HITI Revolution: Mastering Homology-Independent Targeted Integration for Advanced Gene Therapies
This article provides a comprehensive exploration of Homology-Independent Targeted Integration (HITI), a groundbreaking CRISPR/Cas9-based genome editing technology that leverages the non-homologous end joining (NHEJ) pathway.
HITI Revolution: Mastering Homology-Independent Targeted Integration for Advanced Gene Therapies
Abstract
This article provides a comprehensive exploration of Homology-Independent Targeted Integration (HITI), a groundbreaking CRISPR/Cas9-based genome editing technology that leverages the non-homologous end joining (NHEJ) pathway. Tailored for researchers, scientists, and drug development professionals, we examine HITI's core mechanism enabling efficient transgene integration in both dividing and non-dividing cells—a key advantage over homology-directed repair. The content details practical methodologies from vector design to clinical manufacturing, addresses critical troubleshooting for genotoxicity and optimization, and presents validation data and comparative analysis against other editing strategies. Supported by recent preclinical and emerging clinical evidence, this resource underscores HITI's transformative potential in developing durable therapies for dominant genetic disorders, cancer, and beyond.
Beyond HDR: Unveiling the Core Principles and Advantages of HITI Technology
Homology-Independent Targeted Integration (HITI) is a CRISPR/Cas9-mediated genome editing technique that leverages the non-homologous end joining (NHEJ) pathway for targeted transgene insertion. Unlike homology-directed repair (HDR), which requires homologous templates and active cell division, HITI operates throughout the cell cycle, enabling efficient gene editing in both dividing and non-dividing cells. This mechanism provides substantial advantages for therapeutic applications in primary cells, including hematopoietic stem cells and T lymphocytes, offering improved efficiency and simplified manufacturing for next-generation cell therapies.
Homology-Independent Targeted Integration (HITI) represents a paradigm shift in CRISPR-Cas9-mediated genome editing by exploiting the cell's predominant DNA repair pathway—non-homologous end joining (NHEJ). This approach circumvents the major limitation of homology-directed repair (HDR), which is inherently inefficient in many therapeutically relevant primary cells due to its dependence on specific cell cycle phases and complex repair machinery [1] [2].
The fundamental innovation of HITI lies in its engineered donor design, which incorporates Cas9 target sites as reverse complements of the genomic target. This architecture enables bidirectional selection for proper integration orientation: correctly oriented insertions disrupt the Cas9 recognition site, protecting them from repeated cleavage, while reverse integrations maintain functional Cas9 target sites, allowing for repeated cleavage until proper orientation is achieved [3]. This self-correcting mechanism drives high-fidelity integration without requiring homologous recombination machinery.
Molecular Mechanism and Comparative Advantages
HITI Workflow and Key Components
The HITI mechanism employs CRISPR-Cas9 ribonucleoprotein (RNP) complexes to create simultaneous double-strand breaks at both the genomic target locus and the donor DNA template. The cellular NHEJ machinery then ligates these broken ends, resulting in precise integration of the transgene into the designated genomic site [1] [2].
Comparative Analysis: HITI vs. HDR
Table 1: Comparative analysis of HITI versus HDR integration mechanisms
| Parameter | HITI | HDR |
|---|---|---|
| Repair Pathway | NHEJ [1] | Homology-directed repair [2] |
| Cell Cycle Dependence | Cell cycle independent [2] | Requires S/G2 phases [2] |
| Efficiency in Primary Cells | High (~21% in HSPCs) [1] | Limited [1] |
| Template Design | Reverse complement Cas9 target sites [3] | Homology arms [2] |
| Therapeutic Applications | HSPCs, T cells, post-mitotic cells [1] [2] | Limited to dividing cells [2] |
| Manufacturing Simplicity | Compatible with non-activated T cells [4] | Requires T cell activation [4] |
Experimental Implementation and Workflow
HITI Protocol for CAR-T Cell Engineering
The following protocol outlines HITI-mediated CAR knock-in into the TRAC locus for clinical-scale CAR-T cell manufacturing, adaptable to other primary cell types [2]:
Day 0: T Cell Isolation and Preparation
- Isolate primary human T cells from leukopaks using negative selection (EasySep Human T Cell Isolation Kit)
- Optional: For HITI, T cell activation is not required, unlike HDR-based approaches [4]
- Resuspend cells in TexMACS medium supplemented with IL-7 (12.5 ng/mL) and IL-15 (12.5 ng/mL) with 3% human AB serum
Day 2: Electroporation and HITI Knock-in
- Remove Dynabeads if activation was performed
- Wash cells once in electroporation buffer and resuspend at 2×10⁸ cells/mL
- Prepare RNP complex:
- Mix wild-type Cas9 (61 μM) with sgRNA (125 μM) at 2:1 molar ratio
- Incubate 10 minutes at room temperature
- Add nanoplasmid DNA (3 mg/mL) and incubate ≥10 minutes to allow RNP cutting of nanoplasmid
- Electroporate using Maxcyte GTx with Expanded T Cell 4 protocol (activated T cells) or Resting T Cell 14-3 protocol (non-activated T cells)
- Rest cells in electroporation buffer for 30 minutes post-electroporation
- Transfer to final G-Rex vessels for expansion
Days 3-14: Expansion and Enrichment
- Expand cells maintaining concentration at ~1.5×10⁶/mL
- For enrichment, apply CRISPR EnrichMENT (CEMENT) using methotrexate (MTX) selection for cells expressing dihydrofolate reductaseL22F/F31S (DHFR-FS)
- MTX treatment enriches CAR+ T cells to approximately 80% purity [2]
- Harvest cells after 14-day process, yielding 5.5×10⁸–3.6×10⁹ CAR+ T cells from starting population of 5×10⁸ T cells [2]
Critical Reagent Specifications
Table 2: Essential research reagents for HITI-based genome editing
| Reagent | Specification | Function | Optimization Notes |
|---|---|---|---|
| Cas9 Protein | Wild-type (61 μM) [2] | Creates DSBs at target loci | Use high-purity, endotoxin-free grade |
| sgRNA | TRAC-targeting: 5'-GGGAATCAAAATCGGTGAAT-3' [2] | Guides Cas9 to specific genomic locus | Include mismatch base for optimal on-target performance [4] |
| Donor Template | Nanoplasmid (450bp backbone) [2] [4] | Delivers transgene for integration | Single cut site design yields higher KI efficiency vs. 0 or 2 cut sites [4] |
| Electroporation System | Maxcyte GTx [2] | Deliver RNP and donor to cells | Use preset Expanded T-Cell 4 protocol (activated) or Resting T cell 14-3 (non-activated) [2] |
| Selection System | DHFR-FS with methotrexate [2] | Enriches successfully edited cells | Shortened MTX exposure maintains cell viability while achieving ~80% purity [2] |
Therapeutic Applications and Performance Data
Hematopoietic Stem and Progenitor Cells (HSPCs)
HITI-mediated genome editing achieves approximately 21% stable editing efficiency in repopulating human mobilized peripheral blood CD34+ HSPCs after transplantation into immunodeficient mice [1]. This approach, utilizing recombinant AAV serotype 6 vectors for donor delivery, demonstrates robust site-specific transgene integration at clinically relevant genetic loci, offering promising therapeutic potential for inherited blood disorders like leukocyte adhesion deficiency type 1 (LAD-1) [1].
CAR-T Cell Manufacturing
HITI enables non-viral, site-directed integration of chimeric antigen receptor (CAR) transgenes into the TRAC locus, achieving at least 2-fold higher cell yields compared to HDR-based approaches [2] [5]. When combined with CEMENT enrichment, this platform generates therapeutically relevant doses of 5.5×10⁸–3.6×10⁹ CAR+ T cells from a starting population of 5×10⁸ T cells across a 14-day manufacturing process [2]. The resulting CAR-T cells demonstrate functional comparability to virally transduced counterparts while eliminating requirements for viral vector manufacturing [2].
In Vivo Therapeutic Applications
HITI has been successfully applied for in vivo gene correction in Duchenne muscular dystrophy (DMD) models using AAV9 vectors, achieving editing of 1.4% of genomes in heart tissue leading to 30% transcript correction and restoration of 11% normal dystrophin levels [3]. This approach corrects mutations upstream of intron 19, potentially benefiting approximately 25% of DMD patients [3].
Safety Considerations and Limitations
Genotoxicity Assessment
Comprehensive safety profiling is essential for clinical translation of HITI-based therapies. Key considerations include:
- Off-target editing: Utilize in silico tools (COSMID, CCTop) for gRNA design and empirical methods (GUIDE-seq, CIRCLE-seq) for off-target nomination [4]
- Structural variations: Employ long-read sequencing and single primer amplification to detect large deletions, translocations, and chromothripsis not captured by standard sequencing [6] [4]
- On-target integrity: Monitor chromosomal translocations using ddPCR, particularly for loci like TRAC where chromosome 14 aneuploidy has been reported [4]
Technical Limitations and Mitigation Strategies
While HITI offers significant advantages, several limitations require consideration:
- Random integration events: Sequencing reveals occasional integration of fragmentary and recombined AAV genomes at target sites [3]
- Optimization requirements: Maximal efficiency requires careful optimization of Cas9:donor ratios (1:5 optimal for DMD correction) [3]
- Vector design constraints: Donor templates must include appropriately oriented Cas9 target sites to enable the HITI mechanism [3]
HITI represents a significant advancement in CRISPR-Cas9 genome editing technology, particularly for therapeutic applications in non-dividing and primary cells. By leveraging the efficient NHEJ pathway and incorporating a self-correcting mechanism for integration orientation, HITI achieves robust transgene integration efficiencies that surpass traditional HDR-based approaches. The compatibility with non-viral donor templates and clinical-scale manufacturing platforms positions HITI as a transformative technology for next-generation cell and gene therapies, with demonstrated applications across hematopoietic stem cells, CAR-T engineering, and in vivo gene correction. As the field advances, continued refinement of safety assessment protocols and standardization of manufacturing processes will be essential for clinical translation.
The efficacy of Homology-Independent Targeted Integration (HITI) and Homology-Directed Repair (HDR) in non-dividing cells hinges on fundamental differences in the DNA repair pathways they exploit. Non-dividing, or post-mitotic, cells constitute the majority of adult mammalian tissues, presenting a significant barrier for therapeutic genome editing strategies that rely on HDR [7] [8]. The core differentiator lies in the activity levels of their respective DNA repair mechanisms across the cell cycle. HDR is largely restricted to the S and G2 phases, as it requires a sister chromatid template, which is only available after DNA replication [4] [9]. In contrast, the Non-Homologous End Joining (NHEJ) pathway, which facilitates HITI, is active throughout all phases of the cell cycle, including the quiescent G0 phase, making it the predominant repair mechanism in non-dividing cells [4] [8]. This fundamental biological distinction is the primary reason HITI has emerged as a powerful tool for in vivo gene therapy in tissues such as the brain, retina, and airway epithelium [10] [11] [8].
Molecular Mechanisms: A Pathway-Centric View
The HDR Pathway and Its Cell Cycle Dependence
HDR is a high-fidelity process that uses a homologous DNA template to accurately repair double-strand breaks (DSBs).
- Key Steps: The pathway initiates with the MRN complex (MRE11-Rad50-NBS1) binding to the DSB. 5' to 3' resection of the DNA ends creates single-stranded overhangs, which are bound by Replication Protein A (RPA). The essential recombinase Rad51, aided by BRCA2 and PALB2, then displaces RPA to form a nucleoprotein filament. This filament performs a homology search and invades the sister chromatid, forming a D-loop structure. DNA polymerase then uses the sister chromatid as a template to synthesize the missing genetic information, leading to accurate repair [9].
- The Critical Limitation: The requirement for a sister chromatid as a repair template confines HDR activity almost exclusively to the S and G2 phases of the cell cycle [4] [9]. Consequently, in non-dividing cells, which have exited the cell cycle, HDR is inefficient or inactive, severely limiting its application for in vivo therapeutic editing [7] [8].
The NHEJ Pathway and Its Application in HITI
NHEJ is a more error-prone but universally available repair pathway that directly ligates broken DNA ends.
- Key Steps: The Ku70/Ku80 heterodimer binds to the DSB ends, recruiting DNA-PKcs. The ends may be processed by nucleases like Artemis and filled in by polymerases before being ligated by the DNA Ligase IV/XRCC4/XLF complex [9].
- The HITI Strategy: HITI co-opts the NHEJ machinery. A CRISPR-Cas9 system creates simultaneous DSBs in the genomic target and a donor DNA vector containing the transgene flanked by Cas9 cut sites. The linearized donor is then integrated into the genomic break via NHEJ [4] [8]. A key feature is that correct, forward-oriented integration disrupts the original Cas9/gRNA target sequence, preventing re-cutting, while incorrect integrations remain susceptible to further cleavage and repair attempts [8].
Quantitative Comparison of Editing Outcomes
The following tables summarize key experimental data that highlight the efficiency advantage of HITI over HDR, particularly in non-dividing cells.
Table 1: Knock-in Efficiency Comparison in Different Cell Types
| Cell Type / Model | Target Locus | HITI Knock-in Efficiency | HDR Knock-in Efficiency | Citation |
|---|---|---|---|---|
| Mouse Primary Neurons (in vitro) | Tubb3 | ~55.9% (relative in transfected cells) | Minimal to none | [8] |
| HEK293 GFP-Correction Line | GFP-IRES | Significantly higher than HDR | Lower than HITI | [8] |
| Human CD34+ HSPCs (in vivo engraftment) | Clinically relevant locus | ~21% (stable in repopulating cells) | Inefficient (NHEJ is primary pathway) | [12] |
| CAR-T Cell Manufacturing (TRAC Locus) | TRAC | At least 2-fold higher CAR+ cell yield | Lower yield | [2] |
| Adult Mouse Visual Cortex (in vivo) | Tubb3 | Achieved knock-in | Not demonstrated | [8] |
Table 2: Analysis of HITI Editing Outcomes and Fidelity
| Parameter | Finding | Implication | Citation |
|---|---|---|---|
| Indel Frequency at Junction | Majority of forward knock-ins showed no indels | HITI can achieve precise integration | [8] |
| Directional Bias | Strong bias for forward-oriented integration | Strategy enforces correct orientation of the transgene | [8] |
| Therapeutic Protein Restoration | Restored 11% of normal dystrophin levels in mouse heart (DMD model) | Functional protein can be produced from HITI-edited genes | [13] |
| Genomic Toxicity | No evidence of off-target genomic toxicity in CAR-T cells; low-level aneuploidy monitored | Acceptable safety profile for therapeutic development | [4] [2] |
Experimental Protocol: HITI-Mediated Gene Knock-in in Primary T Cells
This protocol details the application of HITI for site-specific integration of a Chimeric Antigen Receptor (CAR) into the TRAC locus of primary human T cells, as described by Balke-Want et al. [2].
Materials and Reagents
- Primary Cells: Human T cells isolated from leukopaks.
- Activation Reagent: Dynabeads Human T-Activator CD3/CD28.
- Cell Culture Media: TexMACS medium supplemented with IL-7 and IL-15 (12.5 ng/mL each).
- CRISPR Components:
- Wild-type Cas9 protein
- sgRNA targeting TRAC locus (sequence: 5'-GGGAATCAAAATCGGTGAAT-3')
- HITI Donor Template: Nanoplasmid DNA (3 mg/mL) containing the anti-GD2 CAR expression cassette flanked by the TRAC sgRNA target sequences. The nanoplasmid backbone is only ~430 bp, which helps reduce transgene silencing and cytotoxicity [4] [2].
- Electroporation System: Maxcyte GTx with appropriate processing assemblies (e.g., OC-25×3 for small scale, CL1.1 for large scale).
- Electroporation Buffer: Proprietary buffer from Maxcyte.
Step-by-Step Procedure
Day 0: T Cell Isolation and Activation
- Isolate T cells from a leukopak using negative selection.
- Activate the T cells using CD3/CD28 activation beads at a 1:1 bead-to-cell ratio.
- Culture cells in supplemented TexMACS medium.
Day 2: Electroporation
- Remove activation beads magnetically and wash cells once in electroporation buffer.
- Resuspend T cells at a concentration of 2 × 10^8 cells/mL.
- Prepare RNP Complex: Mix wild-type Cas9 and TRAC sgRNA at a 2:1 molar ratio (e.g., 61 µM Cas9 with 125 µM sgRNA) and incubate at room temperature for 10 minutes.
- Formulate Electroporation Mix: Add the required amount of nanoplasmid DNA (e.g., 5-10 µg per 5 million cells) to the pre-formed RNP complex. Incubate for at least 10 minutes to allow Cas9 to linearize the nanoplasmid donor.
- Combine the cell suspension with the RNP/nanoplasmid mix.
- Electroporation: Transfer the mixture to a Maxcyte GTx processing assembly and electroporate using the "Expanded T cell 4" protocol.
- Post-Electroporation Recovery: After electroporation, rest the cells in the processing assembly for 30 minutes before transferring them back into culture vessels with fresh, supplemented medium.
Days 3-14: Cell Expansion and Analysis
- Expand the edited T cells, maintaining the cell concentration at approximately 1.5 × 10^6 cells/mL.
- Enrichment (Optional): To enrich for successfully edited cells, implement the CEMENT strategy by incorporating a mutant DHFR-FS gene in the HITI donor. Between days 5 and 10, treat cultures with Methotrexate (MTX) to select for transgene-positive cells, which can achieve ~80% purity [4] [2].
- Analysis: On day 14, harvest cells and analyze CAR integration efficiency via flow cytometry, and assess functionality and safety through appropriate assays (e.g., cytotoxicity, cytokine release, ddPCR for karyotype) [4] [2].
The Scientist's Toolkit: Essential Reagents for HITI
Table 3: Key Research Reagent Solutions for HITI Workflows
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| Nanoplasmid DNA | Minimal plasmid backbone (~430 bp) lacking bacterial antibiotic resistance genes, reducing silencing and improving biosafety [4] [2]. | Preferred donor template for non-viral HITI in T cells. |
| Cas9 RNP Complex | Pre-complexed Ribonucleoprotein of Cas9 and sgRNA; enables rapid, transient nuclease activity with reduced off-target effects. | Standard for electroporation-based delivery in primary cells. |
| NHEJ Inhibitor (e.g., NU7026) | Small molecule inhibitor of DNA-PKcs, a key NHEJ protein. Used to confirm HITI is NHEJ-dependent [8]. | Mechanistic validation in control experiments. |
| Enrichment Marker (DHFR-FS) | Mutant dihydrofolate reductase conferring resistance to methotrexate. Allows for pharmacological selection of edited cells (CEMENT) [4] [2]. | Enrichment of HITI-edited CAR-T cells to high purity. |
| rAAV Vectors (e.g., serotype 9) | Highly efficient delivery vehicle for in vivo gene editing, particularly in tissues like muscle and retina [13] [8]. | In vivo HITI delivery for DMD therapy in mouse models. |
| In Silico Off-Target Prediction Tools (CCTop, COSMID) | Computational tools to screen and select gRNAs with high on-target and minimal off-target activity during design phase [4]. | Pre-experimental gRNA design and risk mitigation. |
Critical Considerations and Concluding Remarks
While HITI overcomes the major limitation of HDR in non-dividing cells, several aspects require careful consideration for experimental and therapeutic application.
- On-Target Genomic Aberrations: CRISPR-Cas9 editing, including HITI, can lead to unintended on-target outcomes such as large deletions, translocations, or chromosomal aneuploidy [4]. It is crucial to employ comprehensive sequencing methods (e.g., long-read sequencing, ddPCR) to monitor these events.
- Donor Design is Critical: The configuration of the donor DNA significantly impacts efficiency and precision. Using a single cut-site (1cs) donor and minimizing bacterial plasmid sequences in the final integrated product (e.g., using minicircles or nanoplasmids) enhances performance and reduces the risk of unwanted immune responses or silencing [4] [8].
- Therapeutic Scope: HITI is exceptionally well-suited for knock-in of large transgenes (e.g., CARs, full-length dystrophin mini-genes) to restore or introduce function [13] [2]. However, it is not suitable for directly correcting point mutations without an accompanying knock-in strategy, a limitation that base or prime editing may address [10] [7].
In conclusion, the core differentiator establishing HITI as a transformative technology is its exploitation of the NHEJ pathway, a ubiquitous DNA repair mechanism active in both dividing and non-dividing cells. This fundamental advantage over the cell cycle-dependent HDR pathway enables robust and precise gene integration in a wide range of therapeutically relevant post-mitotic tissues, opening new avenues for curing genetic diseases.
Homology-independent targeted integration (HITI) is a sophisticated genome-editing technique that facilitates precise DNA insertion into specific genomic loci without the need for homologous templates. Unlike homology-directed repair (HDR), which requires a template with homologous arms and is primarily active in dividing cells, HITI leverages the non-homologous end joining (NHEJ) DNA repair pathway, making it effective in both dividing and non-dividing cells [2] [14]. This capability is particularly valuable for therapeutic applications in post-mitotic cells, such as neurons and photoreceptors.
