combination of CRISPRi screens with other single-cell omics technologies, such as scATAC-seq for chromatin accessibility, provides a systems-level view of how gene perturbations rewire the epigenome and transcriptome [5]. Furthermore, the titratable nature of CRISPRi is essential for studying essential genes, as it allows for the creation of hypomorphic alleles (partial loss-of-function) that can be grown in competition and analyzed to dissect dose-dependent gene functions [1] [4].
Future developments will focus on improving the precision and expanding the scope of CRISPRi. This includes engineering novel Cas variants with altered PAM specificities to access more of the genome, developing more potent repressor domains, and refining computational models to predict gRNA efficacy by integrating genomic, transcriptomic, and epigenomic features [6] [5]. As these tools mature, CRISPRi will remain a cornerstone technology for deriving causal, mechanistic insights from correlative omics datasets.
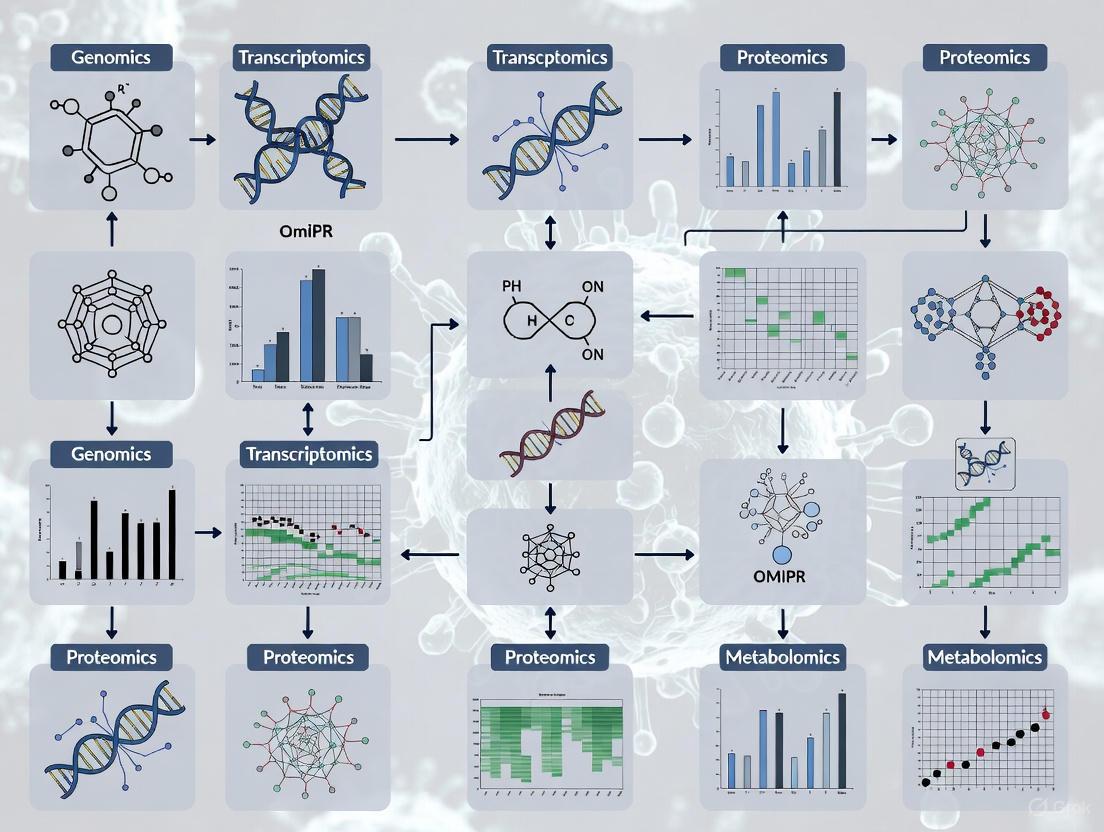
The study of biological systems has evolved from examining single molecular layers to integrating multiple "omics" fields—genomics, transcriptomics, proteomics, and epigenomics—to gain a comprehensive understanding of cellular function and disease mechanisms. While each omic provides valuable data alone, in concert, they can reveal new and valuable insights into cell subtypes, cell interactions, and interactions between different omic layers leading to gene regulatory and phenotypic outcomes [10]. Since each omic layer is causally tied to the next, multi-omics integration serves to disentangle this relationship to properly capture cell phenotype [10].
The integration of these large, complex, multimodal datasets has tremendous potential to reveal intricate biological mechanisms and pathways, but represents a considerable computational challenge for researchers [10]. Multi-omics research is particularly valuable for understanding complex diseases like cancer, where capturing disease complexity requires more than a panel of genomic markers [11]. Unlike rare genetic disorders caused by few genetic variations, complex diseases require a comprehensive understanding of interactions between various cellular regulatory layers [11].
Categories of Omics Technologies
Core Omics Layers
Biological systems are investigated through several core omics technologies, each providing a distinct perspective on cellular function:
Genomics: The study of entire genomes, including the collection, characterization, and quantification of all genes of an organism and their interrelationships [12]. Genome-wide association studies (GWAS) represent a typical application, identifying disease-associated single nucleotide polymorphisms (SNPs) across the genome [12].
Transcriptomics: The study of the expression of all RNAs from a given cell population, providing a global perspective on molecular dynamic changes induced by environmental factors or pathogenic agents [12]. This includes protein-coding RNAs (mRNAs) and various noncoding RNAs such as long noncoding RNAs, microRNAs, and circular RNAs [12].
Proteomics: The maximum identification and quantification of all proteins in cells or tissues [12]. Since RNA analysis often lacks correlation with protein expression due to post-transcriptional modifications, proteomics provides more direct information about cellular responses to environmental changes or disease progression [12].
Epigenomics: The investigation of epigenetic phenomena at genomic and transcriptional levels, encompassing chromatin architecture, chromatin accessibility, histone modifications, transcription factor binding, DNA methylation, and RNA methylation [13]. These modifications regulate gene expression without altering the underlying DNA sequence.
Advanced Omics Technologies
Recent technological advancements have expanded multi-omics research capabilities:
Single-cell omics: Technologies such as single-cell RNA sequencing (scRNA-seq) enable the detection of transcripts in specific cell types, revealing cellular heterogeneity and function [12] [5].
Spatial omics: Methods including spatial transcriptomics provide location context to molecular measurements, preserving architectural relationships within tissues [12].
Metabolomics: The study of small molecule metabolites derived from cellular metabolic processes, providing immediate reflection of dynamic changes in cell physiology [12].
Computational Integration Strategies
Types of Multi-Omics Integration
Integration approaches are broadly categorized based on the relationship between the measured omics data:
Matched (Vertical) Integration: Merges data from different omics within the same set of samples, using the cell as an anchor to bring these omics together [10]. This approach relies on technologies that profile omics data from two or more distinct modalities from within a single cell [10].
Unmatched (Diagonal) Integration: Integrates different omics from different cells or different studies, requiring derivation of anchors through co-embedded spaces where commonality between cells is found [10]. This represents a more substantial computational challenge since the cell or tissue cannot be used as a direct anchor [10].
Mosaic Integration: Used when experimental designs have various combinations of omics that create sufficient overlap across samples [10]. For example, if one sample was assessed for transcriptomics and proteomics, another for transcriptomics and epigenomics, and a third for proteomics and epigenomics, there is enough commonality to integrate the data [10].
Integration Methods and Tools
Multiple computational methods have been developed to address the challenges of multi-omics integration:
Table 1: Multi-Omics Integration Tools and Methodologies
| Tool Name | Year | Methodology | Integration Capacity | Data Types |
|---|---|---|---|---|
| Seurat v4 | 2020 | Weighted nearest-neighbour | Matched | mRNA, spatial coordinates, protein, accessible chromatin [10] |
| MOFA+ | 2020 | Factor analysis | Matched | mRNA, DNA methylation, chromatin accessibility [10] |
| totalVI | 2020 | Deep generative | Matched | mRNA, protein [10] |
| GLUE | 2022 | Variational autoencoders | Unmatched | Chromatin accessibility, DNA methylation, mRNA [10] |
| Cobolt | 2021 | Multimodal variational autoencoder | Mosaic | mRNA, chromatin accessibility [10] |
| MultiVI | 2022 | Probabilistic modelling | Mosaic | mRNA, chromatin accessibility [10] |
| StabMap | 2022 | Mosaic data integration | Unmatched | mRNA, chromatin accessibility [10] |
| Flexynesis | 2025 | Deep learning toolkit | Bulk multi-omics | Multiple modalities for precision oncology [11] |
These tools employ diverse computational approaches:
- Matrix factorization methods (e.g., MOFA+) decompose multi-omics data into latent factors representing shared and specific variations across modalities [10].
- Neural network-based approaches (e.g., scMVAE, DCCA) use deep learning architectures to learn representations that integrate multiple data types [10].
- Network-based methods (e.g., cite-FUSE, Seurat v4) construct biological networks to identify relationships across omics layers [10].
- Probabilistic modeling (e.g., totalVI, MultiVI) uses statistical frameworks to account for uncertainty and technical noise in integrated data [10].
Multi-Omics Integration in CRISPRi Research
Integrated Framework for Functional Genomics
The combination of CRISPR interference (CRISPRi) with multi-omics profiling provides a powerful framework for functional genomics and drug discovery. CRISPRi utilizes a catalytically dead Cas9 (dCas9) fused to repressor domains like KRAB (Krüppel-associated box) for targeted transcriptional repression [5]. When integrated with multi-omics technologies, this enables systematic investigation of gene function and perturbation effects at unprecedented resolution [5].
A notable application combines CRISPRi with metabolomics to create a reference map of metabolic changes from genetic perturbations, enabling de novo predictions of compound functionality [14]. This approach links genetic to drug-induced changes in metabolites, allowing for high-throughput functional annotation of compound libraries [14].
CRISPRi Multi-Omics Integration Workflow
Experimental Protocol: CRISPRi with Metabolomics
A detailed methodology for integrating CRISPRi with metabolomics screening includes the following key steps [14]:
CRISPRi Library Construction:
- Utilize an arrayed strain library with tunable knockdowns of essential genes
- For E. coli studies, target 376 gene loci, including 304 growth-essential proteins in glucose minimal medium
- Include appropriate non-essential gene controls for validation
Strain Culture and Knockdown Induction:
- Grow mutant strains in appropriate medium (e.g., glucose M9 for E. coli) for 12 hours before inoculation
- Induce knockdown with optimized IPTG concentration (e.g., 1 mM for ~10-fold repression)
- Collect samples at multiple time points during mid-log growth phase (3-7 hours post-inoculation)
Metabolomic Profiling:
- Use flow-injection time-of-flight mass spectrometry (FIA-TOFMS) for metabolite detection
- Detect approximately 991 putatively annotated metabolites
- Profile an average of 3 time points per mutant strain
Data Normalization and Processing:
- Correct raw mass spectrometry data for instrumental biases (e.g., plate effects)
- Adjust for systematic changes in cell numbers using optical density measurements
- Calculate relative log2 fold-changes of metabolite levels for each mutant/time point versus wild-type
- Apply Z-score normalization after estimating average and standard deviation of fold-changes across replicates
Similarity Analysis and Functional Prediction:
- Use iterative similarity (iSim) metrics to assess functional associations from metabolome profiles
- Compare metabolic signatures between gene knockdowns and compound treatments
- Employ Cluster of Orthologous Groups (COG) classification and KEGG pathway analysis for functional annotation
Table 2: Research Reagent Solutions for CRISPRi Multi-Omics Studies
| Reagent/Resource | Function | Application Example |
|---|---|---|
| CRISPRi Library | Targeted gene knockdown | Arrayed library with 376 gene targets in E. coli [14] |
| dCas9-KRAB Fusion | Transcriptional repression | CRISPRi system for gene silencing [5] |
| FIA-TOFMS | Metabolite detection | High-throughput metabolome profiling [14] |
| IPTG | Induction of knockdown | Tunable gene repression in CRISPRi system [14] |
| COG Database | Functional classification | Gene function categorization based on metabolic profiles [14] |
| KEGG Pathways | Pathway analysis | Metabolic pathway annotation and enrichment [14] |
| iSim Algorithm | Similarity quantification | Comparing genetic and chemical metabolic profiles [14] |
Integrative Analysis of Epigenomics and Transcriptomics
Methodological Approaches
The integration of epigenomics and transcriptomics data provides powerful insights into gene regulatory mechanisms. Common strategies include [13]:
Identification of Common Genes: Intersecting genes associated with epigenomic data (e.g., from ATAC-seq or ChIP-seq) with differentially expressed genes (DEGs) from transcriptomic analysis using gene IDs, visualized with Venn diagrams or quadrant plots [13].
Functional Enrichment Analysis: Using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment to identify biologically relevant gene sets from integrated data [13].
Genomic Visualization: Employing visualization software to display transcriptional levels and epigenomic peak analysis results simultaneously, enabling direct observation of chromatin accessibility, histone modifications, transcription factor binding sites, and gene expression levels at target loci [13].
Gene Regulatory Network Construction: Building networks using databases like STRING and software such as Cytoscape based on epigenome-associated genes and DEGs from transcriptome analyses [13].
Data Interpretation Framework
Quadrant plots provide enhanced interpretation of integrated epigenomics and transcriptomics data [13]:
- Quadrant 1: Increased chromatin accessibility with upregulated expression (potential transcription factor activation)
- Quadrant 3: Decreased chromatin accessibility with upregulated expression (potential activation by other factors)
- Quadrant 7: Increased chromatin accessibility with downregulated expression (potential transcriptional suppression)
- Quadrant 9: Decreased chromatin accessibility with downregulated expression (potential transcriptional inactivity)
Epigenomics-Transcriptomics Integration Workflow
Advanced Applications and Future Directions
Single-Cell Multi-Omics
Single-cell multi-omics technologies have revolutionized our understanding of cellular heterogeneity by enabling correlated study of specific genomic, transcriptomic, and epigenomic changes in individual cells [15]. The convergence of CRISPR technology with single-cell platforms provides unique opportunities to investigate gene function and perturbation effects at unprecedented resolution [5]. CRISPR pooled screens integrated with single-cell readouts enable identification of gene regulatory networks and cellular responses [5].
AI-Driven Integration and Predictive Modeling
Artificial intelligence (AI) and machine learning are playing increasingly important roles in multi-omics data integration [15] [16]. These technologies can detect intricate patterns and interdependencies across omics modalities, providing insights impossible to derive from single-analyte studies [15]. AI-powered biology-inspired multi-scale modeling frameworks can integrate multi-omics data across biological levels, organism hierarchies, and species to predict genotype-environment-phenotype relationships under various conditions [16].
Deep learning frameworks like Flexynesis streamline data processing, feature selection, hyperparameter tuning, and marker discovery for bulk multi-omics integration [11]. These tools support diverse modeling tasks including regression, classification, and survival analysis, facilitating applications in precision oncology and beyond [11].
Clinical Translation and Therapeutic Discovery
Multi-omics approaches are increasingly applied in clinical settings, particularly in oncology [15]. By integrating molecular data with clinical measurements, multi-omics can help patient stratification efforts by predicting disease progression and optimizing treatment plans [15]. Liquid biopsies exemplify the clinical impact of multi-omics, analyzing biomarkers like cell-free DNA (cfDNA), RNA, proteins, and metabolites non-invasively [15].
In therapeutic discovery, multi-omics integration aids in identifying novel molecular targets, biomarkers, pharmaceutical agents, and personalized medicines for presently unmet medical needs [16]. The combination of CRISPR screening with multi-omics profiling accelerates target identification and validation, particularly for complex diseases [17] [5].
The integration of genomics, transcriptomics, proteomics, and epigenomics provides unprecedented insights into biological systems and disease mechanisms. While computational challenges remain in harmonizing and interpreting these complex datasets, continued development of integration methodologies and AI-driven approaches will further enhance our ability to extract meaningful biological knowledge. The framework of combining CRISPR screening with multi-omics profiling represents a particularly powerful approach for functional genomics and therapeutic discovery, enabling systematic dissection of gene function and regulatory networks across molecular layers. As these technologies mature, they hold tremendous promise for advancing precision medicine and understanding complex biological systems.
Modern biological research, particularly in functional genomics using tools like CRISPR interference (CRISPRi), generates multidimensional data from various molecular layers. This technical guide demonstrates how moving beyond siloed, single-omics analyses to integrated, multi-omics approaches is crucial for elucidating complex biological mechanisms. Through detailed experimental protocols and data analysis frameworks, we illustrate how integrated data provides a systems-level understanding of cellular responses to genetic perturbations, enabling significant advances in basic research and therapeutic discovery for scientific and drug development professionals.
The Critical Role of Data Integration in Functional Genomics
CRISPRi has emerged as a powerful tool for precise gene knockdown, allowing researchers to probe gene function without complete knockout. However, the response to a genetic perturbation is rarely confined to a single molecular layer. Cells activate complex compensatory mechanisms, making it difficult to identify primary from secondary effects using a single data type. Integrated multi-omics analysis addresses this by providing a comprehensive view of the molecular cascade resulting from a perturbation, from epigenetic changes and transcript abundance to protein levels and metabolic states.
Research demonstrates that integrative analysis is indispensable for defining complex regulatory networks. For instance, a multi-omics integrative analysis based on CRISPR screens successfully redefined the pluripotency regulatory network in embryonic stem cells (ESCs). By combining DNA binding, epigenetic modification, chromatin conformation, and RNA expression profiles, the study resolved the network into six functionally independent transcriptional modules (CORE, MYC, PAF, PRC, PCGF, and TBX). This integrated approach revealed that activated CORE/MYC/PAF module activity and repressed PRC/PCGF/TBX module activity was a pattern shared by mouse ESCs, human ESCs, and even cancers, providing novel insights into the molecular basis of pluripotency [17].
Similarly, in studying metabolism, integrating data from CRISPRi-knockdowns with metabolomic and proteomic profiles has identified specific buffering mechanisms that maintain metabolic flux even when key enzymes are repressed. For example, knockdown of carbamoyl phosphate synthetase (CarAB) was buffered by ornithine increasing CarAB activity, and knockdown of homocysteine transmethylase (MetE) was buffered by S-adenosylmethionine de-repressing the methionine pathway [18]. These regulatory insights are only possible through the simultaneous analysis of multiple data types.
Multi-Omics Technologies and Integration Methodologies
Core Omics Technologies for CRISPRi Research
The following table summarizes the key omics technologies that can be integrated with CRISPRi screening to build a systems-level view.
Table 1: Key Omics Technologies for Integrated CRISPRi Studies
| Omics Layer | Technology Examples | Data Output | Primary Application in CRISPRi Studies |
|---|---|---|---|
| Genomics/Epigenomics | ChIP-seq, ATAC-seq, Hi-C | Protein-DNA binding, chromatin accessibility, 3D chromatin conformation [19] | Identifying direct binding targets and epigenetic consequences of perturbations. |
| Transcriptomics | RNA-seq, single-cell RNA-seq (scRNA-seq) | Genome-wide expression profiles, cell-to-cell variation [19] [20] | Measuring gene expression changes and identifying differentially expressed pathways. |
| Proteomics | Mass Spectrometry (MS), CITE-seq | Protein abundance, post-translational modifications [21] | Correlating transcript changes with functional protein levels and activity. |
| Metabolomics | Mass Spectrometry (MS) | Abundance of small molecule metabolites [18] | Assessing the functional output of metabolic pathways following perturbation. |
Experimental Workflow for Integrated CRISPRi Screening
A typical integrated workflow begins with a pooled CRISPRi screen, where cells are transduced with a library of guide RNAs (gRNAs) targeting genes of interest. The phenotypic readout can then be expanded far beyond simple fitness to include multi-omic measurements. The diagram below outlines a comprehensive experimental workflow.
Computational Integration and Analysis Methods
The computational integration of multi-omics data is a critical step. Bioinformatics approaches can be broadly categorized as:
- Unsupervised Integration: Methods like Multi-Omics Factor Analysis (MOFA) identify hidden factors that drive variation across different data modalities without prior knowledge of the experimental design.
- Supervised and Network-Based Integration: These methods use the known perturbation (gRNA identity) as a guide to link changes across omics layers. Tools like MAGeCK [20] can be extended to analyze not just gRNA abundance but also omics-level readouts. For single-cell CRISPR screens (e.g., Perturb-seq, CROP-seq), methods like MIMOSCA and scMAGeCK use linear models and robust rank aggregation to associate gRNAs with transcriptomic changes [20].
The integration process often involves mapping data onto prior biological knowledge, such as known protein-protein interactions, metabolic pathways, or gene regulatory networks, to infer functional relationships and build testable models.
Detailed Experimental Protocol: A Multi-Omics CRISPRi Case Study
This protocol outlines the steps for a CRISPRi screen integrated with transcriptomic and proteomic analysis to identify rate-limiting genes and their downstream effects, based on established methodologies [21] [18].
Research Reagent Solutions
Table 2: Essential Reagents for Integrated CRISPRi Screening
| Reagent / Material | Function and Specification | Critical Notes |
|---|---|---|
| Inducible dCas9-KRAB Cell Line | Expresses a nuclease-dead Cas9 fused to the KRAB transcriptional repressor domain under a doxycycline-inducible promoter [21]. | Enables synchronous, inducible gene knockdown. Integration into a "safe harbor" locus (e.g., AAVS1) ensures consistent expression. |
| CRISPRi sgRNA Library | A pooled lentiviral library targeting genes of interest (e.g., a custom metabolic gene set or genome-wide). Typically includes 3-10 sgRNAs per gene and non-targeting controls [21]. | Use algorithms like CRISPRiaDesign for sgRNA selection. Include a high percentage (e.g., 10%) of non-targeting control sgRNAs. |
| Lentiviral Packaging System | Plasmids (psPAX2, pMD2.G) for producing replication-incompetent lentivirus to deliver the sgRNA library. | Aim for a low MOI (e.g., 0.3-0.5) to ensure most cells receive a single sgRNA. |
| Cell Culture Reagents | Lineage-specific differentiation media for generating relevant cell types (e.g., neurons, cardiomyocytes) from iPSCs [21]. | Maintain consistent culture conditions throughout the screen to minimize technical variability. |
| Single-Cell Partitioning Platform | Equipment for single-cell RNA sequencing, such as the 10x Genomics Chromium Controller. | Essential for Perturb-seq workflows that link sgRNA identity to transcriptomic phenotype in single cells. |
Step-by-Step Workflow
Cell Line Engineering and Validation:
- Generate a clonal cell line (e.g., hiPSC) with doxycycline-inducible dCas9-KRAB stably integrated at the AAVS1 locus [21].
- Validate KRAB-dCas9 expression and repression efficiency via RT-qPCR and immunoblotting upon doxycycline addition. Test with a control sgRNA targeting a highly expressed gene.
Library Transduction and Screening:
- Transduce the target cell line with the sgRNA lentiviral library at a low multiplicity of infection (MOI ~0.3) to ensure most cells receive only one sgRNA.
- After transduction, select with puromycin for 3-5 days to eliminate non-transduced cells.
- Induce CRISPRi knockdown by adding doxycycline to the culture medium. Maintain cells for a defined period (e.g., 10-14 days or several population doublings) to allow phenotypic manifestation [21].
Sample Harvesting for Multi-Omics Analysis:
- For Genomics (gRNA abundance): Harvest a portion of cells for genomic DNA extraction. Amplify the integrated sgRNA cassette via PCR and prepare libraries for next-generation sequencing (NGS) to quantify gRNA abundance [20].
- For Transcriptomics: Harvest cells for total RNA extraction (bulk RNA-seq) or partition cells for single-cell RNA-seq (e.g., using 10x Genomics) to capture cell-to-cell heterogeneity in response to perturbations [21] [20].
- For Proteomics/Metabolomics: Harvest cells, lyse, and process proteins for quantitative mass spectrometry (e.g., TMT or label-free). For metabolomics, perform metabolite extraction and analyze via LC-MS [18].
Data Analysis Pipeline
The logical flow of data from raw sequencing reads to integrated biological insights is summarized in the following diagram.
Primary Screen Analysis:
- Process NGS data from gRNA amplification to count reads for each sgRNA in the population.
- Use specialized algorithms like MAGeCK [20] to compare sgRNA abundance between initial and final time points (for dropout screens) or between different sorted populations. MAGeCK uses a negative binomial model and robust rank aggregation (RRA) to identify genes whose targeting leads to significant enrichment or depletion.
Multi-Omics Data Processing:
- Transcriptomics: Align RNA-seq reads to the reference genome, generate count matrices, and perform differential expression analysis using tools like DESeq2 or edgeR. For scRNA-seq data, tools like scMAGeCK [20] specifically associate each cell's expressed sgRNA with its transcriptome.
- Proteomics/Metabolomics: Process raw MS data to quantify protein or metabolite abundances. Perform statistical analysis to identify significant changes between perturbation and control conditions.
Integrated Pathway and Network Analysis:
- Overrepresentation Analysis: Input lists of significant genes, proteins, and metabolites into tools for Gene Ontology (GO) and KEGG pathway enrichment analysis to identify biological processes affected by the perturbations [22].
- Causal Network Inference: Use the CRISPRi perturbations as causal anchors to build regulatory networks. For example, if knocking down Gene A leads to changes in the expression of Gene B and Protein C, a directed edge from A to B and C can be proposed. Tools like MIMOSCA are designed for this purpose in single-cell data [20].
- Data Visualization: Employ specialized software and custom scripts to create multi-omics visualization plots, such as heatmaps that combine gRNA depletion scores, gene expression changes, and metabolite abundances for a set of related genes [23] [24].
Key Insights from Integrated Studies
Integrated analyses consistently reveal that biological systems are highly interconnected and robust. A CRISPRi screen targeting mRNA translation machinery in hiPSCs and differentiated cells (neural and cardiac) found that while core ribosomal proteins were universally essential, the essentiality of translation-coupled quality control factors was highly cell-type dependent [21]. This underscores that the molecular context, defined by the cell's unique multi-omics landscape, critically determines the outcome of a genetic perturbation.
Furthermore, integrated data helps elucidate specific buffering mechanisms. As noted in E. coli metabolism, CRISPRi knockdown of certain enzymes triggered immediate metabolome and proteome changes that partially compensated for the loss of enzyme function, a insight that would be missed by measuring fitness or a single omics layer alone [18].
The integration of multi-omics data is no longer an optional enhancement but a fundamental requirement for moving from a list of candidate genes to a mechanistic understanding of biological systems. The protocols and frameworks outlined here provide a roadmap for researchers to implement this powerful approach. As single-cell technologies and AI-driven analysis methods—such as machine learning models for predicting on-target/off-target effects and deriving perturbation scores from scRNA-seq data [19]—continue to mature, the resolution and predictive power of integrated models will only increase. This will profoundly accelerate the identification of novel drug targets and the development of personalized therapeutic strategies.
