CRISPRi in Bacteria: Mechanisms, Applications, and Optimization for Transcriptional Repression
This article provides a comprehensive overview of CRISPR interference (CRISPRi), a powerful and programmable tool for transcriptional repression in bacteria.
CRISPRi in Bacteria: Mechanisms, Applications, and Optimization for Transcriptional Repression
Abstract
This article provides a comprehensive overview of CRISPR interference (CRISPRi), a powerful and programmable tool for transcriptional repression in bacteria. We detail the foundational mechanism by which a catalytically dead Cas9 (dCas9) and guide RNA complex sterically blocks RNA polymerase, leading to gene knockdown. The scope extends to methodological implementation, from single-gene repression to genome-scale functional genomics screens, highlighting applications in metabolic engineering and antibiotic target discovery. We systematically address common troubleshooting and optimization challenges, including off-target effects and polarity. Finally, we present a comparative analysis validating CRISPRi against other genetic perturbation technologies like RNAi and TALENs, underscoring its superior specificity and reversibility. This resource is tailored for researchers, scientists, and drug development professionals seeking to leverage CRISPRi for advanced genetic studies and biotechnological applications.
The Core Mechanism of CRISPRi: How dCas9 Achieves Programmable Gene Repression in Bacteria
The repurposing of the bacterial adaptive immune system, CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats), into a programmable genetic tool represents a pivotal advancement in molecular biology. Central to this evolution is the engineering of the catalytically dead Cas9 (dCas9), a key transformation that converted a DNA-cutting enzyme into a precise, RNA-guided DNA-binding platform. This whitepaper details the fundamental engineering of dCas9, its mechanism as a transcriptional repressor in CRISPR interference (CRISPRi), and its application in bacterial research. We provide a technical guide covering core principles, quantitative performance data, and detailed experimental protocols for implementing CRISPRi in bacterial systems, framing this technology within the context of functional genomics and drug discovery.
The native Type II CRISPR-Cas9 system is an adaptive immune mechanism in bacteria and archaea that cleaves invading foreign DNA in a sequence-specific manner [1] [2]. This system requires a Cas9 nuclease and two RNA components, a CRISPR RNA (crRNA) and a trans-activating crRNA (tracrRNA), which have been engineered into a single guide RNA (sgRNA) for simplicity [3] [4]. The sgRNA directs Cas9 to a target DNA sequence complementary to its 5' end, leading to a double-strand break (DSB) adjacent to a Protospacer Adjacent Motif (PAM), typically 5'-NGG-3' for Streptococcus pyogenes Cas9 [1] [4].
The genesis of dCas9 involved the deliberate disruption of Cas9's nuclease activity. Key catalytic residues in its two nuclease domains were mutated: the D10A mutation in the RuvC domain and the H840A mutation in the HNH domain [3] [5] [2]. The resulting dCas9 protein is incapable of cleaving DNA but retains its ability to bind DNA with high specificity based on the sgRNA sequence [2] [6]. This transformation from a "cutting" tool to a "binding" tool laid the foundation for a vast array of new applications, most notably its use as the core component in CRISPRi for programmable transcriptional regulation in bacteria and beyond [3] [7].
The CRISPRi Mechanism for Transcriptional Repression in Bacteria
In bacterial systems, CRISPRi functions through the binding of the dCas9-sgRNA complex to genomic DNA, where it acts as a steric block to the transcriptional machinery [3] [6]. The mechanism and efficiency of repression are fundamentally determined by the genomic location of the dCas9-sgRNA binding site.
Steric Inhibition of Transcription
When the dCas9-sgRNA complex targets a region within the promoter, it can sterically hinder the binding of RNA polymerase (RNAP) or essential transcription factors, thereby preventing transcription initiation [3]. When the complex binds within the coding sequence of a gene, particularly to the non-template strand, it creates a physical roadblock that halts the progression of the elongating RNAP, leading to aborted transcription [3] [7]. Repression is generally stronger when targeting the non-template strand for elongation blocking, as helicase activity can displace dCas9 bound to the template strand [2]. This mechanism allows for repression efficiencies of up to 99.9% in model bacteria like E. coli [3] [2].