The foundational principle of HITI involves using CRISPR/Cas9 to create a double-strand break (DSB) at a predetermined genomic target site. A donor DNA construct, flanked by the same CRISPR/Cas9 target sequences in the same orientation as the genomic target, is provided. When Cas9 cleaves both the genomic locus and the donor DNA, the cellular NHEJ machinery ligates the donor fragment into the DSB, resulting in precise integration [2] [15]. This review details the HITI workflow and protocols, contextualized within the emerging research on CRISPR-associated transposase (CAST) systems for HITI, providing a practical guide for its application in genetic research and therapeutic development [14].
The Core HITI Mechanism: A Comparative Workflow
The following diagram illustrates the key steps and comparative outcomes of the HITI mechanism alongside the traditional HDR pathway.
Key Protocol Steps for HITI-Mediated Knock-in
The successful implementation of HITI requires careful execution of the following protocol, optimized for primary human T cells but adaptable to other cell types [2]:
- sgRNA Design and Complex Formation: Design sgRNAs to target a specific genomic locus (e.g., the TRAC locus for CAR-T cell generation). Synthesize the sgRNA and complex it with wild-type Cas9 protein at a molar ratio of 2:1 (sgRNA:Cas9). Incubate the ribonucleoprotein (RNP) complex for 10 minutes at room temperature [2].
- Donor Template Preparation: Clone the transgene of interest (e.g., a GD2-CAR) into a nanoplasmid donor vector. This vector should be optimized for gene therapy, featuring a minimal backbone (e.g., ~430 bp R6K origin) to prevent transgene silencing. The transgene must be flanked by the same sgRNA target sequences recognized by the RNP complex. Add this nanoplasmid DNA directly to the pre-formed RNP complex and incubate for at least 10 minutes to allow Cas9 to pre-cleave the donor plasmid [2].
- Cell Preparation and Electroporation: Isolate primary human T cells and activate them with CD3/CD28 beads. On day 2 post-activation, remove the beads and wash the cells. Resuspend the cells in electroporation buffer at a concentration of 2 × 10^8 cells/mL. Combine the cell suspension with the RNP/nanoplasmid mixture and electroporate using a system like the Maxcyte GTx. For activated T cells, use the "Expanded T cell 4" protocol; for non-activated cells, use the "Resting T cell 14–3" protocol and stimulate immediately after electroporation [2].
- Post-Electroporation Recovery and Culture: After electroporation, rest the cells in the electroporation buffer for 30 minutes. Then, transfer them to culture vessels containing TexMACS media supplemented with cytokines (e.g., IL-7 and IL-15 at 12.5 ng/mL each) and human serum. Maintain the cells at a concentration of approximately 1.5 × 10^6 cells/mL, expanding the culture volume as needed [2].
- Enrichment of Knock-in Cells (CEMENT): To enrich for successfully edited cells, incorporate a selection marker like dihydrofolate reductaseL22F/F31S (DHFR-FS) into the donor construct. Between days 4 and 10 post-electroporation, add a selection agent such as methotrexate (MTX) to the culture. This enriches the population of CAR-positive T cells to approximately 80% purity [2].
Quantitative Performance of HITI in Practice
The efficiency and yield of HITI have been quantitatively evaluated in pre-clinical studies, demonstrating its potential for clinical application. The table below summarizes key performance metrics from a study generating anti-GD2 CAR-T cells.
Table 1: Quantitative Performance Metrics of HITI-Mediated CAR Knock-in in Primary Human T Cells [2]
| Parameter | Result | Experimental Context |
|---|---|---|
| Knock-in Efficiency | ~80% purity post-CEMENT | HITI into the TRAC locus followed by methotrexate selection [2]. |
| Cell Yield | 5.5 × 10^8 – 3.6 × 10^9 CAR+ T cells | Starting from 5 × 10^8 T cells in a 14-day manufacturing process [2]. |
| Comparison to HDR | ≥2-fold higher yield | HITI yielded at least twice as many CAR-positive T cells as HDR using the same nanoplasmid donor [2]. |
| In Vivo Rodent Efficacy | Effective tumor control | HITI/CEMENT GD2 CAR-T cells mediated control of metastatic neuroblastoma in vivo [2]. |
| Therapeutic Transgene Size | Demonstrated with large constructs | HITI is effective for transgenes >5 kb [2]. |
The application of HITI extends beyond immunology to other fields, such as treating inherited retinal dystrophies. For instance, HITI-mediated gene insertion of a normal RHO gene into rod photoreceptor cells achieved integration in 80% to 90% of transduced cells and effectively restored visual function in mutant mouse models [15].
The Scientist's Toolkit: Essential Reagents for HITI
A successful HITI experiment relies on a suite of specialized reagents and tools. The following table catalogs the essential components of the HITI workflow.
Table 2: Key Research Reagent Solutions for the HITI Workflow [2] [15]
| Reagent / Tool | Function in HITI Workflow | Specifications & Examples |
|---|---|---|
| CRISPR/Cas9 System | Induces a precise DSB at the genomic target and pre-cleaves the donor template. | Wild-type S. pyogenes Cas9 protein complexed with target-specific sgRNA as an RNP complex [2]. |
| HITI Donor Vector | Provides the transgene for integration into the DSB. | Nanoplasmid with minimal backbone (e.g., R6K origin); transgene flanked by gRNA target sites [2]. Can also be delivered via AAV [15]. |
| Electroporation System | Enables efficient co-delivery of RNP and donor DNA into the target cells. | Instruments like the Maxcyte GTx with optimized protocols for specific cell types (e.g., activated or resting T cells) [2]. |
| Selection System | Enriches for cells with successful knock-in. | Integration of a selection marker (e.g., DHFR-FS) allowing for drug-based selection (e.g., with Methotrexate) in the CEMENT protocol [2]. |
| Cell Culture Media & Cytokines | Supports cell viability, recovery, and expansion post-electroporation. | Specialized media (e.g., TexMACS) supplemented with cytokines like IL-7 and IL-15 for T cell culture [2]. |
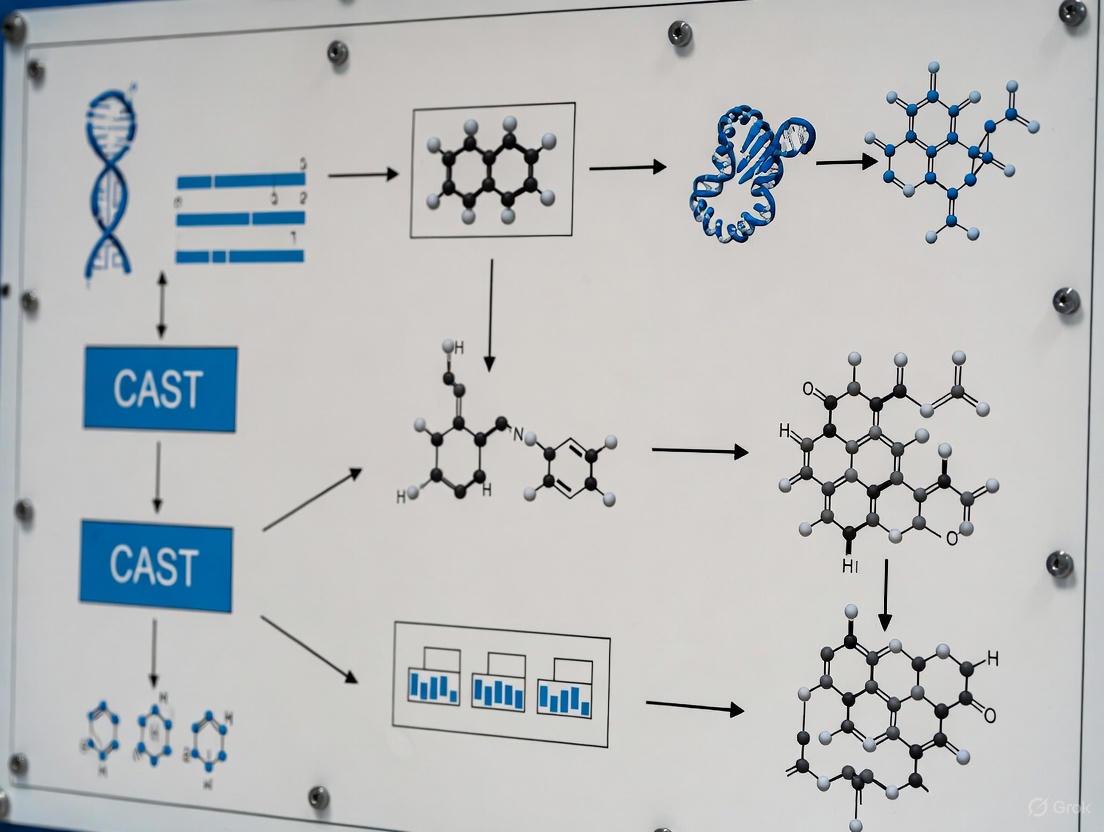
HITI in the Context of CAST Systems
The workflow and logical progression of HITI technology, particularly its relationship with newer CRISPR-associated transposase (CAST) systems, can be visualized as follows. CAST systems represent a cutting-edge evolution in homology-independent integration, leveraging RNA-guided mechanisms for precise DNA insertion.
HITI serves as a robust, well-characterized platform for homology-independent integration. Its development addressed a critical limitation of HDR—dependence on the cell cycle [14]. The principles and workflows established by HITI provide a foundational understanding for exploring and utilizing more advanced systems like CAST, which utilize RNA-guided transposases for targeted DNA insertion without creating double-strand breaks in the recipient genome [14]. This positions HITI as a crucial technological milestone and a reliable protocol for current therapeutic and research applications.
Allele-Independent Editing and Stable Expression in Proliferating Tissues
CRISPR-associated transposase (CAST) systems represent a transformative advance in genome engineering, enabling precise, targeted integration of large DNA sequences without relying on the host cell's DNA repair mechanisms. This application note details the key advantages of CAST systems, focusing on their allele-independent editing capability and capacity for stable transgene expression in proliferating cells. We provide a detailed experimental protocol for implementing a type I-F CAST system for targeted gene integration in human cells, based on the structurally engineered PseCAST system [16].
Key Advantages of CAST Systems
Allele-Independent Gene Editing
Traditional CRISPR-Cas9 editing requires cellular repair pathways (HDR or NHEJ) that are inefficient, cell-type dependent, and can produce heterogeneous editing outcomes [17] [18]. In contrast, CAST systems function independently of these pathways, leveraging a completely different mechanism for DNA integration:
- No Double-Strand Break Dependency: CAST systems catalyze the direct insertion of large DNA fragments through a transposition mechanism that bypasses DSBs, eliminating the associated DNA damage response, p53 activation, and unintended indel mutations [16] [18].
- Homology-Independent Integration: The insertion process does not require homologous sequences or donor DNA templates with extensive homology arms, simplifying vector design and improving reproducibility [18].
Table 1: Comparison of Genome Engineering Platforms
| Feature | CRISPR-Cas9 HDR | HITI | CAST Systems |
|---|---|---|---|
| Editing Mechanism | Homology-Directed Repair | Non-Homologous End Joining | RNA-guided transposition |
| Dependency on DSBs | Yes | Yes | No |
| Payload Capacity | Limited (few kb) | Limited (few kb) | Multi-kilobase (kb-scale) |
| Allele Independence | Limited (requires HDR) | Limited | Yes |
| Product Purity | Low (mixed outcomes) | Low (high indel rate) | High (precise, homogeneous) |
Stable Expression in Proliferating Tissues
CAST systems facilitate long-term transgene expression through specific integration mechanisms suited for dividing cells:
- Faithful Copy Number Integration: CAST systems typically integrate a single, full-length copy of the donor transgene, avoiding the multi-copy, concatemeric insertions common with viral vectors that can lead to gene silencing [18].
- Durable Expression in Proliferating Cells: Once integrated into the host genome, the transgene is replicated and partitioned into daughter cells during cell division, enabling persistent expression [16] [18]. This is critical for therapeutic applications involving hematopoietic stem cells or actively dividing tissues.
Table 2: Quantitative Performance of CAST Systems in Recent Studies
| CAST System | Target Cell Type | Integration Efficiency | Payload Size | Key Outcome |
|---|---|---|---|---|
| PseCAST (I-F) | Human cells [16] | Improved efficiencies with engineered variants | Multi-kilobase | Demonstrated RNA-guided transposition in human cells |
| Type I-F CASTs | E. coli [18] | Highly specific and homogeneous integration | Kilobase-level | Superior product purity compared to other subtypes |
| Type V-K CASTs | Diverse bacteria [18] | Functional in challenging industrial strains | Kilobase-level | Enabled efficient genome editing without homologous recombination |
Experimental Protocol: Targeted Gene Insertion Using Type I-F CAST
This protocol describes the use of the engineered PseCAST system for targeted, DSB-free integration of a gene of interest into the genome of human cells.
Principle
The system consists of two core modules: a DNA targeting module (QCascade complex) that uses a guide RNA to locate a specific genomic site, and an integration module (TnsA, TnsB, TnsC transposase proteins) that catalyzes the excision of a donor gene from a plasmid and its insertion into the target site. This process is independent of the cell's DNA repair machinery [16].
Materials and Reagents
Table 3: Research Reagent Solutions for CAST Experiments
| Reagent / Material | Function / Description | Example or Note |
|---|---|---|
| PseCAST Plasmid System | Provides genes for CAST machinery (QCascade & TnsABC) | Deliver as plasmid DNA or mRNA [16] |
| Donor Plasmid | Contains transgene flanked by necessary recognition sequences | Must include transposon ends recognized by TnsA/B [16] |
| Guide RNA (crRNA) | Directs QCascade complex to specific genomic target | Design guide sequence complementary to target genomic DNA [16] |
| Human Cell Line | Target for gene integration | HEK293T or other relevant cell types |
| Transfection Reagent | For delivery of CAST components into cells | Use method suitable for your cell type (e.g., lipofection, electroporation) |
| Selection Antibiotics | For enriching successfully transfected cells | Optional, depends on donor plasmid design |
Procedure
Guide RNA and Donor Plasmid Design
- gRNA Design: Design a crRNA sequence with a 20-nt spacer complementary to your target genomic locus. For the PseCAST system, ensure the target site is adjacent to a 5'-CC-3' PAM sequence [16].
- Donor Plasmid Construction: Clone your gene of interest (GOI) into a donor plasmid, ensuring it is flanked by the specific transposon end sequences (e.g., attL and attR) recognized by the TnsA and TnsB transposase proteins [16].
Cell Preparation and Transfection
- Culture human cells (e.g., HEK293T) in appropriate medium until they are 60-80% confluent.
- Co-transfect the cells with the following components using a suitable transfection method:
- Plasmids encoding the PseCAST QCascade and TnsABC proteins.
- Plasmid expressing the designed crRNA.
- Donor plasmid containing the GOI.
Incubation and Expression
- Incubate the transfected cells for 48-72 hours under standard conditions (37°C, 5% CO₂) to allow for expression of the CAST components and completion of the integration process.
Analysis and Validation
- Genomic DNA Extraction: Harvest cells and extract genomic DNA.
- Integration Efficiency Assessment: Use junctional PCR with one primer binding within the genomic target site and another binding within the integrated GOI to confirm precise insertion.
- Functional Assays: Perform relevant assays (e.g., flow cytometry, Western blot, enzymatic activity) to confirm stable expression of the integrated transgene.
Critical Steps and Troubleshooting
- Optimizing Expression: The large size of the type I-F CAST system (~8 kb coding sequence) can be a delivery challenge. Consider using mRNA delivery or split systems to improve efficiency [16].
- DNA Binding Bottleneck: The initial DNA binding by the QCascade complex can be a limiting factor. The use of structurally engineered PseCAST variants with enhanced DNA binding can mitigate this issue [16].
- Specificity Confirmation: Always sequence the integration junctions to verify on-target, precise insertion and rule out large-scale deletions or rearrangements.
The following diagram illustrates the core components and mechanism of a CRISPR-associated transposase (CAST) system for targeted gene integration.
CAST systems provide a powerful and precise alternative to DSB-dependent editors, offering a unique combination of allele-independent editing and reliable long-term transgene expression. This makes them particularly suited for therapeutic applications requiring the integration of large genetic elements into dividing cell populations, such as in ex vivo hematopoietic stem cell gene therapy [16] [18]. The continuous engineering of CAST components promises further enhancements in efficiency and specificity, solidifying their role as a next-generation genome engineering tool.
From Bench to Bedside: HITI Workflow, Delivery Systems, and Therapeutic Applications
Within the rapidly advancing field of genome engineering, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-based systems have emerged as powerful tools for targeted DNA modification. For therapeutic applications and sophisticated disease modeling, a paramount goal is the efficient knock-in of large DNA fragments. This application note focuses on the design of donor templates to maximize knock-in efficiency, specifically within the context of the CRISPR-associated transposase (CAST) system, a leading platform for homology-independent targeted integration. CAST systems facilitate the precise insertion of substantial genetic payloads without relying on endogenous DNA repair pathways, thereby offering a versatile solution for large-scale DNA engineering [19]. The following sections provide a detailed examination of CAST system performance, structured protocols for implementation, and key design principles to optimize donor templates for this innovative technology.
CAST systems represent a significant evolution in gene insertion technology by combining the programmability of CRISPR with the DNA integration capabilities of transposases. Unlike methods that create double-strand breaks (DSBs) and harness cellular repair mechanisms like homology-directed repair (HDR) or non-homologous end joining (NHEJ), CAST systems facilitate a "cut-and-paste" transposition mechanism. This process is guided by a CRISPR RNA, which directs the integration complex to a specific genomic locus without inducing DSBs, thereby minimizing unintended on-target indels and off-target effects [19].
These systems are categorized into subtypes, with type I-F and type V-K being the most well-characterized. The performance of a CAST system, particularly its integration efficiency, is highly dependent on the design of the donor DNA template. The table below summarizes the documented performance of different CAST systems across various host organisms, highlighting the critical impact of donor size and design.
Table 1: Performance Metrics of CAST Systems for Large DNA Insertion
| CAST Subtype | Host System | Donor DNA Size | Reported Efficiency | Key Features & Limitations |
|---|---|---|---|---|
| Type I-F | Escherichia coli | ~15.4 kb | Nearly 100% [19] | Stable integration; high efficiency in prokaryotes. |
| Type I-F | HEK293 Cells | ~1.3 kb | ~1% [19] | Low editing efficiency in human cells. |
| Type V-K | Escherichia coli | Up to 30 kb | High [19] | Large cargo capacity; replicative pathway. |
| Type V-K (nAnil-TnsB fusion) | HEK293T (plasmid target) | 2.6 kb | 0.06% [19] | Early-stage development for eukaryotic use. |
| V-K (MG64-1) | HEK293 Cells (AAVS1 locus) | 3.2 kb | ~3% [19] | Identified via metagenomic mining; promising for therapeutics. |
| V-K (MG64-1) | Hep3B Cells | 3.6 kb | <0.05% [19] | Highlights significant cell-type dependency. |
Donor Template Design and Workflow Protocol
A critical component of the CAST system is the donor DNA plasmid, which must be meticulously designed to serve as an effective substrate for the transposase. The following workflow and detailed protocols outline the steps for designing the donor template, delivering the CAST components, and validating successful integration.
Critical Donor Plasmid Design Specifications
The efficiency of transposition is profoundly influenced by the architecture of the donor plasmid. Adherence to the following design principles is essential:
- Essential Flanking Sequences: The donor DNA sequence to be integrated must be flanked by the specific recognition sites for the transposase TnsB. These sites are non-negotiable for the transposition reaction to occur [19].
- Orientation of Recognition Sites: The TnsB recognition sites must be in an inverted repeat orientation relative to each other. This specific configuration is crucial for the assembly of the active transposase complex and subsequent excision and integration of the donor fragment [19].
- Cargo Placement: The genetic cargo of interest (e.g., a therapeutic gene or reporter) must be located between the two TnsB recognition sites. Any sequence outside these sites will not be integrated into the genome.
Delivery and Validation Protocol
This protocol is adapted for human cell lines such as HEK293T, utilizing the type V-K CAST system.
Table 2: Reagent Solutions for CAST Genome Editing
| Reagent / Material | Function / Description | Considerations for Use |
|---|---|---|
| Donor Plasmid | Provides the DNA template for integration, flanked by TnsB sites. | Maximize plasmid quality (e.g., endotoxin-free); confirm inverted repeat orientation of TnsB sites. |
| Cas12k Expression Plasmid | Encodes the Cas protein for type V-K systems. | Alternative: Deliver as mRNA or ribonucleoprotein (RNP) complex to boost speed and reduce off-targets. |
| gRNA Expression Plasmid | Directs Cas12k to the specific genomic target site. | Critical to design gRNA with high on-target efficiency; verify PAM sequence (TTTV for Cas12k) is present. |
| TnsB & TniQ Expression Plasmids | Provide the transposase and accessory proteins for integration. | TniQ recruits TnsC to the Cas complex; optimal stoichiometry of components must be determined. |
| Transfection Reagent | Enables delivery of plasmids/molecules into cells. | For hard-to-transfect cells (e.g., iPSCs), electroporation is preferred [20]. |
Procedure:
- Cell Seeding: Seed HEK293T cells in an appropriate culture vessel to reach 70-90% confluency at the time of transfection.