Clustered Regularly Interspaced Short Palindromic Repeats interference (CRISPRi) has emerged as a transformative technology in functional genomics, enabling precise interrogation of gene function without permanent DNA disruption. This technical guide examines core applications of CRISPRi in essential gene identification and drug resistance mechanism elucidation. By integrating multi-omics data and advanced screening methodologies, CRISPRi provides unprecedented insights into bacterial adaptation, antibiotic action, and genetic networks. We detail experimental frameworks, analytical workflows, and reagent solutions that empower researchers to map genetic landscapes and identify novel therapeutic targets with high precision and scalability.
CRISPRi technology utilizes a catalytically inactive Cas9 (dCas9) protein that binds to target DNA without creating double-strand breaks, enabling reversible gene repression [5]. When fused to transcriptional repressors like the Krüppel-associated box (KRAB) domain, dCas9 blocks transcription initiation or elongation, achieving efficient gene knockdown [20] [25]. Unlike CRISPR knockout that introduces irreversible frameshift mutations through non-homologous end joining, CRISPRi offers tunable and partial gene suppression, making it ideal for studying essential genes where complete knockout would be lethal [26] [20]. This temporal control allows researchers to study gene function under specific conditions, including antibiotic stress, and to decipher complex genotype-phenotype relationships that drive drug resistance evolution.
The integration of CRISPRi with single-cell technologies and other omics data has created powerful frameworks for understanding CRISPRi responses at systems level [5] [25]. This perturbomics approach—systematic analysis of phenotypic changes resulting from gene perturbations—enables comprehensive functional annotation of genes and reveals how genetic networks reorganize under selective pressures [25]. Within drug discovery, CRISPRi screens can identify potential antibiotic targets and resistance mechanisms by pinpointing genes whose knockdown affects bacterial survival under treatment [26].
Identifying Essential Genes with CRISPRi
Experimental Design and Workflow
Essential genes are those required for an organism's survival under specific conditions. CRISPRi enables genome-wide essentiality mapping through pooled screens where knockdown of essential genes results in fitness defects quantified by sgRNA depletion [26] [20]. A robust essential gene screen requires careful design of several key components.
The foundation of a successful screen is a high-quality sgRNA library. A genome-scale approach involves designing multiple sgRNAs targeting each coding sequence at regular intervals. For example, one study designed a high-density library targeting every 100 base pairs of the Escherichia coli coding sequences, representing 39,574 sgRNAs with 99.96% coverage [26]. This high-resolution mapping ensures comprehensive gene coverage and robust hit identification. Library design should incorporate approximately 7 sgRNAs per coding gene and 10 sgRNAs for noncoding genes, supplemented with 350 non-targeting sgRNAs as negative controls to establish background variation and false discovery rates [27].
The experimental workflow begins with library transformation into cells expressing dCas9, followed by cultivation under appropriate conditions. Cells are harvested at multiple time points, and genomic DNA is extracted for sgRNA abundance quantification via next-generation sequencing [26] [20]. Bioinformatic analysis identifies essential genes by detecting sgRNAs that become depleted over time, indicating that their target gene knockdown impaired cellular fitness.
Data Analysis and Quality Control
Fitness effects are quantified using the enrichment ratio (ER), calculated as the median ratio of all sgRNAs targeting a gene, comparing their abundance in the knockdown condition to their abundance in the initial library [26]. Essential genes typically show significantly lower ER values (median ~0.346) compared to non-essential genes (median ~0.989) [26]. Several computational tools have been developed specifically for CRISPR screen analysis:
Table 1: Bioinformatics Tools for CRISPR Screen Analysis
| Tool | Year | Statistical Method | Key Features | Citations |
|---|---|---|---|---|
| MAGeCK | 2014 | Negative binomial distribution, Robust rank aggregation | First workflow designed for CRISPR screens; identifies positively and negatively selected genes simultaneously | 794 [20] |
| BAGEL | 2016 | Reference gene set distribution, Bayes factor | Uses essential gene references for comparison; calculates Bayes factor for essentiality | 130 [20] |
| CRISPRCloud2 | 2019 | Beta binomial distribution, Fisher's test | Web-based platform with visualization capabilities | 16 [20] |
| gscreend | 2020 | Skew-normal distribution, α-RRA | Handles high-variance screens through skewed distribution modeling | 8 [20] |
Quality control metrics should include library representation assessment (aim for >99% sgRNA recovery), uniform sgRNA abundance in the initial library, and high correlation between biological replicates [26] [20]. Positional effects should be evaluated by analyzing whether sgRNAs targeting different gene regions (5′ vs. 3′) show consistent depletion patterns [26].
Application Example: Bacterial Essential Gene Mapping
A genome-wide CRISPRi screen in E. coli exposed to various antibiotics identified conditionally essential genes required for survival under stress [26]. The high-density sgRNA library enabled precise mapping of fitness effects, revealing nuances not detectable in knockout studies. For instance, knockdown of groS and rpoD genes produced varying levels of growth retardation, indicating different fitness contributions that would be masked in all-or-nothing knockout approaches [26]. This approach identified essential membrane proteins and highlighted the importance of transcriptional modulation of essential genes in antibiotic tolerance [26].
Uncovering Drug Resistance Mechanisms
CRISPRi Screening Under Antibiotic Stress
CRISPRi enables systematic dissection of drug resistance mechanisms by identifying genes whose knockdown enhances or reduces susceptibility to antimicrobial agents. The experimental approach involves screening CRISPRi libraries under sub-inhibitory antibiotic concentrations and identifying sgRNAs that become enriched or depleted relative to untreated controls [26].
In a comprehensive study examining E. coli responses to 12 antibiotics with different mechanisms of action, researchers identified 1,085 gene knockdowns that induced significant fitness differences under antibiotic stress [26]. The majority (72.9%) were specific to only one or two antibiotics, while a small subset demonstrated pleiotropic effects across multiple drugs [26]. This approach revealed previously unrecognized genes involved in antibiotic resistance, including essential membrane proteins and key cellular processes.
Table 2: Categories of Drug Resistance Genes Identifiable via CRISPRi
| Resistance Mechanism | CRISPRi Phenotype | Example Genes | Detection Method |
|---|---|---|---|
| Efflux pumps | Enhanced sensitivity when knocked down | ABC transporters | sgRNA depletion under antibiotic treatment [28] |
| Drug inactivation enzymes | Enhanced sensitivity when knocked down | β-lactamases, acetyltransferases | sgRNA depletion under antibiotic treatment [28] |
| Cell wall permeability | Enhanced sensitivity when knocked down | Membrane porins, lipid transporters | sgRNA depletion under antibiotic treatment [28] |
| Stress response pathways | Enhanced sensitivity when knocked down | degP, rpoS | sgRNA depletion under antibiotic treatment [26] |
| Target bypass pathways | Enhanced resistance when knocked down | Alternative metabolic enzymes | sgRNA enrichment under antibiotic treatment [26] |
Advanced Methodologies: CRISPRi-TnSeq for Genetic Interaction Mapping
CRISPRi-TnSeq represents a powerful extension that maps genetic interactions between essential and non-essential genes by combining CRISPRi-mediated essential gene knockdown with transposon-based non-essential gene knockout [29]. This approach identifies synthetic lethal and suppressor relationships on a genome-wide scale.
The methodology involves:
- Constructing CRISPRi strains targeting essential genes
- Generating transposon mutant libraries in each CRISPRi strain
- Culturing libraries with and without CRISPRi induction (e.g., using IPTG)
- Sequencing transposon insertion sites to quantify mutant fitness under essential gene knockdown
- Identifying genetic interactions where combined perturbation produces unexpected fitness effects [29]
In Streptococcus pneumoniae, CRISPRi-TnSeq screened approximately 24,000 gene pairs and identified 1,334 significant genetic interactions (754 negative, 580 positive) [29]. Negative interactions indicate synthetic sickness/lethality, where combined impairment of both genes reduces fitness more than expected. Positive interactions indicate suppression, where impairment of one gene mitigates the fitness cost of impairing the other [29].
Case Study: Universal Stress Response Genes
CRISPRi screening under diverse antibiotic stresses revealed seven genes in E. coli that consistently exhibited fitness changes across 10 or more different antibiotics, indicating universal stress response functions [26]. Among these, degP encoding the protease Do, which degrades abnormal proteins in the periplasm, showed protective roles against multiple antibiotics [26]. Growth profiling confirmed that degP null mutants exhibited weaker growth under antibiotic stress compared to wild-type strains [26]. This universal response gene network represents a core cellular defense system against diverse antimicrobial challenges.
Integration with Omics Technologies
Single-Cell CRISPRi Screening
The integration of CRISPRi with single-cell RNA sequencing (scRNA-seq) enables high-resolution mapping of transcriptional responses to gene perturbations. Technologies such as Perturb-seq, CRISP-seq, and CROP-seq combine pooled CRISPR screening with single-cell transcriptomics, allowing simultaneous analysis of sgRNA identity and whole-transcriptome profiles in individual cells [5] [20].
This multi-modal approach reveals how specific gene perturbations alter cellular states, identifies heterogeneous responses within cell populations, and maps gene regulatory networks [5] [25]. In cancer research, single-cell CRISPRi screens in human gastric organoids have identified genes influencing chemotherapy response and uncovered novel relationships between biological pathways, such as an unexpected link between fucosylation and cisplatin sensitivity [30].
Chemical-Genetic Interaction Mapping
CRISPRi screens can be extended to map chemical-genetic interactions by screening under drug treatments. The DrugZ algorithm specifically analyzes such datasets by normalizing sgRNA counts and computing gene-level z-scores based on the collective behavior of targeting sgRNAs [20]. This approach identifies genes that modulate sensitivity to therapeutic compounds, potentially revealing synthetic lethal interactions that can be exploited for targeted therapies.
In practice, chemical-genetic screens involve:
- Conducting parallel CRISPRi screens under vehicle control and drug treatment
- Calculating differential sgRNA abundance between conditions
- Aggregating sgRNA-level effects to gene-level scores
- Identifying significant modifiers of drug sensitivity [20]
Research Reagent Solutions
Table 3: Essential Research Reagents for CRISPRi Experiments
| Reagent Category | Specific Examples | Function & Importance | Technical Considerations |
|---|---|---|---|
| CRISPRi vectors | dCas9-KRAB, dCas9-VPR | Transcriptional repression/activation | Inducible systems enable temporal control; various promoters allow tissue-specific expression [30] |
| sgRNA libraries | Genome-wide, pathway-specific | Target genes of interest | Library complexity and coverage critical for screen quality; ~1000x coverage per sgRNA recommended [26] [27] |
| Delivery systems | Lentivirus, lipid nanoparticles (LNPs) | Introduce CRISPR components into cells | LNPs preferred for in vivo work; viral vectors efficient for hard-to-transfect cells [31] |
| Selection markers | Puromycin, blasticidin | Enforce stable expression of CRISPR components | Concentration must be optimized for each cell type to ensure complete selection without excessive toxicity [30] |
| Induction systems | Doxycycline, IPTG | Control timing and degree of dCas9 expression | Tight regulation essential for studying essential genes; leakiness can confound results [30] |
CRISPRi technology has revolutionized functional genomics by enabling precise, reversible gene perturbation at scale. Its applications in essential gene identification and drug resistance mechanism elucidation provide powerful insights into genetic networks underlying cellular survival and adaptation. The integration of CRISPRi with other omics technologies, including single-cell transcriptomics and transposon mutagenesis, creates multidimensional perturbomics approaches that reveal system-wide responses to genetic perturbations.
As CRISPRi methodologies continue to evolve, they offer increasingly sophisticated tools for mapping genetic interactions, identifying therapeutic targets, and understanding complex biological systems. The experimental frameworks and reagent solutions outlined in this technical guide provide researchers with robust foundations for implementing these cutting-edge approaches in their own investigations of gene function and drug resistance mechanisms.
Advanced Workflows: Integrating Multi-Omics Datasets with CRISPRi Screening
Experimental Design for Multi-Omics CRISPRi Screens
Clustered Regularly Interspaced Short Palindromic Repeats interference (CRISPRi) has emerged as a powerful tool for functional genomics, enabling precise, programmable gene repression without altering DNA sequences. The CRISPRi system utilizes a catalytically dead Cas9 (dCas9) protein fused to transcriptional repressor domains like the Krüppel-associated box (KRAB), which is guided by a single-guide RNA (sgRNA) to specific genomic loci to sterically hinder transcription [32] [20]. This technology is particularly valuable for pooled screening approaches, allowing researchers to systematically interrogate gene function at scale. When integrated with multi-omics readouts—including transcriptomics, epigenomics, and proteomics—CRISPRi screening enables the comprehensive mapping of gene regulatory networks and their functional outcomes [5].
The integration of CRISPRi with single-cell technologies represents a paradigm shift in functional genomics. This powerful combination allows researchers to not only identify essential genes but also to understand their roles in shaping cellular identities, states, and responses through simultaneous measurement of multiple molecular layers [32] [5]. This approach is particularly valuable for investigating non-coding genomic elements, epigenetic regulators, and genes sensitive to copy number effects that are difficult to study with traditional CRISPR knockout approaches [32]. For drug development professionals, multi-omics CRISPRi screens offer unprecedented insights into therapeutic mechanisms of action, resistance pathways, and potential off-target effects, ultimately accelerating the target validation pipeline.
Core Principles and Technological Foundations
Molecular Mechanisms of CRISPRi
The foundational component of CRISPRi is nuclease-dead Cas9 (dCas9), generated through point mutations (D10A and H840A for Streptococcus pyogenes Cas9) in the RuvC and HNH nuclease domains [32] [5]. This modified protein retains its ability to bind DNA in an RNA-guided manner but cannot introduce double-strand breaks. When targeted to promoter regions or transcription start sites, the dCas9-sgRNA complex physically obstructs RNA polymerase binding or progression, leading to transcriptional repression [33]. The repression efficiency can be enhanced by fusing dCas9 to effector domains such as KRAB, which recruits additional repressive complexes to establish heterochromatin and further silence target gene expression [32] [5].
Unlike CRISPR knockout which introduces irreversible frameshift mutations, CRISPRi offers reversible and tunable gene repression. The degree of repression can be modulated by adjusting sgRNA expression levels, targeting multiple sgRNAs to the same gene, or using truncated sgRNAs with reduced efficacy [33]. This tunability is particularly valuable for studying essential genes where complete knockout would be lethal, and for modeling the partial loss-of-function effects often seen in heterozygous disease states or pharmacological inhibition.
Advancements in CRISPRi Screening Platforms
Recent technological advancements have significantly expanded the capabilities of CRISPRi screening. The development of highly specific sgRNA libraries with minimal off-target effects, combined with improved dCas9 variants with enhanced specificity and efficiency, has increased the reliability of screening results [5]. Furthermore, the integration of CRISPRi with single-cell multi-omics technologies enables high-resolution dissection of transcriptional and epigenetic responses to gene perturbations across diverse cell types and states [34] [5].
Emerging approaches now combine CRISPRi with single-cell RNA sequencing (scRNA-seq), single-cell ATAC-seq (scATAC-seq), and other omics modalities to capture multidimensional responses to genetic perturbations [5]. For instance, Perturb-seq, CRISP-seq, and CROP-seq enable linked readouts of sgRNA identities and transcriptomic profiles in thousands of individual cells [20]. More recently, technologies like SDR-seq (single-cell DNA–RNA sequencing) allow simultaneous profiling of genomic DNA loci and gene expression in the same cells, enabling confident determination of variant zygosity alongside associated expression changes [34].
Diagram 1: CRISPRi Core Mechanism and Multi-Omics Integration. This figure illustrates the fundamental components of the CRISPRi system and its connection to multi-omics readouts.
Experimental Design Framework
A well-designed multi-omics CRISPRi screen requires careful planning at each step to ensure robust, interpretable results. The complete workflow spans from initial library design to final integrated data analysis, with multiple quality control checkpoints throughout the process. The timeline typically ranges from 4-8 weeks for cell culture and perturbation, followed by 2-4 weeks for sample processing and sequencing, and finally 2-6 weeks for computational analysis depending on the scale and complexity of the omics measurements.
Diagram 2: Multi-Omics CRISPRi Screening Workflow. This diagram outlines the key experimental stages and quality control checkpoints.
Research Reagent Solutions
Successful implementation of multi-omics CRISPRi screens depends on carefully selected reagents and tools. The table below summarizes essential materials and their functions in screen implementation.
Table 1: Essential Research Reagents for Multi-Omics CRISPRi Screens
| Reagent Category | Specific Examples | Function | Key Considerations |
|---|---|---|---|
| dCas9 Effectors | dCas9-KRAB, dCas9-DNMT3A, dCas9-HDAC | Transcriptional repression, epigenetic modification | Fusion partners determine repression mechanism and strength |
| sgRNA Libraries | Brunello CRISPRi library, custom libraries | Target-specific gene repression | Library size, sgRNAs per gene, non-targeting controls |
| Delivery Systems | Lentiviral vectors, AAV, lipid nanoparticles | Introduction of CRISPR components into cells | Transduction efficiency, cellular toxicity, delivery efficiency |
| Cell Lines | iPSCs, primary cells, immortalized lines | Biological context for screening | dCas9 stable expression, relevance to disease model |
| Multi-omics Assays | 10x Multiome, SDR-seq, CITE-seq, Perturb-seq | Multiplexed molecular profiling | Compatibility with CRISPRi, single-cell resolution, cost |
| Sequencing Platforms | Illumina NovaSeq, PacBio Revio, Oxford Nanopore | High-throughput readout | Read length, depth, multi-omics compatibility |
sgRNA Library Design and Validation
The design of the sgRNA library is a critical determinant of screening success. For comprehensive coverage, libraries should include 3-6 sgRNAs per target gene, with each sgRNA typically spanning 19-20 nucleotides complementary to the target sequence. Library design should prioritize targeting regions within 50-100 base pairs upstream of the transcription start site (TSS) for optimal repression efficiency [33]. Essential design considerations include minimizing off-target effects through careful specificity scoring, incorporating non-targeting control sgRNAs for background normalization, and including positive control sgRNAs targeting essential genes known to produce strong phenotypes.
Recent advances in library design have enabled more specialized applications, including tiling screens for non-coding regulatory elements, epigenetic modifier screens targeting specific chromatin states, and dual sgRNA approaches for studying genetic interactions [32]. For multi-omics readouts, libraries should be designed with compatible amplification handles and constant regions that do not interfere with single-cell barcode sequences in downstream omics assays.
Validation of library functionality should be performed through pilot experiments measuring: (1) repression efficiency of control sgRNAs via qRT-PCR or fluorescent reporters, (2) library representation throughout the screening process to ensure maintenance of diversity, and (3) specificity assessment through transcriptome-wide profiling to confirm minimal off-target effects [33].
Cell Line Engineering and Culture Conditions
Stable integration of dCas9-effector constructs is preferred over transient expression to ensure consistent performance throughout the screen. Lentiviral transduction at low multiplicity of infection (MOI < 0.3) followed by antibiotic selection generates polyclonal cell populations with uniform dCas9 expression. Single-cell cloning can further ensure homogeneity but may increase clonal variation effects. Critical validation steps include verifying dCas9 expression via Western blot, assessing nuclear localization through immunofluorescence, and confirming functionality using control sgRNAs [35].
For multi-omics screens, cell culture conditions must maintain library representation while providing appropriate experimental contexts. Maintain a minimum of 300-500 cells per sgRNA during expansion to prevent stochastic loss of library elements [35]. For perturbation experiments, consider relevant biological contexts such as disease-relevant stimuli, drug treatments, or differentiation states that align with the research questions. Appropriate control conditions—such as non-targeting sgRNAs or non-induced states—should be included for rigorous comparison.
Multi-Omics Readout Methodologies
Integrating multiple molecular profiling modalities significantly enhances the informational yield from CRISPRi screens. The selection of specific omics technologies should be guided by biological questions, available resources, and computational capabilities.
Table 2: Multi-Omics Technologies for CRISPRi Screen Readouts
| Omics Layer | Technologies | Key Metrics | Data Output | Compatibility with CRISPRi |
|---|---|---|---|---|
| Transcriptomics | scRNA-seq, SDR-seq, Perturb-seq | Gene expression, splicing variants | UMI counts, differential expression | High - direct measurement of perturbation effects |
| Epigenomics | scATAC-seq, CUT&Tag, DNA methylation | Chromatin accessibility, histone marks | Peak counts, differential accessibility | Moderate - reveals mechanistic insights |
| Proteomics | CITE-seq, flow cytometry, mass cytometry | Protein abundance, post-translational modifications | Protein counts, differential abundance | Moderate - closer to functional phenotype |
| Multi-omics | 10x Multiome, SDR-seq, TEA-seq | Linked transcriptome + epigenome | Paired measurements from single cells | High - captures coordinated regulation |
Single-cell DNA-RNA sequencing (SDR-seq) represents a particularly powerful approach for multi-omics CRISPRi screens, as it enables simultaneous profiling of up to 480 genomic DNA loci and gene expression in thousands of single cells [34]. This technology allows accurate determination of variant zygosity alongside associated gene expression changes, providing a comprehensive view of genotype-phenotype relationships. Fixation conditions significantly impact data quality in SDR-seq, with glyoxal-based fixation generally providing superior RNA target detection compared to paraformaldehyde [34].
For CRISPRi screens with single-cell multi-omics readouts, cell multiplexing using lipid-based hashing antibodies or genetic barcodes can significantly reduce costs by processing multiple samples in a single sequencing run. The targeted nature of CRISPRi perturbations makes them particularly compatible with focused multi-omics approaches that prioritize depth over breadth in relevant molecular features.
Computational Analysis and Data Integration
Primary Screen Analysis
The initial analysis of CRISPRi screen data focuses on connecting sgRNA abundances to phenotypic readouts. For multi-omics screens, this process involves both conventional abundance-based analyses and molecular phenotype assessments. The computational workflow typically begins with raw sequencing data processing, including quality control, adapter trimming, and alignment of reads to the reference sgRNA library [20].
For essential gene identification in dropout screens, sgRNA depletion is quantified using tools like MAGeCK (Model-based Analysis of Genome-wide CRISPR/Cas9 Knockout), which employs a negative binomial distribution to model read counts and a robust rank aggregation (RRA) algorithm to identify significantly depleted genes [20]. BAGEL (Bayesian Analysis of Gene EssentiaLity) represents another powerful approach that uses a Bayesian framework to compare sgRNA abundances to a reference set of known essential and non-essential genes [20].
In multi-omics screens, the primary analysis must also account for the specific readout modality. For scRNA-seq-based screens, the analysis typically involves: (1) assigning sgRNA identities to individual cells based on expressed barcodes, (2) quantifying transcriptomic changes in perturbed cells compared to controls, and (3) identifying genes and pathways affected by each perturbation [20]. Tools like MUSIC (Mutation and Expression-based Multi-task Learning for Single-cell Data) employ topic modeling to extract recurrent cellular programs affected by genetic perturbations, while scMAGeCK extends the MAGeCK algorithm to single-cell data using RRA or linear regression approaches [20].
Multi-Omics Data Integration
Integrating multiple omics layers represents both the greatest opportunity and challenge in advanced CRISPRi screening. Effective integration approaches can be categorized as early, intermediate, or late integration based on when different data types are combined [34] [5].
Early integration involves concatenating features from different omics layers before analysis, enabling the detection of complex cross-modality relationships but requiring sophisticated normalization. Intermediate integration uses methods like multi-omics factor analysis (MOFA+) or coupled non-negative matrix factorization to identify latent factors that capture coordinated variation across data types. Late integration analyzes each omics layer separately before combining results, preserving modality-specific characteristics but potentially missing subtle correlations.
For CRISPRi screens specifically, the perturbation dimension provides a natural anchor for integration. By comparing multi-omics profiles across different perturbations, researchers can identify: (1) direct transcriptional targets (immediate transcriptome changes), (2) downstream regulatory consequences (epigenomic adaptations), and (3) functional outcomes (proteomic and phenotypic effects). The recent development of SDR-seq demonstrates how integrated DNA-RNA profiling enables confident linking of genotypes to gene expression changes at single-cell resolution, particularly valuable for studying both coding and non-coding variants [34].
Hit Prioritization and Validation
Following integrated analysis, candidate hits must be prioritized for validation based on multiple criteria: (1) strength and reproducibility of phenotype across biological replicates, (2) consistency across omics layers, (3) specificity of effect (minimal off-target signatures), and (4) biological relevance to the research context. For drug development applications, additional prioritization factors include druggability, safety profiles, and connection to disease mechanisms.
Validation strategies should employ orthogonal approaches to confirm screening results: (1) individual sgRNA validation with dose-response characterization, (2) complementary techniques such as RNAi or pharmacological inhibition, (3) mechanistic follow-up studies to elucidate downstream pathways, and (4) physiological relevance assessment in disease models. For multi-omics hits, validation should confirm consistency across molecular layers and establish causal relationships between observed changes.
Applications in Drug Development and Therapeutic Discovery
Multi-omics CRISPRi screens offer particular value for drug development pipelines by providing comprehensive functional annotation of potential therapeutic targets. In oncology, these approaches have identified novel synthetic lethal interactions, resistance mechanisms, and combination therapy opportunities [32] [5]. For example, CRISPRi screens in primary B cell lymphoma samples have revealed that cells with higher mutational burden exhibit elevated B cell receptor signaling and tumorigenic gene expression, suggesting potential therapeutic vulnerabilities [34].
In immunotherapy development, CRISPRi screens have enabled precise engineering of CAR-T cells, including modulation of endogenous T-cell receptors to improve tumor targeting and overcome immunosuppressive microenvironments [5]. The multi-omics dimension further allows comprehensive assessment of therapeutic effects on cellular states, exhaustion markers, and functional persistence.
Beyond oncology, multi-omics CRISPRi screens are advancing therapeutic discovery for neurological disorders, cardiovascular diseases, and rare genetic conditions by elucidating disease-relevant gene regulatory networks and identifying nodes amenable to pharmacological intervention [32]. The perturbomics approach—systematic analysis of phenotypic changes resulting from gene perturbations—provides a powerful framework for linking genetic targets to disease mechanisms and therapeutic opportunities [32].
Multi-omics CRISPRi screening represents a transformative approach for functional genomics and therapeutic discovery. The integration of precise gene perturbation with multidimensional molecular profiling enables unprecedented resolution in mapping gene function and regulatory networks. As single-cell multi-omics technologies continue to advance in scalability and affordability, and as computational methods for data integration become more sophisticated, these approaches will increasingly become standard tools for both basic research and drug development.