Quantitative Performance of CRISPRi in Bacteria
The performance of CRISPRi is characterized by its high efficiency and specificity. The table below summarizes key quantitative data from foundational and application-based studies in bacteria.
Table 1: Quantitative Performance Metrics of CRISPRi in Bacterial Systems
| Metric | Reported Value/Range | Experimental Context | Source |
|---|---|---|---|
| Max. Repression Efficiency | Up to 99.9% | Target gene repression in E. coli | [3] [2] |
| Dynamic Range | > 100-fold | Tunable repression via inducer titration | [6] |
| Specificity Seed Region | 12 nt + PAM | Critical for determining binding specificity; the 8-10 bases at the 3' end of the gRNA (the "seed") are most critical | [3] [4] |
| Multiplexing Capacity | Up to 12 genes simultaneously | Demonstrated using Extra-Long sgRNA Arrays (ELSAs) in E. coli | [2] |
Experimental Protocol: Implementing CRISPRi in Bacteria
This section provides a detailed methodology for establishing and validating a CRISPRi system for targeted gene repression in a bacterial model like E. coli.
Key Reagent Solutions
The successful deployment of CRISPRi relies on a core set of genetic tools. The following table outlines the essential reagents and their functions.
Table 2: Essential Research Reagents for Bacterial CRISPRi
| Reagent | Function | Key Considerations |
|---|---|---|
| dCas9 Expression Vector | Constitutively or inducibly expresses catalytically dead Cas9 (D10A, H840A mutations). | High-level expression may cause toxicity [7] [6]. Inducible promoters (e.g., P_tet_, P_BAD_) allow temporal control. |
| sgRNA Expression Vector | Expresses the single guide RNA. Contains a scaffold sequence and a customizable 20-nt spacer. | May be on a separate plasmid from dCas9 or combined into a single plasmid [6]. |
| sgRNA Spacer Sequence | The 20-nucleotide sequence defining genomic target. | Must be adjacent to a PAM (NGG for SpCas9). Must be specific to avoid off-target effects [3] [4]. |
| Inducer Molecules | Small molecules to titrate dCas9/sgRNA expression (e.g., aTc, IPTG, Arabinose). | Enables tuning of repression levels and study of essential genes [7] [6]. |
Step-by-Step Workflow
The following diagram and protocol outline the complete process from system design to phenotypic validation.
Step 1: Target Selection and sgRNA Design
- Identify Target Gene(s): Choose the gene(s) for transcriptional repression.
- Locate PAM Sites: Scan the target locus for 5'-NGG-3' PAM sequences.
- Design sgRNA Spacers: Select a 20-nt spacer sequence immediately 5' to the PAM.
- Specificity Check: Use BLAST or similar tools to ensure the 12-nt "seed" sequence plus the PAM is unique in the genome to minimize off-target effects [3].
Step 2: Reagent Cloning
- sgRNA Cloning: Synthesize and clone the designed spacer oligonucleotides into the sgRNA expression plasmid. The sgRNA scaffold is typically under a constitutive promoter [3] [6].
- dCas9 Cloning: If not using a pre-made vector, the dCas9 gene (with D10A and H840A mutations) must be cloned under a controllable promoter.
Step 3: System Delivery and Strain Engineering
- Transformation: Co-transform the dCas9 and sgRNA plasmids into the host bacterial strain. For chromosomal integration, the dCas9 can be integrated into a neutral site on the genome, and the sgRNA delivered via a plasmid [6].
Step 4: Induction and Knockdown
- Culture Induction: Inoculate and grow transformed bacteria. Add the appropriate inducer (e.g., aTc for a P_tet_ promoter) to express dCas9 and the sgRNA.
- Titration: For essential genes, use sub-saturating inducer concentrations to achieve partial knockdown and avoid lethality [7].
Step 5: Validation and Phenotyping
- Transcriptional Validation: Measure repression efficiency by quantifying mRNA levels using RT-qPCR.