- Complex Formation: Prepare the transfection mixture. A suggested starting ratio for the plasmids is Donor:Cas12k:gRNA:TnsB:TniQ = 5:2:2:2:2. This ratio requires empirical optimization for specific experimental conditions [19] [3].
- Transfection: Introduce the plasmid mixture into the cells using a high-efficiency transfection method suitable for the cell type.
- Incubation: Allow the cells to recover and express the integrated DNA for a minimum of 72 hours before analysis.
- Validation:
- Droplet Digital PCR (ddPCR): Design probes to detect the unique junction between the integrated donor and the genomic target site. This method provides absolute quantification of knock-in efficiency with high sensitivity [3] [21].
- Next-Generation Sequencing (NGS): For a comprehensive analysis of editing outcomes, design primers to create an amplicon spanning the integration site. NGS can precisely quantify the rate of correct integration and detect any spurious editing events [20].
Strategic Considerations for Enhancing Efficiency
Achieving high knock-in efficiency requires more than a correctly designed donor template. The following strategic factors are critical for success:
- Component Stoichiometry: The ratio of the delivered CAST components is a major determinant of efficiency. A higher relative amount of the donor plasmid has been shown to enhance knock-in rates in vivo. For instance, in a HITI-based DMD correction study, a Cas9:donor ratio of 1:5 yielded superior results compared to a 1:1 ratio [3]. Systematic titration of the donor, Cas protein, and transposase plasmids is highly recommended.
- Target Site Selection: Not all genomic loci are equally amenable to integration. Local chromatin architecture (e.g., open vs. closed) can dramatically influence efficiency [21] [22]. Furthermore, a study aiming to correct the SLC26A4 c.919-2A>G variant using HITI reported very low efficiency (0.15%), suggesting that the specific genomic context of that region was unfavorable for integration [20]. Preliminary screening of multiple gRNAs is advised.
- Minimizing Off-Target Effects: While CAST systems are more specific than DSB-dependent methods, off-target integration remains a concern [23]. The use of purified ribonucleoprotein (RNP) complexes instead of plasmid-based expression can reduce the duration of nuclease activity and potentially lower off-target events [22]. Comprehensive off-target analysis using unbiased methods like NGS is essential for therapeutic applications.
The following diagram illustrates the core mechanism of the type V-K CAST system, showing how the donor template and gRNA direct integration.
The efficacy of homology-independent targeted integration (HITI) for advanced genome editing is fundamentally constrained by the delivery vehicle efficiency. This application note provides a comparative analysis of two prominent delivery systems: Adeno-Associated Virus (AAV) vectors and non-viral nanoplasmid DNA. Within the context of CRISPR-associated transposon (CAST) system research, the choice of delivery vehicle impacts critical parameters including packaging capacity, integration efficiency, immunogenicity, and scalability for therapeutic development. We present structured quantitative data, detailed protocols for both delivery methods, and key reagent solutions to inform strategic decision-making for research and drug development professionals.
Technical Comparison of Delivery Vehicles
The selection between AAV and nanoplasmid delivery systems requires careful consideration of their fundamental properties, which are summarized in the following table.
Table 1: Technical Comparison of AAV and Nanoplasmid Delivery Vehicles for HITI Applications
| Characteristic | AAV Vectors | Non-Viral Nanoplasmid |
|---|---|---|
| Packaging Capacity | Limited (<4.7 kb) [24] | Significantly larger; can accommodate entire CAST systems and donor templates [2] |
| Backbone Size | N/A (viral capsid) | ~430-500 bp minimal backbone [25] [2] |
| Selection System | N/A | RNA-OUT antibiotic-free selection [25] |
| Integration Mechanism | Primarily episomal; limited integration | Designed for NHEJ/HITI-mediated integration [2] |
| Immunogenicity | Moderate to high; pre-existing immunity common [26] | Lower immunogenicity profile [27] |
| Manufacturing Scalability | Complex and costly viral production [26] [27] | Simplified, cost-effective bacterial fermentation [25] |
| Typical HITI Efficiency | Varies by serotype and tissue (e.g., 20% in retinal cells) [20] | High in primary cells (e.g., 2-fold greater yield than HDR in T-cells) [2] |
Experimental Protocols
Nanoplasmid-Mediated HITI in Primary Human T-Cells
This protocol, adapted from Balke-Want et al. [2], details the knock-in of a therapeutic transgene into the TRAC locus using nanoplasmid DNA and HITI, achieving high yields of engineered cells suitable for clinical-scale manufacturing.
Key Reagents:
- Nanoplasmid DNA: Designed with R6K origin of replication and RNA-OUT selection marker, resuspended at 3 mg/mL in H₂O [2].
- Cells: Primary human T-cells isolated from leukopaks via negative selection.
- Activation: Dynabeads Human T-Activator CD3/CD28 at a 1:1 bead-to-cell ratio.
- Culture Media: TexMACS medium supplemented with IL-7 (12.5 ng/mL) and IL-15 (12.5 ng/mL).
Step-by-Step Procedure:
- Day 0: T-Cell Activation. Isolate T-cells and activate them with CD3/CD28 beads in culture media.
- Day 2: Electroporation Preparation.
- Magnetically remove activation beads.
- Count cells and wash once in electroporation buffer.
- Resuspend cells at a concentration of 2 × 10⁸ cells/mL.
- Prepare RNP complex by mixing wild-type Cas9 (61 µM) and sgRNA (125 µM) at a 2:1 molar ratio and incubating for 10 minutes at room temperature.
- Add the required amount of nanoplasmid DNA (e.g., 5-10 µg per 5×10⁶ cells) to the pre-formed RNP complex and incubate for ≥10 minutes to allow RNP cleavage of the nanoplasmid.
- Day 2: Electroporation.
- Combine cell suspension with the RNP/nanoplasmid mixture.
- Electroporate using the Maxcyte GTx system with the "Expanded T cell 4" protocol for activated T-cells.
- Post-electroporation, rest cells in the electroporation buffer for 30 minutes before transferring them back into culture media.
- Days 3-14: Cell Expansion and Analysis.
- Expand cells in G-Rex vessels, maintaining a density of ~1.5 × 10⁶ cells/mL.
- Assess knock-in efficiency around Day 7-10 via flow cytometry for surface marker expression or genomic analysis.
- For enrichment, apply CRISPR EnrichMENT (CEMENT) using a selection marker like DHFR-FS and methotrexate to achieve >80% purity of knock-in cells [2].
The workflow for this protocol is illustrated below.
AAV-Mediated In Vivo HITI Delivery
This protocol outlines the use of recombinant AAV for delivering HITI components in vivo, a strategy constrained by the vector's limited packaging capacity but valuable for direct in vivo applications [24] [28].
Key Reagents:
Step-by-Step Procedure:
- Vector Design and Packaging:
- All-in-One AAV: Clone a compact Cas ortholog (e.g., Cas12f, IscB, or TnpB) and its guide RNA into a single AAV vector. This is suitable for edits <4.7 kb [24].
- Dual AAV System: Package the Cas nuclease and the HITI donor template (containing the transgene flanked by AAV inverted terminal repeats and CRISPR target sites) into separate AAV vectors. A third vector for sgRNA may be required [24].
- Vector Production and Purification: Produce high-titer rAAV vectors using standard triple-transfection in HEK293 cells or baculovirus-insect cell systems, followed by purification via ultracentrifugation or chromatography.
- In Vivo Administration:
- Dosage: Determine the optimal titer based on the target organ. High doses (e.g., >1e14 vg/kg for systemic delivery) are often needed but carry toxicity risks [26].
- Route of Administration: Inject via a route appropriate for the target tissue (e.g., systemic intravenous for liver, subretinal for retina [24] [20]).
- Efficiency and Safety Assessment:
- After 1-4 weeks, analyze target tissues for HITI integration efficiency using next-generation sequencing (NGS) [20].
- Assess potential off-target effects and immune responses (e.g., hepatotoxicity).
- Vector Design and Packaging:
The logical workflow for implementing an AAV-HITI strategy is as follows.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of HITI strategies relies on a core set of specialized reagents. The following table outlines essential solutions and their functions.
Table 2: Key Research Reagent Solutions for HITI Experiments
| Reagent / Solution | Function / Application | Key Features / Examples |
|---|---|---|
| Nanoplasmid DNA Backbone | Non-viral delivery vector for HITI templates [25] [2] | - ~430-500 bp minimal backbone [2]- R6K origin of replication- RNA-OUT antibiotic-free selection [25] |
| RNA-OUT Selection System | Antibiotic-free plasmid maintenance in bacteria [25] | - 150 bp antisense RNA represses toxic SacB marker- Allows high-yield manufacturing without antibiotic resistance genes |
| Compact Cas Orthologs | Enables all-in-one AAV packaging [24] | - Cas12f, IscB, TnpB, SaCas9, CjCas9- Small molecular size fits AAV capacity |
| Electroporation Systems | Non-viral delivery of RNP and nanoplasmid to primary cells [2] [29] | - Maxcyte GTx with optimized T-cell protocols- High viability and efficiency in sensitive primary cells |
| CRISPR EnrichMENT (CEMENT) | Postsynthetic enrichment of HITI-edited cells [2] | - Uses selection marker (e.g., DHFR-FS) and drug (Methotrexate)- Enriches knock-in cells to >80% purity |
Discussion and Future Perspectives
The quantitative data and protocols presented herein underscore a clear technological trade-off. Nanoplasmids offer superior packaging capacity, simplified manufacturing, and high HITI efficiency in ex vivo settings like CAR-T cell manufacturing [2]. In contrast, AAV vectors provide excellent transduction efficiency for in vivo delivery but are severely limited by packaging constraints and immunogenicity concerns [26] [24].
For CAST system research, which often involves large multi-component assemblies, nanoplasmids currently present a more viable delivery solution for ex vivo applications. However, innovations in AAV technology, such as the development of hypercompact editors and trans-splicing systems, are crucial for advancing in vivo HITI-based therapies [24]. Future developments in lipid nanoparticles (LNPs) and other non-viral carriers may further bridge the gap between delivery efficiency and cargo capacity, opening new avenues for therapeutic genome editing [27].
Application Notes
The CRISPR-associated transposase (CAST) system represents a transformative advancement in genome engineering, enabling homology-independent, RNA-guided integration of large DNA cargo. This technology is particularly suited for addressing loss-of-function genetic disorders, as it allows for the one-time, allele-agnostic installation of therapeutic genes at specific genomic loci without relying on double-strand break (DSB) repair pathways [30] [18]. Its application in vivo for complex tissues such as the retina and liver offers a promising therapeutic pathway for conditions that have been historically challenging to treat.
Key Advantages for In Vivo Therapy
CAST systems offer several distinct benefits for in vivo gene therapy compared to traditional CRISPR-Cas systems or viral gene addition:
- DSB-Free Integration: Unlike CRISPR-Cas9 which induces double-strand breaks, CAST systems facilitate "cut-and-paste" transposition, avoiding the inherent risks of indel formation, chromosomal translocations, and p53 activation associated with DSBs [30] [18].
- Large Cargo Capacity: CAST systems can integrate DNA fragments ranging from 1 kb to over 10 kb, enabling the delivery of full-length cDNA sequences for most genes, which is a significant limitation for prime editing technologies [30] [18].
- Homology- and Cell Cycle-Independence: The integration mechanism does not depend on host homology-directed repair (HDR) machinery, making it effective in both dividing and, crucially, non-dividing cells such as neurons and photoreceptors [19] [30].
- High Product Purity and Unidirectional Insertion: Evolved CAST (evoCAST) systems produce predominantly precise, unidirectional integration products with minimal byproduct formation, a key advantage for therapeutic safety and predictability [30].
Quantitative Profile of Advanced CAST Systems
Table 1: Performance Metrics of Evolved CAST Systems in Human Cells
| System | Average Integration Efficiency | Cargo Size Demonstrated | Indel Formation | Key Feature |
|---|---|---|---|---|
| Wild-type PseCAST | < 0.1% | ~1.3 kb | Undetectable | Minimal baseline activity in human cells [30] |
| PseCAST + ClpX | ~1% | ~1.3 kb | Undetectable | Bacterial unfoldase supplement boosts activity [30] |
| Evolved CAST (evoCAST) | 10-25% (across 14 genomic loci) | Kilobase-scale | Undetectable | Protein-evolved variant; therapeutically relevant efficiency [30] |
| Type V-K CAST (MG64-1) | ~3% (at AAVS1 locus) | 3.2 - 3.6 kb | Data not specified | Identified via metagenomic mining [19] |
Experimental Protocols
The following protocols detail the application of CAST systems for in vivo gene therapy development for retinal degeneration and liver fibrosis. These methodologies are adapted from recent breakthrough studies and are designed for preclinical model systems.
Protocol 1: Targeted Gene Insertion for Retinal Degeneration in a Mouse Model
This protocol outlines the use of an evolved CAST system to install a healthy cDNA copy of a mutated gene into a defined "safe harbor" locus in retinal cells, providing a universal strategy for various loss-of-function mutations causing diseases like retinitis pigmentosa (RP) and Leber hereditary optic neuropathy (LHON) [31] [30].
Research Reagent Solutions
Table 2: Essential Reagents for Retinal Gene Integration
| Reagent | Function | Example or Specification |
|---|---|---|
| evoCAST Ribonucleoprotein (RNP) Complex | Catalyzes RNA-guided transposition | Purified evolved TnsA, TnsB, TnsC, and QCascade complex [30] |
| Target-Specific gRNA | Directs CAST complex to genomic locus | sgRNA targeting the 3' end of the ALB intron 1 or a safe harbor locus [30] |
| ssAAV Donor Template | Carries therapeutic transposon cargo | Single-stranded AAV vector containing therapeutic cDNA (e.g., CNGA1 for RP), flanked by the necessary ~150 bp transposon ends [32] [30] |
| Subretinal Injection Delivery System | Enables localized in vivo delivery | NanoFil syringe with a 36-gauge blunt-end needle [32] |
Detailed Methodology
Vector Design and Production:
- Clone the therapeutic cDNA (e.g., a 2.5 kb wild-type CNGA1 cDNA for a form of RP) into an ssAAV donor vector between the defined ~150 bp transposon ends recognized by the evoCAST system [30].
- Package the donor construct into an AAV serotype with high tropism for photoreceptor cells (e.g., AAV8, as used in retinal gene therapy studies) and purify via iodixanol gradient ultracentrifugation [32].
- Complex the purified evoCAST proteins with the target-specific sgRNA to form the RNP complex.
In Vivo Delivery:
- Anesthetize 2-week-old Cnga1-/- mice using an intraperitoneal injection of pentobarbital sodium (50 mg/kg) [32].
- Dilate pupils with a topical application of 0.5% tropicamide and 0.5% phenylephrine hydrochloride.
- Using a surgical microscope, make a small incision through the sclera behind the iris with a 32-gauge needle.
- Insert a 36-gauge blunt-end needle attached to a NanoFil syringe into the subretinal space.
- Co-inject a total of 1 µL containing the evoCAST RNP complex and the ssAAV donor template (∼1×10^12 vg/mL). A visible bleb confirms successful subretinal delivery [32] [30].
Functional and Structural Validation:
- Electroretinography (ERG): At 1-month and 3-months post-injection, record scotopic (rod-mediated) and photopic (cone-mediated) ERG responses under anesthesia to assess restoration of retinal function [32].
- Optical Coherence Tomography (OCT): Perform in vivo longitudinal imaging to measure the thickness of the outer nuclear layer (ONL), which contains photoreceptor nuclei, to evaluate photoreceptor survival [32].
- Immunohistochemistry: At the study endpoint, analyze retinal sections for correct localization of the expressed therapeutic protein (e.g., CNGA1 protein in rod outer segments) and markers of healthy photoreceptors [32].
Diagram 1: Workflow for retinal gene integration.
Protocol 2: Nanoparticle-Mediated CAST Delivery for Liver Fibrosis
This protocol combines the high payload capacity of the CAST system with the targeted delivery capabilities of nanoparticles (NPs) to deliver a therapeutic transgene specifically to activated hepatic stellate cells (HSCs), the primary effector cells in liver fibrosis [33] [34].
Research Reagent Solutions
Table 3: Essential Reagents for Liver-Targeted Integration
| Reagent | Function | Example or Specification |
|---|---|---|
| Lipid Nanoparticles (LNPs) | In vivo delivery vehicle for CAST components | LNPs composed of ionizable lipid, phospholipid, cholesterol, and PEG-lipid [33] |
| Targeting Ligand-Modified LNPs | Enables cell-specific targeting | LNPs surface-functionalized with a ligand for the PDGFβ receptor, highly expressed on activated HSCs [33] |
| mRNA Encoding CAST System | Provides transient expression of transposase | mRNA encoding evolved TnsA, TnsB, TnsC, and Cas12k/Cascade [30] |
| pDNA Donor Template | Carries therapeutic transposon | Plasmid DNA containing an anti-fibrotic gene (e.g., TGF-β antagonist) flanked by transposon ends [30] |
Detailed Methodology
LNP Formulation and Characterization:
- Formulate LNPs using microfluidic mixing to encapsulate both the mRNA encoding the evoCAST components and the pDNA donor template.
- Incorporate a PDGFβR-targeting peptide onto the LNP surface via PEG-lipid conjugation to achieve active targeting of activated HSCs [33].
- Characterize the final LNP preparation for size (aim for 80-150 nm), polydispersity index, zeta potential, and encapsulation efficiency using dynamic light scattering.
In Vivo Dosing in a Fibrosis Model:
- Induce liver fibrosis in a mouse model via repeated injections of carbon tetrachloride (CCl₄) or a methionine-choline-deficient (MCD) diet.
- Administer the targeted LNPs intravenously via the tail vein at a dose of ~0.5 mg/kg mRNA and ~1.0 mg/kg pDNA when fibrosis is established.
- Repeat dosing if necessary based on pharmacokinetic and efficacy data.
Efficacy and Safety Assessment:
- Histological Analysis: Score liver sections stained with Sirius Red or Masson's Trichrome to quantify collagen deposition and fibrosis stage.
- Biochemical Assays: Measure serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) to monitor liver injury and treatment-related toxicity.
- qPCR and Western Blot: Quantify the integration efficiency of the therapeutic transgene and its expression in isolated HSCs. Assess the downregulation of pro-fibrotic markers (e.g., α-SMA, collagen I).
- Off-Target Analysis: Use GUIDE-seq or similar unbiased methods to profile the genome-wide specificity of integration [30].
Diagram 2: Nanoparticle delivery for liver therapy.
Chimeric Antigen Receptor (CAR)-T cell therapy has revolutionized the treatment of hematological malignancies, yet its widespread adoption faces significant challenges related to manufacturing complexity, cost, and scalability. Homology-Independent Targeted Insertion (HITI) has emerged as a powerful CRISPR-Cas9-based genome editing strategy that leverages the non-homologous end joining (NHEJ) DNA repair pathway to enable efficient transgene integration. Unlike homology-directed repair (HDR), which is active only in specific cell cycle phases, NHEJ operates throughout the cell cycle, making HITI particularly suitable for engineering primary human T cells [2] [4].
The CAST system (CRISPR-associated transposase systems) for homology-independent targeted integration represents a paradigm shift in cell engineering methodologies. By eliminating the reliance on viral vectors and homology arms, HITI streamlines the manufacturing process while maintaining precision. This application note details the implementation of HITI for CAR insertion into the T Cell Receptor Alpha Constant (TRAC) locus, enabling simultaneous CAR expression and endogenous TCR disruption [2].
Theoretical Foundation: HITI Mechanism and Advantages
Molecular Mechanism of HITI
HITI utilizes CRISPR-Cas9 to create double-strand breaks (DSBs) at both the genomic target site and the donor DNA vector. The repair of these breaks via NHEJ results in the integration of the transgene cargo into the genome. The strategic design of the donor plasmid is crucial—it contains the CAR transgene flanked by Cas9 guide RNA (gRNA) target sequences that are reverse complements of the genomic target sites. This design enables re-cleavage and correction of reverse integrations, ensuring high efficiency and directional accuracy [2] [4] [35].
Comparative Advantages of HITI for CAR-T Manufacturing
Cell cycle independence represents a fundamental advantage of HITI over HDR-based approaches. Since NHEJ is active throughout all phases of the cell cycle, HITI enables efficient gene editing in both activated and non-activated T cells, providing greater flexibility in manufacturing workflows [4]. This characteristic is particularly valuable for clinical-scale production where cell synchronization is impractical.