Future directions in the field include: (1) spatial multi-omics integration to contextualize perturbations within tissue architecture, (2) longitudinal perturbation tracking to capture dynamic responses, (3) enhanced base editing and prime editing screens for modeling specific disease variants, and (4) machine learning approaches to predict combinatorial perturbation effects. For drug development professionals, embracing these integrated approaches will accelerate target identification, enhance understanding of mechanism of action, and ultimately improve success rates in therapeutic development.
The convergence of single-cell multi-omics technologies and CRISPR interference (CRISPRi) screening represents a paradigm shift in functional genomics, enabling the systematic deconvolution of cellular heterogeneity and gene regulatory networks. This powerful integration allows researchers to move beyond population-averaged measurements and instead observe how precise genetic perturbations manifest in individual cells across multiple molecular layers. The programmability of CRISPRi—a catalytically dead Cas9 (dCas9) fused to repressive domains like KRAB—enables targeted transcriptional repression without altering DNA sequence, making it ideal for probing gene function in native contexts [5]. When combined with single-cell readouts that capture transcriptomic, epigenomic, and proteomic states simultaneously, CRISPRi screening transitions from measuring singular phenotypes to mapping multidimensional cellular responses [36] [37]. This technical synergy is particularly transformative for understanding complex biological systems where heterogeneous cell populations drive physiological and disease processes, from cancer development to immune responses [38] [5].
Framed within the broader challenge of omics data integration for understanding CRISPRi responses, this approach addresses a fundamental limitation in biomedical research: the inability to connect genetic perturbations to molecular phenotypes while accounting for cellular heterogeneity. Recent computational advances, including foundation models pretrained on millions of cells and novel integration algorithms, now provide the analytical framework needed to interpret these complex datasets and extract biologically meaningful insights [37]. This technical guide explores the current methodologies, analytical frameworks, and practical implementations at the intersection of single-cell multi-omics and CRISPRi screening, providing researchers with the tools to dissect cellular heterogeneity with unprecedented resolution.
Technological Foundations
CRISPRi Systems for Precision Perturbation
CRISPR interference (CRISPRi) utilizes a nuclease-dead Cas9 (dCas9) mutant that retains DNA-binding capability but lacks cleavage activity. When fused to transcriptional repressor domains such as the Krüppel-associated box (KRAB), dCas9 efficiently silences target genes by recruiting chromatin-modifying complexes that establish repressive epigenetic states [5]. Unlike CRISPR knockout approaches that cause permanent DNA damage, CRISPRi offers reversible, tunable repression that more closely mimics pharmacological inhibition—a particular advantage for studying essential genes and dose-dependent effects [39].
Key advantages of CRISPRi for single-cell screening include:
- Precise targeting to transcription start sites (TSS) for efficient transcriptional repression
- Reduced toxicity compared to nuclease-based approaches since no DNA damage occurs
- Compatibility with multiplexing enabling simultaneous targeting of multiple genomic loci
- Reversible effects allowing studies of gene function recovery
- Minimal confounding effects from DNA damage response pathways
The specificity of CRISPRi depends on guide RNA (gRNA) design, with optimal targeting typically within -50 to +300 bp relative to the TSS [39]. Recent Cas9 variants with altered PAM specificities (e.g., SpCas9-NG, xCas9) have expanded the targeting range, while engineered gRNA scaffolds with MS2 or other RNA aptamers enable enhanced recruitment of repressive complexes for increased efficacy [5].
Single-Cell Multi-Omics Profiling Platforms
Single-cell multi-omics technologies simultaneously measure multiple molecular layers from individual cells, capturing the interconnected nature of cellular regulation. These platforms have evolved from measuring just transcriptomes to comprehensively profiling epigenomic, proteomic, and spatial information from the same cells [40].
Table 1: Major Single-Cell Multi-Omics Technologies
| Technology | Measured Modalities | Key Applications | Considerations |
|---|---|---|---|
| ECCITE-seq | Transcriptome, surface proteins, CRISPR gRNAs | Immune cell profiling, Perturb-seq | 5' capture; direct gRNA capture [36] |
| CITE-seq | Transcriptome, surface proteins | Cell type identification, surface marker quantification | Requires antibody conjugation [40] |
| Perturb-ATAC | Chromatin accessibility, CRISPR perturbations | Epigenetic regulation, enhancer mapping | DNA tagmentation-based [36] |
| TAP-seq | Targeted transcriptome, gRNAs | High-sensitivity gene expression | Custom primer panels [36] |
| SPEAR-ATAC | Chromatin accessibility, gRNAs | Chromatin landscape changes | Combines Nextera adapters with gRNAs [36] |
These platforms differ in their gRNA capture strategies, with direct capture methods (e.g., ECCITE-seq) providing more accurate gRNA-to-cell assignment by avoiding barcode swapping issues that plagued earlier indirect capture approaches [36]. The choice of platform depends on the biological questions, with targeted approaches like TAP-seq offering higher sensitivity for specific gene panels while untargeted methods provide discovery-based insights.
Experimental Design and Workflows
Integrated CRISPRi Screening with Single-Cell Multi-Omics
The successful integration of CRISPRi screening with single-cell multi-omics requires careful experimental planning from library design through data generation. A typical workflow encompasses several critical stages that must be optimized for specific research applications.
Guide RNA Library Design and Validation
Effective CRISPRi screens begin with comprehensive gRNA library design targeting genes of interest with multiple gRNAs per gene to ensure statistical robustness. For non-coding screens, tiling approaches across regulatory elements are employed. Library size considerations balance comprehensive coverage with maintaining sufficient cell coverage per gRNA (typically 500-1,000 cells per gRNA) [36]. Controls should include:
- Non-targeting gRNAs with scrambled sequences
- Positive controls targeting essential genes
- Targeting controls for known functional elements
- Labeling gRNAs for cell tracking in pooled formats
Lentiviral Delivery and Multiplicity of Infection (MOI) Optimization
Lentiviral delivery remains the most efficient method for introducing CRISPRi components into diverse cell types. Critical parameters include:
- Low MOI (≤0.3) to ensure most cells receive only one gRNA
- Stable dCas9-KRAB expression before gRNA delivery
- Selection markers (e.g., puromycin) to enrich for transduced cells
- Titration experiments to determine viral potency across cell models
For sensitive cell types, inducible dCas9 systems or transient expression approaches may be preferable to minimize toxicity from prolonged KRAB expression [5].
Single-Cell Multi-Omics Capture and Library Preparation
Cells are harvested after perturbation and processed through appropriate single-cell multi-omics platforms. The 10x Genomics Multiome ATAC + Gene Expression platform simultaneously profiles chromatin accessibility and transcriptomes from the same nuclei, while CITE-seq approaches add surface protein measurements [40]. Key considerations include:
- Cell viability >90% to ensure high-quality data
- Cell number sufficient to maintain gRNA representation
- Multiplexing strategies (e.g., CellPlex, MULTI-seq) to pool conditions
- Spike-in controls for technical variation monitoring
Research Reagent Solutions
Table 2: Essential Research Reagents for Single-Cell Multi-omics CRISPRi Screening
| Reagent Category | Specific Examples | Function | Technical Considerations |
|---|---|---|---|
| CRISPRi Effectors | dCas9-KRAB, dCas9-Mxi1, dCpf1-KRAB | Transcriptional repression | Varying repression efficacy; cell-type dependent performance [5] |
| gRNA Delivery Vectors | Lentiviral transfer plasmids (lentiGuide, lentiSAM), All-in-one dCas9-KRAB+gRNA vectors | gRNA expression and delivery | MOI critical for single-copy delivery; titer monitoring essential [36] |
| Single-Cell Barcoding | 10x Barcoded Beads, MULTI-seq barcodes, CellPlex antibodies | Cell multiplexing and identification | Barcode balance affects demultiplexing efficiency [40] |
| Antibody Conjugates | CITE-seq antibodies (TotalSeq), Feature Barcoding antibodies | Protein surface marker quantification | Titration required to minimize background [40] |
| Library Prep Kits | 10x Multiome ATAC + Gene Expression, Parse Biosciences kits | Sequencing library construction | Protocol optimization for cell type; cost considerations [36] |
Analytical Frameworks and Computational Integration
Data Processing and Quality Control
Raw sequencing data from single-cell CRISPRi screens requires specialized processing pipelines to accurately assign gRNAs to cells while handling multi-modal data. The analytical workflow begins with demultiplexing and quality control before advancing to more sophisticated integrative analyses.
Quality control must address both single-cell data quality and CRISPRi-specific metrics:
- Cells per gRNA: Minimum 500 cells per gRNA for statistical power
- gRNA diversity: Even distribution across targeted genes
- Multimodal data quality: Correlation between modalities should reflect biology
- Doublet rates: Typically <10% depending on platform
- Mitochondrial reads: <20% for healthy cells
Direct gRNA capture methods significantly improve assignment accuracy compared to early indirect approaches that suffered from high barcode-swapping rates (up to 50% in Perturb-seq) [36].
Perturbation Response Quantification
A critical advancement in single-cell CRISPRi analysis is the move beyond binary perturbation detection toward continuous quantification of perturbation strength. The Perturbation-response Score (PS) framework addresses this by modeling perturbation responses as a continuous variable ranging from 0 (no effect) to 1 (maximal effect) based on expression changes in downstream target genes [39].
Table 3: Computational Methods for Single-Cell CRISPR Screen Analysis
| Method | Statistical Approach | Key Features | Applicability to CRISPRi |
|---|---|---|---|
| PS (Perturbation-response Score) | Constrained quadratic optimization | Quantifies partial perturbations; enables dosage analysis | Excellent for graded CRISPRi responses [39] |
| sceptre | Negative binomial with resampling framework | High sensitivity for element-gene pairs; efficient computation | Compatible with CRISPRi screens [41] |
| mixscape | Gaussian mixture modeling | Identifies complete vs. incomplete knockouts | Limited for partial perturbations [39] |
| MUSIC | Matrix factorization | Deconvolves multiple perturbations | Works with combinatorial screens [39] |
| scMAGeCK | Linear modeling | Identifies enriched/depleted gRNAs | Best for growth-based screens [39] |
The PS framework particularly excels with CRISPRi data where partial repression is common, outperforming methods like mixscape that assume bimodal (on/off) perturbation effects [39]. In benchmark analyses using K562 CROP-seq data, PS correctly estimated CRISPRi efficiency in >40% of gene perturbations compared to <5% for mixscape in high-MOI conditions [39].
Multi-Omics Data Integration Strategies
Integrating multiple data modalities from single-cell CRISPRi screens requires specialized computational approaches that account for the different statistical properties and biological meanings of each data type.
Matched integration approaches like MOFA+ and Seurat v4 are used when all modalities are measured from the same cells, leveraging the cell itself as a natural anchor [10]. Unmatched integration methods like GLUE employ graph-based variational autoencoders to align cells measured across different modalities [10]. Mosaic integration tools including StabMap are particularly valuable for combining datasets with partially overlapping modality measurements [37] [10].
Recent foundation models like scGPT, pretrained on over 33 million cells, demonstrate exceptional capabilities in cross-modal prediction and zero-shot cell type annotation, significantly accelerating the analysis of single-cell multi-omics perturbation data [37].
Applications in Biological Research
Dissecting Context-Specific Gene Functions
Single-cell multi-omics CRISPRi screening has revealed how gene functions are shaped by cellular context, moving beyond the concept of static gene essentiality. In T cell activation studies, PS analysis of genome-scale CRISPRi Perturb-seq in Jurkat cells identified transcription factors whose perturbation effects depended strongly on stimulation state [39]. This context-dependency explains why traditional bulk screens often miss functionally important genes that only operate in specific cellular states.
The technology has been particularly powerful for studying dosage-sensitive genes where partial repression by CRISPRi reveals graded phenotypic effects. PS analysis distinguishes "buffered" genes (where moderate perturbation has minimal downstream effects) from "sensitive" genes (where even slight reduction causes strong phenotypic consequences) [39]. This dosage resolution provides insights into network robustness and identifies potential therapeutic targets where partial inhibition might achieve desired effects.
Mapping Gene Regulatory Networks
By coupling CRISPRi perturbations with simultaneous transcriptomic and epigenomic profiling, researchers can reconstruct causal gene regulatory networks rather than just correlation-based associations. For example, applying the GLiMMIRS framework to single-cell CRISPR data revealed that enhancer pairs typically act multiplicatively rather than synergistically, with only 31 of 46,166 tested enhancer pairs showing significant interactions [42]. This finding challenges models of strong enhancer synergy and demonstrates how multi-omics perturbation data can test fundamental regulatory principles.
In pancreatic islet biology, integrating single-cell heterogeneity analysis with CRISPR screening identified novel insulin regulators including the cohesin loading complex (MAU2-NIPBL) and the NuA4/Tip60 histone acetyltransferase complex [38]. These findings emerged from connecting disease-associated gene signatures from human islet single-cell data with functional insulin regulation through CRISPR screening, demonstrating the power of integrative approaches for complex disease modeling.
Identifying Therapeutic Targets in Heterogeneous Environments
Cancer and immune cells exist in dynamically changing microenvironments where cellular heterogeneity drives therapy response and resistance. Single-cell multi-omics CRISPRi screening can identify targets that specifically affect subpopulations responsible for treatment failure. In latent HIV research, PS analysis revealed differential cellular responses to perturbations of key genes involved in viral reactivation, identifying potential combination strategies to address heterogeneous reservoir cells [39].
Similarly, in pancreatic differentiation models, CCDC6 was identified as a previously unrecognized regulator of liver versus pancreatic cell fate decisions through heterogeneous response analysis [39]. These applications demonstrate how accounting for cellular heterogeneity through single-cell multi-omics can reveal therapeutic opportunities invisible to bulk approaches.
Future Perspectives and Challenges
The integration of single-cell multi-omics with CRISPRi screening continues to evolve with several emerging directions and persistent challenges. Foundation models pretrained on massive single-cell datasets are enabling zero-shot perturbation prediction and in silico screening, potentially reducing experimental burden [37]. Cross-species models like scPlantFormer demonstrate the potential for generalizable representations that transfer knowledge across biological contexts [37].
Technical challenges remain in improving gRNA capture efficiency, especially for high-throughput screens with thousands of perturbations. Multi-modal foundation models that incorporate protein structures, gene networks, and perturbation effects show promise for better predicting CRISPRi efficacy and off-target effects [37]. As spatial multi-omics matures, incorporating spatial context into CRISPRi screens will reveal how cellular neighborhoods shape perturbation responses.
Computational methods must continue advancing to handle the increasing scale and complexity of multi-omics perturbation data, with emphasis on interpretable models that provide mechanistic insights rather than black-box predictions [37]. Methods that explicitly model technical confounders like gRNA efficiency variation and capture bias will improve reproducibility across laboratories and platforms.
The trajectory points toward increasingly comprehensive cellular atlases that map gene function across diverse cell states, environments, and genetic backgrounds, ultimately enabling predictive models of cellular behavior that accelerate therapeutic development and fundamental biological discovery.
Network integration represents a pivotal advancement in systems biology, enabling researchers to interweave multiple omics datasets into unified biochemical networks for enhanced mechanistic understanding. This approach moves beyond simple correlative analyses by mapping various molecular entities—genes, transcripts, proteins, and metabolites—onto shared networks based on known biological interactions [15]. The fundamental premise is that disease states and cellular responses originate from perturbations across multiple molecular layers, and by measuring multiple analyte types within a pathway, biological dysregulation can be precisely pinpointed to specific reactions and regulatory events [15].
In the context of CRISPRi response research, network integration provides a powerful framework for interpreting functional genomics screens. By superimposing CRISPRi perturbation data onto established pathway maps, researchers can identify not only primary targets but also compensatory mechanisms and network-wide effects that might be missed when examining individual omics layers in isolation [17]. This holistic perspective is particularly valuable for understanding complex cellular responses to transcriptional repression, where pathway context often determines phenotypic outcomes.
Methodological Approaches for Multi-Omics Data Integration
Core Computational Frameworks
Multi-omics integration employs diverse computational strategies, each with distinct strengths for particular research applications. The table below summarizes the primary methodological approaches:
Table 1: Computational Methods for Multi-Omics Data Integration
| Model Approach | Key Strengths | Typical Applications | Limitations |
|---|---|---|---|
| Correlation/Covariance-based | Captures relationships across omics; interpretable; flexible sparse extensions | Disease subtyping; detection of co-regulated modules | Limited to linear associations; requires matched samples |
| Matrix Factorisation | Efficient dimensionality reduction; identifies shared and omic-specific factors | Disease subtyping; biomarker discovery; shared pattern identification | Assumes linearity; does not explicitly model uncertainty |
| Probabilistic-based | Captures uncertainty in latent factors; probabilistic inference | Latent factor discovery; biomarker discovery; disease subtyping | Computationally intensive; may require strong model assumptions |
| Network-based | Robust to missing data; represents sample or omics relationships as networks | Identification of regulatory mechanisms; patient similarity analysis | Sensitive to similarity metrics choice; may require extensive tuning |
| Deep Generative Learning | Learns complex nonlinear patterns; supports missing data and denoising | High-dimensional omics integration; data augmentation; disease subtyping | High computational demands; limited interpretability |
Directional integration methods represent a significant advancement in addressing the challenges of biological interpretation. Methods like Directional P-value Merging (DPM) incorporate user-defined directional constraints based on established biological relationships—for instance, expecting that promoter DNA methylation typically correlates negatively with gene expression, or that mRNA expression should positively correlate with protein abundance [43]. This approach prioritizes genes and pathways with consistent directional changes across omics datasets while penalizing those with conflicting signals, thereby reducing false positives and providing more mechanistically plausible insights [43].
A critical component of network integration is the utilization of comprehensive pathway databases that provide the scaffold for mapping multi-omics data. The table below summarizes essential pathway resources:
Table 2: Key Pathway Databases for Multi-Omics Integration
| Database Name | Pathway Count | Primary Focus | Supported Formats |
|---|---|---|---|
| KEGG | >500 pathways | Metabolic and signaling pathways across diverse organisms | BioPAX, PNG, KGML |
| Reactome | N/A | Curated pathways for model organisms | BioPAX, PNG, PDF |
| WikiPathways | >2,800 pathways | Community-curated pathways for multiple organisms | BioPAX, SVG, PNG, PDF, GPML |
| BioCyc | >3,800 pathways (MetaCyc) | Metabolic and regulatory pathways for ~5,500 organisms | BioPAX, PNG, SBML |
| PANTHER Pathway | 176 pathways | Primarily signaling pathways with user curation | BioPAX, SBML |
| Pathway Commons | N/A | Meta-database integrating multiple sources | BioPAX, SIF, PNG |
These resources enable the mapping of experimental data onto established biological pathways, though they differ in scope and specialization. KEGG provides broad coverage across diverse organisms, while MetaCyc offers extensive organism-specific metabolic pathways [44]. WikiPathways stands out for its community-driven approach, allowing researchers to contribute and curate pathways [44]. For novel or incompletely understood pathways, tools like MetaboMAPS offer a platform for sharing customized pathway maps beyond common knowledge, supporting ongoing research on emerging biological systems [45].
Experimental Workflow for Network Integration in CRISPRi Research
Integrated Multi-Omics Analysis Pipeline
The following diagram illustrates the comprehensive workflow for integrating multi-omics data in CRISPRi response studies:
Detailed Methodological Protocols
CRISPRi Functional Genomics Screen Protocol
The foundation of network integration in CRISPRi studies begins with a rigorously executed functional genomics screen, based on established methodologies from pluripotency research [17]:
Cell Line Preparation: Utilize Cas9-expressing embryonic stem cells (or cell line relevant to your research question) cultured under defined conditions. For pluripotency studies, LIF/serum conditions are commonly used to maintain naïve state pluripotency [17].
CRISPR Library Design and Delivery:
- Select a genome-wide CRISPR library such as the Brie library (targeting ~19,674 genes) with high coverage across the genome [17].
- Infect Cas9-expressing cells with lentiviruses containing the sgRNA library at appropriate MOI to ensure single integration events.
- Include non-targeting sgRNAs as negative controls and essential gene-targeting sgRNAs as positive controls.
Screen Execution and Sample Collection:
- Propagate infected cells for 14 days under appropriate selection pressure.
- Collect cells at day 0 (baseline) and day 14 (endpoint) post-infection for sequencing analysis.
- Maintain adequate cell numbers throughout (typically >500 cells per sgRNA) to maintain library representation [17].
Sequencing and Data Analysis:
- Extract genomic DNA and amplify sgRNA regions for sequencing.
- Sequence pre-transfected plasmid library, P.Sc0d, and P.Sc14d samples.
- Use analysis tools like MAGeCK to identify sgRNAs with significantly depleted or enriched abundance [17].
- Validate screen quality by confirming high concordance between biological replicates (r > 0.7) and high sgRNA representation (>99%) [17].
Multi-Omics Data Acquisition and Preprocessing
Following CRISPR screening, comprehensive molecular profiling generates the multi-omics data for network integration:
Transcriptomic Profiling:
- Extract total RNA using quality-controlled methods (RIN > 8.0 recommended).
- Perform RNA sequencing with appropriate depth (typically 30-50 million reads per sample for bulk RNA-seq).
- Process raw sequencing data through standard pipelines: quality control (FastQC), alignment (STAR), and quantification (featureCounts).
Proteomic Profiling:
- Prepare protein extracts using appropriate lysis buffers with protease inhibitors.
- Perform data-independent acquisition (DIA) mass spectrometry or TMT-based quantitation.
- Process raw mass spectrometry data using tools like MaxQuant or Spectronaut for identification and quantification.
Epigenomic Profiling:
- Conduct ATAC-seq or ChIP-seq for histone modifications relevant to your biological question.
- Process sequencing data through appropriate pipelines for peak calling and quantification.
Data Preprocessing and Normalization:
- Apply appropriate normalization methods for each data type (e.g., TMM for RNA-seq, median normalization for proteomics).
- Perform batch effect correction using methods like ComBat when integrating data from multiple cohorts or batches.
- Ensure proper data quality metrics are met for each omics dataset.
Directional Multi-Omics Integration Protocol
The directional integration of multi-omics data follows a structured analytical workflow:
Data Matrix Preparation:
- Compile gene-level P-values and directional changes (e.g., fold changes) for each omics dataset into separate matrices.
- Align features across datasets using standard gene identifiers.
Constraints Vector Definition:
- Define expected directional relationships between datasets based on biological knowledge. For example:
- Transcriptomics and proteomics: positive association (+1)
- DNA methylation and transcriptomics: negative association (-1)
- CRISPRi essentiality scores and transcriptomics: context-dependent [43]
- Define expected directional relationships between datasets based on biological knowledge. For example:
Directional P-value Merging:
- Apply the DPM method to integrate P-values across datasets using the formula: [ X{DPM} = -2(-|\Sigma{i=1}^{j} \ln(Pi) oi ei| + \Sigma{i=j+1}^{k} \ln(Pi)) ] where (Pi) represents P-values, (oi) represents observed directional changes, and (ei) represents expected directional relationships [43].
- Compute merged P-values using the empirical Brown's method to account for gene-to-gene covariation [43].
Pathway Enrichment Analysis:
Successful implementation of network integration for multi-omics data requires specific computational tools, biological reagents, and analytical resources:
Table 3: Essential Research Resources for Multi-Omics Network Integration
| Resource Category | Specific Tool/Reagent | Function and Application |
|---|---|---|
| CRISPR Screening | Brie CRISPR Library (or similar) | Genome-wide sgRNA collection for functional genomics screens [17] |
| Pathway Databases | KEGG, Reactome, WikiPathways | Curated biological pathways for data mapping and interpretation [44] |
| Integration Algorithms | ActivePathways with DPM | Directional multi-omics data fusion and pathway enrichment [43] |
| Visualization Tools | PathVisio, Cytoscape with plugins | Pathway visualization and data mapping [44] |
| Network Analysis | Pathway Commons, ConsensusPathDB | Integrated biological networks combining multiple resources [44] |
| Data Repositories | TCGA, ICGC, ProCan | Reference multi-omics datasets for comparison and validation [46] |
Application to CRISPRi Response Research
Case Study: Defining Pluripotency Regulatory Networks
A exemplary application of network integration in CRISPRi research comes from studies redefining the pluripotency gene regulatory network (PGRN) in embryonic stem cells (ESCs). Through CRISPR/Cas9-based functional genomics screens integrated with transcriptomic, proteomic, and epigenomic data, researchers constructed an expanded PGRN with nine sub-classes resolved into six functionally independent transcriptional modules: CORE, MYC, PAF, PRC, PCGF, and TBX [17].
The analysis revealed that activated CORE/MYC/PAF module activity and repressed PRC/PCGF/TBX module activity represent a fundamental pattern shared by mouse ESCs, human ESCs, and even cancer cells [17]. This systems-level understanding of pluripotency regulation demonstrates how network integration of multi-omics data can elucidate fundamental biological principles with broad applicability across different cellular contexts and species.
Analyzing CRISPRi Perturbation Responses
The following diagram illustrates the network-level analysis of CRISPRi perturbation responses:
In practice, analyzing CRISPRi responses through network integration involves:
Primary Target Identification: Mapping direct molecular changes to the targeted gene and its immediate network neighbors.
Compensatory Mechanism Detection: Identifying pathway-level responses and alternative routes that cells employ to bypass the targeted perturbation.
Network Rewiring Analysis: Characterizing how regulatory relationships change in response to the perturbation, potentially revealing new functional connections not apparent in unperturbed cells.
This approach is particularly powerful when combined with single-cell multi-omics technologies, which enable the correlation of specific genomic, transcriptomic, and epigenomic changes within individual cells [15]. The development of artificial intelligence-based computational methods further enhances our ability to understand how each multi-omic change contributes to the overall state and function of cells following CRISPRi perturbations [15].
Future Directions and Concluding Remarks
Network integration of multi-omics data represents a paradigm shift in how we approach functional genomics and CRISPRi response research. As single-cell multi-omics technologies continue to advance, we will gain unprecedented resolution in understanding cellular heterogeneity and response dynamics [15]. The integration of both extracellular and intracellular protein measurements, including cell signaling activity, will provide additional layers for understanding tissue biology and drug responses [15].
The future of this field will be shaped by several key developments. First, purpose-built analysis tools specifically designed for multi-omics data integration will become increasingly important, moving beyond siloed analytical workflows [15]. Second, appropriate computing and storage infrastructure, along with federated computing specifically designed for multi-omic data, will be essential for handling the massive data outputs [15]. Finally, collaborative efforts among academia, industry, and regulatory bodies will be crucial for establishing standards and creating frameworks that support the clinical application of multi-omics insights [15].