- Functional/Phenotypic Validation: Assess the effect of gene knockdown by measuring growth (for essential genes), metabolite production, antibiotic susceptibility, or other relevant phenotypic assays [7] [6].
Advanced Applications and Engineering in Bacterial Research
The simplicity and programmability of dCas9 have enabled its use in sophisticated genetic applications.
- High-Throughput Functional Genomics: CRISPRi is ideal for genome-wide screens in bacteria. Arrayed or pooled sgRNA libraries can be used to systematically knockdown every non-essential and essential gene, identifying genes critical for growth under specific conditions (e.g., antibiotic treatment, nutrient limitation) [7]. This is particularly valuable for identifying new drug targets in pathogenic species.
- Tunable and Multiplexed Repression: Repression levels can be finely tuned not only by inducer concentration but also by designing sgRNAs with mismatches to their target, which results in partial repression [6]. Furthermore, the expression of multiple sgRNAs enables the simultaneous repression of several genes, allowing for the study of synthetic lethality and complex genetic interactions [7] [2].
- Metabolically-Targeted dCas9 Expression: Recent innovations have integrated biosensors with dCas9 expression. For example, a glucuronide-responsive promoter has been used to drive dCas9 expression, restricting its activity to specific bacterial subpopulations within a complex community that possess glucuronide-utilization enzymes [8]. This offers a strategy for precision microbiome editing.
The engineering of catalytically dead Cas9 was a transformative step that unlocked the potential of CRISPR technology beyond irreversible genome editing. As a precise, programmable, and reversible tool for transcriptional repression in bacteria, CRISPRi has become an indispensable component of the functional genomics toolkit. Its ability to generate hypomorphic alleles, conduct high-throughput screens, and be dynamically controlled provides researchers and drug development professionals with a powerful means to dissect gene function, validate therapeutic targets, and engineer novel bacterial phenotypes. Continued optimization of the system, including the development of novel Cas orthologs with different PAM specificities and reduced size, will further expand its utility across diverse bacterial species.
CRISPR interference (CRISPRi) has emerged as a powerful genetic perturbation technique that allows for sequence-specific repression of gene expression in prokaryotic and eukaryotic cells [2]. This technology repurposes the bacterial adaptive immune system for targeted transcriptional regulation. The core mechanism relies on steric hindrance, where a catalytically dead Cas9 (dCas9) protein, guided by a single-guide RNA (sgRNA), binds to specific DNA sequences and physically obstructs the transcription machinery [6]. Unlike CRISPR-Cas9 which introduces permanent DNA breaks, CRISPRi offers reversible gene knockdown without altering the DNA sequence, making it invaluable for functional genomics research and drug discovery [9] [2].
The dCas9 protein is generated by introducing point mutations (D10A and H840A) into the nuclease domains of the native Cas9, eliminating its DNA cleavage activity while preserving DNA-binding capability [6] [2]. When complexed with sgRNA, dCas9 can be programmed to target any genomic locus with a protospacer adjacent motif (PAM) sequence, typically NGG for Streptococcus pyogenes Cas9 [10]. This minimal system achieves highly specific gene repression by exploiting the fundamental principle of steric hindrance—physically blocking RNA polymerase (RNAP) progression along the DNA template [6].
Molecular Mechanism of Transcriptional Blockade
Strand-Specific Blockage of Transcription Elongation
The dCas9-sgRNA complex achieves transcriptional repression primarily by obstructing the elongating RNAP. The efficiency of this blockade depends critically on which DNA strand is targeted and the location of the binding site relative to the transcription machinery.
When the dCas9-sgRNA complex binds within an open reading frame, it creates a physical barrier that prevents RNAP from transcribing through the region [6]. Research confirms this repression is strand-specific [2]. For dCas9 systems, stronger repression occurs when the sgRNA is complementary to the non-template strand (the coding strand). This strand-specific effect is attributed to the activity of helicase, which unwinds the RNA:DNA heteroduplex ahead of RNAP when the sgRNA binds to the template strand, potentially reducing the complex's obstructive efficiency [2].