Additional benefits include:
- Reduced manufacturing complexity: Elimination of homology arms simplifies vector design
- Higher yield: HITI generates at least 2-fold more CAR-T cells compared to HDR in primary human T cells [2]
- Versatile cargo capacity: Successful integration of large transgenes (>5 kb) including promoter elements and multiple exons [13] [3]
- Reduced pre-stimulation requirements: Potential for engineering non-activated T cells, potentially enhancing safety profiles [4]
Experimental Protocol: HITI-Mediated CAR-T Cell Generation
T Cell Isolation and Culture
Begin with fresh leukopaks from human donors. Isplicate T cells using negative selection with the EasySep Human T Cell Isolation Kit. Activate isolated T cells with Dynabeads Human T-Activator CD3/CD28 at a 1:1 bead-to-cell ratio. Culture cells in TexMACS medium supplemented with 12.5 ng/mL human IL-7 and 12.5 ng/mL IL-15, plus 3% human male AB serum. Maintain cells at approximately 1.5 × 10^6 cells/mL using appropriate culture vessels such as G-Rex plates [2].
gRNA Design and RNP Complex Formation
Design gRNAs targeting the TRAC locus (e.g., 5'-GGGAATCAAAATCGGTGAAT-3') [2]. For optimal results:
- Utilize bioinformatics tools (COSMID, CCTop) for gRNA selection and off-target prediction [4]
- Include a mismatch base in the gRNA sequence to enhance specificity
- Form ribonucleoprotein (RNP) complexes by mixing wild-type Cas9 (61 µM) with sgRNA (125 µM) at a 2:1 molar ratio (sgRNA:Cas9)
- Incubate the mixture for 10 minutes at room temperature to allow RNP complex formation [2]
Nanoplasmid Donor Design and Preparation
Employ nanoplasmid vectors optimized for gene therapy applications. Key features include:
- R6K origin of replication and antibiotic-free selection system
- Minimal backbone size (~430 bp) to reduce cytotoxicity and prevent transgene silencing [2] [4]
- CAR expression cassette flanked by single gRNA cut sites (not two) for optimal efficiency [4]
- Anti-GD2 CAR transgene followed by enrichment markers (DHFR-FS, tEGFR, or tNGFR)
Clone the CAR construct into the nanoplasmid backbone using NheI and KpnI restriction sites. Produce high-quality nanoplasmid DNA at concentrations of 3 mg/mL in sterile water [2].
Electroporation Process
On day 2 post-activation, magnetically remove Dynabeads and count cells. Wash cells once in electroporation buffer and resuspend at 2 × 10^8 cells/mL. Add the predetermined amount of nanoplasmid DNA (typically 1-2 µg per 10^6 cells) to the pre-formed RNP complex and incubate for at least 10 minutes to allow RNP-mediated linearization of the nanoplasmid. Electroporate using the Maxcyte GTx system with the Expanded T Cell 4 protocol for activated T cells or the Resting T Cell 14-3 protocol for non-activated T cells. After electroporation, rest cells in the electroporation buffer for 30 minutes before transferring to final culture vessels [2] [4].
Post-Electroporation Culture and CRISPR EnrichMENT (CEMENT)
Following electroporation, continue culturing cells in cytokine-supplemented media. For enrichment of successfully edited cells, implement the CEMENT system using integrated selection markers:
Table 1: Enrichment Strategies for HITI-Edited CAR-T Cells
| Selection Method | Mechanism | Efficiency | Advantages | Limitations |
|---|---|---|---|---|
| DHFR-FS + Methotrexate | Metabolic selection with methotrexate-resistant dihydrofolate reductase | ~80% CAR+ purity [2] | Scalable, cost-effective, compatible with closed systems | Requires optimization of timing/dosage |
| tEGFR | Surface marker detection with anti-EGFR antibodies | Variable | Rapid detection, magnetic separation | Additional processing steps, yield loss |
| tNGFR | Surface marker detection with anti-NGFR antibodies | Variable | Rapid detection, magnetic separation | Additional processing steps, yield loss |
For DHFR-FS-based enrichment, add methotrexate (MTX) to the culture media during the expansion phase. Optimize MTX concentration and exposure duration to achieve effective selection while maintaining cell viability [2] [4].
Analytical and Functional Validation
Comprehensive characterization of HITI-edited CAR-T cells should include:
- Flow cytometry to quantify CAR expression and T cell phenotype
- ddPCR to assess vector copy number and chromosomal integrity
- Next-generation sequencing (GUIDE-seq, rhAMPSeq) to evaluate on-target efficiency and off-target effects [4]
- Functional assays including cytokine release, cytotoxicity, and exhaustion markers
- In vivo models to assess antitumor efficacy and persistence [2]
Performance Data and Benchmarking
Quantitative Assessment of HITI Efficiency
Rigorous evaluation of HITI-edited CAR-T cells from multiple donors demonstrates robust manufacturing outcomes:
Table 2: HITI Performance Metrics for Clinical-Scale CAR-T Manufacturing
| Parameter | HITI Performance | HDR Benchmark | Significance |
|---|---|---|---|
| Cell Yield | 5.5 × 10^8 – 3.6 × 10^9 CAR+ cells from 5 × 10^8 input T cells [2] | ~2-fold lower than HITI [2] | Meets clinical dosing requirements |
| Purity Post-CEMENT | ~80% CAR+ cells [2] | Variable without selection | Reduces need for additional purification |
| TRAC Disruption | Efficient knockout | Similar efficiency | Ensures endogenous TCR disruption |
| Off-target Editing | Minimal with optimized gRNA [2] [4] | Comparable | Acceptable safety profile |
| Functional Potency | Equivalent to viral transduced CAR-T cells [2] | Similar when achieved | Therapeutically relevant |
Process Workflow and Timeline
The complete HITI-based CAR-T manufacturing process requires 14 days from leukopak to final product, comparing favorably with viral manufacturing processes that typically require longer durations [2].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of HITI for CAR-T cell generation requires the following key reagents and systems:
Table 3: Essential Research Reagents for HITI CAR-T Cell Generation
| Reagent/System | Function | Specifications | Alternative/Notes |
|---|---|---|---|
| Nanoplasmid DNA | Donor vector for CAR transgene | R6K origin, ~430 bp backbone, antibiotic-free selection [2] [4] | Superior to conventional plasmids for reduced cytotoxicity |
| High-fidelity Cas9 | CRISPR nuclease for DSB generation | Wild-type, 61 µM working concentration [2] | Can be substituted with other precise nucleases (e.g., Cas12a) |
| TRAC-specific gRNA | Targets TRAC locus for integration | Sequence: 5'-GGGAATCAAAATCGGTGAAT-3' [2] | Mismatch base included for enhanced specificity |
| Electroporation System | Delivery of RNP and donor DNA | Maxcyte GTx with Expanded T Cell protocol [2] | CL1.1 assembly for GMP-compatible scale-up |
| Enrichment Marker | Selection of successfully edited cells | DHFR-FS, tEGFR, or tNGFR [2] [4] | DHFR-FS enables methotrexate-based selection |
| Cell Culture Platform | T cell expansion and maintenance | G-Rex vessels with IL-7/IL-15 supplementation [2] | Enables gas-permeable rapid expansion |
Troubleshooting and Optimization Guidelines
Common Challenges and Solutions
- Low Knock-in Efficiency: Optimize RNP:donor ratio (typically 2:1 molar ratio sgRNA:Cas9), verify nanoplasmid quality, and ensure proper electroporation parameters [2]
- Poor Cell Viability Post-Electroporation: Reduce DNA concentration, optimize electroporation buffer, and ensure proper post-electroporation recovery conditions [4]
- Incomplete TCR Disruption: Verify gRNA activity using T7E1 assay, optimize RNP concentration, and consider using multiple gRNAs targeting TRAC [2]
- Variable Enrichment Efficiency: Titrate methotrexate concentration (for DHFR-FS) and optimize timing of selection pressure application [2] [4]
Safety Assessment and Mitigation Strategies
Implement comprehensive genotoxicity screening:
- Utilize ddPCR to monitor chromosomal translocations and aneuploidy [4]
- Perform genome-wide insertion site analysis (LAM-PCR, TLA) to identify potential genotoxic events [2]
- Employ GUIDE-seq or CIRCLE-seq for unbiased off-target nomination [4]
- Conduct long-term persistence studies in relevant animal models to assess functional safety [2]
HITI technology represents a significant advancement in non-viral CAR-T cell engineering, offering a streamlined manufacturing process that addresses critical bottlenecks in current cell therapy production. The protocol outlined herein enables researchers to consistently generate therapeutically relevant doses of CAR-T cells with functional properties equivalent to virally transduced products [2].
The integration of HITI with the CAST system framework provides a versatile platform that may be extended to other therapeutic cell engineering applications beyond CAR-T cells, including CAR-NK and CAR-macrophage therapies [36]. Future developments may focus on further enhancing the specificity and efficiency of integration through novel computational design tools [37] and potentially adapting the system for in vivo applications [36] [38] [39].
As the field advances, HITI-based manufacturing is poised to increase accessibility to CAR-T cell therapies by reducing costs and complexity while maintaining the high quality standards required for clinical applications [2].
Navigating Challenges: A Guide to Enhancing HITI Efficiency and Safety
The advent of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated transposases (CASTs) represents a significant leap forward for homology-independent targeted integration of large DNA cargos. Unlike traditional methods that rely on homology-directed repair (HDR), CAST systems facilitate precise, one-step integration of kilobase-scale transgenes without requiring donor DNA templates or cellular replication phases. This technology is particularly transformative for therapeutic applications, including gene correction therapies and the engineering of chimeric antigen receptor (CAR) T-cells, where it enables mutation-agnostic treatments for loss-of-function genetic diseases. The efficiency of these systems, however, hinges on two critical experimental pillars: the rational design of guide RNAs (gRNAs) and the optimization of electroporation parameters for delivery. This application note provides a detailed framework for maximizing editing efficiency in CAST-based experiments through optimized gRNA design and electroporation protocols.
Comprehensive gRNA Design for CAST Systems
Designing a highly efficient gRNA is the foremost step for successful CAST system application. While CASTs use a nuclease-deficient Cas, the gRNA remains paramount for directing the complex to the specific genomic target. The principles of gRNA design for CAST systems share similarities with traditional CRISPR-Cas9 systems but require special considerations for homology-independent integration.
Fundamental gRNA Design Parameters
The foundational parameters for effective gRNA design focus on ensuring on-target activity and minimizing off-target effects. The target sequence must be unique within the genome to ensure specificity, a consideration especially critical in polyploid organisms or genomes with high repetitive content [40]. The target site must also be adjacent to a compatible Protospacer Adjacent Motif (PAM) sequence. While the canonical PAM for Streptococcus pyogenes Cas9 is 5'-NGG-3', CAST systems may utilize different Cas proteins with distinct PAM requirements [41]. The seed sequence (the 8–10 bases at the 3' end of the gRNA targeting sequence) requires perfect homology to the target DNA, as mismatches in this region are known to significantly inhibit target binding and complex activity [41].
Advanced Design Strategies for Enhanced Specificity
To enhance the specificity of genomic integration, leverage multiplexed gRNA strategies. Using two or more gRNAs targeting the same locus can significantly increase the efficiency of large cargo integration and is particularly useful for defining the boundaries of large genomic deletions or inversions [41]. Furthermore, a thorough in silico off-target analysis is non-negotiable. Utilize BLAST and specialized gRNA design tools to scan the entire genome for sequences with partial homology to your gRNA, particularly those with mismatches in the 5' region distal to the PAM, which are more permissive of cleavage in nuclease-active systems [40].
The physical properties of the gRNA itself also contribute to its efficiency. Assess the secondary structure and Gibbs free energy of the synthesized gRNA. Stable secondary structures can occlude the spacer region, impairing its ability to bind the target DNA. Tools that predict secondary structure and calculate binding stability are essential for selecting gRNAs with optimal physical characteristics [40].
Table 1: Key Parameters for Efficient gRNA Design
| Parameter | Description | Optimal Characteristic |
|---|---|---|
| Specificity | Uniqueness of the target sequence within the genome [40] | Unique match with no or minimal off-target sites |
| PAM Sequence | Short sequence adjacent to the target site required for Cas binding [41] | Matches the requirement of the specific Cas protein in use |
| Seed Sequence | 8-10 nucleotides at the 3' end of the gRNA spacer [41] | Perfect homology to the target DNA sequence |
| GC Content | Proportion of guanine and cytosine nucleotides in the spacer | 40-60% |
| Polymerase | RNA polymerase used for gRNA expression (e.g., U6) [42] | Avoids a guanine (G) at the first position of the spacer |
gRNA Design Workflow
The following diagram illustrates a systematic workflow for designing and validating efficient gRNAs for CAST-based experiments.
Optimizing Electroporation for CAST Delivery
Electroporation is a cornerstone technique for delivering CAST components into target cells. Optimizing this process is critical for achieving high editing efficiency while maintaining superior cell viability.
Core Electroporation Principles
Electroporation uses a controlled electric pulse to create transient pores in the cell membrane, allowing payloads like CAST plasmids, ribonucleoproteins (RNPs), or nucleic acids to enter the cell [43]. The key to success lies in balancing the electric parameters to achieve sufficient membrane permeability without causing irreversible damage that leads to cell death. The composition of the electroporation buffer is also vital; its conductivity and osmolarity must be optimized to protect cell integrity during the procedure. Commercially available, cell-type-specific buffers often provide the most consistent results [42] [43].
Critical Electroporation Parameters
The electric pulse characteristics—including waveform, voltage, duration, and number of pulses—are the most critical variables. Different cell types have distinct membrane properties, necessitating tailored protocols. For example, primary T cells and delicate stem cells require gentler protocols compared to hardy cell lines like HEK293T [42] [2]. Utilizing pre-optimized protocols from instrument manufacturers can drastically reduce optimization time.
The form of the CAST payload significantly influences outcomes. Delivering pre-assembled Cas RNP complexes (where the Cas protein is complexed with the gRNA) is often superior to plasmid DNA, as it leads to faster activity, reduced off-target effects, and higher editing efficiency in primary cells [2] [44]. When using DNA payloads, such as the transposon donor, nanoplasmid or minicircle DNA are advanced options. These are characterized by a minimized bacterial backbone, which reduces cytotoxicity and can improve knock-in efficiency compared to standard plasmids [2] [44].
Finally, proper post-electroporation handling is crucial for cell recovery. After pulsing, cells should be rested in the electroporation buffer for a short period (e.g., 30 minutes) before being transferred to pre-warmed, nutrient-rich culture media supplemented with appropriate cytokines and growth factors to support recovery and proliferation [2].
Table 2: Key Parameters for Electroporation Optimization
| Parameter | Impact on Efficiency | Optimization Guidance |
|---|---|---|
| Cell Health & Viability | Primary determinant of post-electroporation recovery | Use cells in log-phase growth; ensure high pre-electroporation viability (>90%) [2] |
| Payload Form | Affects delivery efficiency, timing, and cytotoxicity | Use RNP for Cas/gRNA and nanoplasmid/minicircle for donor DNA [2] [44] |
| Electric Pulse | Creates membrane pores for payload entry | Use cell-type-specific pre-optimized protocols (e.g., "Expanded T cell" for T cells) [2] [43] |
| Electroporation Buffer | Maintains cell viability during procedure | Use specialized, low-conductivity buffers; avoid standard phosphate-buffered saline [42] [43] |
| Cell Concentration | Influences pulse efficiency and payload delivery | Typically 1-2 x 10^8 cells/mL for primary T cells [2] |
| Post-Transfection Rest | Allows membrane resealing and cell recovery | Rest cells for 30 min in electroporation buffer before transferring to culture media [2] |
Integrated Electroporation Workflow
The following diagram outlines a standardized workflow for the electroporation of CAST components into primary human T cells, a key therapeutic cell type.
Integrated Application Protocol: HITI in Primary T Cells
This protocol details the application of optimized gRNA design and electroporation for homology-independent targeted integration (HITI) of a CAR transgene into the TRAC locus of primary human T cells using the CAST system, based on a validated clinical-scale manufacturing process [2].
Materials and Reagents
Table 3: Research Reagent Solutions for CAST-based HITI
| Reagent/Kit | Function/Description | Example/Source |
|---|---|---|
| T Cell Isolation Kit | Negative selection to purify primary human T cells from leukopaks. | EasySep Human T Cell Isolation Kit [2] |
| T Cell Activator | Activates T cells via CD3/CD28 receptors to induce proliferation. | Dynabeads Human T-Activator CD3/CD28 [2] |
| Cell Culture Media | Supports growth and expansion of primary T cells. | TexMACS medium with IL-7 & IL-15 cytokines [2] |
| Wildtype Cas9 Nuclease | The DNA-binding module of the CAST system. | Integrated DNA Technologies (IDT) [2] |
| Synthetic sgRNA | Guides the CAST complex to the genomic target site. | TRAC-targeting sequence: GGGAATCAAAATCGGTGAAT [2] |
| Nanoplasmid Donor DNA | Carries the transgene cargo for integration; minimal backbone reduces toxicity. | Nature Technology Corp. [2] |
| Electroporation System | Instrument for delivering payload via electrical pulse. | Maxcyte GTx with "Expanded T cell" protocol [2] [43] |
Step-by-Step Procedure
- T Cell Preparation: Isolate T cells from a leukopak using a negative selection isolation kit. Activate the isolated T cells with CD3/CD28 activator beads at a 1:1 bead-to-cell ratio. Culture cells in TexMACS medium supplemented with 12.5 ng/mL IL-7 and 12.5 ng/mL IL-15 for 48 hours [2].
- Payload Complex Formation: On the day of electroporation, magnetically remove the activator beads. For each electroporation, mix wildtype Cas9 protein (61 µM) and TRAC-targeting sgRNA (125 µM) at a 1:1 volume ratio to form the RNP complex. Incubate for 10 minutes at room temperature. Add the nanoplasmid donor DNA (e.g., 15 µg) to the RNP mixture and incubate for an additional 10 minutes to allow the RNP to pre-cleave the donor plasmid [2].
- Electroporation: Wash the T cells and resuspend them in electroporation buffer at a concentration of 2 × 10^8 cells/mL. Combine the cell suspension with the pre-formed RNP/donor payload. Transfer the mixture to an appropriate electroporation processing assembly and electroporate using a pre-optimized protocol, such as the "Expanded T cell 4" protocol on the Maxcyte GTx system [2] [43].
- Post-Transfection Recovery: After electroporation, rest the cells in the processing assembly for 30 minutes. Then, transfer the cells to fresh G-Rex vessels containing pre-warmed TexMACS medium supplemented with IL-7 and IL-15. Culture the cells for expansion and analysis [2].
- Enrichment (Optional): To further purify successfully edited cells, employ a selection strategy such as CEMENT (CRISPR EnrichMENT). This can involve co-integrating a selectable marker like dihydrofolate reductaseL22F/F31S (DHFR-FS), which confers resistance to methotrexate, allowing for drug-based enrichment of CAR-positive T cells [2].
Expected Outcomes and Analysis
Using this optimized protocol, researchers can expect targeted integration efficiencies of 10-25% for kilobase-sized cargos in human cells [30]. The resulting CAR T-cell products are typically highly functional, demonstrating tumor control efficacy equivalent to or better than virally transduced CAR-T cells in pre-clinical models [2]. Genomic safety analyses, including ddPCR-based copy number assays and genome-wide insertion site profiling, should show low levels of off-target integration and an acceptable safety profile [2].
The synergistic optimization of gRNA design and electroporation parameters is fundamental to harnessing the full potential of CAST systems for homology-independent targeted integration. By adhering to the detailed guidelines and protocols outlined in this document—from in silico gRNA selection to post-electroporation cell handling—researchers can achieve high-efficiency integration of large DNA cargos. This paves the way for advanced applications in gene therapy and the streamlined production of engineered cell therapeutics, making complex genomic manipulations more accessible, efficient, and clinically relevant.
Within the broader scope of homology-independent targeted integration research, particularly concerning the CRISPR-associated transposase (CAST) system, obtaining a pure population of successfully edited cells is a fundamental challenge. The CAST system represents a significant advancement in third-generation gene editing tools, enabling the precise insertion of large DNA fragments without relying on DNA double-strand breaks (DSBs) [45]. This system utilizes a deactivated CRISPR complex for target site recognition, which then recruits a transposase complex to insert the donor DNA cargo at a specific site downstream of the target [45].
While the precision of CAST is high, the intrinsic efficiency of any gene editing delivery method, including viral vectors commonly used for such applications, is never 100% [46]. Consequently, a heterogeneous mixture of edited and unedited cells is invariably produced. This heterogeneity introduces significant noise into experimental readouts and is a major barrier to developing effective cell-based therapies. Therefore, robust post-editing enrichment strategies are not merely beneficial but essential. This application note details two powerful selection methodologies: one based on a drug-selectable marker, Dihydrofolate Reductase Fused to a Fluorescent Protein (DHFR-FS), and another based on a Surface Marker. Both strategies enable the isolation of a highly pure cell population following CAST-mediated targeted integration.