For CRISPRi research specifically, network integration provides the contextual framework necessary to distinguish driver effects from passenger effects, identify synthetic lethal interactions, and understand compensatory network adaptations. This comprehensive understanding ultimately accelerates the translation of basic CRISPR research into therapeutic applications, particularly in oncology and immunotherapy where network context often determines treatment efficacy and resistance mechanisms.
Leveraging AI and Machine Learning for Holistic Data Analysis and Pattern Recognition
The integration of artificial intelligence (AI) with multi-omics data represents a transformative approach for deciphering complex biological systems, particularly in the analysis of CRISPR interference (CRISPRi) responses. CRISPRi enables precise gene knockdown, allowing researchers to probe gene function at scale. However, interpreting the resulting multifaceted datasets requires advanced computational strategies. AI and machine learning (ML) provide the essential framework for integrating diverse omics layers—genomics, transcriptomics, proteomics, and epigenomics—to construct predictive models and uncover regulatory mechanisms that buffer genetic perturbations [47]. This holistic integration is crucial for moving beyond single-omics snapshots to a systems-level understanding of cellular behavior, ultimately accelerating therapeutic discovery and development.
The challenge in multi-omics data integration lies in the heterogeneous nature of the data. Each omics layer provides a unique but interconnected view of cellular state. For instance, genomic variants influence transcriptional regulation, which subsequently impacts protein abundance and metabolic activity [47]. Machine learning excels at identifying complex, non-linear patterns across these disparate data types, revealing the emergent properties that define cellular responses to genetic perturbations such as CRISPRi knockdowns [48] [47]. This capability is foundational for progressing from correlation to causation in biological research.
AI and ML Foundations for Multi-Omics Data Integration
Machine learning provides a suite of adaptive algorithms that learn functional relationships from complex training data. In the context of multi-omics, three primary learning paradigms are employed:
- Supervised Learning: This approach uses labeled datasets to train models that can predict outcomes, such as classifying cancer subtypes based on integrated omics profiles or predicting drug response [49] [47]. It is particularly valuable when established phenotypic outcomes are available for training.
- Unsupervised Learning: These algorithms discover hidden patterns and intrinsic structures in unlabeled data. Techniques such as clustering are used to identify novel molecular subtypes from multi-omics data without pre-existing labels [49] [47].
- Deep Learning (DL): Utilizing multi-layered neural networks, DL automatically extracts high-level features from raw data, capturing non-linear dependencies without manual feature engineering [50]. This is particularly powerful for processing high-dimensional omics data and has been successfully applied to predict CRISPR guide RNA efficacy and editing outcomes [50] [51].
Multi-Omics Integration Strategies
The integration of diverse omics data can be computationally approached through three principal strategies, each with distinct advantages:
- Early Integration: Also known as feature concatenation, this method merges raw datasets from multiple omics layers into a single combined matrix before analysis. While straightforward, it can be challenged by the high dimensionality and heterogeneous nature of the data [47].
- Intermediate Integration: This approach employs machine learning models to consolidate and extract latent features from multiple omics datasets simultaneously, preserving the unique characteristics of each data type while identifying cross-omics patterns [47].
- Late Integration: Analyses are performed separately on each omics dataset, with results merged at the final stage to form an integrated conclusion. This preserves data-specific analysis but may miss underlying interactions between omics layers [47].
Table 1: Machine Learning Approaches for Multi-Omics Data Analysis
| ML Category | Key Algorithms | Primary Applications in Multi-Omics | Considerations |
|---|---|---|---|
| Supervised Learning | Random Forest, Support Vector Machines (SVM), Regression Models | Cancer type classification, drug response prediction, phenotype forecasting | Requires high-quality labeled data; prone to overfitting with small datasets |
| Unsupervised Learning | K-means Clustering, Principal Component Analysis (PCA), Autoencoders | Novel subtype discovery, data dimensionality reduction, pattern recognition | Discovery-oriented; results may require experimental validation |
| Deep Learning | Convolutional Neural Networks (CNNs), Recurrent Neural Networks (RNNs), Transformers | Guide RNA design, protein structure prediction, cross-omics feature extraction | Computationally intensive; requires large datasets; enables automatic feature learning |
AI-Enhanced CRISPR Technology for Functional Genomics
CRISPRi has emerged as a powerful tool for functional genomics, enabling precise gene knockdown without complete gene knockout. When combined with multi-omics readouts and AI analysis, it provides unprecedented insights into gene function and regulatory networks. The application of AI to CRISPR technology addresses several fundamental challenges:
Optimizing Guide RNA Design and Efficacy
A critical challenge in CRISPRi experiments is designing guide RNAs (gRNAs) with high on-target efficiency and minimal off-target effects. AI models have dramatically improved gRNA design by learning from vast experimental datasets:
- DeepCRISPR: This pioneering deep learning platform employs unsupervised pre-training on billions of guide RNA sequences followed by supervised fine-tuning. It simultaneously predicts on-target knockout efficacy and off-target profiles while automatically incorporating epigenetic features such as histone modifications and chromatin accessibility [51].
- CRISPR-GPT: Developed by Stanford Medicine and collaborators, this large language model acts as a conversational AI assistant for designing CRISPR experiments. Trained on 11 years of scientific literature and expert discussions, it provides experimental designs, troubleshooting guidance, and explanations for researchers at all skill levels [52].
- CRISPRon: This approach integrates high-quality experimental data from 23,902 guide RNAs, combining sequence composition with thermodynamic properties and gRNA-DNA binding energy calculations to predict editing outcomes with superior accuracy [49] [51].
These AI tools have demonstrated remarkable performance, with some models achieving over 95% prediction accuracy in specific applications, significantly reducing the trial-and-error approach that traditionally characterizes CRISPR experimental design [51].
Predicting and Minimizing Off-Target Effects
Off-target effects remain a significant concern in CRISPR applications. AI-based approaches have substantially advanced off-target prediction:
- CRISPR-M: A multi-view deep learning architecture that combines convolutional neural networks (CNNs) and bidirectional long short-term memory (LSTM) networks to predict off-target sites, including those with insertions, deletions, and mismatches. Its novel encoding scheme captures multiple perspectives of gRNA-DNA interactions [51].
- CALITAS: An integrated off-target search algorithm that provides comprehensive off-target profiling across entire genomes [50].
These tools leverage diverse features including sequence composition, epigenetic context, chromatin accessibility, and cellular environment to provide increasingly accurate off-target predictions, enhancing the safety profile of CRISPR-based therapies [49] [50].
Diagram 1: AI-Driven CRISPRi Multi-Omics Analysis Workflow. This workflow illustrates the iterative process of integrating multi-omics data with AI analysis to derive biological insights from CRISPRi screens.
Methodologies for Multi-Omics Analysis of CRISPRi Responses
Experimental Design for CRISPRi Multi-Omics Studies
A well-designed CRISPRi multi-omics experiment requires careful planning across several dimensions:
- CRISPRi Library Design: Utilize AI-based tools such as CRISPR-GPT or DeepCRISPR to design high-efficacy gRNAs with minimal off-target effects [52] [51]. For genome-wide screens, the Brie library (targeting ~19,674 genes) provides comprehensive coverage [17].
- Multi-Omics Data Collection: Plan for coordinated collection of genomic, transcriptomic, proteomic, and epigenomic data from the same biological samples to enable robust integration.
- Controls and Replication: Include appropriate controls (non-targeting gRNAs, positive essential genes) and biological replicates to account for technical and biological variability [17].
Data Integration and Analysis Workflow
The analysis of multi-omics data from CRISPRi screens follows a structured pipeline:
Quality Control and Preprocessing:
- Process raw sequencing data (trimming, alignment, quantification)
- Perform normalization and batch effect correction
- Confirm gRNA representation and distribution across samples [17]
Identification of Hit Genes:
- Use specialized algorithms (e.g., MAGeCK) to identify genes whose knockdown significantly impacts phenotype [17]
- Distinguish between core essential genes and context-specific dependencies
Multi-Omics Data Integration:
- Employ intermediate integration approaches to combine transcriptomic, proteomic, and epigenomic data
- Utilize dimensionality reduction techniques to visualize relationships
- Apply network analysis to identify regulatory modules [17]
Table 2: Quantitative Data Analysis Methods for Multi-Omics Studies
| Analysis Method | Primary Application | Key Metrics | AI/ML Enhancement |
|---|---|---|---|
| Cross-Tabulation | Analyzing relationships between categorical variables | Frequency counts, proportions | Automated pattern detection through association rule learning |
| Gap Analysis | Comparing actual vs. expected performance | Difference measures, ratio analysis | Anomaly detection algorithms to identify significant deviations |
| MaxDiff Analysis | Identifying most preferred items from option sets | Preference scores, utility values | Neural networks for ranking and preference prediction |
| Text Analysis | Extracting insights from unstructured textual data | Word frequencies, sentiment scores | Natural language processing (NLP) for concept extraction |
| Regression Analysis | Modeling relationships between variables | Coefficients, p-values, R-squared | Regularization methods (LASSO, Ridge) for high-dimensional data |
Diagram 2: Multi-Omics Data Integration Strategies for CRISPRi Response Analysis. This diagram illustrates how different omics layers are integrated through machine learning approaches to derive biological insights.
Case Study: Multi-Omics Analysis of CRISPRi-Knockdowns in E. coli Metabolism
A landmark study demonstrated the power of integrating multi-omics data with CRISPRi knockdowns to identify metabolic buffering mechanisms [48]. The methodology included:
Pooled CRISPRi Screening:
- Created 7,177 E. coli strains with individual gene knockdowns
- Measured fitness defects hours after CRISPRi induction
- Identified genes where metabolism buffered fitness defects
Multi-Omics Profiling:
- Conducted metabolomic and proteomic analysis on 30 selected CRISPRi strains
- Quantified metabolite abundance and protein expression changes
- Integrated these measurements with fitness data
Identification of Buffering Mechanisms:
- Discovered gene-specific regulatory mechanisms that compensate for enzyme deficiencies
- Examples included ornithine buffering CarAB knockdown, S-adenosylmethionine buffering MetE knockdown, and 6-phosphogluconate buffering Gnd knockdown [48]
This approach revealed that metabolic networks contain sophisticated regulatory circuits that maintain homeostasis despite enzymatic deficiencies, providing insights into metabolic robustness with implications for therapeutic development.
Table 3: Research Reagent Solutions for AI-Enhanced CRISPRi Multi-Omics Studies
| Resource Category | Specific Tools/Platforms | Function | Access |
|---|---|---|---|
| CRISPRi Design Tools | CRISPR-GPT, DeepCRISPR, CRISPRon | AI-powered guide RNA design, efficiency prediction, and off-target assessment | Web-based platforms, standalone software [52] [51] |
| Multi-Omics Databases | TCGA, DepMap, COSMIC, ICGC | Provide comprehensive multi-omics datasets for model training and validation | Public data portals [47] |
| Machine Learning Frameworks | TensorFlow, PyTorch, Scikit-learn | Develop and implement custom ML models for data integration | Open-source libraries |
| Bioinformatics Pipelines | MAGeCK, CALITAS, CRISPRDirect | Process CRISPR screening data, identify hits, analyze off-target effects | Open-source tools [17] [50] |
| Data Visualization Platforms | ChartExpo, Ajelix BI, R/Shiny | Create interactive visualizations for exploring complex multi-omics datasets | Commercial and open-source tools |
The integration of AI and machine learning with multi-omics data represents a paradigm shift in our ability to understand and interpret CRISPRi responses. By moving beyond single-omics approaches to holistic data integration, researchers can uncover the complex regulatory networks and buffering mechanisms that maintain cellular homeostasis. The methodologies and resources outlined in this technical guide provide a framework for designing, executing, and analyzing multi-omics CRISPRi studies that leverage the latest advances in AI.
As these technologies continue to evolve, we anticipate increasingly sophisticated models that can not only interpret but predict cellular responses to genetic perturbations, accelerating the development of novel therapeutic strategies and advancing our fundamental understanding of biological systems. The convergence of AI-driven CRISPR optimization with comprehensive multi-omics profiling marks the beginning of a new era in functional genomics and personalized medicine.
Navigating Challenges: Data Harmonization, Analysis, and Computational Infrastructure
Overcoming Data Harmonization Issues from Disparate Cohorts and Platforms
The functional characterization of non-coding cis-regulatory elements (CREs) using CRISPR interference (CRISPRi) has emerged as a powerful approach for understanding gene regulatory landscapes. However, the integration of data from disparate cohorts and experimental platforms presents significant harmonization challenges that can compromise data interpretation and scientific validity. The ENCODE Consortium's efforts, which involved analyzing 108 CRISPRi screens comprising over 540,000 perturbations across 24.85 megabases of the human genome, highlight both the scale of this data integration challenge and the potential insights gained from overcoming it [53]. Such large-scale multicenter analyses have revealed that only 4.0% of perturbed bases displayed regulatory function, and merely 4.79% of candidate CREs that were perturbed directly overlapped with confirmed functional CREs, underscoring the critical need for rigorous harmonization to distinguish true biological signals from technical artifacts [53].
The foundational challenge in multi-platform CRISPRi research lies in the substantial technical variability introduced by differing experimental conditions, screening methodologies, and analytical pipelines. CRISPRi employs a deactivated Cas9 (dCas9) fused to transcriptional repressor domains like KRAB to silence gene expression without editing DNA, but efficiency varies considerably based on guide RNA design, delivery methods, and cellular context [20] [54]. Without proper harmonization, these technical differences can obscure biological insights, particularly when integrating data from diverse biological samples ranging from cancer cell lines like K562 to induced pluripotent stem cells (iPSCs) and their derivatives [53]. This technical guide outlines comprehensive strategies and best practices to overcome these harmonization barriers, enabling more robust integration of CRISPRi data across platforms and cohorts.
Methodological Standardization for Cross-Platform Harmonization
Experimental Design and Platform Selection
Establishing consistent experimental designs is the first critical step in ensuring data harmonization. For CRISPRi screens, this begins with selecting appropriate perturbation approaches based on research objectives. Tiling screens that include sgRNAs targeting both candidate CREs and non-cCRE regions within specific loci can identify novel regulatory elements lacking conventional epigenetic marks, while cCRE-targeted approaches that focus sgRNAs specifically on putative regulatory elements enable screening of more elements with the same number of sgRNAs [53]. The ENCODE analysis revealed that 99.7% of confirmed CREs were within ±500 base pairs of open chromatin regions or enhancer-like signature cCREs, providing guidance for targeted screen design [53].
Platform selection must also consider the delivery method for CRISPRi components, as this significantly impacts data comparability. Table 1 outlines the primary delivery methods and their appropriate applications. For extended assays lasting more than 120 hours, lentiviral sgRNA delivery is recommended, while synthetic sgRNAs typically provide more robust repression for short-term assays [54]. Creating stable dCas9-expressing cell lines through lentiviral transduction of dCas9-repressor fusions (e.g., dCas9-KRAB or dCas9-SALL1-SDS3) prior to sgRNA delivery ensures consistent repression efficiency across experiments and platforms [55] [54]. For rapid, transient repression in lentiviral-free workflows, co-transfection of dCas9-SALL1-SDS3 mRNA with synthetic sgRNA represents an effective alternative [54].
Table 1: CRISPRi Delivery Methods and Applications
| Delivery Method | Application Context | In vivo/Ex vivo | Human/Non-human | Benefits and Limitations |
|---|---|---|---|---|
| Lentiviral vectors | Gene therapy, experimental and clinical use | Ex vivo/In vivo | Human/Animal | Stable expression, suitable for difficult-to-transfect cells; potential insertional mutagenesis |
| Electroporation | Preclinical research, clinical trials | Ex vivo/In vivo | Human/Animal | Effective for hard-to-modify cell types; can cause tissue damage and sensitivity issues |
| Lipid-based nanoparticles | Human cells, clinical trials | Ex vivo/In vivo | Human | High efficiency, minimal immunogenicity; limited packaging capacity |
| Microinjection | Animal models, embryonic editing | Ex vivo | Non-human | Precise control over delivery; technical complexity and low throughput |
Library Design and Reference Standards
Standardized library design is crucial for cross-platform harmonization. The ENCODE Consortium's analysis of 53 noncoding CRISPR screens in K562 cells revealed that CREs showed greatest enrichment for H3K27ac, RNA polymerase II, and H3K4me3 peaks (OR = 22.1, 14.5, and 10.8, respectively) [53]. These epigenetic features should inform guide RNA design to maximize functional targeting. Additionally, the discovery of a subtle DNA strand bias for CRISPRi in transcribed regions has direct implications for guide RNA design and screening analysis [53].
Each CRISPRi screen should include multiple negative control sgRNAs (non-targeting controls) and positive control sgRNAs targeting genes with known essential functions or well-characterized regulatory elements [54]. The ENCODE Consortium provides predesigned sgRNAs for targeting 3,275,697 candidate CREs, offering a valuable resource for standardizing library design across studies [53]. For transporter studies, as exemplified in nutrient transport screens, custom libraries targeting all annotated members of solute carrier (SLC) and ATP-binding cassette (ABC) transporter families (typically with 10 sgRNAs per gene and 730 non-targeting controls) enable consistent cross-study comparisons [55].
Phenotyping Standardization
Phenotyping strategies must be standardized to ensure comparability across platforms. For fitness-based screens, consistent culture conditions and passage protocols are essential. The application of CRISPRi/a screening to study cellular nutrient transport highlights the importance of modeling diverse microenvironments, from standard culture media to conditions that mimic tumors [55]. When screening under nutrient-limited conditions, it is critical to use amino acid concentrations that reduce proliferation by approximately 50% for growth-limiting amino acids, as this sensitive yet sublethal threshold maximizes detection of transporter dependencies [55].
Functional validation of CRE-gene links should follow standardized protocols. The ENCODE Consortium established 332 functionally confirmed CRE-gene links in K562 cells, providing a benchmark set for validating new screening approaches [53]. For gene expression phenotyping, single-cell RNA sequencing methods like Perturb-seq, CRISP-seq, and CROP-seq enable high-dimensional phenotyping but require careful standardization of cell processing, sequencing depth, and analytical pipelines to ensure cross-platform comparability [20].
Computational Harmonization Strategies
Data Processing and Normalization
Raw data processing must address platform-specific technical artifacts while preserving biological signals. Initial quality control should assess sequencing depth, sgRNA representation, and sample-level quality metrics. For read count normalization, methods that adjust for library sizes and count distributions are essential, as sgRNA abundance data typically exhibits over-dispersion similar to other high-throughput sequencing experiments [20]. The MAGeCK workflow incorporates such normalization approaches and has been widely adopted for CRISPR screen analysis [20].
Batch effect correction represents a critical step in harmonizing data across platforms. The growing volume and complexity of omics data has created a need for standardized approaches to detect and correct for batch effects, systematic drift, or outlier runs early in analysis [56]. Principal component analysis (PCA) and quality control (QC) trend visualization should be integrated into data preprocessing to flag problematic samples or batches before downstream analysis [56]. Computational methods like the mixed-effect random forest model, which separates features affecting guide efficiency from gene-specific effects, have demonstrated particular utility in learning from multiple independent CRISPRi screens while accounting for platform-specific biases [57].
Analysis Tool Selection and Benchmarking
Selecting appropriate analysis tools is paramount for effective data harmonization. The ENCODE Consortium benchmarked five screen analysis tools and found that CASA produces the most conservative CRE calls and is robust to artifacts of low-specificity sgRNAs [53]. Table 2 provides a comprehensive overview of computational tools for CRISPR screen analysis, their methodologies, and applications.
Table 2: Computational Tools for CRISPR Screen Data Analysis
| Tool | Year | Statistical Approach | sgRNA/Gene Ranking | Key Applications | FDR Control |
|---|---|---|---|---|---|
| MAGeCK | 2014 | Negative binomial distribution, robust rank aggregation | Both | Genome-wide knockout/interference screens | Yes |
| BAGEL | 2016 | Reference gene set distribution, Bayes factor | Gene | Essential gene identification | Yes |
| CASA | 2024 | Conservative calling, artifact resistance | Gene | Non-coding CRISPRi screens | Yes |
| CRISPRcloud2 | 2019 | Beta binomial distribution, Fisher's test | Both | Web-based analysis platform | Yes |
| JACKS | 2019 | Bayesian hierarchical modeling | Gene | Pooled screen analysis | Yes |
| DrugZ | 2019 | Normal distribution, sum z-score | Gene | Chemogenetic interaction screens | Yes |
For different screening modalities, specific tools may be preferred. MAGeCK was the first workflow specifically designed for CRISPR/Cas9 screen analysis and uses a negative binomial distribution to test for significant differences between treatment and control groups, followed by robust rank aggregation (RRA) to identify enriched genes [20]. BAGEL employs a Bayes factor approach based on reference gene sets and is particularly effective for essential gene identification [20]. For chemogenetic screens investigating drug-gene interactions, DrugZ implements a normal distribution-based sum z-score approach that specifically addresses this application [20].
Machine Learning and Data Integration
Advanced machine learning approaches offer powerful solutions for data harmonization challenges. Mixed-effect random forest models have demonstrated particular utility for predicting CRISPRi guide efficiency from depletion screens by separating guide-specific effects from gene-specific effects [57]. This approach is especially valuable when only indirect measurements of guide activity are available, as is common in genome-wide essentiality screens [57].
Explainable AI methods, including SHapley Additive exPlanation (SHAP) values, provide interpretable insights into factors influencing guide efficiency across platforms [57]. Application of these methods to E. coli CRISPRi essentiality screens revealed that maximal RNA expression had the largest effect on depletion (~1.6-fold difference), followed by the number of downstream essential genes (~1.3-fold difference), indicating presence of polar effects [57]. Interestingly, guide-specific features like distance to transcriptional start site had relatively small effects (~1.07-fold) compared to gene-specific features [57].
Data fusion across multiple independent screens significantly improves prediction accuracy. Integration of data from three E. coli CRISPRi screens (E75 Rousset, E18 Cui, and Wang libraries) demonstrated that models trained on combined datasets generalized better across platforms than those trained on individual datasets [57]. This multi-dataset approach also facilitates identification of consistent biological signals while filtering platform-specific technical artifacts.
Visualization of the Harmonization Workflow
The following diagram illustrates the integrated computational and experimental workflow for overcoming data harmonization challenges in multi-platform CRISPRi studies:
Integrated Workflow for CRISPRi Data Harmonization
This workflow emphasizes the continuous interaction between experimental and computational harmonization approaches, with iterative validation ensuring robust data integration across disparate platforms and cohorts.
Analytical Framework for Multi-Cohort Data Integration
Statistical Processing Best Practices
Statistical processing of integrated CRISPRi data must address the unique characteristics of omics datasets, including missing values, heteroscedasticity, and non-normal distributions. Missing values in omics data can be categorized as missing completely at random (MCAR), missing at random (MAR), or missing not at random (MNAR), with each requiring different imputation strategies [58]. For MNAR data common in lipidomics and metabolomics (where values fall below detection limits), k-nearest neighbors (kNN)-based imputation or substitution with a percentage of the lowest concentration value have proven effective [58]. For MCAR and MAR data, random forest-based imputation often provides superior performance [58].
Data normalization should address both analytical variation (batch effects, signal intensity fluctuations) and biological variation (sample amount differences) [58]. Pre-acquisition normalization based on cell count, protein amount, or DNA amount is preferred over post-acquisition statistical normalization [58]. Quality control samples (QCs) obtained by pooling aliquots of all biological samples or using standard reference materials like NIST SRM 1950 for plasma samples enable evaluation of technical variability and facilitate normalization to remove batch effects [58].
Multi-Omics Data Integration
Integrating CRISPRi screening data with other omics layers (genomics, epigenomics, transcriptomics, proteomics) enables more comprehensive understanding of gene regulatory mechanisms but introduces additional harmonization challenges. The ENCODE Consortium's integrated analysis revealed that while most functional CREs overlapped either accessible chromatin regions or H3K27ac peaks (95.2%), some exhibited distinct epigenetic signatures, with 24 CREs marked by H3K27ac but not overlapping DHSs, and 18 overlapping DHSs but lacking H3K27ac peaks [53]. In stem cells, a greater proportion of CREs overlap repressive histone marks (H3K9me3 and H3K27me3), consistent with the presence of poised and bivalent regulatory elements [53].
Five primary objectives guide successful multi-omics integration in translational medicine applications: (1) detecting disease-associated molecular patterns, (2) subtype identification, (3) diagnosis/prognosis, (4) drug response prediction, and (5) understanding regulatory processes [59]. Intermediate integration approaches that learn joint representations of separate datasets have proven particularly effective for addressing these objectives [59]. Publicly available multi-omics resources like The Cancer Genome Atlas (TCGA), Answer ALS, and DevOmics provide valuable reference datasets for method development and validation [59].
Essential Research Reagents and Tools
Table 3: Key Research Reagent Solutions for CRISPRi Studies
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| dCas9-Repressor Fusions | dCas9-KRAB, dCas9-SALL1-SDS3 | Transcriptional repression without DNA cleavage |
| sgRNA Formats | Synthetic sgRNA, lentiviral sgRNA | Target-specific guidance; format choice depends on assay duration and cell type |
| Delivery Systems | Lentiviral particles, lipid nanoparticles, electroporation | Efficient cellular delivery of CRISPRi components |
| Control Reagents | Non-targeting sgRNAs, positive control sgRNAs | Assessment of baseline response and system functionality |
| Screening Libraries | Genome-wide libraries, custom cherry-pick libraries | Targeted perturbation of specific gene sets or regulatory elements |
| Validation Tools | Antibodies for FACS, qPCR assays, scRNA-seq | Confirmation of perturbation efficiency and phenotypic effects |
Implementation of these reagent systems requires careful consideration of experimental goals and cell models. For extended timepoint assays (exceeding 120 hours), lentiviral sgRNA delivery is recommended, while synthetic sgRNAs typically provide more robust repression for short-term assays [54]. Creating stable dCas9-expressing cell lines through lentiviral transduction of dCas9-repressor fusions prior to sgRNA delivery ensures consistent repression efficiency across experiments [55] [54]. Commercial CRISPRi systems such as the Dharmacon CRISPRmod CRISPRi system provide optimized, pre-validated components that enhance reproducibility across laboratories and platforms [54].
Overcoming data harmonization challenges in CRISPRi research requires integrated experimental and computational approaches that address variability at each stage of the research pipeline. Methodological standardization in library design, delivery methods, and phenotyping strategies establishes a foundation for comparable data generation. Computational harmonization through appropriate tool selection, batch effect correction, and advanced machine learning enables robust integration of disparate datasets. The implementation of standardized statistical processing practices and multi-omics integration frameworks further enhances the biological insights derived from multi-platform CRISPRi studies. As CRISPRi technologies continue to evolve and scale, the harmonization strategies outlined in this technical guide will play an increasingly critical role in enabling robust, reproducible functional genomics research that effectively bridges diverse experimental platforms and biological systems.