Table 1: Strand-Specific Repression Efficiency in Bacteria
| Targeted DNA Strand | Repression Efficiency | Molecular Rationale |
|---|---|---|
| Non-template strand | Strong repression (~99.9%) [2] | Optimal steric blockage of RNA polymerase progression |
| Template strand | Moderate repression (~50%) [10] | Helicase activity may partially overcome the barrier |
Inhibition of Transcription Initiation
The dCas9-sgRNA complex can also suppress transcription by targeting the promoter region or transcription start site (TSS), thereby preventing transcription initiation [2]. When bound to these regulatory regions, the complex can block the binding of RNAP or essential transcription factors to the DNA, effectively shutting down transcription before it begins.
Unlike elongation blockage, initiation inhibition is independent of the targeted DNA strand when targeting the transcriptional start site [2]. The binding of dCas9-sgRNA to promoter elements creates a steric shield that makes key DNA sequences inaccessible to the transcription machinery, providing an alternative strategy for gene silencing that can be equally effective as targeting the coding region.
Quantitative Aspects and Efficiency of CRISPRi Repression
The repression efficiency of CRISPRi varies across organisms but consistently demonstrates high efficacy in bacterial systems. In prokaryotes, this steric inhibition can repress transcription of the target gene by almost 99.9%, while in archaea, more than 90% repression has been achieved [2]. The level of transcriptional repression depends on several factors, including the concentration of dCas9-sgRNA complexes, the accessibility of the target site, and the binding affinity determined by the sgRNA sequence [10].
In bacteria, it is possible to saturate the target with a high enough level of dCas9 complex. Under such conditions, the repression strength primarily depends on the probability that dCas9 is ejected upon collision with the RNA polymerase, which is determined by the guide sequence [2]. Higher temperatures are also associated with higher ejection probability, thus resulting in weaker repression [2].
Table 2: CRISPRi Repression Efficiency Across Organisms
| Organism | Repression Efficiency | Key Factors Influencing Efficiency |
|---|---|---|
| Bacteria | Up to 99.9% [2] | dCas9-sgRNA concentration, target location, temperature |
| Archaea | >90% [2] | PAM availability, chromatin accessibility |
| Mammalian Cells | Up to 90-99% [9] [2] | Chromatin state, repressor domains (e.g., KRAB) |
The following diagram illustrates the core mechanism of steric hindrance by the dCas9-sgRNA complex during transcription elongation:
Optimizing CRISPRi Efficiency Through Experimental Design
Strategic sgRNA Design for Enhanced Repression
The effectiveness of CRISPRi-mediated steric hindrance depends significantly on strategic sgRNA design. For optimal repression efficiency, the 20-nucleotide base pairing region of the sgRNA should bind to the non-template DNA strand of the coding region [10]. Targeting the template DNA strand of the coding sequence is generally ineffective, yielding at most mild repression (~50%) [10].
Choosing a target closer to the 5' end of the gene generally results in greater repression efficiency, as it prevents RNAP from initiating productive transcription [10]. The target site must be adjacent to a protospacer adjacent motif (PAM) sequence, which for S. pyogenes dCas9 is NGG or NAG (where N is any nucleotide) [10]. Thus, the targetable sites are restricted to 20-nt regions 5' to NGG in the genome.
To ensure specificity, potential off-target DNA-binding sites with partial complementarity to the sgRNA should be evaluated using BLAST searches against the complete genome of the target organism [10]. Mismatches, particularly in the PAM-adjacent 12-nt "seed" region, can significantly reduce binding efficiency and repression capability [10].
Advanced CRISPRi System Configurations
Several system configurations can enhance CRISPRi efficiency:
Inducible Systems: When dCas9 is under the control of inducible promoters (e.g., anhydrotetracycline-inducible promoter), the knockdown can be induced by adding the inducer or reversed by removing it from the culture, enabling temporal and dynamic regulation of target genes [6].