The CAST system is a prime example of the shift from "break-and-repair" to "copy-and-paste" gene editing paradigms [45]. Its core mechanism involves:
- Targeting: A catalytically dead Cas (dCas) protein, guided by a crRNA, binds to a specific genomic locus without creating a DSB.
- Recruitment: The bound dCas complex recruits a associated transposase.
- Integration: The transposase catalyzes the excision of a donor DNA fragment from a delivered plasmid and its subsequent integration into the genome at a precise location, typically 60-66 base pairs downstream of the target site [45].
This homology-independent integration is highly efficient for large fragments, but as with all editing tools, delivery and efficiency vary between cell types and experiments, necessitating the enrichment protocols described below.
Enrichment Strategy 1: DHFR-FS Selection
Principle and Mechanism
The DHFR-FS system leverages a mutant form of the dihydrofolate reductase enzyme that is resistant to the anti-folate drug methotrexate (MTX). By fusing this mutant DHFR to a fluorescent protein (e.g., GFP), a combined selectable and screenable marker is created. The CAST system is programmed to co-integrate the gene of interest (GOI) with the DHFR-FS expression cassette. Successfully edited cells express the MTX-resistant DHFR, allowing them to proliferate in the presence of the drug, while unedited cells die. The fluorescent tag enables parallel monitoring via flow cytometry.
Detailed Experimental Protocol
This protocol begins after the delivery of the CAST system and the donor plasmid containing your GOI and the DHFR-FS cassette into your target cell population.
Step 1: Post-Transfection Recovery
- After transfection (e.g., via electroporation or viral transduction), culture the cells in complete growth medium for 48 hours. This allows for the expression of the integrated DHFR-FS gene.
Step 2: Antibiotic Selection Initiation
- Begin applying selection pressure 48-72 hours post-transfection.
- Prepare culture medium containing the appropriate concentration of Methotrexate (MTX). A common starting concentration is 50 nM, but this must be titrated for each cell line using a kill curve assay.
- Note: For non-integrated vector clearance, some protocols first use a different antibiotic (e.g., Puromycin) if the donor plasmid contains a corresponding resistance gene for a brief selection period (1-2 days).
Step 3: Sustained Selection and Expansion
- Replace the MTX-containing medium every 2-3 days.
- Monitor cell death and the emergence of stable, resistant colonies. This selection process typically takes 7-14 days.
- Once a confluent, resistant population is established, the cells can be analyzed by flow cytometry to assess the purity based on the fluorescent signal of the DHFR-FS fusion.
Table 1: Key Reagents for DHFR-FS Selection
| Research Reagent | Function/Explanation |
|---|---|
| CAST System Plasmids | Donor plasmid containing GOI and DHFR-FS cassette; plasmids encoding dCas and transposase. |
| Methotrexate (MTX) | Selective agent that inhibits wild-type DHFR, eliminating non-edited cells. |
| Transfection Reagent | For intracellular delivery of CAST system components (e.g., lipofection, electroporation kit). |
| Cell Culture Media | Optimized media for the target cell line, without components that antagonize MTX. |
| Flow Cytometer | For quantifying the percentage of fluorescent (edited) cells pre- and post-selection. |
Enrichment Strategy 2: Surface Marker Selection
Principle and Mechanism
This strategy involves the co-integration of a compact, non-immunogenic cell surface protein (e.g., a truncated human EGFR, CD34, or a similar marker) along with the GOI. The expressed surface marker serves as a physical tag on successfully edited cells. This population can then be isolated with high purity using Fluorescence-Activated Cell Sorting (FACS) or Magnetic-Activated Cell Sorting (MACS). This method is faster than drug selection as it does not rely on cell proliferation and death.
Detailed Experimental Protocol
This protocol covers the steps from cell preparation to sorting after CAST-mediated editing.
Step 1: Cell Harvest and Staining
- Harvest the edited cell population 72-96 hours post-transfection/enfection. This allows sufficient time for surface marker expression.
- Wash the cells with a cold (4°C) FACS buffer (e.g., PBS with 1-2% FBS).
- Resuspend the cell pellet in cold FACS buffer and incubate with a primary antibody conjugated to a fluorophore (for FACS) or magnetic beads (for MACS) against the selected surface marker (e.g., anti-EGFR-APC for FACS, anti-CD34 microbeads for MACS). Perform this incubation on ice for 20-30 minutes, protected from light.
Step 2: Cell Washing and Resuspension
- Wash the cells twice with a generous volume of cold FACS buffer to remove unbound antibody.
- Resuspend the cells in an appropriate volume of cold, sterile FACS buffer for sorting. Pass the cell suspension through a cell strainer (e.g., 40 µm) to remove clumps that could clog the sorter.
Step 3: Cell Sorting and Analysis
- For FACS: Use a high-speed cell sorter to collect the population of cells that are positive for the surface marker fluorescence. Establish gating parameters using a non-transfected control sample stained with the same antibody.
- For MACS: Pass the cell-antibody-bead complex through a magnetic column. The labeled cells are retained in the column. After washing, remove the column from the magnet and elute the positively selected cells.
- After sorting, a small aliquot of the purified population should be re-analyzed by flow cytometry to confirm purity, which should typically exceed 95%.
Table 2: Key Reagents for Surface Marker Selection
| Research Reagent | Function/Explanation |
|---|---|
| Surface Marker Gene | Compact cell surface protein (e.g., tEGFR, CD34) encoded in the donor plasmid for expression on edited cells. |
| Fluorescent/Magnetic Antibody | Antibody against the surface marker, conjugated to a fluorophore (FACS) or magnetic bead (MACS). |
| Cell Sorting Instrument | FACS sorter or MACS separation setup for isolating marker-positive cells. |
| FACS Buffer | Protein-rich, cold buffer (e.g., PBS + 2% FBS) to maintain cell viability and reduce non-specific binding. |
| Sterile Cell Strainer | To ensure a single-cell suspension for efficient and sterile sorting. |
Comparative Analysis and Workflow Integration
The choice between DHFR-FS and surface marker selection depends on the experimental goals, available tools, and timeline.
Table 3: Quantitative Comparison of Enrichment Strategies
| Feature | DHFR-FS Selection | Surface Marker Selection |
|---|---|---|
| Primary Mechanism | Drug-based positive selection | Physical separation based on surface tag |
| Time to Pure Population | ~10-14 days | ~3-4 days |
| Typical Purity Yield | >99% (clonal outgrowth) | 95-99% (post-sort analysis dependent) |
| Key Equipment Needed | Standard cell culture incubator | Flow cytometer / Cell sorter |
| Cost Factor | Cost of methotrexate | Cost of antibodies and sorting services |
| Impact on Cell Physiology | Subject to drug stress; may influence phenotype | Rapid, no drug stress; immediate downstream use |
| Suitability for Sensitive Cells | Lower, due to cytotoxic kill of neighbors | Higher, as it is a gentle physical process |
The following workflow diagram illustrates how these enrichment strategies are integrated into a CAST-mediated gene integration experiment.
Troubleshooting and Best Practices
- Low Purity Post-Enrichment: For DHFR-FS, ensure MTX concentration is lethal to wild-type cells by performing a kill curve. For surface marker selection, optimize antibody titration and sorting gates using stringent controls.
- Low Cell Viability Post-Sorting: Always use cold buffers and process cells quickly. For MACS, use the gentlest possible magnets and columns designed for the specific cell type.
- Validation: Regardless of the method, validate the purity and the correct targeted integration of the GOI. Use techniques such as flow cytometry (for surface marker or fluorescence), PCR-based genotyping, and Sanger sequencing across the integration junctions to confirm on-target insertion and sequence fidelity.
The combination of the precise CAST system for targeted gene integration with a robust enrichment strategy is critical for generating high-quality, reliable data and therapeutic cell products. The DHFR-FS system provides a powerful means of selecting for stable integrants over time, while surface marker selection offers a rapid, high-purity isolation method independent of cell proliferation. The choice between them should be guided by the specific requirements of the experiment, including timeline, available equipment, and the nature of the target cells. Both methods significantly enhance the stringency and reproducibility of homology-independent targeted integration research.
The advent of clustered regularly interspaced short palindromic repeats (CRISPR)-associated transposase (CAST) systems represents a paradigm shift in homology-independent targeted integration research, offering DSB-free, programmable insertion of large DNA fragments. Unlike conventional CRISPR-Cas systems that rely on double-strand breaks (DSBs) and host repair mechanisms, CAST systems leverage a CRISPR-associated DNA targeting module coupled with a transposase effector module, enabling highly specific, multi-kilobase integrations without DSB intermediates [16]. This application note provides a comprehensive framework for assessing both on-target and off-target genotoxicity risks associated with CAST system applications, outlining standardized protocols and analytical methodologies essential for therapeutic development.
CAST systems, particularly type I-F variants like PseCAST and VchCAST, demonstrate superior product purity and specificity compared to earlier gene editing tools [16]. However, their clinical translation necessitates rigorous genotoxicity assessment to characterize unintended genomic consequences. Genotoxicity encompasses any detrimental alteration to DNA structure, information content, or segregation, including point mutations, chromosomal aberrations, and translocations [47]. For DNA-reactive substances, the widely accepted "one-hit" hypothesis suggests that exposure to a single genotoxic molecule could theoretically trigger a harmful mutation [47]. This underscores the critical importance of comprehensive genotoxicity profiling for CAST-based therapies, particularly as they advance toward clinical applications.
Genotoxicity Risk Landscape for CAST Systems
Mechanisms and Manifestations of Genotoxicity
Genotoxicity in genome engineering manifests through multiple mechanisms. DNA-reactive effects primarily involve covalent DNA adduct formation and cross-linking, while non-DNA-reactive effects include reactive oxygen species generation or interference with components maintaining genomic stability [47]. CAST systems, despite their DSB-free mechanism, still present potential genotoxicity risks requiring systematic evaluation:
- Off-target activity: Unintended DNA modifications at sites with sequence similarity to the target locus [48]
- Collateral cleavage activity: Non-specific nuclease activity observed in some CRISPR systems like Cas12a [48]
- Large deletions and rearrangements: Chromosomal abnormalities potentially arising from aberrant DNA repair [48]
- Vector integration events: Unintended incorporation of delivery vehicle components [49]
The clinical significance of genotoxic events depends on multiple factors, including the specific genetic locus affected, cell type, and mutation type. Mutations in "cancer driver genes" are particularly concerning, as even single mutations in certain contexts can initiate malignant transformation [47].
CAST-Specific Risk Considerations
While CAST systems mitigate DSB-associated risks, they introduce unique considerations. Type I-F CAST systems (PseCAST, VchCAST) demonstrate highly specific integration but exhibit variable DNA binding efficiencies that may influence editing outcomes [16]. Structural analyses reveal that PseCAST QCascade complexes exhibit distinct conformational dynamics, particularly in the TniQ dimer region, which may impact targeting fidelity [16]. Understanding these molecular nuances is essential for accurate risk assessment.
Table 1: Quantitative Genotoxicity Assessment Metrics for Genome Editing Tools
| Assessment Metric | CRISPR-Cas9 | CAST Systems | Clinical Threshold Considerations |
|---|---|---|---|
| Off-target mutation frequency | Variable (0.1-50%) depending on gRNA design and delivery [48] | Not fully characterized; predicted lower due to DSB-free mechanism | Case-by-case evaluation based on therapeutic context [50] |
| Large deletion frequency | Up to several kilobases reported [48] | Limited data; theoretically reduced | Risk-benefit analysis relative to disease severity [50] |
| On-target efficiency | Highly variable (5-90%) depending on cell type and target locus [20] | Currently low (single-digit % in human cells) but improvable through engineering [16] | Therapeutic thresholds depend on specific application |
| Product purity | Heterogeneous mixtures common [16] | Highly homogeneous integration products [16] | Critical for predictable therapeutic outcomes |
Experimental Framework for Genotoxicity Assessment
Comprehensive Off-Target Analysis Workflow
A robust off-target assessment strategy employs complementary computational and empirical approaches to identify potential unintended editing events throughout the genome.
In silico Prediction Methods
Initial off-target prediction utilizes computational tools that identify genomic loci with sequence similarity to the intended target site:
- CRISPOR: Incorporates multiple off-target scoring algorithms and considers genetic variants [50]
- Cas-OFFinder: Genome-wide search for potential off-target sites with bulges or mismatches [50]
- CCTop: User-friendly platform with comprehensive specificity analysis [48]
These tools typically evaluate factors including PAM sequence compatibility, mismatch tolerance, and genomic context to generate a prioritized list of potential off-target sites for experimental validation.
In vitro Screening Methods
Cell-free biochemical methods provide sensitive, unbiased off-target profiling:
- CIRCLE-seq: Highly sensitive in vitro method that circularizes genomic DNA for comprehensive off-target identification [50]
- CHANGE-seq: Library preparation method that captures CRISPR cleavage events in vitro [50]
- DIG-seq: Utilizes chromatin DNA to account for epigenetic influences on accessibility [50]
These approaches offer broad detection capabilities but may identify sites not relevant in cellular contexts due to chromatin structure or temporal factors.
In cellula Verification Methods
Cell-based methods validate predicted off-targets in relevant biological systems:
- GUIDE-seq: Uses oligonucleotide integration to mark DSB sites genome-wide [50]
- DISCOVER-Seq: Leverages MRE11 recruitment to identify CRISPR cuts in cells and tissues [50]
- ONE-seq: Population-specific off-target profiling that accounts for genetic variation [50]
These methods provide critical context-specific data but may have sensitivity limitations for detecting low-frequency events.
Functional Validation Methods
Definitive off-target characterization employs comprehensive genomic analysis:
- Whole Genome Sequencing (WGS): Unbiased detection of structural variants and unexpected modifications [49]
- S-EPTS/LM-PCR: Sensitive method for identifying vector integration sites and DNA breaks [49]
A recent study of AAV-delivered CRISPR-Cas9 in mouse liver demonstrated the utility of these approaches, revealing efficient on-target editing (36.45% ± 18.29%) with rare off-target events below WGS detection limits [49].
On-Target Genotoxicity Assessment
On-target assessment focuses on characterizing intended editing outcomes and associated unintended consequences at the target locus:
Editing Efficiency Quantification
- Next-generation sequencing (NGS): Amplify target region (e.g., 150-300bp flanking cut site) using high-fidelity polymerase. Sequence with minimum 10,000x coverage and analyze for precise integration using tools like CRISPResso2 [20]
- Digital PCR: Absolute quantification of edited versus wild-type alleles using target-specific probes
For CAST systems, specifically evaluate correct orientation integration and full cargo insertion using junctional PCR and long-read sequencing.
Product Purity Analysis
- ICE Analysis (Inference of CRISPR Edits): Deconvolute Sanger sequencing chromatograms to quantify indel percentages [48]
- TIDE Decomposition (Tracking of Indels by Decomposition): Web tool for rapid assessment of editing efficiency and pattern from sequence traces [48]
These methods are particularly important for evaluating CAST system performance, as product purity represents a key advantage over traditional CRISPR-based integration [16].
Structural Variant Detection
- Long-range PCR: Amplify large regions (5-20kb) flanking the target site to detect major deletions or rearrangements
- Karyotyping/G-banding: Macroscopic chromosomal analysis to identify gross abnormalities
- Optical genome mapping: High-resolution structural variant detection across the genome
Studies have shown that CRISPR-Cas9 can induce large deletions spanning several kilobases [48], though CAST systems may reduce this risk through their DSB-free mechanism.
Quantitative Genotoxicity Assessment Protocols
Benchmark Dose (BMD) Analysis for Genotoxicity
Quantitative dose-response analysis represents a paradigm shift from traditional binary genotoxicity assessment. The International Workshop on Genotoxicity Testing (IWGT) recommends benchmark dose (BMD) modeling as the preferred approach for defining points of departure (PoDs) for genotoxicity risk assessment [51].
Table 2: Point of Departure Metrics for Quantitative Genotoxicity Risk Assessment
| Metric | Definition | Application | Advantages | Limitations |
|---|---|---|---|---|
| Benchmark Dose (BMD) | Statistical lower confidence limit on dose corresponding to a specified increase in effect (e.g., 10% increase over background) | Primary PoD for risk assessment [51] | Utilizes all dose-response data; accounts for experimental variability | Requires multiple dose levels with adequate response data |
| No Observed Genotoxic Effect Level (NOGEL) | Highest experimental dose without statistically significant genotoxic effect | Secondary PoD when BMD modeling not feasible [51] | Simple to determine; conservative estimate | Depends on study design and statistical power |
| Break Point Dose (BPD) | Dose at which response significantly increases from background, derived from bilinear models | When mechanistic data support threshold approach [51] | Reflects apparent thresholds for some mechanisms | Theoretical possibility of effects below BPD cannot be excluded |
BMD Modeling Protocol
Experimental Design:
- Test a minimum of 5 dose concentrations plus vehicle control
- Include positive control for assay validation
- Ensure adequate sample size (n ≥ 3 per group for in vitro, n ≥ 5 for in vivo)
Dose-Response Modeling:
- Fit family of exponential models (e.g., Hill, power, linear) to response data
- Select best-fitting model using statistical criteria (Akaike Information Criterion)
- Calculate BMD corresponding to benchmark response (BMR) of 10% extra risk
Uncertainty Factor Application:
- Apply appropriate uncertainty factors (typically 10-1000x) based on data quality and relevance
- Consider interspecies differences, intraspecies variability, and database deficiencies
The quantitative interpretation of in vivo genotoxicity data for prioritization purposes represents a promising opportunity for routine application [47].
In Vitro to In Vivo Extrapolation (IVIVE) Protocol
IVIVE modeling translates in vitro genotoxicity concentrations to human equivalent doses for risk assessment:
- In vitro concentration-response testing: Determine potency in relevant cell systems
- Physiologically-based pharmacokinetic (PBPK) modeling: Estimate human blood concentrations equivalent to in vitro effective concentrations
- Point of departure (PoD) derivation: Calculate health-protective doses based on in vitro BMDs
A recent evaluation of 31 reference chemicals demonstrated that IVIVE-derived PODs were protective for most chemicals (20/31) relative to in vivo PODs from animal studies [52].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for CAST System Genotoxicity Assessment
| Reagent Category | Specific Products/Tools | Application | Key Considerations |
|---|---|---|---|
| CAST Systems | PseCAST, VchCAST, Type I-F variants [16] | DSB-free targeted integration | PseCAST shows higher efficiency in human cells; engineering efforts ongoing |
| Off-target Detection | GUIDE-seq, CIRCLE-seq, DISCOVER-Seq kits [50] | Genome-wide off-target mapping | Method selection depends on sensitivity requirements and cellular context |
| Sequence Analysis | CRISPOR, CRISPResso2, Cas-OFFinder [50] | gRNA design and sequencing analysis | Consider genetic variation in target population when designing gRNAs |
| Vector Delivery | AAV vectors, lipid nanoparticles [49] | In vivo delivery of editing components | AAV integration patterns require specific assessment [49] |
| DNA Break Mapping | S-EPTS/LM-PCR [49] | Sensitive detection of integration events and DNA breaks | Unbiased approach that doesn't rely on prediction algorithms |
| Quantitative Analysis | TIDE, ICE Analysis [48] | Rapid efficiency and specificity assessment | Suitable for initial screening but complemented by NGS for comprehensive analysis |
Comprehensive genotoxicity assessment is indispensable for translating CAST system technologies into safe therapeutic applications. The framework presented herein enables researchers to systematically evaluate both on-target and off-target effects, incorporating state-of-the-art computational predictions, empirical validation methods, and quantitative risk assessment approaches.
As CAST systems evolve through protein engineering and structural optimization [16], parallel advances in genotoxicity assessment methodologies will be essential. The field is moving toward increasingly sensitive detection methods capable of identifying rare genotoxic events, while quantitative risk assessment paradigms facilitate more nuanced benefit-risk determinations [50]. By implementing robust genotoxicity assessment protocols early in development, researchers can accelerate the translation of CAST-based therapies while ensuring rigorous safety standards.
The clinical and commercial success of cell and gene therapies, including those utilizing homology-independent targeted integration, hinges on overcoming a critical bottleneck: scalable manufacturing. The regenerative medicine field faces a sobering reality; without scalable, efficient manufacturing, these revolutionary treatments will never reach the hundreds of millions of patients who need them [53]. Behind promising scientific milestones lies a troubling trend—a significant portion of approved therapies face market withdrawal not due to safety or efficacy concerns, but due to lack of commercial viability. Specifically, 8 of the 28 authorized Advanced Therapy Medicinal Products (ATMPs) in the EU have been pulled from the market for these reasons [53]. The cost of goods for cell and gene therapies remains among the highest in biopharma, often reaching hundreds of thousands or even millions of dollars per dose, creating unsustainable economic models for widespread patient access [53].