Selecting and Developing Purpose-Built Analytical Pipelines for Multi-Modal Data
The integration of multi-modal omics data has become a cornerstone for advancing functional genomics, particularly in the study of CRISPR interference (CRISPRi) responses. Modern CRISPRi experiments have evolved beyond simple knockouts to probe complex gene regulatory networks, generating diverse data types including transcriptomic, proteomic, epigenomic, and high-content imaging readouts [60] [61]. This multi-modal approach is essential for capturing the full complexity of cellular responses to targeted perturbations. Where single-modal analyses provide fragmented insights, multi-modal integration enables researchers to simultaneously investigate the effects of genomic perturbations on transcription, translation, and epigenetic regulation within the same biological system [61]. This technical guide provides a comprehensive framework for selecting and developing analytical pipelines specifically designed for multi-modal data, with particular emphasis on applications in CRISPRi response research for drug discovery and functional genomics.
The analytical challenge lies not merely in processing individual data streams but in creating unified analytical frameworks that can extract biologically meaningful insights from these interconnected data layers. As highlighted in recent surveys of the field, "single-cell mono-omics results in fragmentation of information and could not provide complete cell states" [61]. This guide addresses this challenge by presenting purpose-built solutions for multi-modal data integration, experimental design considerations, and computational strategies tailored to the specific requirements of CRISPRi studies in both basic research and drug development contexts.
Analytical Framework: Pipeline Architectures for Multi-Modal Data
Specialized Analytical Platforms
The selection of an appropriate analytical pipeline depends on the specific multi-modal data types being generated and the research questions being addressed. Several specialized platforms have emerged to handle the distinct computational requirements of different CRISPR screening modalities.
Table 1: Analytical Platforms for Multi-Modal CRISPR Data
| Platform Name | Primary Data Modality | Key Features | Application in CRISPRi Studies |
|---|---|---|---|
| nf-core/crisprseq [62] | Targeted sequencing & pooled screening | Modular workflow; supports KO, KI, base editing, CRISPRa/i; includes QC, alignment, UMI processing | Evaluation of editing quality; discovery of hits from CRISPRi screens |
| CRISPRmap [60] | Optical phenotyping + immunofluorescence + RNA | In situ barcode readout; spatial phenotypes; compatible with primary cells and tissues | Investigating morphology, protein localization, cell-cell interactions post-CRISPRi |
| Flexynesis [11] | Bulk multi-omics (transcriptome, epigenome, genome) | Deep learning toolkit; multi-task modeling; supports classification, regression, survival analysis | Predicting drug response; identifying biomarkers from multi-omics CRISPRi data |
The nf-core/crisprseq pipeline represents a robust, community-supported framework for analyzing CRISPR editing data from both targeted sequencing and pooled screening approaches [62]. Its modular architecture allows researchers to process diverse data types through standardized workflow steps including read QC, adapter trimming, UMI clustering, and read mapping. For CRISPRi screening data specifically, the pipeline utilizes MAGeCK count for read mapping and quantification, followed by comprehensive statistical analysis to rank sgRNAs and identify candidate genes [62].
For spatial multi-modal phenotyping, CRISPRmap enables "in situ barcode readout in cell types and contexts that were elusive to conventional optical pooled screening, including cultured primary cells, embryonic stem cells, induced pluripotent stem cells, derived neurons and in vivo cells in a tissue context" [60]. This platform combines CRISPR guide-identifying barcode readout with multiplexed immunofluorescence and RNA detection, allowing researchers to correlate genetic perturbations with spatial phenotypes including cell morphology, protein subcellular localization, and tissue organization.
When predictive modeling from integrated bulk multi-omics data is required, Flexynesis provides "a deep learning framework for multi-omics data integration designed to overcome limitations of transparency, modularity, and deployability" [11]. This toolkit streamlines data processing, feature selection, and hyperparameter tuning for tasks including drug response prediction, cancer subtype classification, and survival modeling - all highly relevant for interpreting CRISPRi screening outcomes in translational research contexts.
Workflow Integration Strategies
Effective multi-modal data integration requires strategic approaches to combine information across analytical domains. The workflow below illustrates how these specialized platforms can be incorporated into a comprehensive analytical strategy for multi-modal CRISPRi studies:
Experimental Design for Multi-Modal CRISPRi Studies
Library Design and sgRNA Selection
The foundation of any successful CRISPRi study begins with optimized library design. Recent advances in machine learning have significantly improved sgRNA design algorithms by incorporating multiple feature types. As demonstrated in the development of highly active next-generation CRISPRi libraries, "nucleosomes directly block access of CRISPR/Cas9 to DNA" [63], highlighting the importance of chromatin accessibility in sgRNA efficacy.
A comprehensive machine learning approach that integrated nucleosome positioning, sequence features, and refined sgRNA design rules resulted in libraries where "the large majority of sgRNAs are highly active" [63]. This integrated model strongly weighted both positional features relative to the transcription start site (TSS) and sequence characteristics, with the nucleosome-deprived region immediately downstream of the TSS yielding the strongest predicted activity for CRISPRi applications. These design principles are crucial for researchers developing custom CRISPRi libraries for multi-modal studies.
Specialized tools like CRISPy-web 3.0 provide "a unified platform for multi-modal guide RNA design for CRISPR and TnpB genome editing applications" [64]. This platform extends beyond classical Cas9 systems to support diverse editing modalities including CRISPRi, enabling researchers to "toggle between multiple editing modes, select target regions such as ORFs or 5' UTRs, and visualize strand orientation, off-targets, and predicted mutation outcomes" [64].
Multi-Modal Profiling Technologies
Recent technological advances have dramatically expanded the possibilities for multi-modal profiling in CRISPRi studies. Various experimental methods now enable joint profiling of multiple molecular modalities from the same single cells [61]:
- CITE-seq enables simultaneous measurement of mRNA and over 100 surface proteins from the same single cells [61]
- ECCITE-seq extends this capability to include mRNA, V(D)J regions, perturbation sgRNA, and surface protein profiling [61]
- SHARE-seq jointly profiles mRNA and chromatin accessibility from over 10,000 cells or nuclei [61]
- scNOMeRe-seq enables measurement of single-cell nucleosome occupancy, DNA methylome, and RNA expression [61]
These technologies provide the experimental foundation for comprehensive multi-modal CRISPRi studies by enabling researchers to capture complementary data types from the same biological samples.
Computational Methodologies and Workflow Specification
Pipeline Architecture Specifications
The nf-core/crisprseq pipeline implements a comprehensive workflow for CRISPR data analysis, with specific modules for different screening modalities [62]. For targeted editing analysis, the pipeline includes:
- Read Processing: Merge paired-end reads (Pear), quality control (FastQC), adapter trimming (Cutadapt), and quality filtering (Seqtk)
- UMI Processing: UMI extraction (Python script), UMI clustering (Vsearch), consensus sequence generation (minimap2, racon, Medaka)
- Variant Calling: Read mapping followed by CIGAR parsing for edit calling (R)
For CRISPR screening data analysis, the workflow includes:
- Read QC (FastQC)
- Read mapping and quantification (MAGeCK count)
- Optional CNV correction and normalization (CRISPRcleanR)
- Statistical analysis to rank sgRNAs and genes
- Hit selection with controlled false discovery rates
The pipeline is built using Nextflow, ensuring portability across different compute infrastructures, and uses Docker/Singularity containers for reproducibility [62].
Multi-Modal Data Integration Techniques
The computational integration of multi-modal data requires specialized approaches that can handle the distinct statistical characteristics of different data types. Multi-omics data integration methods have rapidly developed to address this challenge [61]. These include:
- Matrix factorization approaches that identify shared latent factors across modalities
- Deep learning architectures that learn joint representations across omics layers
- Graph-based methods that model interactions between different molecular entities
Flexynesis implements a flexible deep learning approach that can handle "a mixture of such tasks" including regression, classification, and survival analysis [11]. The platform enables both single-task and multi-task modeling, where "more than one MLPs are attached on top of the sample encoding networks, thus the embedding space can be shaped by multiple clinically relevant variables" [11]. This approach is particularly valuable for CRISPRi studies aiming to predict multiple phenotypic outcomes from multi-modal molecular data.
Implementation Framework and Research Reagents
Essential Research Reagent Solutions
Table 2: Key Research Reagents for Multi-Modal CRISPRi Studies
| Reagent / Tool | Function | Application Notes | Source |
|---|---|---|---|
| CRISPRi v2 Libraries [63] | Optimized sgRNA collections for transcriptional repression | Designed using integrated algorithm incorporating chromatin, position, and sequence features | Addgene |
| dCas9-KRAB Fusion Protein | Engineered CRISPR effector for transcriptional repression | Core component of CRISPRi system; can be cell line-engineered or delivered via lentivirus | Multiple commercial sources |
| Multiplexed Antibody Panels (CITE-seq) | Surface protein quantification alongside transcriptomics | Enables paired transcriptome and proteome analysis in single cells | BioLegend, TotalSeq |
| Chromatin Accessibility Reagents (ATAC-seq) | Profiling open chromatin regions | Can be combined with transcriptomics in multi-ome protocols | Commercial kits available |
| DNA Barcode Libraries (CRISPRmap) | In situ perturbation identification | Enables spatial mapping of CRISPR perturbations with phenotypic readouts | Custom design required [60] |
| Single-Cell Multi-ome Kits | Simultaneous profiling of transcriptome and epigenome | Commercial solutions for coordinated multi-modal profiling | 10x Genomics, Parse Biosciences |
Experimental Protocol for Multi-Modal CRISPRi Screening
A comprehensive protocol for multi-modal CRISPRi screening integrates the following key steps, adapted from established methodologies [60] [63]:
Stage 1: Library Design and Validation
- Select target genes based on research objectives and prior knowledge
- Design sgRNAs using CRISPy-web 3.0 or similar platform, focusing on regions within 50-100bp downstream of FANTOM-annotated TSSs [64] [63]
- Apply filtering for off-target effects using updated genomic annotations
- Synthesize library as pooled oligonucleotide pool with appropriate barcoding strategies
Stage 2: Cell Engineering and Screening
- Lentivirally deliver CRISPRi library at low MOI (≤0.3) to ensure single integration events
- Select successfully transduced cells with appropriate antibiotics (e.g., puromycin)
- Validate knockdown efficiency for a subset of targets via qRT-PCR
- Apply experimental conditions (e.g., drug treatments, differentiation protocols)
- Harvest cells for multi-modal profiling, preserving viability for single-cell analyses
Stage 3: Multi-Modal Profiling (following CRISPRmap methodology [60])
- For spatial phenotyping: Fix cells and perform combinatorial barcode detection via hybridization chain reaction
- For transcriptomics: Perform single-cell RNA sequencing using appropriate platform (10x Genomics, Parse, etc.)
- For proteomics: Incubate with antibody-conjugated markers for CITE-seq
- For chromatin accessibility: Process samples for scATAC-seq using commercial kits
- Image samples using high-content microscopy systems for morphological analyses
The experimental workflow for multi-modal profiling, based on the CRISPRmap approach [60], can be visualized as follows:
Validation and Quality Control Framework
Rigorous quality control is essential throughout multi-modal CRISPRi experiments to ensure data quality and interpretability. The nf-core/crisprseq pipeline incorporates multiple QC checkpoints including [62]:
- Sequencing Quality: FastQC reports for read quality, adapter contamination, and complexity metrics
- Editing Efficiency: Assessment of successful editing rates across the library
- Screen Quality: Assessment of replicate correlation and negative control distributions
For optical CRISPR screens using CRISPRmap, quality control includes [60]:
- Barcode Decoding Precision: Monitoring of allowed versus unallowed barcode combinations (≥98% precision expected)
- Cell Segmentation Accuracy: Visual inspection of nuclear and cellular segmentation
- Registration Accuracy: Alignment of images across multiple imaging cycles
Multi-modal data integration requires additional validation to ensure biological consistency across modalities. This includes:
- Correlation analysis between transcriptomic and proteomic measurements for the same targets
- Chromatin accessibility confirmation at regulatory elements of differentially expressed genes
- Morphological correlation with molecular phenotypes where expected
The field of multi-modal CRISPRi analytics is rapidly evolving, with several emerging trends likely to shape future methodological developments. Artificial intelligence approaches are playing an increasingly important role, as demonstrated by the successful application of large language models to design novel CRISPR effectors "with comparable or improved activity and specificity relative to SpCas9" [65]. These AI-designed editors, along with the continued expansion of single-cell multi-ome technologies, will further enhance our ability to probe gene function across multiple molecular layers.
The integration of spatial information represents another frontier, with methods like CRISPRmap enabling "in situ barcode readout in cell types and contexts that were elusive to conventional optical pooled screening" [60]. As these technologies mature, they will increasingly enable researchers to contextualize CRISPRi responses within tissue architecture and cellular communities - essential for understanding gene function in physiological contexts.
For researchers embarking on multi-modal CRISPRi studies, the selection and development of purpose-built analytical pipelines requires careful consideration of experimental goals, data types, and computational resources. By leveraging the frameworks and methodologies outlined in this guide, researchers can implement robust, reproducible analytical strategies to extract maximum biological insight from complex multi-modal datasets, ultimately advancing both basic science and drug discovery efforts.
The integration of multi-omics data represents a transformative approach in biological research, particularly for elucidating complex cellular responses to perturbations such as CRISPR interference (CRISPRi). The simultaneous analysis of genomics, transcriptomics, proteomics, and epigenomics provides unprecedented opportunities for understanding hierarchical gene regulatory networks and their functional outcomes [66]. However, this comprehensive approach generates datasets of extraordinary volume and complexity, creating substantial computational bottlenecks that can impede research progress.
The scalability challenge manifests in two primary dimensions: storage infrastructure and computational capacity. Next-generation sequencing technologies now generate terabytes of data per instrument run, while multi-omic studies incorporating single-cell resolution and temporal profiling can easily reach petabyte-scale [67] [68]. This data explosion is particularly acute in CRISPRi functional genomics screens, which combine gene perturbation data with multiple molecular readouts across thousands of experimental conditions [17]. Without specialized computational strategies, the storage, processing, and integration of these massive datasets becomes computationally prohibitive, limiting the scope and translational potential of multi-omics research.
High-performance computing (HPC) infrastructure has emerged as an essential solution to these challenges, providing the specialized architecture needed to handle data- and compute-intensive problems that conventional desktops cannot process [67]. The parallelized, high-throughput computational environment offered by HPC systems enables researchers to apply sophisticated artificial intelligence (AI) and machine learning (ML) approaches to multi-omics data at biologically meaningful scales, opening new avenues for discovery in precision medicine and functional genomics [69].
The Data Deluge: Quantifying Multi-Omics Data Generation
The scalability challenge begins at the data generation stage, where technological advancements across multiple omics layers are producing data at an unprecedented rate and scale. Understanding the magnitude of this data generation is crucial for designing appropriate computational infrastructure.
Data Volume by Technology and Application
Table 1: Data Generation Scales Across Omics Technologies
| Technology Type | Data Per Sample | Typical Study Scale | Total Data Volume |
|---|---|---|---|
| Whole Genome Sequencing (WGS) | 100-200 GB | 1,000-100,000 samples [70] | 100 TB - 20 PB |
| Single-Cell RNA-seq | 50-100 GB | 10,000-1,000,000 cells | 5 TB - 100 TB |
| Proteomics (Mass Spectrometry) | 10-50 GB | 100-10,000 samples | 1 TB - 500 TB |
| Spatial Transcriptomics | 100-500 GB | 10-1,000 samples | 1 TB - 500 TB |
| Epigenomics (ATAC-seq, ChIP-seq) | 20-100 GB | 100-10,000 samples | 2 TB - 1 PB |
The integration of these technologies in multi-omics studies creates a multiplicative effect on data requirements. For example, a comprehensive CRISPRi multi-omics study investigating pluripotency networks—similar to the approach described in the Communications Biology study—might incorporate genome-scale CRISPR screens alongside transcriptomic, proteomic, and epigenomic profiling [17]. Such a study could easily generate 500-1000 TB of raw data before any processing or integration occurs.
Data Characteristics Intensifying Storage Challenges
Beyond sheer volume, several data characteristics specific to multi-omics research intensify storage challenges:
- Heterogeneity: Multi-omics datasets comprise wildly diverse data types, each with unique formats, scales, and biases [68]. Genomic variant call format (VCF) files, transcriptomic count matrices, proteomic mass spectrometry raw files, and epigenomic alignment files all require different storage strategies and processing approaches.
- Multi-dimensionality: Single-cell technologies add dimensional complexity by measuring multiple parameters across thousands to millions of individual cells. The DNBelab C-YellowR 16, for example, enables parallel processing of 16 single-cell samples, dramatically increasing data complexity while reducing hands-on time [70].
- Temporal resolution: Time-series experiments capturing dynamic molecular responses to CRISPRi perturbations generate sequential data snapshots that multiply storage requirements while creating complex data dependency structures.
- Metadata complexity: Comprehensive experimental metadata—including perturbation parameters, sample processing details, and analytical provenance—must be stored and linked with primary data, adding substantial overhead.
The storage infrastructure for multi-omics must therefore address both scale and complexity, providing solutions for diverse data types while maintaining accessibility for computational processing.
High-Performance Computing Architectures for Multi-Omics Data
High-performance computing provides specialized infrastructure to address the computational demands of large-scale multi-omics studies. HPC systems combine parallel processing capabilities, high-speed interconnects, and specialized hardware to reduce processing time from weeks to hours for complex analytical workflows [67].
Core HPC Components for Multi-Omics Research
Table 2: HPC System Components and Their Functions in Multi-Omics Analysis
| HPC Component | Technical Specification | Function in Multi-Omics |
|---|---|---|
| Compute Nodes | CPUs with high core counts (64-128 cores) | Coordinate tasks, preprocessing, serial workloads |
| GPU Accelerators | NVIDIA A100, H100; 80GB VRAM | Data-parallel workloads, neural-network training |
| High-Speed Interconnects | InfiniBand HDR (200 Gb/s) | Minimize latency for tightly coupled simulations |
| Parallel File Systems | Lustre, Spectrum Scale; 100 GB/s+ I/O | High throughput for large genomic files |
| Hierarchical Storage | Flash (TB), disk (PB), tape (archive) | Cost-effective data lifecycle management |
| Job Schedulers | Slurm, PBS Pro | Workload distribution across cluster |
These components create an integrated system where computational capability is matched to data intensity. For example, GPU-accelerated nodes can perform inference on deep learning models for variant calling or pattern recognition across omics layers, while high-speed interconnects enable efficient communication between nodes during distributed genome assembly or network analysis [67].
HPC Deployment Models for Multi-Omics Research
Different deployment options offer flexibility for institutions with varying resources and requirements:
- On-premises clusters: Dedicated institutional resources providing maximum control over data security and customization, ideal for sensitive genomic data or specialized analytical pipelines.
- Cloud HPC: Services like Amazon Web Services (AWS) and Google Cloud Genomics offer scalable, pay-as-you-go access to HPC resources, particularly valuable for projects with fluctuating computational demands [71] [68].
- Hybrid approaches: Combining on-premises resources for sensitive data with cloud bursts for peak workloads, as exemplified by platforms like Anvil, which provides a user-friendly HPC interface for biomedical researchers [72].
The UC Irvine BigCARE training program leverages Anvil to introduce researchers to HPC-based analysis of diverse omics datasets, demonstrating how accessible interfaces can lower barriers to high-performance computing in life sciences [72].
Computational Strategies for Multi-Omics Data Integration
Effective integration of multi-omics data requires specialized computational strategies that address both the scale and heterogeneity of the data. These approaches can be categorized by when integration occurs in the analytical workflow.
Integration Approaches and Their Computational Characteristics
Table 3: Computational Strategies for Multi-Omics Data Integration
| Integration Strategy | Timing | Key Algorithms | Computational Demand | Best Suited Applications |
|---|---|---|---|---|
| Early Integration | Before analysis | Simple concatenation | High (curse of dimensionality) | Capturing all cross-omics interactions |
| Intermediate Integration | During analytical transformation | Network fusion, Matrix factorization | Medium | Incorporating biological context |
| Late Integration | After individual analysis | Ensemble methods, Stacking | Low to medium | Projects with missing data types |
| Deep Learning Approaches | Flexible | VAEs, GCNs, Transformers | Very high | Large-scale nonlinear pattern detection |
AI and Machine Learning for Scalable Multi-Omics Integration
Artificial intelligence approaches have become indispensable for large-scale multi-omics integration, with different model architectures offering distinct advantages:
- Variational Autoencoders (VAEs): These deep generative models learn compressed representations of high-dimensional omics data in a lower-dimensional latent space, enabling efficient integration while preserving biological patterns [46] [68]. VAEs have proven particularly valuable for tasks such as data imputation, denoising, and creating joint embeddings of multi-omics data.
- Graph Convolutional Networks (GCNs): These network-based approaches represent biological entities (genes, proteins) as nodes and their interactions as edges, allowing integration of multi-omics data onto biological networks [68]. GCNs aggregate information from a node's neighbors to make predictions, proving effective for clinical outcome prediction.
- Transformers: Originally developed for natural language processing, transformer architectures adapt effectively to biological sequences and omics profiles through self-attention mechanisms that weigh the importance of different features and data types [68].
- Similarity Network Fusion (SNF): This method constructs patient-similarity networks from each omics layer and iteratively fuses them into a single comprehensive network, enabling more accurate disease subtyping [68].
These AI methods typically require distributed GPU clusters and optimized software libraries to achieve practical runtime for large datasets. The training of these models is computationally intensive, but enables analytical capabilities far beyond traditional statistical approaches [69].
Experimental Framework: Multi-Omics Analysis of CRISPRi Responses
The following section outlines a comprehensive experimental framework for applying scalable computational approaches to multi-omics analysis of CRISPRi responses, based on methodologies demonstrated in recent literature.
Research Reagent Solutions for CRISPRi Multi-Omics Studies
Table 4: Essential Research Reagents and Platforms for CRISPRi Multi-Omics
| Reagent/Platform | Function | Application in CRISPRi Multi-Omics |
|---|---|---|
| CRISPRi Library | Targeted gene repression | Introduction of specific perturbations |
| DNBelab C-YellowR 16 | Automated single-cell library prep | Parallel processing of 16 single-cell samples [70] |
| DNBSEQ-T1+ Sequencer | Mid-throughput sequencing | Flexible sequencing for multi-omics profiling [70] |
| Stereo-seq Technology | Spatial transcriptomics | Mapping gene expression in tissue context |
| Full-length Transcriptome Kit | RNA library preparation | Sensitive profiling from limited input (10 cells) [70] |
Computational Workflow for CRISPRi Multi-Omics Integration
The analytical workflow for CRISPRi multi-omics studies involves sequential stages of data processing, quality control, and integration, with scalability considerations at each step.
Workflow for CRISPRi multi-omics data analysis, showing parallel integration strategies.
Detailed Methodologies for Key Analytical Steps
CRISPRi Functional Genomics Screen Analysis
The foundational step in understanding CRISPRi responses involves comprehensive functional screening, as demonstrated in the pluripotency regulatory network study [17]. The methodology includes:
- Library Design and Coverage: Utilize a genome-scale CRISPR library (e.g., Brie library targeting 19,674 genes) with high guide RNA coverage (mean of 166 reads per sgRNA recommended).
- Infection and Selection: Infect Cas9-expressing cells with lentiviral library at appropriate MOI to ensure single-copy integration, then culture under experimental conditions for 14 days.
- Sequencing and Quantification: Sequence plasmid library and cell samples at day 0 and day 14 post-infection using high-throughput platforms like DNBSEQ-T7 [70].
- Essential Gene Identification: Apply analytical tools such as MAGeCK to identify genes under negative selection based on sgRNA depletion, comparing to negative controls (low-expression and non-essential genes) [17].
Multi-Omics Data Integration Protocol
For integration of CRISPR screening data with other omics layers, a structured protocol ensures reproducible and scalable analysis:
- Data Harmonization: Normalize each omics dataset using technology-specific approaches (TPM/FPKM for RNA-seq, intensity normalization for proteomics) to enable cross-dataset comparison [68].
- Batch Effect Correction: Apply statistical methods like ComBat to remove technical variation from different processing batches while preserving biological signals [68].
- Missing Data Imputation: Implement robust imputation methods (k-NN or matrix factorization) to handle missing omics measurements that commonly occur in multi-modal studies.
- Multi-view Clustering: Apply integrative non-negative matrix factorization (intNMF) or similar approaches to identify molecular subtypes that span multiple omics layers [46].
- Network Integration: Map multiple omics datasets onto shared biochemical networks to improve mechanistic understanding of CRISPRi effects, connecting analytes based on known interactions [15].
Implementation Guide: Storage and Computing Infrastructure
Deploying appropriate infrastructure requires careful planning across both storage and computational dimensions. The following recommendations provide guidance for establishing scalable solutions.
Tiered Storage Architecture for Multi-Omics Data
A tiered storage approach balances performance requirements with cost considerations across the data lifecycle:
Tiered storage architecture for managing multi-omics data through its lifecycle.
Implementation Specifications:
- Tier 1 (Active Processing): Deploy all-flash storage arrays with parallel file systems (Lustre, Spectrum Scale) capable of 100+ GB/s I/O throughput for active computational workloads.
- Tier 2 (Active Archive): Implement large-scale disk arrays (10+ PB) with automated tiering policies for datasets requiring occasional access but not active computation.
- Tier 3 (Long-term Archive): Utilize tape libraries or cloud cold storage services for data preservation, with appropriate indexing and retrieval systems.
Computational Resource Provisioning
Based on the analysis of multi-omics workflows and their computational demands, the following provisioning guidelines ensure adequate capacity:
- CPU Cluster: Minimum of 1000 compute cores with high-clock-speed processors for alignment, variant calling, and standard bioinformatics workflows.
- GPU Resources: 8-16 high-end GPUs (NVIDIA A100 or equivalent) with 80GB VRAM for deep learning approaches to multi-omics integration.
- Memory Configuration: Nodes with 1-2TB RAM for large-scale genome assembly and population-level analyses; standard nodes with 256-512GB for routine analyses.
- Job Scheduler: Implement Slurm or similar workload manager with container support (Singularity/Apptainer) for reproducible pipeline execution.
The partnership between UC Irvine and Purdue's Rosen Center for Advanced Computing exemplifies this approach, providing researchers with access to Anvil, an HPC platform featuring a user-friendly interface that lowers barriers to big data analysis [72].
The scalability challenge in multi-omics research represents both a formidable obstacle and a transformative opportunity. As CRISPRi studies increasingly incorporate multiple molecular layers to comprehensively map gene regulatory networks, the computational infrastructure supporting this research must evolve in parallel. The integration of high-performance computing architectures, AI-driven analytical methods, and scalable storage solutions creates a foundation for discoveries that were previously computationally infeasible.
The field is rapidly advancing toward even more data-intensive approaches, with emerging technologies like spatial multi-omics and single-cell proteomics further expanding data dimensions. Success in this environment will require continued innovation in computational methods, particularly in federated learning approaches that enable analysis across distributed datasets while preserving privacy [68]. Additionally, the development of more purpose-built analysis tools specifically designed for multi-omics data will be essential for maximizing the scientific return from these complex datasets [15].
By implementing the storage and computing strategies outlined in this technical guide, research institutions can position themselves to not only manage the current multi-omics data deluge but also leverage these rich datasets for transformative insights into gene function and regulatory networks. The sophisticated integration of computational and biological approaches will ultimately accelerate the translation of CRISPRi research into clinical applications and therapeutic innovations.
Optimizing sgRNA Design and Delivery for Specific Cell Types and In Vivo Models
The efficacy of CRISPR-based functional genomics, particularly within complex experimental systems such as in vivo models and primary human organoids, hinges on two interdependent pillars: the rational design of single-guide RNAs (sgRNAs) and the selection of a delivery strategy that is precisely tailored to the target cell type. An optimized sgRNA ensures high on-target activity while minimizing off-target effects, but its potential is only realized if it can be efficiently delivered to the nucleus of the cell of interest. As research moves beyond transformed cell lines to more physiologically relevant but challenging models, the integration of omics data—from genomics to transcriptomics—is becoming critical for informing both sgRNA design and delivery choices. This guide synthesizes current methodologies and best practices for navigating this complex landscape, providing a technical foundation for robust and reproducible CRISPR screening.
Optimizing sgRNA Design
The selection of a highly efficient and specific sgRNA is the foundational step of any CRISPR experiment. This process has been greatly enhanced by computational tools and empirical validation protocols.
Algorithm Selection and In Silico Design
Several algorithms exist to predict sgRNA efficiency. A recent systematic evaluation compared three widely used scoring algorithms in an optimized doxycycline-inducible Cas9 human pluripotent stem cell (hPSC) system. The study found that Benchling provided the most accurate predictions for sgRNA cleavage activity compared to other tested algorithms [73]. This highlights the importance of selecting a well-validated in-silico tool for the initial design phase.
Beyond efficiency, predicting off-target risk is paramount. Tools like CCTop can be used to search for potential off-target sites across the genome, allowing researchers to prioritize sgRNAs with unique target sequences [73].
Chemical Modifications for Enhanced Stability
The intrinsic stability of sgRNA within cells can be significantly improved through chemical synthesis. Using chemical synthesized and modified (CSM) sgRNA that incorporates 2’-O-methyl-3'-thiophosphonoacetate at both the 5’ and 3’ ends enhances sgRNA stability, leading to more consistent and potent editing outcomes [73].
Experimental Validation of sgRNA Efficacy
A critical, often overlooked step is the experimental confirmation that a high INDEL frequency actually results in a loss of protein function. Some sgRNAs can induce high INDEL rates but fail to eliminate the target protein—these are termed "ineffective sgRNAs" [73].
Key Validation Workflow:
- Measure INDEL Efficiency: After editing, use Sanger sequencing of the target locus and analyze chromatograms with tools like ICE (Inference of CRISPR Edits) or TIDE (Tracking of Indels by Decomposition) to quantify the percentage of insertions and deletions [73].
- Confirm Protein Knockout: Integrate Western blotting to verify the loss of target protein expression. For example, a study identified an sgRNA targeting exon 2 of ACE2 that produced 80% INDELs but retained ACE2 protein expression, underscoring the necessity of this confirmatory step [73].
Table 1: Key Algorithms and Reagents for sgRNA Design and Validation
| Tool/Reagent | Type | Primary Function | Key Feature |
|---|---|---|---|
| Benchling | Software Algorithm | Predicts sgRNA on-target cleavage efficiency | Identified as providing the most accurate predictions in a comparative study [73] |
| CCTop | Software Algorithm | Identifies potential sgRNA off-target sites | Helps in selecting sgRNAs with minimal off-target risk [73] |
| ICE & TIDE | Analysis Algorithm | Quantifies INDEL efficiency from Sanger sequencing data | Provides a quantitative measure of editing efficiency without cloning [73] |
| CSM-sgRNA | Research Reagent | Chemically synthesized guide RNA with enhanced stability | 2’-O-methyl-3'-thiophosphonoacetate modifications reduce degradation [73] |
Selecting and Optimizing Delivery Methods
The choice of delivery method is dictated by the target cell type, the cargo format, and the experimental context (in vitro, in vivo, or ex vivo).
Cargo Formats: DNA, mRNA, or RNP?
The form in which the CRISPR machinery is delivered has significant implications for editing efficiency, timing, and off-target effects.
- DNA (Plasmid): An early standard, but can lead to cytotoxicity, variable editing efficiency, and prolonged Cas9 expression that increases off-target risks [74].
- mRNA: Offers transient expression and reduced off-target effects compared to DNA plasmids. Delivery of Cas9 mRNA via lipid nanoparticles (LNPs) has been successfully used in vivo, for example, to edit the MYOC gene in a mouse glaucoma model [75].
- Ribonucleoprotein (RNP): The complex of Cas9 protein and sgRNA. RNP delivery is immediate and short-lived, leading to high precision and reduced off-target effects. It is highly suitable for ex vivo applications and is considered a gold standard for many knockout experiments [74].
Delivery Vehicles for In Vivo and Complex Models
Achieving efficient delivery in vivo or in primary 3D organoids remains a major challenge. The following table summarizes the primary viral delivery vectors.
Table 2: Comparison of Viral Delivery Vectors for CRISPR Components
| Vector | Packaging Capacity | Integration | Best Suited For | Key Advantages | Key Challenges |
|---|---|---|---|---|---|
| Adeno-Associated Virus (AAV) | ~4.7 kb [74] | No (Episomal) | In vivo delivery to non-dividing cells (e.g., CNS, muscle) [76] | Low immunogenicity; well-suited for in vivo use; broad tissue tropism [74] | Small payload size; transient expression in dividing cells [76] |
| Lentivirus (LV) | 8-10 kb [76] | Yes (Genomic) | In vitro screens; ex vivo cell engineering; in vivo targeting of hepatocytes [76] | Stable, long-term expression; large cargo capacity; can infect dividing & non-dividing cells [74] | Safety concerns due to genomic integration; lower efficiency for most extrahepatic in vivo targets [74] [76] |
| Adenovirus (AdV) | Up to 36 kb [74] | No (Episomal) | Models requiring large cargo or Cas9/sgRNA expression in vivo | Very large packaging capacity; high transduction efficiency [74] | Can induce strong immune responses [74] |
Innovative Strategies for Challenging Models:
- In Vivo Genome-Wide Screening: Delivering a genome-wide sgRNA library in vivo requires immense coverage. New approaches include:
- AAV-Transposon Hybrid Systems: Combining AAV delivery with a transposon system enables stable genomic integration of sgRNAs, overcoming the transient expression limitation of AAV in proliferating cells [76].
- Transgenic Effector Mice: Using mice that constitutively or inducibly express Cas9 (or other effectors like Cas12a, dCas9-KRAB, dCas9-VPR) simplifies delivery to only the sgRNA, making genome-wide screens in specific tissues more feasible [76] [30].
- Primary Human 3D Organoids: These models preserve tissue architecture but are difficult to edit. Success has been achieved by first generating stable Cas9-expressing organoid lines via lentiviral transduction, then introducing pooled sgRNA libraries. This approach has enabled knockout, interference (CRISPRi), and activation (CRISPRa) screens in gastric organoids to study gene-drug interactions [30].
- Non-Viral Delivery: Lipid Nanoparticles (LNPs) are a promising non-viral method, particularly for in vivo delivery of mRNA or RNP cargoes. They have minimal immunogenicity and are being engineered for organ-specific targeting (e.g., using SORT molecules) [75] [74].
Diagram 1: Decision workflow for selecting CRISPR cargo and delivery vehicles.
Integrated Experimental Protocols
This section outlines detailed protocols for setting up a CRISPR screen in a complex 3D organoid model and for validating sgRNA efficacy.
Protocol: CRISPR Knockout Screen in Human Gastric Organoids
This protocol is adapted from a study that successfully performed large-scale genetic screens in primary human 3D gastric organoids [30].
Research Reagent Solutions:
- Organoid Line: TP53/APC double knockout (DKO) human gastric organoids.
- Cas9 Source: Lentivirus for stable Cas9 expression.
- sgRNA Library: Pooled lentiviral library (e.g., 12,461 sgRNAs targeting membrane proteins).
- Culture Medium: PGM1 (Pluripotency Growth Master 1) Medium or equivalent on Matrigel-coated plates.
- Selection Agent: Puromycin.
Methodology:
- Generate Stable Cas9-Expressing Organoids:
- Lentivirally transduce TP53/APC DKO organoids with a construct containing a Cas9 expression cassette.
- Select transduced cells with an appropriate antibiotic (e.g., puromycin) and validate Cas9 activity using a GFP-reporter assay (>95% GFP loss indicates robust activity) [30].
Transduce with Pooled sgRNA Library:
- Transduce the Cas9-expressing organoids with the pooled lentiviral sgRNA library at a low Multiplicity of Infection (MOI) to ensure most cells receive only one sgRNA.
- Maintain a cellular coverage of >1000 cells per sgRNA throughout the transduction and screening process to ensure library representation [30].
- Harvest a subset of cells 2 days after puromycin selection as a reference baseline (T0).
Phenotypic Selection and Screening:
- Culture the remaining organoids under the condition of interest (e.g., normal growth or drug treatment like cisplatin) for a defined period (e.g., 28 days), maintaining the >1000x coverage.
- Harvest the final cell population (T1).
Genomic DNA Extraction and Sequencing:
- Extract genomic DNA from both T0 and T1 samples.
- Amplify the integrated sgRNA sequences by PCR and subject them to next-generation sequencing.
Data Analysis:
- Map sequenced reads to the sgRNA library to determine the abundance of each guide in T0 and T1 populations.
- Compare sgRNA abundance between T0 and T1. sgRNAs that are significantly depleted (drop-out genes) indicate a growth defect upon gene knockout, while enriched sgRNAs indicate a growth advantage [30].
Protocol: Validating sgRNA Editing Efficiency and Protein Knockout
This protocol is crucial for confirming the functional impact of selected sgRNAs before scaling up to a full screen [73].
Research Reagent Solutions:
- Cells: Target cell line (e.g., hPSCs with inducible Cas9).
- Nucleofection System: 4D-Nucleofector with appropriate buffer and program.
- sgRNA: Chemically synthesized and modified (CSM-sgRNA).
- Lysis Buffers: For genomic DNA and protein extraction.
- Antibodies: Target-specific for Western blot.
Methodology:
- Deliver sgRNA: Electroporate the CSM-sgRNA into the target cells using an optimized nucleofection protocol (e.g., program CA137 for hPSCs) [73].
- Harvest Genomic DNA: After 3-7 days, extract genomic DNA from a portion of the edited cell pool.
- Amplify and Sequence Target Locus: PCR-amplify the genomic region surrounding the sgRNA target site. Subject the PCR product to Sanger sequencing.
- Quantify INDEL Efficiency: Analyze the Sanger sequencing chromatograms using the ICE algorithm to calculate the percentage of INDELs in the cell population [73].
- Verify Protein Loss: In parallel, lyse another portion of the edited cells and perform Western blotting with an antibody against the target protein. Confirm the loss of protein signal despite a high INDEL percentage to rule out ineffective sgRNAs [73].
Integration with Omics Data
The future of optimized CRISPR screening lies in the deep integration of multi-omics data. Large language models (LLMs) and other AI-driven approaches are emerging as powerful tools to address the high dimensionality and noise inherent in omics datasets [77] [78]. These models can capture complex patterns to uncover disease mechanisms, identify therapeutic targets, and, critically, inform CRISPR experimental design.
The convergence of CRISPR with single-cell technologies (e.g., scRNA-seq) creates a powerful feedback loop. Single-cell CRISPR screens can profile perturbation effects at unprecedented resolution, generating vast datasets on gene regulatory networks [5]. This functional data can then be used to train predictive models that improve sgRNA design rules and anticipate cell-type-specific responses to genetic perturbations, thereby refining future screen design and interpretation within the context of a broader research thesis on omics-integrated CRISPR research [5] [78].
Diagram 2: Omics data integration creates a cycle for refining CRISPR screen design and biological interpretation.
Ensuring Rigor: Validating Findings and Comparative Analysis Across Biological Contexts
Benchmarking Against Established Datasets and Alternative Technologies
In the rapidly evolving field of functional genomics, robust benchmarking strategies are indispensable for validating new methodologies against established standards. For researchers investigating CRISPR interference (CRISPRi) responses through multi-omics integration, benchmarking provides the critical framework for assessing analytical performance, technological limitations, and biological relevance. The integration of omics data—genomics, transcriptomics, and proteomics—with CRISPR screening data presents unique computational and experimental challenges that necessitate systematic validation approaches [10] [79]. This technical guide outlines comprehensive strategies for benchmarking against established datasets and alternative technologies, specifically framed within omics data integration for understanding CRISPRi responses.
Benchmarking in this context serves multiple purposes: it validates the performance of genetic interaction scoring methods, assesses the efficiency and specificity of CRISPR systems compared to alternative gene-editing technologies, and evaluates the effectiveness of multi-omics integration pipelines. By establishing standardized benchmarking protocols, the research community can accelerate the identification of synthetic lethal interactions, enhance the reproducibility of CRISPR-based functional genomics studies, and ultimately advance the development of targeted therapies [80] [81].
Benchmarking Genetic Interaction Scoring Methods
Established Benchmarks for Synthetic Lethality
The identification of synthetic lethality (SL), where simultaneous disruption of two genes leads to cell death, has significant therapeutic implications, particularly in oncology. Pooled combinatorial CRISPR screens have become the predominant method for SL discovery, but varying analytical approaches necessitate rigorous benchmarking against established reference sets.
Two benchmark datasets have emerged as community standards for evaluating SL detection methods:
- De Kegel Benchmark: A curated set of known synthetic lethal pairs, particularly focused on paralog genes with validated interactions [80].
- Köferle Benchmark: Another comprehensive compilation of genetic interactions that provides a ground truth for method validation [80].
These benchmarks enable quantitative assessment of scoring methods using standardized metrics including Area Under the Receiver Operating Characteristic Curve (AUROC) and Area Under the Precision Recall Curve (AUPR) [80] [81].
Performance Comparison of Scoring Algorithms
Recent systematic evaluations of five prominent genetic interaction scoring methods across five different combinatorial CRISPR double knock-out (CDKO) datasets reveal important performance characteristics. The table below summarizes the key algorithms and their performance:
Table 1: Genetic Interaction Scoring Methods for Synthetic Lethality Detection
| Scoring Method | Key Algorithmic Approach | Performance Characteristics | Implementation |
|---|---|---|---|
| zdLFC | Z-transformed difference between expected and observed double mutant fitness (DMF) | Moderate performance; sensitive to data distribution | Python notebooks [80] |
| Gemini-Strong | Coordinate ascent variational inference (CAVI) comparing combination effect to individual effects | Identifies interactions with "high synergy" | R package [80] |
| Gemini-Sensitive | CAVI approach comparing total effect to most lethal individual effect | Captures "modest synergy"; consistently high performance across datasets | R package with comprehensive documentation [80] |
| Orthrus | Additive linear model comparing expected to observed LFC for each orientation | Good performance with flexible orientation handling | R package [80] |
| Parrish Score | Not fully detailed in available literature | Performs reasonably well across multiple screens | Custom implementation [80] |
No single method performs best across all screening datasets, which highlights the context-dependent nature of genetic interaction scoring. However, Gemini-Sensitive demonstrates consistently strong performance across most datasets and benchmarks, making it a recommended starting point for researchers new to this field [80]. The availability of its R package with comprehensive user documentation further enhances its practical utility.
Experimental Protocol for Method Benchmarking
To implement a rigorous benchmarking pipeline for genetic interaction scoring methods:
- Data Acquisition: Obtain CDKO datasets from published studies (e.g., Dede, CHyMErA, Ito, Parrish, or Thompson screens) [80].
- Benchmark Selection: Download the De Kegel and Köferle benchmark datasets of known synthetic lethal pairs.
- Method Application: Apply each scoring algorithm to the CDKO datasets using standardized parameters.
- Performance Quantification:
- Calculate AUROC values for each method-benchmark combination
- Compute AUPR scores to account for class imbalance in SL pairs
- Assess consistency across different screening platforms
- Result Interpretation: Identify optimal methods for specific experimental contexts and screen designs.
Table 2: Example Performance Metrics Across Screening Datasets
| Screening Dataset | Cell Lines | Number of Gene Pairs | Top Performing Methods | AUROC Range |
|---|---|---|---|---|
| Dede | A549, HT29, OVCAR8 | 400 | Gemini-Sensitive, Parrish | 0.72-0.89 [80] |
| CHyMErA | HAP1, RPE1 | 672 | Gemini-Sensitive, zdLFC | 0.68-0.85 [80] |
| Ito | Multiple cancer lines | 5065 | Gemini-Sensitive, Orthrus | 0.75-0.91 [80] |
| Parrish | PC9, HeLa | 1030 | Gemini-Strong, Parrish | 0.71-0.87 [80] |
| Thompson | MEWO, A375, RPE | 1191 | Gemini-Sensitive, Orthrus | 0.70-0.84 [80] |
Benchmarking Against Alternative Gene Editing Technologies
Comparative Analysis of Editing Platforms
While CRISPR-Cas systems have revolutionized functional genomics, benchmarking against established gene-editing technologies provides critical insights into relative strengths and limitations. The comparison is particularly relevant for CRISPRi studies where alternative technologies may offer complementary capabilities.
Table 3: Gene Editing Platforms Comparison
| Feature | CRISPR-Cas9 | CRISPR-Cas12 | TALENs | ZFNs |
|---|---|---|---|---|
| Targeting Mechanism | gRNA-DNA complementarity [82] | gRNA-DNA complementarity [83] | Protein-DNA recognition [82] | Protein-DNA recognition [82] |
| Ease of Design | Simple gRNA design [82] | Simple gRNA design [83] | Complex protein engineering [82] | Complex protein engineering [82] |
| Cost Efficiency | Low [82] | Low to moderate [83] | High [82] | High [82] |
| Scalability | High (ideal for high-throughput) [82] | High [83] | Limited [82] | Limited [82] |
| Precision | Moderate to high [82] | High (hfCas12Max variant) [83] | High [82] | High [82] |
| Multiplexing Capacity | High (multiple gRNAs) [82] | Moderate to high [83] | Low [82] | Low [82] |
| Primary Applications | Functional genomics, therapeutics [82] | Therapeutics, diagnostics [83] | Niche precision edits [82] | Niche precision edits [82] |
CRISPR Variants and Their Applications
Beyond standard Cas9, engineered CRISPR variants offer specialized functionalities that may be preferable for specific benchmarking contexts:
- eSpOT-ON (ePsCas9): Delivers high on-target precision with extremely low off-target editing, creating staggered-end cuts that minimize translocation risks [83].
- hfCas12Max: Sets a new standard in clinical gene editing with compact design, staggered-end cut outcomes, and broad PAM recognition [83].
- dCas9: Catalytically dead Cas9 enables gene regulation without DNA cleavage (CRISPRi) and serves as a platform for base editing and epigenome editing [83].
- Cas13: Targets RNA instead of DNA, expanding CRISPR applications to transcriptome editing and RNA-mediated diseases [83].
Experimental Protocol for Technology Comparison
To benchmark CRISPR systems against alternative technologies:
- Target Selection: Identify identical genomic targets for head-to-head comparison.
- Delivery Optimization: Standardize delivery methods (e.g., lipofection, electroporation) across platforms.
- Efficiency Assessment:
- Quantify editing rates using next-generation sequencing
- Compare knock-in and knock-out efficiencies
- Specificity Evaluation:
- Genome-wide off-target analysis using GUIDE-seq or CIRCLE-seq
- Compare specificity scores across platforms
- Functional Impact: Assess phenotypic concordance using relevant assays for gene function.
Multi-Omis Integration Strategies for CRISPR Data
Computational Frameworks for Data Integration
Integrating CRISPR screening data with multi-omics datasets requires specialized computational approaches that can handle the unique characteristics of each data modality. The integration strategies can be categorized based on the nature of the input data:
Table 4: Multi-Omics Integration Tools for CRISPR and Omics Data
| Tool Name | Integration Type | Methodology | Compatible Data Types | Reference |
|---|---|---|---|---|
| Seurat v4 | Matched | Weighted nearest-neighbour | mRNA, protein, accessible chromatin, spatial coordinates [10] | [10] |
| MOFA+ | Matched | Factor analysis | mRNA, DNA methylation, chromatin accessibility [10] | [10] |
| totalVI | Matched | Deep generative | mRNA, protein [10] | [10] |
| GLUE | Unmatched | Graph variational autoencoders | Chromatin accessibility, DNA methylation, mRNA [10] | [10] |
| LIGER | Unmatched | Integrative non-negative matrix factorization | mRNA, DNA methylation [10] | [10] |
| CellOracle | Matched | Gene regulatory network modeling | mRNA, CRISPR screening, chromatin accessibility [10] | [10] |
Workflow for Multi-Omics and CRISPR Data Integration
The integration of multi-omics data with CRISPR screening results follows a structured workflow that enables comprehensive biological insights. The following diagram illustrates the key steps in this process:
Diagram 1: Multi-omics and CRISPR Data Integration Workflow. This workflow outlines the process for integrating diverse omics datasets with CRISPR screening data to derive biological insights.
Types of Multi-Omics Integration
The computational strategy for integration depends largely on whether the multi-omics data originates from the same or different cells:
- Matched (Vertical) Integration: Merges data from different omics modalities within the same set of samples or cells. The cell itself serves as the anchor, making this approach more straightforward [10].
- Unmatched (Diagonal) Integration: Combines different omics data from different cells, requiring more sophisticated computational methods to find commonality through co-embedded spaces or manifolds [10].
- Mosaic Integration: Employed when experimental designs have various combinations of omics that create sufficient overlap across samples, requiring specialized tools like COBOLT or MultiVI [10].
Experimental Protocols and Controls
Essential Controls for CRISPR Experiments
Proper experimental controls are fundamental for generating reliable, interpretable data in CRISPR studies. The table below outlines critical control types and their applications:
Table 5: Essential Controls for CRISPR Experiments
| Control Type | Components | Purpose | Interpretation |
|---|---|---|---|
| Transfection Control | Fluorescence reporter (e.g., GFP mRNA) | Assess delivery efficiency of CRISPR components | Low fluorescence indicates poor delivery efficiency [84] |
| Positive Editing Control | Validated gRNA (e.g., targeting TRAC, RELA) + Cas nuclease | Verify optimized editing conditions under workflow parameters | High editing efficiency confirms properly optimized system [84] |
| Negative Editing Control (Scramble) | Scramble gRNA (no genomic target) + Cas nuclease | Establish baseline for non-specific effects | Phenotype indicates off-target effects or transfection stress [84] |
| Guide RNA Only | Target-specific gRNA without Cas nuclease | Control for gRNA-specific effects without editing | Phenotype suggests gRNA-mediated effects independent of editing [84] |
| Cas Nuclease Only | Cas nuclease without gRNA | Control for Cas protein effects | Phenotype indicates Cas protein toxicity or non-specific effects [84] |
| Mock Control | Transfection reagents only (no CRISPR components) | Assess cellular response to transfection stress | Phenotype reveals transfection-induced artifacts [84] |
Protocol for Control Implementation
Implementing a comprehensive control strategy requires systematic planning throughout the experimental workflow:
Experimental Design Phase:
- Include all relevant controls in experimental layout
- Determine appropriate replication for each control condition
- Plan for resource allocation (cells, reagents) for control samples
Transfection Optimization:
- Use transfection controls to validate delivery efficiency
- Titrate CRISPR components to determine optimal concentrations
- Test multiple delivery methods if efficiency is suboptimal
Editing Validation:
- Employ positive editing controls to confirm system functionality
- Use ICE (Inference of CRISPR Edits) analysis or next-generation sequencing to quantify editing efficiency [84]
- Compare results to negative controls to establish significance thresholds
Phenotypic Analysis:
- Include all controls in downstream assays and readouts
- Normalize experimental results to appropriate controls
- Use statistical methods to account for control-based variance
Research Reagent Solutions
Successful benchmarking studies require access to reliable, high-quality reagents. The following table outlines essential research tools for CRISPR-omics investigations:
Table 6: Essential Research Reagents for CRISPR-Omics Studies
| Reagent Category | Specific Examples | Key Function | Considerations |
|---|---|---|---|
| CRISPR Nucleases | hfCas12Max, eSpOT-ON, SaCas9, dCas9 | Target DNA (or RNA) cleavage or binding | Size constraints, PAM requirements, specificity [83] |
| Delivery Systems | Lipid Nanoparticles (LNPs), AAVs, Lentiviruses, Electroporation | Deliver CRISPR components to cells | Packaging capacity, cell type specificity, efficiency [85] |
| Control Reagents | Validated gRNAs (TRAC, RELA), Scramble gRNAs, Fluorescence reporters | Experimental validation and standardization | Species compatibility, cell line validation [84] |
| Omics Profiling | RNA-seq kits, ATAC-seq kits, Mass spectrometry panels, Antibody panels | Molecular profiling of CRISPR perturbations | Sensitivity, multiplexing capacity, cost [10] |
| Bioinformatics Tools | Gemini, Orthrus, Seurat, MOFA+, GLUE | Data analysis and multi-omics integration | Computational requirements, usability, documentation [80] [10] |
Benchmarking against established datasets and alternative technologies provides the foundation for rigorous, reproducible research in CRISPR-omics. The rapid advancement of CRISPR technologies, coupled with increasingly sophisticated multi-omics integration methods, demands continuous evaluation and validation against community standards. By implementing the benchmarking strategies outlined in this technical guide—including standardized genetic interaction scoring, comparative analysis of editing platforms, systematic control implementation, and robust data integration—researchers can enhance the reliability and impact of their investigations into CRISPRi responses.