Traditional manufacturing processes for cell-based therapies are often manual, bespoke, and difficult to scale cost-effectively. These processes typically rely on disconnected units of operation—separate technologies for cell culture, intracellular delivery, cell expansion, fill and finish, cryopreservation, and quality control. This fragmented approach creates "islands of automation" that depend on manual interventions between critical steps, increasing the risk of process failures, contamination, and data loss [53]. For therapies based on CRISPR-Cas9 homology-independent targeted integration (HITI), such as those being developed for Duchenne muscular dystrophy [13] and CAR-T cells [2], these manufacturing challenges are particularly acute due to the complexity of the gene editing components and the need for precise quality control.
Implementing closed-system production from early development stages represents a paradigm shift essential for clinical translation and commercial viability. As emphasized by regulatory agencies like the FDA, early implementation of closed, automated systems not only reduces costs but significantly enhances batch consistency [53]. For the CAST (CRISPR-associated transposase) systems currently under investigation for homology-independent targeted integration, scalable manufacturing considerations must be integrated from the earliest research phases to enable successful clinical translation [54].
Quantitative Analysis of Manufacturing Platforms and Methods
Comparison of Automated Cell Manufacturing Platforms
Table 1: Automated Bioreactor Systems for Scalable Cell Therapy Manufacturing
| Platform Name | Manufacturer | Technology Type | Culture Surface Area | Reported MSC Yield | Key Applications |
|---|---|---|---|---|---|
| Quantum Cell Expansion System | Terumo BCT | Hollow fiber bioreactor | 21,000 cm² (equivalent to 120 T-175 flasks) | 100-276 × 10⁶ BM-MSCs in 7-day expansion | BM-MSCs, AT-MSCs, UC-MSCs; clinical trials for GVHD, type 2 diabetes, Parkinson's disease |
| CliniMACS Prodigy | Miltenyi Biotec | Integrated automated cell processing system | 1-layer CellSTACK | 29-50 × 10⁶ MSCs (equine model, P0) | Automated isolation, cultivation, and harvesting of BM-MSCs, AT-MSCs, UC-MSCs |
| Xuri Cell Expansion System W25 | Cytiva | Wave-mixed bioreactor | System dependent | Not specified in results | Large-scale expansion of adherent cells |
| Cocoon Platform | Lonza | Automated, closed cell manufacturing | Platform dependent | Not specified in results | Personalized cell therapies, including CAR-T cells |
| NANT001/XL System | VivaBioCell | Not specified | Not specified | Not specified in results | Not specified in results |
| CellQualia | Sinfonia Technology | Not specified | Not specified | Not specified in results | Not specified in results |
Performance Metrics for Homology-Independent Integration Methods
Table 2: Comparison of Homology-Independent Targeted Integration Approaches
| Method | Therapeutic Application | Efficiency | Payload Capacity | Key Advantages | Limitations |
|---|---|---|---|---|---|
| HITI (Homology-Independent Targeted Integration) | CAR-T cell manufacturing [2] | 2-fold higher cell yields compared to HDR; 80% purity post-CEMENT enrichment | Large transgenes (>5 kb) [2] | Cell cycle independent; uses predominant NHEJ pathway; works in dividing and resting cells | Requires careful optimization of RNP:donor DNA ratios |
| HITI with AAV9 delivery | Duchenne muscular dystrophy gene correction [13] | 1.4% genome editing in heart; 30% transcript correction; 11% dystrophin restoration | Exons 1-19 mega-exon | Enables correction of mutations upstream of intron 19 (25% of DMD patients) | Lower efficacy in skeletal muscles; fragmentary AAV genome integration |
| CAST (CRISPR-associated transposase) systems | Large DNA insertions in human cells [54] | Reached single-digit efficiencies at genomic target sites | Multi-kilobase insertions | DSB-free integration; highly specific and homogeneous integration products | DNA binding identified as critical bottleneck limiting efficiency |
Experimental Protocols for Closed-System Manufacturing
Protocol 1: HITI-Mediated CAR-T Cell Manufacturing in Closed Systems
Principle: This protocol outlines a homology-independent targeted integration approach for generating CAR-T cells using fully closed, automated systems, eliminating reliance on viral vectors and enabling scalable production [2].
Materials:
- Primary human T cells from leukopaks
- Nanoplasmid DNA containing CAR transgene (3 mg/mL in H₂O)
- Wildtype Cas9 (61 µM) and TRAC-targeting sgRNA (125 µM)
- Electroporation system (Maxcyte GTx)
- GMP-compatible closed cell processing assembly (CL1.1 for large scale)
- TexMACS media with IL-7 and IL-15 (12.5 ng/mL each)
- Human male AB Serum (3%)
- Dynabeads Human T-Activator CD3/CD28
- G-Rex culture vessels
Procedure:
- T Cell Isolation and Activation: Isolate T cells from leukopaks using negative selection. Activate cells with CD3/CD28 Dynabeads at 1:1 ratio in TexMACS media supplemented with cytokines and serum [2].
- RNP Complex Formation: Mix wildtype Cas9 and sgRNA at 2:1 molar ratio (61 µM Cas9:125 µM sgRNA), incubate 10 minutes at room temperature. Add nanoplasmid DNA (3 mg/mL) and incubate additional 10 minutes to allow RNP cleavage of nanoplasmid [2].
- Closed-System Electroporation: On day 2, magnetically remove Dynabeads and wash cells once in electroporation buffer. Resuspend cells at 2 × 10⁸/mL. Transfer to appropriate closed processing assembly and electroporate using "Expanded T cell 4" protocol for activated T cells [2].
- Post-Electroporation Processing: Rest cells in electroporation buffer within closed processing assembly for 30 minutes. Transfer to final G-Rex vessels within closed system [2].
- CEMENT Enrichment: Implement CRISPR EnrichMENT using drug-selection system (dihydrofolate reductaseL22F/F31S) with methotrexate to enrich CAR+ T cells to approximately 80% purity [2].
- Expansion and Harvest: Expand cells in closed bioreactor system (e.g., Cocoon Platform) for 14-day process, maintaining cell concentration at ~1.5 × 10⁶/mL [2].
Quality Control:
- Perform ddPCR-based copy number analysis
- Conduct genome-wide insertion site analysis
- Validate immunomodulatory function through T-cell suppression assays
- Confirm absence of microbial contamination
Protocol 2: Closed-System MSC Expansion for EV Production
Principle: This protocol describes the automated, large-scale expansion of mesenchymal stem/stromal cells (MSCs) in closed bioreactor systems for production of extracellular vesicles (EVs), maintaining GMP compliance throughout the process [55] [56].
Materials:
- Quantum Cell Expansion System (Terumo BCT) or equivalent closed bioreactor
- MSC-Brew GMP medium or human platelet lysate (hPL)-supplemented media
- Fibronectin, vimentin, or cryoprecipitate for hollow fiber coating
- Bone marrow, adipose tissue, or umbilical cord-derived MSCs
- GMP-compatible closed tubing sets
- Controlled-rate freezing system
Procedure:
- Bioreactor Preparation: Coat hollow fibers with adhesive substrate (fibronectin, vimentin, or cryoprecipitate) using closed-system fluid paths [56].
- Cell Seeding: Load thawed MSCs (P2) into Quantum system at density of 20 × 10⁶ cells using closed transfer techniques [56].
- Automated Expansion: Culture for 7-10 days with continuous medium exchange optimized for MSC expansion. Maintain hypoxic microenvironment (3% O₂) if enhanced productivity is desired [56].
- Process Monitoring: Monitor glucose and lactate levels automatically through integrated sensors. Adjust feeding parameters to support optimal growth and maintain cell viability [56].
- Harvesting: Automatically harvest cells using integrated system, minimizing open manipulation steps (reduction from ~54,400 to 133 steps compared to manual processes) [56].
- EV Collection and Purification: Collect conditioned medium through closed-system transfer for downstream EV isolation using tangential flow filtration or size exclusion chromatography [55].
- Cryopreservation: Implement controlled-rate freezing within closed system for final product preservation [56].
Quality Control:
- Verify MSC phenotype (CD105+, CD73+, CD90+, CD45-, CD34-, CD14-, CD11b-, CD79α-, HLA-DR-)
- Assess differentiation potential (osteogenic, chondrogenic, adipogenic)
- Evaluate immunomodulatory activity through T-cell suppression assays
- Test genome stability
- Characterize EV markers (CD63, CD81, CD9) and functionality
Visualization of Manufacturing Workflows
Closed-System CAR-T Cell Manufacturing Workflow
Closed-System CAR-T Manufacturing
CAST System Mechanism for Targeted Integration
CAST System Integration Mechanism
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Research Reagent Solutions for Homology-Independent Integration Manufacturing
| Reagent/Material | Function | Application Notes | Closed-System Compatibility |
|---|---|---|---|
| Nanoplasmid DNA | Non-viral vector for transgene delivery | R6K origin, antibiotic-free selection; resuspend at 3 mg/mL in H₂O [2] | High - compatible with closed electroporation |
| Cas9 RNP Complex | Genome editing machinery | Wildtype Cas9 (61 µM) + sgRNA (125 µM) at 2:1 molar ratio; 10 min pre-incubation [2] | High - stable at room temperature |
| Maxcyte GTx Electroporator | Non-viral nucleic acid delivery | GMP-compatible; multiple scale options (OC-25×3 to CL1.1) [2] | High - closed processing assemblies |
| Quantum Cell Expansion System | Automated cell expansion | 21,000 cm² surface area; requires matrix coating [56] | High - fully closed system |
| Human Platelet Lysate (hPL) | Serum-free culture supplement | GMP-compatible alternative to FBS; enhances MSC expansion [56] | High - available in GMP grade |
| MSC-Brew GMP Medium | Defined culture medium | Serum-free, xeno-free formulation for clinical MSC expansion [56] | High - optimized for closed systems |
| CEMENT Selection System | Enrichment for edited cells | DHFR-FS with methotrexate resistance; achieves ~80% purity [2] | Medium - requires closed transfer |
| G-Rex Culture Vessels | Scalable cell expansion | Gas-permeable membrane technology; multiple sizes available [2] | Medium - requires closed connections |
The implementation of closed-system production methodologies represents a critical path forward for the clinical translation of therapies based on homology-independent targeted integration, including CAST systems and HITI approaches. The quantitative data presented in this application note demonstrates that automated, closed systems can achieve cell yields sufficient for clinical applications while maintaining quality attributes—Quantum systems producing 100-276 × 10⁶ BM-MSCs in 7-day expansions [56] and HITI/CEMENT approaches generating 5.5 × 10⁸–3.6 × 10⁹ CAR-T cells across a 14-day process [2].
Successful translation requires forward-thinking manufacturing strategies that align with regulatory expectations. As emphasized by regulatory agencies, early implementation of automation during R&D and process development is essential rather than attempting to retrofit scalable processes onto established manual methods [53]. This is particularly crucial for emerging CAST systems, where DNA binding has been identified as a critical bottleneck limiting efficiency [54]. By applying structure-guided engineering to components like the PseCAST QCascade complex, researchers can optimize both the fundamental editing efficiency and the manufacturing scalability concurrently.
The future of scalable manufacturing for advanced therapies will increasingly leverage integrated approaches combining closed-system hardware with digitalization and predictive models. Implementing process analytical technologies that monitor critical quality attributes in real-time, coupled with the accumulation of process data to create digital twins, will enable rapid optimization without time-consuming experimental iterations [53]. For homology-independent integration technologies specifically, continuing advances in reagent delivery, editing efficiency, and cell enrichment will further enhance the commercial viability of these promising therapeutic approaches. Through the adoption of these comprehensive scalable manufacturing strategies, researchers can accelerate the translation of CAST systems and other homology-independent targeted integration technologies from research tools to transformative clinical therapies.
Proof and Perspective: Validating HITI and Comparing it to Alternative Gene Editing Tools
The translation of gene editing technologies from experimental platforms to clinical therapies is contingent upon robust preclinical validation in animal models. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated transposase (CAST) systems have emerged as a powerful class of gene editing tools capable of performing homology-independent targeted integration of large DNA sequences without creating double-strand breaks (DSBs), addressing a critical limitation of earlier CRISPR-based techniques [57] [19]. This application note details the experimental protocols and validation frameworks for evaluating CAST system efficacy in mouse and pig models of human disease, providing researchers with standardized methodologies for preclinical therapeutic development.
CAST systems, particularly type I-F and V-K variants, leverage a CRISPR-guided DNA targeting module coupled with a transposase effector module to enable precise "cut-and-paste" integration of multi-kilobase DNA fragments at specific genomic loci [16] [19]. Unlike DSB-dependent approaches such as homology-directed repair (HDR), CAST-mediated integration occurs through a DSB-free mechanism that minimizes indel formation and other undesirable editing byproducts, making it particularly suitable for therapeutic applications in post-mitotic tissues [16].
CAST System Mechanisms and Advantages
Molecular Architecture and Mechanisms
CAST systems comprise two primary functional modules: the DNA targeting complex and the transposase integration machinery. In type I-F systems, the targeting module consists of a multi-subunit Cascade complex (Cas6, Cas7, and Cas8 proteins) guided by CRISPR RNA (crRNA) that identifies specific genomic target sites [19]. This complex recruits TniQ, which in turn orchestrates the assembly of the transposase proteins (TnsA, TnsB, TnsC) that catalyze the excision and integration of donor DNA [16] [19].
Table 1: Comparison of CAST System Subtypes
| Feature | Type I-F CAST | Type V-K CAST |
|---|---|---|
| Targeting Module | Multi-subunit Cascade complex (Cas6/7/8) | Single-effector Cas12k protein |
| Integration Proteins | TnsA, TnsB, TnsC, TniQ | TnsB, TnsC, TniQ |
| PAM Requirement | 5'-CC-3' [16] | Variable |
| Insertion Site | ~50 bp downstream of target site [19] | 60-66 bp downstream of PAM [19] |
| Donor Capacity | Up to ~15.4 kb in prokaryotes [19] | Up to ~30 kb in prokaryotes [19] |
| Mammalian Efficiency | ~1% in HEK293 cells (1.3 kb donor) [19] | Up to ~3% in HEK293 cells (3.2 kb donor) [19] |
The RNA-guided DNA recognition mechanism enables programmable targeting without requiring pre-engineered recognition sequences, distinguishing CAST systems from traditional recombinase-based approaches like Cre-lox [57] [19]. Structural studies of the PseCAST QCascade complex have revealed subtype-specific interactions and RNA-DNA heteroduplex features that can be engineered to enhance DNA binding affinity and editing efficiency [16].
Advantages Over Alternative Genome Editing Technologies
CAST systems offer several distinct advantages for preclinical therapeutic development:
- DSB-Free Integration: By avoiding double-strand breaks, CAST systems minimize the introduction of indel mutations that commonly plague CRISPR-Cas9 approaches [16] [19].
- Large Cargo Capacity: CAST systems can deliver DNA fragments exceeding 10 kb, enabling the integration of full-length therapeutic genes and complex regulatory elements [19].
- High Product Purity: Type I-F CAST systems exhibit highly specific and homogeneous integration products compared to other integration technologies [16].
- Theoretical Single-Copy Integration: The transposition mechanism favors precise single-copy integration events, reducing concerns about concatemer formation [16].
The following diagram illustrates the core mechanism of type I-F CAST systems:
Preclinical Models for CAST Validation
Murine Disease Models
Mouse models remain indispensable for initial proof-of-concept studies due to their well-characterized genetics, relatively low maintenance costs, and the availability of sophisticated genetic engineering tools [58]. Successful CAST validation requires careful consideration of model selection criteria:
- Genetic Fidelity: Models with orthologous human disease-causing mutations provide the most clinically relevant testing platforms.
- Disease Phenotype: Models should recapitulate key pathological features of the human condition with measurable endpoints.
- Immunocompetence: Models with intact immune systems better predict therapeutic responses in human patients.
The Dmd exon 2 duplication (Dup2) mouse model of Duchenne muscular dystrophy exemplifies an effective validation platform for CAST systems. This model carries an out-of-frame duplication of exon 2 in the Dmd gene, mirroring a mutation found in approximately 1% of DMD patients [3]. CAST-mediated correction can be quantified through multiple endpoints: genomic integration efficiency, transcript correction, and dystrophin protein restoration [3].
Similarly, mouse models of Alternating Hemiplegia of Childhood (AHC) with specific ATP1A3 mutations (D801N and E815K) enable neurological disease modeling with distinct phenotypic manifestations, allowing researchers to evaluate CAST-mediated correction of point mutations in the nervous system [59].
Porcine Disease Models
Pigs (Sus scrofa domesticus) offer significant advantages for translational research due to their physiological similarity to humans in terms of anatomy, metabolism, immunology, and organ size [58]. The annotation of the pig genome has facilitated the development of sophisticated porcine disease models that better recapitulate human disease progression compared to rodent models.
Table 2: Comparative Analysis of Preclinical Model Organisms
| Parameter | Mouse Models | Pig Models | Human Relevance |
|---|---|---|---|
| Genetic Synteny | High | Moderate | Reference |
| Epigenetic Similarity | Moderate | High [58] | Critical for regulation |
| Physiological Scale | Low | High [58] | Impacts dosing & delivery |
| Reproductive Cycle | Short (~9 weeks) | Moderate (~12 months) | Affects study timeline |
| Heterogeneity | Low (often inbred) | High (outbred) [58] | Mimics human diversity |
| Tumor Pathology | Homogeneous | Heterogeneous [58] | Closer to human cancer |
| Operational Costs | Low | High | Resource consideration |
Comparative genomic analyses reveal that pigs share approximately 90 homologously shared elements with humans, with evolutionary divergence occurring approximately 1 million years ago, compared to 1.28 million years for mice [58]. This closer evolutionary relationship is reflected in enhanced epigenetic conservation, particularly in tissue-specific enhancers and promoters associated with complex human diseases [58].
Porcine models have been successfully developed for a wide range of human conditions, including craniofacial disorders, ophthalmological diseases, reproductive conditions, wound healing, musculoskeletal disorders, and various cancer types [58]. The "Oncopig" model exemplifies this approach, providing a comprehensive platform for studying human cancer pathophysiology and therapeutic interventions [58].
Experimental Protocols for CAST Validation
CAST Delivery and Validation in Mouse Models
Protocol: HITI-Mediated Correction in Dmd Dup2 Mice
This protocol adapts homology-independent targeted integration (HITI) principles for CAST system delivery, based on methodology validated for DMD correction [3].
Materials:
- CAST components: PseCAST system (or equivalent type I-F CAST)
- Donor template: MHCK7 promoter + DMD exons 1-19 mega-exon + splice donor
- Delivery vector: AAV9 serotype
- Neonatal Dup2 mice (postnatal day 1-3)
Procedure:
- Vector Preparation:
- Package CAST components and donor template in separate AAV9 vectors.
- Determine optimal vector ratio through preliminary titration (typically 1:5 Cas9:donor for HITI [3]).
- Purify vectors and resuspend in PBS at working concentration (e.g., 2.0×10¹⁴ vg/kg total dose).
Neonatal Injection:
- Anesthetize neonatal pups on ice for 3-5 minutes.
- Administer intraperitoneal injection using 31G insulin syringes.
- Maintain pups at 37°C until fully recovered from anesthesia.
Tissue Collection and Analysis (4-8 weeks post-injection):
- Euthanize animals and collect target tissues (heart, diaphragm, tibialis anterior).
- Process tissues for genomic, transcriptomic, and proteomic analyses.
Efficiency Quantification:
- Genomic Integration: Detect knockin via droplet digital PCR (ddPCR) using primers flanking the integration site [3].
- Transcript Correction: Quantify corrected mRNA using ddPCR assays targeting the junction between synthetic mega-exon and native exon 20 [3].
- Protein Restoration: Assess dystrophin expression via quantitative immunofluorescence and capillary western immunoassay [3].
The experimental workflow for preclinical validation is summarized below:
Large Animal Validation in Porcine Models
Protocol: SAGE-Mediated Integration in Porcine Models
Serine recombinase-Assisted Genome Engineering (SAGE) provides a framework for CAST validation in porcine models, leveraging recombinase-assisted integration for efficient editing in large animals [57] [58].
Materials:
- CAST system with optimized PseCAST variants [16]
- Bxb1 integrase or phiC31 integrase for recombinase-assisted delivery
- Donor construct with landing pad sequence
- Porcine fibroblasts or in vivo delivery system
- Pig embryos or juvenile pigs
Procedure:
- Landing Pad Installation:
- Design landing pad containing recombinase recognition sites (e.g., attB/attP for Bxb1).
- Introduce landing pad into porcine genome via zygotic injection or somatic cell nuclear transfer.
- Validate landing pad integration and specificity.
CAST Delivery:
- For ex vivo approach: Transfect porcine fibroblasts with CAST components and donor template.
- For in vivo approach: Administer via systemic or local injection to target tissues.
- Utilize optimal promoter systems (e.g., SPc5-12 for enhanced expression [3]).
Efficiency Assessment:
- Quantify integration efficiency via next-generation sequencing of target loci.
- Assess functional correction through disease-relevant physiological endpoints.
- Evaluate specificity through whole-genome sequencing for off-target analysis.