As the field evolves, emerging technologies such as base editing, prime editing, and CRISPR-based epigenome editing will introduce new benchmarking challenges and opportunities. The framework presented here establishes a methodological approach for evaluating these future technologies within the context of multi-omics data integration, ultimately accelerating the translation of CRISPR discoveries into therapeutic applications.
Comparative CRISPRi Screens Across Cell Types and Differentiation States
Clustered Regularly Interspaced Short Palindromic Repeats Interference (CRISPRi) has emerged as a powerful platform for functional genomics, enabling researchers to systematically probe gene function across diverse cellular contexts. The development of inducible CRISPRi systems has been particularly transformative for studying essential biological processes in sensitive model systems, including human induced pluripotent stem cells (hiPS cells) and their differentiated derivatives [3]. Comparative CRISPRi screening represents a methodological advance that moves beyond single-cell-type analysis to reveal how genetic dependencies shift during cellular differentiation and lineage specification.
The fundamental principle underlying comparative CRISPRi screens is the systematic perturbation of gene expression across multiple related but distinct cell states, followed by quantitative assessment of how these perturbations affect cellular fitness and function. This approach has revealed that core components of essential biological pathways often remain indispensable across cell types, while regulatory elements and quality control factors frequently exhibit cell-state-specific essentiality [21]. These differential genetic dependencies reflect the unique proteomic and functional demands of specialized cell types, providing insight into how fundamental biological processes are rewired during development and disease.
When framed within the broader context of omics data integration, comparative CRISPRi screens generate functional genomic datasets that can be correlated with transcriptional, epigenetic, and proteomic profiles to build comprehensive models of cellular regulation. The integration of these multi-modal datasets is essential for understanding how genetic perturbations propagate through molecular networks to produce phenotypic outcomes [17] [46].
Key Experimental Models and Systems
Pluripotent Stem Cells and Differentiation Systems
Human induced pluripotent stem cells (hiPS cells) serve as a foundational model for comparative CRISPRi studies due to their capacity for self-renewal and differentiation into virtually any cell type. The inducible CRISPRi system integrated at the AAVS1 safe harbor locus has been successfully implemented in hiPS cells, enabling controlled and reversible gene repression without triggering p53-mediated toxicity, which historically hampered genetic screening in pluripotent stem cells [21] [3]. This technical advancement has opened the door to functional genomics in previously intractable cell types, including hiPS cell-derived neural progenitor cells (NPCs), neurons, and cardiomyocytes [21].
The differentiation capacity of hiPS cells enables researchers to model developmental processes and examine how genetic dependencies emerge during lineage specification. For example, a comparative screen examining genes involved in mRNA translation revealed that human stem cells critically depend on pathways that detect and rescue slow or stalled ribosomes, with particular reliance on the E3 ligase ZNF598 for resolving ribosome collisions at translation start sites [21]. These dependencies were not uniformly essential across all cell types, highlighting the value of comparative approaches.
Specialized Somatic Cell Models
Beyond stem cell systems, comparative CRISPRi screens have been implemented in specialized somatic cells to investigate tissue-specific functions. The HL-60 human neutrophil-like cell line has been particularly valuable for studying immune cell biology, enabling genome-wide assessment of molecular factors critical to proliferation, differentiation, and cell migration [86]. These screens have identified distinct genetic requirements for directed migration (chemotaxis), undirected migration (chemokinesis), and 3D amoeboid migration through extracellular matrix [86].
The immortalized HL-60 cell line can be differentiated into neutrophil-like cells (dHL-60) using all-trans retinoic acid (ATRA) or dimethylsufoxide (DMSO), providing a tractable system for comparing genetic dependencies between proliferative precursor cells and their terminally differentiated counterparts [86]. This model has revealed how mTORC1 signaling influences neutrophil abundance, survival, and migratory behavior, demonstrating how core signaling pathways are repurposed across cellular states [86].
Table: Representative Experimental Models for Comparative CRISPRi Screens
| Cell System | Key Features | Differentiated Cell Types | Applications |
|---|---|---|---|
| hiPS Cells | Self-renewal, multilineage differentiation potential, AAVS1 safe harbor integration | Neural progenitor cells, neurons, cardiomyocytes | Developmental biology, disease modeling, mRNA translation studies [21] |
| HL-60 Cells | Myeloid progenitor line, differentiation into neutrophil-like cells | dHL-60 neutrophil-like cells | Immune cell function, migration studies, chemotaxis [86] |
| HEK293 Cells | Rapid growth, high transfection efficiency, aberrant gene expression | Not typically differentiated | Comparison with normal cells, essential gene identification [21] |
Experimental Design and Methodologies
CRISPRi System Configuration and Validation
The core CRISPRi system employs a nuclease-deactivated Cas9 (dCas9) fused to a KRAB repression domain that enables programmable transcriptional repression without introducing DNA double-strand breaks [3]. For comparative studies, an inducible system regulated by doxycycline provides temporal control over dCas9-KRAB expression, allowing researchers to propagate cells without selection pressure before inducing gene repression [21] [3]. This is particularly important when working with slow-growing differentiated cells or when studying essential genes that would otherwise be depleted from the population.
System validation must include demonstration of efficient knockdown across all cell types included in the comparative study. Quantitative reverse transcription PCR (RT-qPCR) and immunoblot analysis should confirm target gene repression exceeding 70-80% in hiPS cells, differentiated progeny, and any comparator cell lines [21]. Additionally, single-guide RNA (sgRNA) validation is essential, with correlation between individual sgRNA effects and pooled screen results (Spearman's R = 0.51-0.85 reported in published studies) [21]. Protein-level validation using quantitative mass spectrometry can confirm that observed phenotypic differences are not simply due to differential protein stability or turnover rates across cell types [21].
Screening Workflow and Differentiation Protocols
A standardized comparative screening workflow begins with the design and cloning of a focused sgRNA library targeting genes of interest alongside non-targeting controls [21]. The library is transduced at low multiplicity of infection (MOI ≤ 0.3) to ensure most cells receive a single sgRNA, followed by selection and expansion. Cells are then divided into differentiation cohorts or maintained in their original state before screen execution.
For hiPS cell differentiation, established protocols generate highly pure populations of target cell types. Neural differentiation typically involves dual-SMAD inhibition followed by neural induction, resulting in NPCs that can be further differentiated into neurons expressing characteristic markers like MAP2 and CHAT [21]. Cardiac differentiation often employs directed differentiation using growth factors or small molecules, producing cardiomyocytes that express CTNT and ACTN2 [21]. Quality control at each stage should include flow cytometry for lineage-specific markers and functional assessments where appropriate.
During the screen itself, cells are cultured with doxycycline to induce CRISPRi-mediated knockdown, with samples collected at multiple time points to monitor sgRNA abundance changes. The screening timeline must be optimized for each cell type, considering differences in doubling time (for proliferative cells) and protein half-life (for post-mitotic cells) [21]. For migration screens, specialized assays like transwell systems or 3D matrix invasion are employed to separate migratory from non-migratory cells before sgRNA quantification [86].
Diagram Title: Comparative CRISPRi Screen Workflow
Data Analysis and Hit Identification
Essential Gene Calling and Comparative Analysis
The analysis of comparative CRISPRi screens begins with quantification of sgRNA abundance through next-generation sequencing. Read counts are normalized and analyzed using specialized algorithms like MAGeCK or CRISPRiScreenAnalysis pipelines to calculate gene-level enrichment or depletion scores [21] [17]. Essential genes are typically identified as those showing significant depletion of targeting sgRNAs compared to non-targeting controls after multiple population doublings.
In comparative analyses, the essentiality of each gene is assessed across all tested cell types, with cell-type-specific hits identified as genes that are essential in one context but dispensable in others. Analysis of principal components often reveals clustering by both cell type and differentiation state, confirming that genetic requirements are rewired during cellular specialization [21]. The stringency of hit calling must be balanced against the need to identify subtle but biologically important differences, with false discovery rate (FDR) control appropriate for each experimental context.
Validation Strategies and Specificity Assessment
Rigorous validation is particularly important in comparative screens, where technical artifacts could be misinterpreted as biologically meaningful differences. Validation approaches include:
- Individual sgRNA validation: The phenotype of the top scoring sgRNAs should be recapitulated in individual transduction experiments, with correlation between pooled screen scores and individual guide effects (reported Spearman's R = 0.51-0.85) [21].
- Cell type specificity scoring: Quantitative scores can be calculated to represent the degree of phenotype specificity between cell types, with significant positive correlation between screen-derived and validation-derived specificity scores (Spearman's R = 0.67) [21].
- Orthogonal functional assays: Migration screens can be validated using transwell assays or live-cell imaging; differentiation screens can employ flow cytometry for lineage markers; translation factor screens can use puromycin incorporation to measure global protein synthesis rates [21] [86].
Table: Representative Results from Comparative CRISPRi Screens
| Screen Context | Total Genes Targeted | Essential Genes Identified | Cell-Type-Specific Hits | Key Biological Insights |
|---|---|---|---|---|
| hiPS Cells | 262 | 200 (76%) | 27 genes essential in kucg-2 but not WTC11 hiPS cells | Stem cells show exceptional sensitivity to mRNA translation perturbations [21] |
| hiPS-Derived Neurons | 262 | 148 during differentiation, 118 for survival | 1 gene (NAA11) specifically essential for neuron survival | Distinct genetic requirements during differentiation versus maintenance [21] |
| Neutrophil Migration | Genome-wide | 344 genes reduced migration, 31 increased migration | Different gene sets for chemotaxis vs. chemokinesis vs. 3D migration | mTORC1 signaling influences differentiation, survival, and migration [86] |
| HEK293 Cells | 262 | 176 (67%) | 4 genes (CARHSP1, EIF4E3, EIF4G3, IGF2BP2) specifically essential | Lower overall essentiality compared to hiPS cells [21] |
Multi-Omics Data Integration Approaches
Integration Frameworks and Methodologies
The true power of comparative CRISPRi screens emerges when functional genomic data is integrated with other molecular profiling datasets. Multi-omics integration methods can be broadly categorized into correlation/covariance-based approaches, matrix factorization methods, probabilistic models, and deep learning frameworks [46]. Each approach offers distinct strengths for different integration scenarios.
Canonical Correlation Analysis (CCA) and its sparse extensions (sGCCA) are particularly valuable for identifying relationships between different omics data types collected from the same samples [46]. These methods find linear combinations of variables that maximize correlation between datasets, effectively identifying shared patterns across transcriptional, epigenetic, and functional genomic dimensions. For more complex nonlinear relationships, multiple kernel learning and deep generative models like variational autoencoders (VAEs) can capture higher-order interactions that linear methods might miss [46].
Supervised integration methods like DIABLO extend these approaches to simultaneously maximize common information between multiple omics datasets and minimize prediction error for a response variable, effectively linking molecular patterns to phenotypic outcomes [46]. This is particularly relevant for CRISPRi screens, where the response variable might be differentiation efficiency, migration capacity, or cellular fitness.
Application to CRISPRi Data
In practice, multi-omics integration of CRISPRi data involves combining genetic dependency information with complementary datasets such as:
- RNA-seq transcriptomics: To determine whether genetic essentiality correlates with basal gene expression levels across cell types
- ATAC-seq or ChIP-seq epigenomics: To investigate whether chromatin accessibility or histone modifications predict genetic dependencies
- Proteomic profiling: To examine relationships between protein abundance and essentiality
- Metabolic profiling: To connect genetic perturbations with functional metabolic consequences
A compelling example comes from a study that integrated CRISPR/Cas9-based functional genomics with multi-omics datasets to redefine the pluripotency regulatory network in embryonic stem cells (ESCs) [17]. This integrative analysis resolved the network into six functionally independent transcriptional modules (CORE, MYC, PAF, PRC, PCGF, and TBX) with distinct activity patterns during development [17]. Such integrated models provide a more comprehensive understanding of how genetic perturbations disrupt coordinated regulatory programs.
Diagram Title: Multi-Omics Data Integration Framework
The Scientist's Toolkit: Essential Research Reagents
Table: Key Research Reagent Solutions for Comparative CRISPRi Screens
| Reagent/Catalog Number | Function | Application Notes |
|---|---|---|
| Inducible dCas9-KRAB System | Doxycycline-regulated transcriptional repressor | Integrated at AAVS1 safe harbor locus; minimal leaky expression; compatible with differentiation protocols [21] [3] |
| Focused sgRNA Libraries | Target-specific gene repression | Typically include 3-5 sgRNAs per gene + 10% non-targeting controls; designed with CRISPRiaDesign or similar algorithms [21] |
| Lentiviral Packaging System | sgRNA library delivery | Second-generation systems (psPAX2, pMD2.G); low MOI transduction critical for screen quality |
| Lineage-Specific Differentiation Kits | Cell type generation | Commercial kits available for neural, cardiac, hepatic lineages; quality control with marker expression essential [21] |
| Cell Recovery Reagents | Migratory cell isolation | Nattokinase for fibrin degradation in 3D migration screens; collagenase for matrix dissociation [86] |
| Barcoded Expression Reporters | Phenotypic profiling | CiBER-Seq reporters enable massive parallel reporter assays; link guides to molecular phenotypes [87] |
Biological Insights and Applications
Cell-Type-Specific Regulation of Fundamental Processes
Comparative CRISPRi screens have revealed that even the most fundamental cellular processes are subject to cell-type-specific regulation. A striking example comes from studies of mRNA translation machinery, where core ribosomal proteins and translation factors showed broad essentiality across hiPS cells, neural progenitors, and cardiomyocytes, but quality control factors exhibited striking cell-type-specific requirements [21]. Human stem cells showed particular dependence on mRNA translation-coupled quality control pathways, especially those detecting and rescuing slow or stalled ribosomes [21].
The E3 ligase ZNF598, which resolves ribosome collisions, was identified as critically important in hiPS cells but less essential in other cell types [21]. Further investigation revealed that ZNF598 functions in stem cells to resolve a distinct type of ribosome collision occurring at translation start sites on endogenous mRNAs with highly efficient initiation [21]. This discovery underscores how comparative approaches can reveal specialized implementations of core processes in different cellular contexts.
Developmental Transitions and Lineage Commitment
CRISPRi screens conducted across differentiation timecourses have provided insight into how genetic requirements shift during developmental transitions. In studies of neutrophil differentiation from HL-60 cells, screens identified distinct gene sets important for proliferation, differentiation, and migratory behaviors [86]. The mTORC1 signaling pathway emerged as a key regulator influencing multiple aspects of neutrophil biology, including differentiation, survival, and migration capacity [86].
Similar approaches in hiPS cell differentiation have begun to map how genetic dependencies are rewired as cells transition from pluripotent states to lineage-committed progenitors and terminally differentiated cells. These studies have practical implications for regenerative medicine, as they identify potential barriers to efficient differentiation and maintenance of differentiated cell types [21].
Disease Modeling and Therapeutic Opportunities
The application of comparative CRISPRi screens to disease modeling has begun to reveal context-specific genetic vulnerabilities that could be exploited therapeutically. By comparing genetic dependencies in healthy and disease states, researchers can identify disease-specific essential genes while avoiding targets that would also disrupt normal tissue function.
In cancer research, comparisons between malignant cells and their normal counterparts have identified cancer-specific dependencies, though the genetic heterogeneity and aberrant gene expression in cancer cell lines can complicate interpretation [21] [46]. The use of isogenic disease models derived from hiPS cells may provide cleaner experimental systems for identifying bona fide disease vulnerabilities while accounting for genetic background effects.
Future Directions and Concluding Perspectives
The field of comparative CRISPRi screening is rapidly evolving, with several promising directions emerging. Technologically, the development of CRISPRi-ART using RNA-binding dCas13d rather than DNA-targeting dCas9 may expand the range of applicable systems, particularly for organisms with modified genomes or non-standard genetic codes [88]. The demonstration that dCas13d targeting near ribosome-binding sites efficiently represses protein translation suggests this system could complement existing approaches [88].
Methodologically, the integration of comparative CRISPRi with single-cell omics technologies represents a particular opportunity. While current pooled CRISPRi screens typically rely on bulk readouts, emerging approaches like Perturb-Seq could enable single-cell resolution of genetic perturbation effects across heterogeneous cell populations [87]. This would be particularly valuable for studying rare cell types or continuous differentiation processes where bulk measurements might obscure important biology.
From an analytical perspective, improved methods for multi-omics data integration will be essential for extracting maximum biological insight from comparative screens. Deep generative models, particularly variational autoencoders (VAEs), show promise for integrating high-dimensional multi-omics data while handling missing values and technical noise [46]. Foundation models pretrained on large-scale molecular datasets may eventually provide context-aware representations that enhance our ability to predict how genetic perturbations will affect different cell states.
In conclusion, comparative CRISPRi screens across cell types and differentiation states represent a powerful approach for understanding how cellular context shapes genetic dependencies. When integrated with other omics data types, these functional genomic profiles provide unprecedented insight into the molecular logic of cell identity and specialization. As the technologies and analytical methods continue to mature, comparative CRISPRi approaches will undoubtedly yield fundamental discoveries in developmental biology, disease mechanisms, and therapeutic opportunities.
Integrating Molecular Data with Clinical Outcomes for Patient Stratification
The convergence of multi-omics profiling, advanced computational tools, and CRISPR-based technologies is revolutionizing patient stratification in biomedical research and therapeutic development. This technical guide provides a comprehensive framework for integrating genomic, transcriptomic, proteomic, and epigenomic data with clinical outcomes to identify molecularly-defined patient subgroups. Focusing specifically on applications in CRISPR research, we detail experimental methodologies, computational pipelines, and visualization approaches that enable precise correlation of molecular signatures with therapeutic responses. By establishing standardized protocols for data integration and analysis, this guide aims to equip researchers with the tools necessary to advance personalized medicine and accelerate the development of targeted therapies.
Patient stratification represents a fundamental paradigm shift from population-based to personalized medicine, moving beyond one-size-fits-all treatment approaches toward precisely targeted interventions based on individual molecular profiles. This approach is particularly critical in oncology, where tumor heterogeneity remains a major obstacle in clinical trials. Differences between tumors and even within a single tumor can drive drug resistance by altering treatment targets or shaping the tumor microenvironment [89].
Multi-omics approaches have transformed cancer research by providing a comprehensive view of tumor biology, with each omics layer offering distinct insights. Genomics examines the full genetic landscape, identifying mutations, structural variations, and copy number variations that drive tumor initiation and progression. Transcriptomics analyzes gene expression, providing a snapshot of pathway activity and regulatory networks. Proteomics investigates the functional state of cells by profiling proteins, including post-translational modifications, interactions, and subcellular localization [89].
The integration of artificial intelligence with pharmacogenomics and CRISPR has further refined precision medicine by improving drug-gene interaction predictions, optimizing gene-editing specificity, and advancing predictive modeling for therapeutic responses. AI algorithms enhance CRISPR guide RNA design, reducing off-target effects and improving editing precision, while pharmacogenomic insights inform the selection of CRISPR-based interventions for personalized disease management [78].
Core Principles of Multi-Omics Data Integration
Multi-Omics Data Layers and Their Clinical Relevance
Table 1: Core Multi-Omics Data Types and Their Applications in Patient Stratification
| Data Type | Key Technologies | Biological Insights | Clinical Applications |
|---|---|---|---|
| Genomics | Whole Genome/Exome Sequencing, SNP arrays | Mutations, CNVs, structural variations | Driver mutation identification, inherited risk assessment |
| Transcriptomics | RNA-seq, single-cell RNA-seq, spatial transcriptomics | Gene expression, pathway activity, regulatory networks | Disease subtyping, drug response prediction |
| Proteomics | Mass spectrometry, multiplex immunofluorescence | Protein expression, post-translational modifications, signaling activity | Therapeutic target validation, resistance mechanism elucidation |
| Epigenomics | ChIP-seq, ATAC-seq, methylation arrays | Chromatin accessibility, histone modifications, DNA methylation | Gene regulation analysis, cellular memory characterization |
| Spatial Omics | Multiplex IHC/IF, spatial transcriptomics | Cellular organization, tissue architecture, cell-cell interactions | Tumor microenvironment characterization, immune context analysis |
Computational Frameworks for Data Integration
The scale and complexity of multi-omics data require standardized pipelines and robust bioinformatics frameworks. Emerging tools like Flexynesis, a deep learning toolkit for bulk multi-omics data integration, demonstrate the potential for robust stratification even with partial data. Flexynesis streamlines data processing, feature selection, hyperparameter tuning, and marker discovery, supporting both deep learning architectures and classical supervised machine learning methods with a standardized input interface for single/multi-task training and evaluation for regression, classification, and survival modeling [11].
Frameworks like NMFProfiler identify biologically relevant signatures across omics layers, improving biomarker discovery and patient subgroup classification. Other approaches include IntegrAO, which integrates incomplete multi-omics datasets and classifies new patient samples using graph neural networks [89].
Experimental Methodologies for CRISPR Response Research
CRISPR Screening in Physiologically Relevant Models
Large-scale CRISPR-based genetic screens, including knockout, interference (CRISPRi), activation (CRISPRa), and single-cell approaches, can be applied in primary human 3D gastric organoids to systematically identify genes that affect drug sensitivity. This approach enables comprehensive dissection of gene-drug interactions in a system that preserves tissue architecture, stem cell activity, multilineage differentiation, genomic alterations, and pathology of primary tissues [30].
Protocol 1: CRISPR Screening in 3D Organoids
- Organoid Generation: Establish organoid lines from patient-derived normal or tumor tissues
- CRISPR System Integration: Generate stable Cas9-expressing organoid lines using lentiviral transduction
- Library Delivery: Transduce validated pooled lentiviral sgRNA library with >1000 cells per sgRNA coverage
- Selection and Expansion: Apply puromycin selection 2 days post-transduction and continue culture maintaining cellular coverage
- Phenotypic Assessment: Harvest subpopulations at multiple time points (e.g., T0, T7, T14, T28 days) for sequencing
- Hit Identification: Measure relative sgRNA abundance by next-generation sequencing to identify genes affecting cellular growth or drug response
Protocol 2: Inducible CRISPRi/CRISPRa in Organoids
- System Engineering: Create organoid lines with doxycycline-inducible dCas9-KRAB (iCRISPRi) or dCas9-VPR (iCRISPRa) using sequential two-vector lentiviral approach
- Line Establishment: Generate organoid lines expressing rtTA, then introduce doxycycline-inducible dCas9 fusion protein with fluorescent reporter
- Cell Sorting: Sort fluorescent-positive cells after induction to establish stable lines
- Functional Validation: Design sgRNAs targeting gene promoters and analyze target population changes via antibody staining and flow cytometry
- Dose-Response Testing: Apply varying doxycycline concentrations to titrate repression/activation levels
DNA Repair Characterization in Clinically Relevant Cells
Understanding DNA repair outcomes is crucial for therapeutic genome editing, particularly in nondividing cells like neurons where repair mechanisms differ significantly from dividing cells [90].
Protocol 3: Characterizing CRISPR Repair in Nondividing Cells
- Cell Differentiation: Differentiate human iPSCs into postmitotic cortical-like excitatory neurons (≥95% NeuN-positive by Day 4)
- VLP Delivery: Produce virus-like particles (VLPs) containing Cas9 ribonucleoprotein pseudotyped with VSVG and/or BaEVRless for efficient transduction
- DSB Confirmation: Validate Cas9-induced double-strand breaks via immunocytochemistry for γH2AX and 53BP1 co-localization
- Kinetic Analysis: Track indel accumulation over extended time courses (up to 16+ days) via next-generation sequencing
- Pathway Manipulation: Apply chemical or genetic perturbations to direct DNA repair toward desired outcomes
Table 2: Research Reagent Solutions for CRISPR Response Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Delivery Systems | Virus-like particles (VLPs), Lentiviral vectors, Lipid nanoparticles (LNPs) | Efficient delivery of CRISPR components to target cells |
| CRISPR Enzymes | Cas9, Cas12f, Cas12a, Cas13a, Base editors, Prime editors | Genome editing, epigenome editing, diagnostics |
| Screening Libraries | Pooled sgRNA libraries, CRISPRi/a libraries, Single-guide RNAs | High-throughput functional genomics screens |
| Model Systems | Patient-derived organoids (PDOs), iPSC-derived neurons, Primary T cells | Physiologically relevant experimental models |
| Analytical Tools | Flexynesis, CRISPR-GPT, Single-cell RNA-seq, Flow cytometry | Data integration, experimental design, outcome assessment |
Data Integration and Analytical Approaches
Multi-Omics Integration for Predictive Modeling
Accurate decision making in precision oncology depends on integration of multimodal molecular information. Flexynesis enables both single-task and multi-task modeling, accommodating regression, classification, and survival modeling within a unified framework [11].
Single-task modeling predicts one outcome variable:
- Regression: Predict continuous outcomes (e.g., drug sensitivity levels)
- Classification: Categorical outcomes (e.g., microsatellite instability status)
- Survival modeling: Time-to-event outcomes with Cox Proportional Hazards
Multi-task modeling jointly predicts multiple outcome variables, allowing the embedding space to be shaped by multiple clinically relevant variables simultaneously, even with missing labels for some variables.
AI-Enhanced Experimental Design
CRISPR-GPT, a large language model developed at Stanford Medicine, accelerates gene-editing processes by helping researchers generate designs, analyze data, and troubleshoot design flaws. The system uses 11 years' worth of expert discussions and published scientific papers to create an AI model that "thinks" like a scientist [52].
Workflow for AI-Assisted CRISPR Design:
- Input Experimental Goals: Researcher provides objectives, context, and gene sequences via text chat
- Plan Generation: CRISPR-GPT suggests experimental approaches and identifies potential problems
- Iterative Refinement: System explains rationale and adjusts designs based on researcher feedback
- Safety Screening: Incorporated safeguards prevent unethical applications (e.g., embryo editing)
Applications in Therapeutic Development and Clinical Translation
CRISPR Clinical Trials and Patient Stratification
The landscape of CRISPR-based therapies has expanded significantly, with applications across genetic disorders, oncology, and infectious diseases. Current clinical trials demonstrate the critical importance of patient stratification for therapeutic success [31] [91].
Table 3: Selected CRISPR Clinical Trials and Stratification Approaches
| Therapy | Condition | Approach | Stratification Method | Development Phase |
|---|---|---|---|---|
| Casgevy | Sickle cell disease, β-thalassemia | Ex vivo HSC editing | Genetic mutation status | Approved (2023) |
| NTLA-2001 | Transthyretin amyloidosis | In vivo LNP delivery | TTR mutation status, cardiomyopathy vs neuropathy | Phase III |
| VERVE-101/102 | Familial hypercholesterolemia | In vivo base editing | LDL-C levels, ASCVD status | Phase Ib |
| FT819 | Systemic lupus erythematosus | Off-the-shelf CAR T-cell | Renal involvement, autoantibody profile | Phase I |
| HG-302 | Duchenne Muscular Dystrophy | In vivo AAV delivery | DMD mutation location | Phase I |
Spatial Biology and Tumor Microenvironment Characterization
Spatial biology preserves tissue architecture, showing how cells interact and how immune cells infiltrate tumors. Key technologies include spatial transcriptomics, spatial proteomics, multiplex immunohistochemistry, and mass spectrometry imaging [89].