Therapeutic Validation:
- Monitor disease-specific biomarkers (e.g., neurofilament light chain for neurological disorders [59]).
- Conduct longitudinal assessment of pathological progression.
- Perform histological analysis of target tissues.
Quantitative Assessment and Data Analysis
Efficiency Metrics and Success Criteria
Rigorous quantification of editing outcomes is essential for meaningful preclinical validation. The following metrics should be reported across multiple biological replicates:
Table 3: Efficacy Benchmarks for CAST Systems in Preclinical Models
| Efficiency Metric | Mouse Models | Pig Models | Analytical Method |
|---|---|---|---|
| Genomic Integration | 0.11-1.1% (HITI in heart vs. muscle [3]) | Target: >1% | ddPCR, NGS |
| Transcript Correction | Up to 10% (HITI in heart [3]) | Target: >5% | RT-ddPCR, RNA-seq |
| Protein Restoration | 11% of normal dystrophin levels [3] | Target: >10% | Western blot, immunofluorescence |
| Functional Improvement | Disease-dependent | Disease-dependent | Physiological assays |
| Therapeutic Threshold | Mutation-dependent | Mutation-dependent | Clinical endpoint alignment |
Troubleshooting and Optimization
Low integration efficiency necessitates systematic optimization:
- Promoter Selection: The SPc5-12 promoter demonstrates enhanced activity in murine models compared to CMV variants [3].
- Vector Ratio Titration: Empirical determination of optimal CAST:donor ratios is critical, with 1:5 Cas9:donor ratio showing maximal efficiency in mouse heart tissue [3].
- Target Site Validation: Comprehensive assessment of genomic accessibility is essential, as some regions (e.g., SLC26A4 c.919-2) exhibit inherently low HITI efficiency (<0.15%) regardless of guide RNA design [20].
- CAST Engineering: Structure-guided engineering of PseCAST QCascade complexes can enhance DNA binding affinity and increase integration efficiencies in human cells [16].
Research Reagent Solutions
Table 4: Essential Reagents for CAST Preclinical Validation
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| CAST Systems | PseCAST, VchCAST, engineered variants [16] | RNA-guided DNA integration | Type I-F shows higher specificity; efficiency varies by variant |
| Delivery Vectors | AAV9, AAV-DJ, lentiviral | In vivo delivery of editing components | AAV9 effective for muscular and neurological targets [3] |
| Promoter Systems | MHCK7, SPc5-12, CAG | Tissue-specific or ubiquitous expression | SPc5-12 shows enhanced activity in mice [3] |
| Detection Tools | ddPCR assays, NGS panels, immunofluorescence | Quantification of editing efficiency | Multiplex ddPCR enables precise efficiency measurement [3] |
| Animal Models | Dup2 mice, Oncopig, AHC mouse models [3] [59] | Disease-specific context | Select models with clinical relevance and measurable endpoints |
The preclinical validation framework outlined in this application note provides a standardized approach for evaluating CAST system efficacy in mouse and pig models of human disease. The homology-independent integration mechanism of CAST systems represents a significant advancement over conventional CRISPR-based editors, offering enhanced precision for therapeutic gene integration without double-strand breaks. Through rigorous implementation of the protocols and assessment metrics described herein, researchers can generate clinically predictive data to support the translation of CAST technologies from preclinical validation to human therapeutic applications.
As CAST engineering continues to evolve, with structure-guided optimization yielding variants with improved efficiency and specificity [16], these systems hold exceptional promise for addressing the unmet need for precise large-scale genome editing in diverse therapeutic contexts. The integration of quantitative efficacy assessment in both small and large animal models remains essential for establishing the therapeutic potential of these innovative genome editing platforms.
The precision of CRISPR-based genome insertion is paramount for both basic research and therapeutic applications. While the CRISPR-Associated Transposase (CAST) system represents a promising new approach for homology-independent integration, its performance must be contextualized against established methods. This application note provides a systematic, quantitative comparison of three leading DNA repair pathway-mediated knock-in techniques: Homology-Independent Targeted Integration (HITI), Homology-Directed Repair (HDR), and Homology-Mediated End Joining (HMEJ). Understanding their relative performance across key metrics enables researchers to select the optimal strategy for their specific experimental or therapeutic goals, whether working with dividing cells, non-dividing cells, or pursuing in vivo applications.
Performance Metrics: Quantitative Comparison of Knock-In Strategies
The table below summarizes the key performance characteristics of HITI, HDR, and HMEJ based on current literature and experimental data.
Table 1: Key Performance Metrics of HITI, HDR, and HMEJ
| Performance Metric | HITI | HDR | HMEJ |
|---|---|---|---|
| Primary Repair Pathway | NHEJ [4] | HDR [10] | MMEJ [60] |
| Cell Cycle Dependence | Independent (works in dividing & non-dividing cells) [4] [2] | Dependent (S/G2 phase only) [10] [61] | Primarily G1/early S phase [61] |
| Typical Integration Efficiency | Variable by locus (e.g., ~21% in HSPCs; [12] as low as 0.15% in SLC26A4) [20] | Generally low in non-dividing cells (<10%) [20] | Highly efficient (e.g., 12.7% in chicken PGCs vs 6.25% for HDR) [60] |
| Optimal Cargo Size | Large transgenes (>1 kb); demonstrated for CAR genes [2] | 1-10 kb [61] | ≤5 kb [61] |
| Junction Precision | Low; prone to indels [4] [37] | High; seamless integration [62] | Moderate; some micro-deletions [37] |
| Key Advantage | Works in non-dividing cells; simplified donor design [4] | High-fidelity, precise integration [62] | High efficiency in relevant cell types (e.g., PGCs, HSPCs) [62] [60] |
| Main Limitation | Unpredictable junctional indels [37] | Inefficient in quiescent cells [62] | Complex donor design [60] |
Experimental Protocols: Detailed Methodologies for Knock-In
Protocol for HITI-Mediated CAR Knock-In in Primary Human T Cells
This protocol, adapted from Balke-Want et al. (2023), details the efficient insertion of a Chimeric Antigen Receptor (CAR) into the TRAC locus of primary human T cells using HITI, achieving high yields for clinical-scale manufacturing [2].
Day -1: T Cell Isolation and Activation
- Isolate primary human T cells from a leukopak using negative selection (e.g., EasySep Human T Cell Isolation Kit).
- Activate T cells using Dynabeads Human T-Activator CD3/CD28 at a 1:1 bead-to-cell ratio.
- Culture cells in TexMACS medium supplemented with IL-7 (12.5 ng/mL) and IL-15 (12.5 ng/mL), plus 3% human AB serum.
Day 0: RNP Complex and Donor Preparation
- Design a HITI-optimized nanoplasmid donor containing the CAR transgene flanked by Cas9 cut sites and the R6K origin of replication [4] [2].
- Form ribonucleoprotein (RNP) complexes by mixing wild-type Cas9 protein (61 µM) and TRAC-targeting sgRNA (125 µM) at a 1:1 volume ratio (2:1 molar ratio). Incubate at room temperature for 10 minutes.
- Add the nanoplasmid donor DNA (e.g., 3 mg/mL) to the pre-formed RNP complex and incubate for an additional 10 minutes to allow pre-cutting of the donor.
Day 2: Electroporation
- Magnetically remove activation beads and wash cells once in electroporation buffer.
- Resuspend T cells at a concentration of 2 × 10^8 cells/mL.
- Combine the cell suspension with the RNP + donor mixture and electroporate using a clinical-scale electroporator (e.g., Maxcyte GTx) using the "Expanded T cell 4" protocol for activated T cells.
- Post-electroporation, rest cells in the electroporation assembly for 30 minutes before transferring back to culture vessels with fresh, cytokine-supplemented medium.
Days 3-14: Post-Transfection Culture and Enrichment (CEMENT)
- Expand cells in G-Rex culture vessels, maintaining a density of ~1.5 × 10^6 cells/mL.
- For enrichment of successfully edited cells, implement the CRISPR EnrichMENT (CEMENT) strategy. This involves using a selection marker like the methotrexate (MTX)-resistant DHFR-FS gene included in the knock-in cassette.
- Around day 4-5 post-electroporation, add MTX to the culture medium. Optimize the concentration and duration of exposure to enrich CAR-positive cells to ~80% purity without significant yield loss [4] [2].
- Continue culture and expansion for a total of 14 days to achieve clinically relevant cell numbers (e.g., 5.5 × 10^8 to 3.6 × 10^9 CAR+ T cells).
Protocol for HMEJ-Mediated Gene Integration in Primordial Germ Cells (PGCs)
This protocol, based on work in chicken PGCs, demonstrates the high efficiency of HMEJ for site-specific gene integration in challenging primary cells [60].
Step 1: Donor Vector Construction
- Clone the gene of interest (e.g., a fluorescent protein reporter) into an HMEJ donor vector. The critical feature is that the cassette must be flanked by ~800 bp homology arms that themselves contain the target sgRNA sequence. This creates long homology arms with an internal cut site [60].
Step 2: Cell Transfection
- Co-transfect chicken PGCs with two plasmids: (1) a CRISPR/Cas9 plasmid expressing both Cas9-EGFP and the specific sgRNA, and (2) the HMEJ donor plasmid.
- Use a suitable transfection reagent (e.g., Lipofectamine 3000) and culture the transfected cells for 3 days.
Step 3: Isolation and Expansion of Edited Cells
- Three days post-transfection, use Fluorescence-Activated Cell Sorting (FACS) to isolate EGFP-positive cells, which indicate successful transfection with the CRISPR/Cas9 plasmid.
- Culture the sorted cells for an additional 4 days to allow for transgene expression and stabilization.
Step 4: Validation of Knock-In
- Analyze the cells for the expression of the knock-in reporter (e.g., mCherry) via flow cytometry. Efficiencies of 12.7% at the DAZL locus and 12.5% at the Pou5f3 locus have been reported, outperforming HDR in the same system [60].
- Confirm precise integration at the target locus using PCR amplification across the integration junctions and Sanger sequencing.
Visualizing the Molecular Workflows
The following diagrams illustrate the core molecular mechanisms and experimental workflows for each knock-in strategy.
HITI (Homology-Independent Targeted Integration)
HDR (Homology-Directed Repair)
HMEJ (Homology-Mediated End Joining)
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Knock-In Experiments
| Reagent / Tool | Function | Example Use Case & Notes |
|---|---|---|
| Nanoplasmid DNA | Non-viral donor template with minimal bacterial backbone (~450 bp) to reduce cytotoxicity and prevent transgene silencing [4] [2]. | Ideal for clinical-scale HITI in T cells; offers higher expression and easier GMP manufacturing than traditional plasmids or viral vectors [4]. |
| Cas9 RNP Complex | Pre-complexed Cas9 protein and sgRNA for high-efficiency editing with reduced off-target effects and rapid degradation [2]. | Standard for electroporation-based delivery in primary cells (e.g., T cells, HSPCs). |
| Electroporation Systems (e.g., Maxcyte GTx) | Enables efficient, closed-system delivery of RNP and donor DNA into sensitive primary cells [4] [2]. | Critical for clinical translation; use optimized protocols (e.g., "Expanded T cell 4" for activated T cells). |
| CEMENT Selection System | Post-editing enrichment using a selection marker (e.g., drug-resistant DHFR-FS) to increase purity of knock-in cells [4] [2]. | Achieves ~80% CAR+ T cell purity using methotrexate (MTX) selection without complex physical separation steps [2]. |
| In Silico Off-Target Prediction Tools (e.g., COSMID, CCTop, CRISPRme) | Computational sgRNA design to minimize off-target effects by accounting for mismatches and human genetic diversity [4]. | Essential first step in therapeutic development; should be followed by empirical off-target assessment (e.g., GUIDE-seq) [4]. |
The choice between HITI, HDR, and HMEJ is not one of absolute superiority but of strategic alignment with experimental goals. HITI is the definitive choice for applications involving non-dividing or slowly dividing cells, such as in vivo neuronal correction or engineering non-activated T cells, where its cell-cycle independence is critical [4] [63]. HDR remains the gold standard for applications where seamless, precise integration is the highest priority and when working with readily dividable cell types [62]. HMEJ has emerged as a powerful hybrid, often demonstrating superior knock-in efficiency in challenging primary cells like PGCs and HSPCs, making it a robust choice for ex vivo cell engineering where its higher efficiency outweighs the more complex donor design [62] [60].
For researchers exploring next-generation systems like CAST, this comparative analysis provides a foundational performance baseline. The ideal knock-in tool balances efficiency, precision, and practical applicability, and the optimal choice is ultimately dictated by the target cell type, the required cargo size, and the tolerance for junctional indels in the final product.
The advent of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated transposase (CAST) systems represents a paradigm shift in genome engineering, offering a pathway for homology-independent targeted integration of large DNA fragments. Unlike conventional CRISPR-Cas systems that rely on double-strand breaks (DSBs) and endogenous repair mechanisms, CAST systems utilize a RNA-guided DNA targeting module complexed with a transposase enzyme to catalyze precise, DSB-free integration of genetic cargo [16]. This mechanism theoretically bypasses the error-prone non-homologous end joining (NHEJ) and homology-directed repair (HDR) pathways, which are major sources of unintended genomic alterations in traditional editing.
However, the implementation of any genome editing technology necessitates rigorous safety profiling. Recent studies reveal that even advanced systems can generate unintended structural variations (SVs), including large deletions, insertions, and chromosomal rearrangements [64] [6]. For drug development professionals and researchers leveraging CAST systems, a comprehensive protocol for analyzing these unintended outcomes is not merely optional—it is critical for therapeutic safety and regulatory approval. This application note provides a detailed framework for the rigorous analysis of unintended genomic alterations in CAST-based homology-independent targeted integration research, ensuring that the field can advance with both efficiency and safety.
Understanding Unintended Genomic Alterations in Genome Editing
The Spectrum of Genomic Alterations
Traditional CRISPR-Cas9 editing is notorious for generating a spectrum of unintended genetic outcomes beyond simple on-target small insertions or deletions (indels). These can be broadly categorized as follows:
- On-Target Structural Variations (SVs): These occur at the intended editing site and include kilobase- to megabase-scale deletions, inversions, chromosomal truncations, and complex rearrangements such as chromothripsis [64] [6]. For instance, studies in HEK293T cells have reported kilobase-sized deletions and inversions at frequencies of ~3% and intra-chromosomal translocations making up to 6.2–14% of editing outcomes [64].
- Off-Target Mutations: These are edits at genomic loci with sequence similarity to the guide RNA. While often low in frequency, they can pose significant risks if they occur in tumor suppressor genes or oncogenes [6].
- Vector Integration-Related Aberrations: The unintended integration of partial or full-length DNA templates, whether viral or plasmid-based, at both on-target and off-target sites can disrupt normal gene function and regulatory networks [6].
Table 1: Types and Frequencies of Unintended Genomic Alterations Documented in CRISPR-Cas Editing
| Alteration Type | Reported Size Range | Example Frequencies | Primary Detection Methods |
|---|---|---|---|
| Large Deletions | 0.1 kb - >1 Mb | ~3% (0.1-5 kb) [64] | Long-range PCR, WGS |
| Chromosomal Truncations | Entire chromosome arms | 10–25.5% in HEK293T clones [64] | Karyotyping, FISH |
| Translocations | Inter-/Intra-chromosomal | Up to 14% of outcomes [64] | CAST-Seq, LAM-HTGTS [6] |
| Complex SVs (e.g., Chromothripsis) | Multiple Mb, complex rearrangements | Reported in multiple studies [64] [6] | WGS |
Why CAST Systems Require Rigorous SV Analysis
CAST systems, particularly type I-F systems like PseCAST and VchCAST, are engineered for DSB-free integration [16]. This is a fundamental safety advantage. However, the process still involves the introduction of a large nucleoprotein complex and the integration of a foreign DNA sequence into the host genome. Potential residual nuclease activity in the transposase module or aberrations during the integration process could theoretically lead to genomic stress and SVs. Furthermore, the efficiency of CAST systems in human cells, while improving through engineering (e.g., PseCAST variants with increased integration efficiencies [16]), is still a subject of optimization. Any manipulation to boost efficiency, akin to the use of DNA-PKcs inhibitors in HDR editing, must be accompanied by stringent SV monitoring to avoid aggravation of genomic aberrations [6]. Therefore, assuming CAST systems are inherently safe without empirical validation is a perilous approach.
Experimental Protocol for SV Analysis in CAST System Workflows
The following protocol provides a step-by-step methodology for profiling unintended genomic alterations following CAST-mediated targeted integration. The workflow is divided into three core phases: the CAST integration experiment, targeted SV analysis, and genome-wide analysis.
Phase 1: CAST System Delivery and Cell Culture
Materials and Reagents
Table 2: Key Research Reagent Solutions for CAST Experiments
| Reagent / Solution | Function / Description | Example / Note |
|---|---|---|
| Type I-F CAST System | Engineered CRISPR-associated transposase for DSB-free integration. | PseCAST system demonstrates higher efficiency in human cells than VchCAST [16]. |
| Nanoplasmid Donor DNA | Optimized plasmid backbone for gene therapy; reduces transgene silencing. | Contains R6K origin and antibiotic-free selection marker [2]. |
| Electroporation System | For co-delivery of CAST ribonucleoprotein (RNP) and donor DNA into cells. | Maxcyte GTx with "Expanded T cell" or "Resting T cell" protocols [2]. |
| Cell Culture Media | Supports growth and viability of edited cells. | TexMACS media with IL-7 and IL-15 for T-cells [2]. |
| Selection Agents | Enriches for successfully edited cells. | Methotrexate for DHFR-FS selection in CEMENT protocol [2]. |
Procedure
CAST Component Design and Preparation:
- Design guide RNA (gRNA) targeting the desired genomic locus (e.g., TRAC locus for T-cell engineering).
- Clone the transgene of interest (e.g., a Chimeric Antigen Receptor or CAR) into a nanoplasmid donor vector. Ensure the donor contains the necessary elements for integration (e.g., RNP cut sites within the nanoplasmid) [2].
- Formulate the CAST RNP complex by incubating purified Cas proteins (e.g., wildtype Cas9 at 61 µM) with the synthesized gRNA (125 µM) at a molar ratio of 2:1 (Cas:gRNA) for 10 minutes at room temperature [2].
- Add the purified nanoplasmid donor DNA (e.g., 3 mg/ml) to the pre-formed RNP complex and incubate for an additional 10 minutes to allow the RNP to pre-cut the donor plasmid.
Cell Electroporation and Culture:
- Harvest and wash the target cells (e.g., primary human T-cells). Resuspend cells in electroporation buffer at a high concentration (e.g., 2 × 10^8 cells/ml) [2].
- Combine the cell suspension with the RNP/donor mixture and electroporate using an optimized protocol (e.g., the "Expanded T cell 4" protocol on a Maxcyte GTx for activated T-cells).
- Post-electroporation, rest cells for 30 minutes before transferring them to pre-warmed culture media supplemented with appropriate cytokines.
- Culture cells for several days to allow for integration and transgene expression. If using a selection system like CEMENT, add the selection agent (e.g., Methotrexate) to enrich for edited cells [2].
Phase 2: Targeted Analysis of On-Target and Local SVs
Materials and Reagents
- Genomic DNA Extraction Kit (e.g., DNeasy Blood & Tissue Kit, Qiagen)
- PCR Reagents: Long-range PCR enzyme mix, standard Taq polymerase, dNTPs.
- Primers: Designed to flank the on-target integration site (for long-range PCR) and internal control primers.
- CAST-Seq Library Preparation Kit or components for LAM-HTGTS [6].
Procedure
Genomic DNA Extraction:
- Harvest edited cells after a sufficient expansion period (e.g., 7-14 days post-editing). Extract high-molecular-weight genomic DNA according to the manufacturer's instructions. Quantify DNA purity and concentration using a spectrophotometer.
On-target Integration Efficiency:
- Perform initial quality control using standard amplicon sequencing. Design primers that amplify a short region spanning the integration site.
- Prepare libraries and sequence on an Illumina MiSeq or similar platform. Analyze sequences for precise integration and small indels. Caution: This method can overestimate precise integration if large deletions remove primer binding sites [6].
Detection of Local Structural Variations:
- Long-range PCR: Design primers several kilobases upstream and downstream of the integration site.
- Set up a long-range PCR reaction with an enzyme mix capable of amplifying large fragments (e.g., 5-20 kb).
- Analyze the PCR products by agarose gel electrophoresis. The presence of multiple or larger-than-expected bands suggests heterogeneous SVs, including large deletions or rearrangements.
- Targeted SV Assays (CAST-Seq):
- For a more sensitive and comprehensive detection of SVs and translocations emanating from the on-target site, employ methods like CAST-Seq [6].
- Briefly, this involves a two-step PCR strategy to circularize fragmented DNA, followed by amplification and sequencing of junctions that involve the target site and distant genomic regions.
- Analyze sequencing data with dedicated bioinformatic pipelines to identify and quantify specific translocation partners and deletion boundaries.
- Long-range PCR: Design primers several kilobases upstream and downstream of the integration site.
Phase 3: Genome-wide Analysis of Unintended Alterations
Materials and Reagents
- Whole Genome Sequencing (WGS) Service or Platform (e.g., Illumina NovaSeq, PacBio).
- Bioinformatic Analysis Tools: SV-callers (e.g., Manta, DELLY), alignment software (e.g., BWA).
Procedure
Whole Genome Sequencing (WGS):
- Submit high-quality genomic DNA from edited cells and a non-edited control from the same batch for WGS. A coverage of >30x is recommended for robust SV detection.
- This allows for an unbiased survey of the entire genome for large-scale deletions, insertions, inversions, and translocations.
Bioinformatic Analysis:
- Align sequencing reads to the reference genome (e.g., GRCh38).
- Use multiple structural variant callers to identify SVs with high confidence.
- Filter SVs present in the edited sample against the control to identify editing-associated variants.
- Annotate the SVs to determine if they affect coding regions, regulatory elements, or known cancer-associated genes.
Visualization and Data Interpretation
The data generated from these protocols must be synthesized to assess the safety profile of the CAST editing process.
Key Interpretation Metrics:
- Variant Allele Frequency (VAF): The proportion of sequencing reads supporting an SV. A high VAF indicates a clonal event, which may pose a greater safety risk.
- SV Size and Genomic Impact: Categorize SVs by their size and whether they disrupt known oncogenes, tumor suppressors, or essential genomic regulatory domains.
- Comparison to Controls: Any SV also detected in the non-edited control population is likely not a result of the editing process and can be filtered out.
The therapeutic potential of CAST systems for targeted gene insertion is immense. However, responsible translation into clinical applications demands a thorough and rigorous assessment of unintended genomic alterations. The multi-tiered experimental protocol outlined here—combining targeted assays like long-range PCR and CAST-Seq with unbiased whole-genome sequencing—provides a comprehensive framework for profiling these hidden risks. By integrating this rigorous analysis into the standard development workflow, researchers and drug developers can advance CAST-based therapies with greater confidence, ensuring that the benefits of precise genome engineering are not undermined by unforeseen genomic instability.
Homology-Independent Targeted Integration (HITI) represents a pivotal advancement in the field of precision genome editing, enabling the precise insertion of therapeutic transgenes without reliance on homology-directed repair (HDR) pathways. Unlike HDR-based approaches, which are active only in dividing cells during the S and G2 phases of the cell cycle, HITI leverages the non-homologous end joining (NHEJ) pathway, a dominant DNA repair mechanism functional in both dividing and non-dividing cells [2] [4]. This fundamental characteristic renders HITI particularly valuable for therapeutic applications in post-mitotic tissues such as skeletal and cardiac muscle, as well as for engineering difficult-to-transfect primary cells like resting T-lymphocytes. The core innovation of HITI lies in its engineered donor design, which incorporates Cas9 target sites as reverse complements of the genomic target site, enabling re-cleavage of reverse-integration products and thereby driving correct directional knockin [3]. This technology is now being leveraged across diverse therapeutic areas, from monogenic disorders to engineered cell therapies, positioning it as a cornerstone of the rapidly evolving CAST (CRISPR-Assisted System for Transgene integration) system for next-generation genetic medicine.
Current Clinical and Preclinical Applications
HITI for Duchenne Muscular Dystrophy (DMD)
Duchenne muscular dystrophy, an X-linked disorder caused by mutations disrupting the reading frame of the dystrophin gene, has emerged as a prime candidate for HITI-mediated intervention. A groundbreaking preclinical study demonstrated the viability of HITI for restoring full-length dystrophin expression in a mouse model carrying a Dmd exon 2 duplication, a mutation found in approximately 1% of DMD patients [13] [3]. Researchers designed a system delivered via paired AAV9 vectors that targeted insertion of a "mega-exon" encoding DMD exons 1-19 into intron 19, effectively restoring the full coding sequence when spliced to endogenous exon 20 [3]. This approach achieved editing of 1.4% of genomes in cardiac tissue, resulting in 30% correction at the transcript level and restoration of 11% of normal dystrophin protein levels [3]. The system was further optimized by evaluating different Cas9:donor vector ratios, with a 1:5 ratio demonstrating maximal efficiency [3]. This proof-of-concept work establishes that HITI can restore therapeutically meaningful levels of dystrophin and could potentially benefit approximately 25% of DMD patients carrying mutations upstream of exon 19 [3].
Table 1: HITI Performance in DMD Mouse Model Across Tissues
| Tissue | Genome Editing Efficiency | Transcript Correction | Dystrophin Restoration |
|---|---|---|---|
| Heart | 1.4% | 30% | 11% of normal levels |
| Diaphragm | 0.26% | 1% | Not significant |
| Tibialis Anterior | 0.11% | 1% | Not significant |
HITI for CAR-T Cell Engineering
In the realm of adoptive cell therapy, HITI has demonstrated remarkable potential for streamlining the manufacturing of chimeric antigen receptor (CAR) T-cells. A comprehensive study comparing HITI with HDR-mediated knock-in for integration of an anti-GD2 CAR into the T-cell receptor alpha constant (TRAC) locus revealed that HITI yielded at least two-fold more CAR-T cells than HDR-based approaches [2]. This enhanced efficiency is particularly valuable for clinical-scale manufacturing, where cell yields directly impact therapeutic applicability. The HITI platform utilized a fully non-viral workflow involving electroporation of CRISPR/Cas9 ribonucleoprotein (RNP) complexes together with nanoplasmid DNA containing the CAR transgene [44] [4]. A critical innovation in this system was the implementation of post-editing enrichment using the CRISPR EnrichMENT (CEMENT) strategy, which employed a mutant dihydrofolate reductase (DHFR-FS) conferring resistance to methotrexate to enrich edited cells to approximately 80% purity [2]. This integrated HITI/CEMENT approach generated therapeutically relevant cell doses ranging from 5.5×10^8 to 3.6×10^9 CAR+ T-cells from a starting population of 5×10^8 cells across multiple donors, meeting the required doses for commercial CAR-T products [2].
Table 2: HITI-CAR-T Cell Manufacturing Yields from 5×10^8 Starting T-Cells
| Donor | CAR+ T-Cell Yield | Purity After CEMENT | Therapeutic Relevance |
|---|---|---|---|
| Donor 1 | 5.5×10^8 | ~80% | Meets clinical dose range |
| Donor 2 | 3.6×10^9 | ~80% | Meets clinical dose range |
| Donor 3 | Intermediate yield | ~80% | Meets clinical dose range |
Detailed Experimental Protocols
HITI Protocol for CAR-T Cell Manufacturing
Day 0: T-Cell Isolation and Culture
- Isolate primary human T-cells from leukopaks using negative selection with the EasySep Human T Cell Isolation Kit.
- Activate T-cells using Dynabeads Human T-Activator CD3/CD28 at a 1:1 ratio.
- Culture cells in TexMACS media supplemented with IL-7 (12.5 ng/ml) and IL-15 (12.5 ng/ml), plus 3% human male AB Serum.
- Maintain cells at approximately 1.5×10^6/ml using appropriate culture vessels.
Day 2: Electroporation and HITI Knock-in
- Magnetically remove Dynabeads and count cells.
- Wash cells once in electroporation buffer and resuspend at 2×10^8 cells/ml.
- Prepare RNP complex by mixing wildtype Cas9 (61 µM) with sgRNA (125 µM) at a 1:1 volume ratio (2:1 molar ratio) and incubate for 10 minutes at room temperature.
- Add nanoplasmid DNA (3 mg/ml) to the RNP complex and incubate for at least 10 minutes to allow RNP-mediated linearization of the donor DNA.
- Electroporate using the Maxcyte GTx system with the "Expanded T cell 4" protocol for activated T-cells or "Resting T cell 14-3" protocol for non-activated T-cells.
- Post-electroporation, rest cells in electroporation buffer for 30 minutes before transferring to final culture vessels.
- For non-activated T-cells, stimulate with Dynabeads after electroporation.
Days 3-14: Expansion and CEMENT Enrichment
- Expand cell culture volume progressively to maintain optimal density.
- Initiate methotrexate (MTX) selection for DHFR-FS enriched cultures between days 5-7 post-electroporation.
- Maintain MTX exposure for optimized duration (typically 72-96 hours) to enrich transgene-positive cells without excessive toxicity.
- Monitor CAR expression by flow cytometry and continue expansion until target cell numbers are achieved.
- Harvest cells for final formulation when CAR+ purity reaches approximately 80% or desired therapeutic dose is obtained.
HITI Protocol for In Vivo DMD Correction
Vector Design and Production
- Design two AAV9 vectors: one expressing SaCas9 under the MHCK7 or SPc5-12 promoter, and a donor vector containing the mega-exon encoding DMD exons 1-19 with appropriate Cas9 target sites in reverse orientation.
- Produce high-titer AAV9 vectors (≥10^13 vg/ml) using standard manufacturing methods.
- Determine optimal Cas9:donor vector ratio through preliminary experiments; a 1:5 ratio has demonstrated highest efficiency in murine models.
In Vivo Administration
- Utilize neonatal Dup2 mice (postnatal day 1-3) for intervention prior to peak muscle degeneration.
- Administer vectors via intraperitoneal injection at a total dose of 2.0×10^14 vg/kg, with Cas9 and donor vectors mixed at the predetermined optimal ratio.
- For larger animals or eventual clinical translation, consider limb perfusion or systemic intravenous delivery.
Analysis and Validation
- Harvest tissues 4-8 weeks post-injection for molecular and histological analysis.
- Quantify knockin efficiency by droplet digital PCR (ddPCR) using primers specific to the donor-genome junction.
- Assess transcript correction by RT-ddPCR or RNA sequencing targeting the splice junction between the mega-exon and endogenous exon 20.
- Evaluate dystrophin restoration by quantitative immunofluorescence, capillary western blot, and immunohistochemistry.
- Monitor for potential off-target effects and vector integration patterns by next-generation sequencing.
Visualization of HITI Workflows
HITI Mechanism and CAR-T Cell Manufacturing Workflow
HITI for DMD Gene Correction
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for HITI Experiments
| Reagent/Category | Specific Examples | Function/Purpose | Considerations |
|---|---|---|---|
| Nuclease System | S. aureus Cas9 (SaCas9), C. jejuni Cas9 | Creates double-strand breaks at target genomic loci and donor DNA | Smaller Cas9 variants preferred for AAV packaging; specificity must be validated |
| Delivery Vectors | AAV9 (in vivo), Nanoplasmid DNA (ex vivo) | Delivers editing components to target cells | AAV serotypes affect tropism; nanoplasmid reduces cytotoxicity vs. traditional plasmids |
| Guide RNA Design | TRAC-targeting sgRNA, DMD intron 19 sgRNAs | Directs Cas9 to specific genomic loci | Must include reverse complement sites in donor; off-target potential requires assessment |
| Donor Template | MHCK7-promoted mega-exon, CAR expression cassette | Provides therapeutic transgene for integration | HITI design requires internal cut sites; size constraints apply for viral delivery |
| Enrichment Systems | DHFR-FS (methotrexate resistance), tEGFR, tNGFR | Selects successfully edited cells from population | DHFR-FS enables pharmacological selection; surface markers allow magnetic sorting |
| Analytical Tools | ddPCR, rhAMPSeq, GUIDE-seq, TLA | Quantifies editing efficiency and detects off-target effects | Multiple orthogonal methods needed for comprehensive safety profiling |
Future Perspectives and Regulatory Considerations
The therapeutic landscape for HITI is rapidly evolving, with regulatory frameworks adapting to accommodate bespoke genetic therapies. The FDA's newly proposed "plausible mechanism pathway" represents a significant advancement, potentially enabling accelerated approval for personalized therapies that demonstrate targeting of specific molecular abnormalities and improvement in clinical outcomes, even without traditional clinical trial data in some cases [65]. This pathway is particularly relevant for HITI-based approaches targeting rare genetic mutations, where conventional trial designs are not feasible. As the technology progresses, key challenges remain, including optimization of delivery efficiency across different tissue types, minimization of unintended genomic alterations, and development of robust manufacturing processes for clinical-scale production. The ongoing integration of HITI with emerging enrichment strategies like CEMENT and advanced delivery platforms positions this technology to substantially impact the treatment of monogenic disorders, cancer, and other diseases amenable to genetic correction. Future directions will likely focus on enhancing the specificity and efficiency of HITI through novel Cas variants, improved donor designs, and combined therapeutic approaches that address both genetic correction and functional recovery.
Conclusion
Homology-Independent Targeted Integration (HITI) represents a paradigm shift in genome editing, effectively overcoming the limitation of HDR by utilizing the ubiquitous NHEJ pathway. This enables therapeutic gene integration in both non-dividing cells, such as photoreceptors, and proliferating tissues, such as hepatocytes, allowing for allele-independent correction of gain-of-function mutations and stable long-term expression. While challenges in optimizing efficiency and ensuring absolute safety remain, the robust preclinical proof-of-concept in retina, liver, and T-cell engineering underscores HITI's immense therapeutic potential. Future directions will focus on refining delivery systems for enhanced specificity, conducting comprehensive long-term safety studies, and advancing the promising in vivo and ex vivo applications into clinical trials, ultimately paving the way for a new class of durable genetic medicines.
References
- 1. Genome Editing in Human Hematopoietic Stem and ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/33309880/]
- 2. Homology-independent targeted insertion (HITI) enables ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-023-01799-7]
- 3. CRISPR-Cas9 homology-independent targeted integration ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10495553/]
- 4. Homology-Independent Targeted Insertion (HITI) for ... [https://www.scientificarchives.com/article/homology-independent-targeted-insertion-hiti-for-therapeutic-t-cell-engineering]
- 5. Homology-independent targeted insertion (HITI) enables ... [https://pubmed.ncbi.nlm.nih.gov/37365642/]
- 6. The hidden risks of CRISPR/Cas: structural variations and ... [https://www.nature.com/articles/s41467-025-62606-z]
- 7. Strategies for In Vivo Genome Editing in Nondividing Cells [https://www.sciencedirect.com/science/article/abs/pii/S016777991830088X]
- 8. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5331785/]
- 9. strategies to enhance the frequency of homology-directed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6177507/]
- 10. Precise in vivo genome editing via single homology arm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6796851/]
- 11. and NHEJ-Based Genome Editing in Airway Epithelial Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12197413/]
- 12. Genome editing in human hematopoietic stem and progenitor cells via CRISPR-Cas9-mediated homology-independent targeted integration - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S1525001620306675]
- 13. CRISPR-Cas9 homology-independent targeted integration of ... [https://pubmed.ncbi.nlm.nih.gov/37706184/]
- 14. Precise genome-editing in human diseases [https://www.nature.com/articles/s41392-024-01750-2]
- 15. Optimization of HITI-Mediated Gene Insertion for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11578159/]
- 16. Structure-guided engineering of type I-F CASTs for ... [https://www.nature.com/articles/s41467-025-63164-0]
- 17. Targeted gene editing and near-universal cDNA insertion ... [https://www.nature.com/articles/s41467-025-62738-2]
- 18. Expanding the frontiers of genome engineering [https://www.sciencedirect.com/science/article/abs/pii/S0734975024001757]
- 19. Advances in large-scale DNA engineering with the ... [https://www.nature.com/articles/s12276-025-01530-0]
- 20. Low Efficiency of Homology-Independent Targeted ... [https://www.mdpi.com/1422-0067/26/11/4980]
- 21. Comparative Analysis of Methods for Assessing On-Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11932317/]
- 22. Comprehensive protocols for CRISPR/Cas9-based gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4988528/]
- 23. Off-target effects in CRISPR-Cas genome editing for ... [https://www.sciencedirect.com/science/article/pii/S2162253125001908]
- 24. innovations and challenges in rAAV vector-based CRISPR ... [https://www.nature.com/articles/s41434-025-00573-2]
- 25. Nanoplasmid™ Vector System [https://www.aldevron.com/custom-manufacturing/nanoplasmid]
- 26. Bringing New Ideas to AAV Gene Therapy [https://themedicinemaker.com/issues/2025/articles/november/bringing-new-ideas-to-aav-gene-therapy/]
- 27. Non-Viral Gene Delivery Systems: Current Advances and ... [https://pubmed.ncbi.nlm.nih.gov/40874945/]
- 28. Trends and challenges of AAV-delivered gene editing ... [https://www.sciencedirect.com/science/article/pii/S2162253125001891]
- 29. Viral and Non-Viral Systems to Deliver Gene Therapeutics ... [https://www.mdpi.com/1422-0067/25/13/7333]
- 30. Continuous evolution of CRISPR-associated transposases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12326709/]
- 31. Targeted gene repair [https://www.nature.com/subjects/targeted-gene-repair/ncomms]
- 32. Gene augmentation therapy restores vision and preserves ... [https://www.nature.com/articles/s43856-025-01108-x]
- 33. A promising approach for the treatment of liver fibrosis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12528922/]
- 34. Nanoparticle-based therapeutic strategies for chronic liver ... [https://www.sciencedirect.com/science/article/pii/S2542568425000273]
- 35. Therapeutic homology-independent targeted integration in ... [https://www.nature.com/articles/s41467-022-29550-8]
- 36. From ex vivo to in vivo chimeric antigen T cells ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-06052-3]
- 37. Precise, predictable genome integrations by deep-learning ... [https://www.nature.com/articles/s41587-025-02771-0]
- 38. Direct in vivo CAR T cell engineering - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S016561472400052X]
- 39. In vivo chimeric antigen receptor (CAR)-T cell therapy [https://www.nature.com/articles/s41573-025-01291-5]
- 40. Frontiers | Comprehensive approaches to design efficient gRNA for... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1579165/full]
- 41. CRISPR Guide [https://www.addgene.org/guides/crispr/]
- 42. An Efficient Electroporation Protocol for the Genetic ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2016.00099/full]
- 43. Electroporation Process [https://maxcyte.com/our-technology/guide-to-electroporation/]
- 44. Efficient Homology-Independent Solution for Precise Targeted ... [https://techfinder.stanford.edu/technology/efficient-homology-independent-solution-precise-targeted-dna-insertions-human-t-cells]
- 45. 综述解读| 基因编辑进入3.0时代:更精准、更安全、更灵活 [https://bydrug.pharmcube.com/news/detail/beeb0df62aabe46935a4c16e35ef594a]
- 46. 使用病毒组分靶向障碍和疾病的crispr-cas系统和组合物的 ... [https://patents.google.com/patent/CN105793425B/zh]
- 47. Genotoxicity assessment: opportunities, challenges and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10404208/]
- 48. Gene Editing and Genotoxicity: Targeting the Off-Targets [https://pmc.ncbi.nlm.nih.gov/articles/PMC8525370/]
- 49. Comprehensive analysis of off-target and on-target effects ... [https://www.sciencedirect.com/science/article/pii/S2329050124001815]
- 50. Measurement and clinical interpretation of CRISPR off- ... [https://www.nature.com/articles/s41588-025-02428-3]
- 51. IWGT report on quantitative approaches to genotoxicity risk ... [https://www.sciencedirect.com/science/article/pii/S1383571814002605]
- 52. Quantitative in vitro to in vivo extrapolation of genotoxicity ... [https://hesiglobal.org/publication/quantitative-in-vitro-to-in-vivo-extrapolation-of-genotoxicity-data-provides-protective-estimates-of-in-vivo-dose/]
- 53. Scalable Manufacturing: The Make-or-Break Factor for Cell ... [https://cellinobio.com/press/scalable-manufacturing-the-make-or-break-factor-for-cell-gene-and-tissue-therapies]
- 54. Structure-guided engineering of type I-F CASTs for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11429998/]
- 55. Strategies for scalable manufacturing and translation of ... [https://www.sciencedirect.com/science/article/pii/S1873506120302798]
- 56. Automated Manufacturing Processes and Platforms for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11872983/]
- 57. Advances in large-scale DNA engineering with the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12508223/]
- 58. Pigs: Large Animal Preclinical Cancer Models - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10965265/]
- 59. Mouse models for ultra-rare disorder could pave the way ... [https://www.jax.org/news-and-insights/2025/july/mouse-models-for-ultra-rare-disorder-could-pave-the-way-for-nervous-system-gene-editing-therapies]
- 60. HMEJ-mediated efficient site-specific gene integration in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6868705/]
- 61. Recent advances in CRISPR-Cas9-based genome insertion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10878794/]
- 62. A comparison of DNA repair pathways to achieve a site ... [https://www.sciencedirect.com/science/article/pii/S2162253121003176]
- 63. In vivo genome editing via CRISPR/Cas9-mediated ... [https://www.nature.com/articles/s41467-024-48092-9]
- 64. Unintended CRISPR-Cas9 editing outcomes [https://pmc.ncbi.nlm.nih.gov/articles/PMC10182114/]
- 65. FDA unveils new pathway to usher bespoke therapies ... [https://www.clinicaltrialsarena.com/news/fda-unveils-new-pathway-to-usher-bespoke-therapies-to-market/]