Integrated Analysis Workflow:
- Tissue Sectioning: Prepare thin sections from patient tumor samples
- Multiplex Staining: Apply cyclic immunofluorescence or spatial transcriptomics panels
- Image Acquisition: Capture high-resolution multichannel images
- Cell Segmentation: Identify individual cells and assign spatial coordinates
- Phenotype Assignment: Classify cell types based on marker expression
- Neighborhood Analysis: Quantify cell-cell interactions and spatial relationships
Real-world examples demonstrate the power of integrated multi-omics to uncover actionable biology. Integrated single-cell RNA and spatial transcriptomics analyses in gastric cancer revealed B-cell subpopulations and tumor B-cell interactions as key modulators of the immune microenvironment. Targeting CCL28 in mouse models enhanced CD8+ T cell activity, demonstrating how multi-omics integration can identify actionable biomarkers and therapeutic strategies [89].
The integration of molecular data with clinical outcomes for patient stratification represents a transformative approach in precision medicine. By combining multi-omics profiling, CRISPR-based functional genomics, and advanced computational methods, researchers can identify molecularly-defined patient subgroups with distinct therapeutic responses and clinical outcomes.
Future developments in this field will likely focus on several key areas: (1) improved single-cell and spatial multi-omics technologies providing higher resolution views of cellular heterogeneity; (2) enhanced AI and machine learning algorithms for better predictive modeling; (3) standardized frameworks for data integration and sharing across institutions; and (4) expanded applications of CRISPR-based screening in physiologically relevant model systems.
As these technologies mature, the systematic integration of molecular data with clinical outcomes will become increasingly central to therapeutic development, clinical trial design, and ultimately, routine clinical care, enabling truly personalized treatment approaches based on comprehensive molecular profiling.
The integration of multi-omics data is revolutionizing our ability to decipher complex biological systems, including the molecular mechanisms underlying CRISPR interference (CRISPRi) responses. A central challenge in functional genomics lies in understanding how genetic dependencies vary across different cellular contexts, particularly between pluripotent stem cells and their differentiated progeny. While core housekeeping genes are universally essential, a growing body of evidence suggests that specialized cellular functions create context-specific genetic vulnerabilities [5]. This case study examines how comparative CRISPRi screens coupled with multi-omics data integration can identify cell-type-specific essential genes, with particular focus on human induced pluripotent stem cells (hiPS cells) and their differentiated neural and cardiac counterparts [21].
The fundamental premise is that cellular identity dictates how cells respond to genetic perturbation. hiPS cells possess exceptionally high global protein synthesis rates and unique regulatory networks to maintain pluripotency, potentially creating distinct genetic dependencies compared to differentiated cells [21]. Advances in CRISPRi technology now enable precise, reversible gene repression without introducing DNA double-strand breaks, making it particularly suitable for functional genomics in sensitive stem cell models where DNA damage-induced toxicity could confound results [92]. By combining CRISPRi screening with multi-omics approaches, researchers can systematically map genetic requirements across cellular states, providing insights into basic biology and revealing novel therapeutic targets for regenerative medicine and disease modeling.
Experimental Design and Workflow
Core Experimental Platform
The foundational methodology for identifying cell-type-specific essential genes employs an inducible CRISPRi system integrated into the AAVS1 safe harbor locus of a reference hiPS cell line [21]. This system utilizes a doxycycline-inducible KRAB-dCas9 construct that remains silent until induction, preventing unintended gene expression effects during differentiation. The platform enables direct comparison of genetic dependencies across hiPS cells, neural progenitor cells (NPCs), neurons, cardiomyocytes (CMs), and control HEK293 cells [21].
A custom-designed sgRNA library targeting 262 genes encoding core and regulatory mRNA translation machinery components was deployed, along with cell-specific marker genes as controls. The library contained 3,000 sequences (including 10% non-targeting controls) delivered via lentiviral transduction at a low multiplicity of infection to ensure single-guide integration per cell [21]. This focused approach on translation machinery enables deep investigation of a fundamental cellular process while controlling for experimental complexity.
Key Research Reagents and Solutions
Table 1: Essential Research Reagents for Comparative CRISPRi Screens
| Reagent/Solution | Function/Application | Technical Specifications |
|---|---|---|
| Inducible KRAB-dCas9 hiPS Cell Line | Engineered platform for CRISPRi screens | AAVS1-safe harbor integration; doxycycline-inducible; mCherry reporter [21] |
| Custom sgRNA Library | Targeted gene repression | 3,000 sgRNAs targeting 262 translation machinery genes + controls; designed via CRISPRiaDesign [21] |
| Neural Differentiation Media | Directed differentiation to neural lineages | Generates uniform neural progenitor cells (NPCs) and neurons expressing PAX6, NES, CHAT, MAP2 [21] |
| Cardiac Differentiation Media | Directed differentiation to cardiac lineages | Generates cardiomyocytes expressing CTNT and ACTN2 [21] |
| dCas9-ZIM3(KRAB)-MeCP2(t) | Enhanced CRISPRi repressor | Next-generation repressor fusion with improved knockdown efficiency and reduced guide-dependent variability [92] |
Screening and Validation Workflow
The experimental workflow encompasses several critical phases: cell line development, differentiation, screening, and validation. First, the inducible CRISPRi hiPS cell line is established and validated for pluripotency markers (NANOG, POU5F1) and tight control of KRAB-dCas9 expression [21]. Next, parallel differentiations generate neural progenitor cells, neurons, and cardiomyocytes, with lineage confirmation through immunostaining and flow cytometry for cell-type-specific markers.
For the essentiality screens, each cell type is transduced with the sgRNA library and cultured with or without doxycycline induction for approximately ten population doublings. sgRNA abundance is quantified through sequencing at multiple time points to calculate gene-level enrichment or depletion scores using established CRISPRi analysis pipelines [21]. Hit validation employs individual sgRNAs against candidates with differential essentiality, followed by functional assays including reverse transcription quantitative PCR (RT-qPCR), immunoblotting, and quantitative mass spectrometry to confirm target knockdown and phenotypic consequences [21].
Diagram 1: CRISPRi screening workflow for identifying cell-type-specific essential genes.
Key Findings and Quantitative Analysis
Global Essentiality Patterns Across Cell Types
The comparative CRISPRi screens revealed both conserved and cell-type-specific genetic dependencies. hiPS cells demonstrated exceptional sensitivity to perturbations in mRNA translation, with 200 of 262 (76%) targeted genes scoring as essential, compared to 175 (67%) in neural progenitor cells and 176 (67%) in HEK293 cells [21]. This heightened sensitivity in stem cells correlates with their exceptionally high global protein synthesis rates, suggesting that pluripotent cells have reduced buffering capacity for translational perturbations [21].
Strikingly, genetic dependencies specific to a single cell type were rare. Only one gene was exclusively essential for neuronal survival (NAA11), and one for cardiomyocyte survival (CPEB2), while four genes were uniquely essential in HEK293 cells (CARHSP1, EIF4E3, EIF4G3, and IGF2BP2) [21]. This pattern suggests that most essential genes function in core cellular processes, with cell-type-specific vulnerabilities emerging from specialized functions rather than fundamental differences in essential pathways.
Cell-Type-Specific Dependencies in Translation-Coupled Quality Control
A particularly significant finding was the divergent essentiality of genes involved in translation-coupled quality control pathways. While core ribosomal proteins and translation factors were broadly essential across all cell types, quality control factors displayed strong cell-type-specific effects [21]. Human stem cells critically depended on pathways that detect and rescue slow or stalled ribosomes, especially the E3 ligase ZNF598, which resolves ribosome collisions at translation start sites on endogenous mRNAs with highly efficient initiation [21].
Table 2: Quantitative Essentiality Scores for Selected Genes Across Cell Types
| Gene | Function | hiPS Cells | Neural Progenitors | Neurons | Cardiomyocytes | HEK293 |
|---|---|---|---|---|---|---|
| ZNF598 | Ribosome quality control | Essential | Non-essential | Non-essential | Non-essential | Non-essential |
| NAA11 | N-terminal acetylation | Non-essential | Non-essential | Essential | Non-essential | Non-essential |
| CPEB2 | Translation regulation | Non-essential | Non-essential | Non-essential | Essential | Non-essential |
| EIF4G3 | Translation initiation | Non-essential | Non-essential | Non-essential | Non-essential | Essential |
| RPS25 | Ribosomal protein | Essential | Essential | Essential | Essential | Essential |
The specialized dependence of hiPS cells on ZNF598-mediated ribosome collision resolution points to unique translational control mechanisms in stem cells. This pathway appears particularly important for handling mRNAs with high initiation efficiency, which may be enriched in the stem cell transcriptome [21]. These findings underscore how basic cellular processes are tuned to meet the specific demands of different cell states.
Integration with Multi-Omics Data
The CRISPRi screen findings gain additional significance when integrated with multi-omics data. Quantitative mass spectrometry revealed that most targeted proteins were expressed at similar levels across cell types, with ZNF598 being a notable exception (~2-fold higher in HEK293 cells) [21]. This suggests that differential essentiality often reflects functional rewiring rather than simple abundance differences.
Advanced computational tools like Flexynesis can further enhance the integration of CRISPR screening data with transcriptomic, proteomic, and epigenomic datasets [11]. This deep learning framework enables multi-omics integration for various prediction tasks, including classification, regression, and survival modeling, allowing researchers to build comprehensive models of how genetic perturbations propagate through molecular networks in different cellular contexts [11].
Detailed Methodologies
CRISPRi Screen Implementation
Cell Culture and Differentiation: Maintain inducible CRISPRi hiPS cells in feeder-free conditions with appropriate pluripotency-supporting media. For differentiation, use established protocols to generate highly pure populations of neural progenitor cells (through dual-SMAD inhibition), neurons (through neurotrophic factor support), and cardiomyocytes (via Wnt modulation) [21]. Confirm differentiation efficiency through immunocytochemistry and flow cytometry for lineage-specific markers before screening.
Library Transduction and Screening: Transduce cells at a low multiplicity of infection (MOI ≈ 0.3) to ensure most cells receive a single sgRNA. Include non-targeting control sgRNAs (10% of library) for normalization. After puromycin selection, split cells into induced (+doxycycline) and non-induced controls, maintaining a minimum of 500 cells per sgRNA to prevent bottleneck effects [21]. Culture cells for approximately ten population doublings, collecting samples at multiple time points for sgRNA abundance quantification by sequencing.
Essentiality Analysis: Process sequencing data through established CRISPRi analysis pipelines (e.g., CRISPRiaDesign) to calculate gene-level scores. Normalize read counts using non-targeting controls and compare sgRNA depletion in induced versus non-induced conditions. Apply statistical tests (e.g., Mann-Whitney U test) to identify significantly depleted genes (P ≤ 0.1) [21]. Compute cell-type specificity scores by comparing essentiality profiles across cell types.
Next-Generation CRISPRi Enhancements
Recent advances in CRISPRi technology offer significant improvements for future studies. The novel repressor fusion dCas9-ZIM3(KRAB)-MeCP2(t) demonstrates enhanced repression efficiency and reduced guide-dependent variability compared to conventional KRAB-based repressors [92]. This improved platform achieves more complete knockdown, particularly valuable when targeting genes with low-to-moderate essentiality where partial repression might miss true dependencies.
Engineering considerations for optimal CRISPRi performance include:
- Repressor Design: Bipartite and tripartite repressor fusions combining KRAB domains with additional repressor modules (e.g., MAX, MeCP2(t)) show synergistic effects [92].
- Delivery Optimization: Both direct dCas9-repressor fusions and scaffold-recruited repressors can be effective, with the optimal approach potentially varying by cell type [92].
- Expression Tuning: Moderate dCas9-repressor expression levels typically outperform both very low and very high expression, likely due to balance between efficacy and cellular toxicity [92].
Diagram 2: Mechanism of cell-type-specific genetic dependencies in mRNA translation.
Single-Cell CRISPR Validation Methods
For comprehensive validation of screening hits, emerging single-cell DNA sequencing methods enable precise quantification of CRISPR editing outcomes across multiple loci simultaneously [93]. This approach can interrogate >100 loci per cell, detecting both on-target and off-target editing with sensitivity comparable to bulk sequencing (∼0.1%) but with the added advantage of revealing co-editing patterns and translocation events [93].
The single-cell validation workflow includes:
- Multiplexed Target Amplification: Design PCR primers for all potential off-target sites identified in silico plus confirmed on-target sites.
- Single-Cell Partitioning: Use microfluidic platforms to partition individual cells with barcoded amplification reagents.
- Library Preparation and Sequencing: Construct sequencing libraries with cell-specific barcodes to track editing events back to individual cells.
- Analysis of Editing Patterns: Quantify mutation rates, zygosity, and co-editing frequencies across the cell population [93].
This method provides unprecedented resolution for understanding how genetic perturbations distribute across cell populations, particularly valuable for detecting rare off-target events and understanding how heterogeneous editing outcomes might influence phenotypic analyses.
Discussion and Future Perspectives
The integration of comparative CRISPRi screens with multi-omics data represents a powerful framework for understanding how cellular context shapes genetic dependencies. The finding that hiPS cells exhibit unique vulnerability to perturbations in translation-coupled quality control, particularly ZNF598-mediated ribosome collision resolution, reveals how fundamental processes are specialized to meet the demands of distinct cell states [21]. This has important implications for both basic biology and therapeutic development, suggesting that targeting context-specific essential genes could enable selective manipulation of specific cell types.
Future directions in this field will likely focus on several key areas. First, expanding screening approaches to encompass more diverse cell types and developmental timepoints will provide a more comprehensive map of genetic dependencies across human biology. Second, tighter integration of single-cell multi-omics technologies with CRISPR screening will enable deconvolution of cellular heterogeneity and reveal how genetic networks operate within individual cells [5] [93]. Third, advanced computational methods like Flexynesis will enhance our ability to integrate diverse data modalities and build predictive models of how genetic perturbations manifest differently across cellular contexts [11].
From a therapeutic perspective, these approaches are already driving advances in disease modeling and drug discovery. The combination of CRISPR editing with hiPS cell technology enables creation of more accurate human disease models, particularly for neurodegenerative disorders like Alzheimer's disease where species differences have hampered progress [94]. As both editing and differentiation technologies continue to mature, we can anticipate increasingly sophisticated models that better recapitulate human disease pathophysiology and enable more effective therapeutic development [95].
In conclusion, the case study presented here demonstrates how comparative functional genomics approaches can reveal the molecular logic of cell-type-specific genetic dependencies. By combining precise genetic perturbation technologies with multi-omics data integration, researchers are building comprehensive maps of how cellular context determines genetic essentiality, providing fundamental insights into biology and creating new opportunities for therapeutic intervention.
Conclusion
The integration of multi-omics data with CRISPRi screening represents a paradigm shift in functional genomics, transforming our ability to move from simple gene-phenotype correlations to a nuanced understanding of complex biological networks. This powerful combination allows for the precise identification of genetic dependencies, the discovery of novel therapeutic targets, and a deeper insight into cell-type-specific responses, as demonstrated in models from stem cells to pathogens. Future progress hinges on overcoming key challenges in data standardization, computational tool development, and the ethical application of these technologies. As artificial intelligence continues to refine data analysis and emerging single-cell technologies provide ever-higher resolution, this integrated approach is poised to accelerate the development of personalized therapies and solidify its role as a cornerstone of modern biomedical research and drug discovery.
References
- 1. CRISPR interference [https://en.wikipedia.org/wiki/CRISPR_interference]
- 2. CRISPRa and CRISPRi: Why You Need These Powerful ... [https://bitesizebio.com/47575/crispra-and-crispri/]
- 3. CRISPR Interference Efficiently Induces Specific and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4830697/]
- 4. CRISPRi functional genomics in bacteria and its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11332340/]
- 5. Unraveling the future of genomics: CRISPR, single-cell ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1565387/full]
- 6. CRISPR guide RNA design for research applications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5014588/]
- 7. CRISPRi Versus CRISPR-Cut [https://oxfordglobal.com/nextgen-biomed/resources/when-to-cut-and-when-to-inhibit-a-low-throughput-hands-on-investigation-of-crispr-cut-and-crispri-efficiencies-in-ipsc-and-neural-cells]
- 8. RNAi vs. CRISPR: Guide to Selecting the Best Gene ... [https://www.synthego.com/blog/rnai-vs-crispr-guide/]
- 9. Systematic Comparison of CRISPR and shRNA Screens to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11475473/]
- 10. A Guide to Multi-omics Integration Strategies [https://frontlinegenomics.com/a-guide-to-multi-omics-integration-strategies/]
- 11. Flexynesis: A deep learning toolkit for bulk multi-omics data ... [https://www.nature.com/articles/s41467-025-63688-5]
- 12. Applications of multi‐omics analysis in human diseases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10390758/]
- 13. Integration Analysis of Epigenomics and Transcriptomics [https://bioinfo.cd-genomics.com/resource-multi-omics-integration-analysis-epigenomics-transcriptomics.html]
- 14. Combining CRISPRi and metabolomics for functional ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7612681/]
- 15. 2025 Trends: Multiomics [https://www.genengnews.com/topics/omics/2025-trends-multiomics/]
- 16. AI-driven multi-omics integration for multi-scale predictive ... [https://www.sciencedirect.com/science/article/pii/S2001037024004513]
- 17. A multi-omics integrative analysis based on CRISPR ... [https://www.nature.com/articles/s42003-023-04700-w]
- 18. Multi-omics Analysis of CRISPRi-Knockdowns Identifies ... [https://www.sciencedirect.com/science/article/pii/S240547122030418X]
- 19. Unraveling the future of genomics: CRISPR, single-cell omics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12021818/]
- 20. Bioinformatics approaches to analyzing CRISPR screen data [https://pmc.ncbi.nlm.nih.gov/articles/PMC10019185/]
- 21. Comparative CRISPRi screens reveal a human stem cell ... [https://www.nature.com/articles/s41594-025-01616-3]
- 22. How to Accurately Interpret CRISPR Screening Data [https://www.cd-genomics.com/biomedical-ngs/resource/crispr-screening-data-interpretation-genomics.html]
- 23. CrisprVi: a software for visualizing and analyzing CRISPR ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04716-9]
- 24. Data Visualization of CRISPR-Cas9 Guide RNA Design ... [https://pubmed.ncbi.nlm.nih.gov/38269913/]
- 25. Perturbomics: CRISPR–Cas screening-based functional ... [https://www.nature.com/articles/s12276-025-01487-0]
- 26. Rapid identification of key antibiotic resistance genes in E. ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12063145/]
- 27. Genome-scale CRISPRi and base-editing libraries for ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925004123]
- 28. CRISPR for detection of drug resistance genes [https://www.sciencedirect.com/science/article/abs/pii/S0009898125005054]
- 29. CRISPRi–TnSeq maps genome-wide interactions between ... [https://www.nature.com/articles/s41564-024-01759-x]
- 30. Large-scale CRISPR screening in primary human 3D ... [https://www.nature.com/articles/s41467-025-62818-3]
- 31. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 32. Perturbomics: CRISPR–Cas screening-based functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12322284/]
- 33. Multistable and dynamic CRISPRi-based synthetic circuits [https://www.nature.com/articles/s41467-020-16574-1]
- 34. Functional phenotyping of genomic variants using joint ... [https://www.nature.com/articles/s41592-025-02805-0]
- 35. Introduction to CRISPR screen analysis [https://training.galaxyproject.org/training-material/topics/genome-annotation/tutorials/crispr-screen/slides.html]
- 36. Uniting High-Throughput Perturbations with Single-Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10872607/]
- 37. Transformative advances in single-cell omics - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12560279/]
- 38. Single-Cell Heterogeneity Analysis and CRISPR Screen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6573026/]
- 39. Decoding heterogeneous single-cell perturbation responses [https://www.nature.com/articles/s41556-025-01626-9]
- 40. Single-cell sequencing to multi-omics - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11438019/]
- 41. Single-cell CRISPR screen analysis with sceptre [https://www.10xgenomics.com/analysis-guides/single-cell-crispr-screen-analysis-with-sceptre]
- 42. Analysis of single-cell CRISPR perturbations indicates that ... [https://www.sciencedirect.com/science/article/pii/S2666979X2400291X]
- 43. Directional integration and pathway enrichment analysis ... [https://www.nature.com/articles/s41467-024-49986-4]
- 44. Tools for visualization and analysis of molecular networks ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4461095/]
- 45. MetaboMAPS: Pathway sharing and multi-omics data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7383707/]
- 46. A technical review of multi-omics data integration methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC12315550/]
- 47. Machine learning for multi-omics data integration in cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC8829812/]
- 48. Multi-omics Analysis of CRISPRi-Knockdowns Identifies ... [https://pubmed.ncbi.nlm.nih.gov/33238135/]
- 49. Revolutionizing CRISPR technology with artificial intelligence [https://www.nature.com/articles/s12276-025-01462-9]
- 50. Advancing CRISPR with deep learning: A comprehensive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12476096/]
- 51. AI & CRISPR 2025: How AI Is Revolutionizing Genome Editing [https://aichronicle.co/how-artificial-intelligence-is-revolutionizing-precision-genome-editing-in-2025/]
- 52. AI-powered CRISPR could lead to faster gene therapies ... [https://med.stanford.edu/news/all-news/2025/09/ai-crispr-gene-therapy.html]
- 53. Multicenter integrated analysis of noncoding CRISPRi ... [https://www.nature.com/articles/s41592-024-02216-7]
- 54. CRISPR interference reagents [https://horizondiscovery.com/en/crisprmod/crispri]
- 55. A CRISPRi/a screening platform to study cellular nutrient ... [https://www.nature.com/articles/s41556-024-01402-1]
- 56. Standardizing Statistical Tools for “Omics': Best Practices ... [https://www.chromatographyonline.com/view/standardizing-statistical-tools-for-omics-best-practices-using-r-and-python]
- 57. Improved prediction of bacterial CRISPRi guide efficiency from ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03153-y]
- 58. Best practices and tools in R and Python for statistical ... [https://www.nature.com/articles/s41467-025-63751-1]
- 59. A guide to multi-omics data collection and integration for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9747357/]
- 60. Mapping multimodal phenotypes to perturbations in cells ... [https://www.nature.com/articles/s41587-024-02386-x]
- 61. Progress in single-cell multimodal sequencing and multi- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10937857/]
- 62. nf-core/crisprseq [https://seqera.io/pipelines/crisprseq--nf-core/]
- 63. Compact and highly active next-generation libraries for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5094855/]
- 64. CRISPy-web 3.0: A unified platform for multi-modal guide ... [https://pubmed.ncbi.nlm.nih.gov/41230494/]
- 65. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 66. Multiomics Research: Principles and Challenges in ... [https://www.sciencedirect.com/science/article/pii/S2693125725000019]
- 67. High-Performance Computing Revolutionizes Biological ... [https://riveraxe.com/high-performance-computing-revolutionizes-biological-research/]
- 68. Integrating Multi-Omics for Precision Medicine: 2025 Guide [https://lifebit.ai/blog/integrating-multi-modal-genomic-and-multi-omics-data-for-precision-medicine/]
- 69. High Performance Computing for AI-driven Omics and ... [https://www.frontiersin.org/research-topics/73866/high-performance-computing-for-ai-driven-omics-and-health-data-processing]
- 70. MGI Tech Showcases Comprehensive Multi-Omics ... [https://en.mgi-tech.com/news/537/]
- 71. 2025 and Beyond: The Future of Genomic Data Analysis ... [https://blog.crownbio.com/2025-and-beyond-the-future-of-genomic-data-analysis-and-innovations-in-genomics-services]
- 72. High-performance computing is expanding access to ... [https://publichealth.uci.edu/2025/11/12/high-performance-computing-is-expanding-access-to-cancer-big-data-training/]
- 73. Optimization of gene knockout approaches and sgRNA ... [https://www.nature.com/articles/s41598-025-24184-4]
- 74. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 75. CMN Weekly (17 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-17-october-2025-your-weekly-crispr-medicine-news/]
- 76. A roadmap toward genome-wide CRISPR screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11960495/]
- 77. Omics-based large language models: A new engine for ... [https://www.sciencedirect.com/science/article/pii/S2211383525007129]
- 78. Transforming Pharmacogenomics and CRISPR Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114816/]
- 79. Omics and CRISPR-Cas9 Approaches for Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7866018/]
- 80. Benchmarking genetic interaction scoring methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12464814/]
- 81. Benchmarking Genetic Interaction Scoring Methods for ... [https://cancerdata.ucd.ie/publication/ajmal-2025-evaluating/]
- 82. Comparing Gene Editing Platforms: CRISPR vs. Traditional ... [https://blog.crownbio.com/comparing-gene-editing-platforms-crispr-vs-traditional-methods]
- 83. Alternatives to CRISPR-Cas9: Nucleases for Next-Gen ... [https://www.synthego.com/blog/alternatives-to-crispr-cas9-nucleases-for-next-gen-therapy/]
- 84. Ensure Proper Controls in Your CRISPR Experiments [https://www.synthego.com/blog/controls-crispr-experiments/]
- 85. The CRISPR therapeutics landscape in 2025 [https://www.kilburnstrode.com/knowledge/european-ip/the-crispr-therapeutics-landscape-in-2025]
- 86. Whole-genome screens reveal regulators of differentiation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10507112/]
- 87. CiBER-Seq dissects genetic networks by quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7819735/]
- 88. CRISPRi-ART enables functional genomics of diverse ... [https://www.nature.com/articles/s41564-025-01935-7]
- 89. The Future of Clinical Trials: Incorporating 'Omics for ... [https://blog.crownbio.com/the-future-of-clinical-trials-incorporating-omics-for-enhanced-stratification]
- 90. Characterizing and controlling CRISPR repair outcomes in ... [https://www.nature.com/articles/s41467-025-66058-3]
- 91. CRISPR Clinical Trials to Follow [https://www.synthego.com/blog/crispr-clinical-trials/]
- 92. Engineering novel CRISPRi repressors for highly efficient ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03640-4]
- 93. Improving resolution with single-cell detection of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12179590/]
- 94. Stem cell and CRISPR/Cas9 gene editing technology in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12417523/]
- 95. The Promising Potential of CRISPR Editing in Stem Cells [https://www.synthego.com/blog/crispr-ipsc-bill-skarnes/]