Titratable Repression: The degree of gene repression can be controlled by titrating the concentration of dCas9 or sgRNA from an inducible promoter [6]. This allows for fine-tuning of repression levels, which is particularly useful for studying essential genes where complete knockdown may be lethal.
Multiplexing: Multiple sgRNAs can be used to simultaneously target different genes or enhance repression of a single gene target [2]. Technologies like Extra-Long sgRNA Arrays (ELSAs) enable direct synthesis of 12-sgRNA arrays that can be integrated into bacterial genomes without homologous recombination [2].
Research Reagent Solutions for CRISPRi Experiments
Table 3: Essential Research Reagents for Bacterial CRISPRi Experiments
| Reagent/Solution | Function/Application | Example Specifications |
|---|---|---|
| dCas9 Expression Plasmid | Expresses catalytically dead Cas9 protein | Addgene ID no. 44249; chloramphenicol resistance; anhydrotetracycline-inducible promoter [10] |
| sgRNA Expression Plasmid | Expresses sequence-specific guide RNA | Addgene ID no. 44251; ampicillin resistance; strong constitutive promoter J23119 [10] |
| Anhydrotetracycline | Inducer for dCas9 expression in inducible systems | Enables temporal control of CRISPRi activation [10] |
| Competent E. coli Cells | Host for plasmid propagation and experimentation | One Shot TOP10 chemically competent cells [10] |
| qPCR Reagents | Quantifies repression efficiency at transcript level | Includes RNA purification kits, reverse transcription systems, and SYBR Green Master Mix [10] |
Experimental Protocol for Implementing CRISPRi in Bacteria
sgRNA Design and Cloning Workflow
The following diagram outlines the key steps for implementing CRISPRi in bacterial systems:
Step-by-Step Methodology
sgRNA Design: Identify a 20-nt target sequence in the non-template strand of the gene of interest, ensuring it is adjacent to a PAM (NGG) motif [10]. For example, to target the mRFP gene, a valid target site on the non-template strand is AGACCGCTAACTGAAAGTT with PAM CCC. The sgRNA base pairing sequence would be the reverse complement: AACTTTCAGTTTAGCGGTCT [10].
Specificity Validation: Perform BLAST (blastn with default settings) searches with the designed sgRNA base pairing region against the complete genome of the target organism to ensure no exact 20-nt matches with adjacent PAM sites exist [10].
Single-sgRNA Cloning Using Inverse PCR:
- Use forward primers containing the 20-nt base pairing region unique to each sgRNA (e.g., 5'-N20 GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGC-3' for Addgene ID no. 44251) [10].
- Pair with a universal reverse primer (e.g., 5'-ACTAGTATTATACCTAGGACTGAGCTAGC-3' for Addgene ID no. 44251) [10].
- Perform PCR amplification, DpnI digestion to eliminate template DNA, and ligation with Quick Ligase [10].
- Transform into competent E. coli cells (e.g., One Shot TOP10) and plate on LB plates with 100 μg/mL ampicillin [10].
CRISPRi Repression Assays:
- Co-transform E. coli test strain (e.g., K12-strain MG1655) with both dCas9 expression plasmid (Addgene ID no. 44249) and sgRNA expression plasmid [10].
- Plate on LB plates with 100 μg/mL ampicillin and 20 μg/mL chloramphenicol for selection [10].
- Induce dCas9 expression with anhydrotetracycline [10].
- Quantify repression using RNA purification (RNeasy Kit), cDNA synthesis (Superscript III System), and qPCR analysis (SYBR Green Master Mix) [10].
Applications in Bacterial Research and Drug Development
The steric hindrance mechanism of CRISPRi has enabled numerous applications in basic research and therapeutic development:
Functional Genetic Screening: CRISPRi allows high-throughput identification of gene essentiality and functional annotation in bacteria [6]. Genome-wide CRISPRi libraries enable systematic interrogation of gene function at scale [11].
Bacterial Physiology Studies: Essential genes can be probed without lethal knockout, enabling study of their functions through titratable repression [6]. This is particularly valuable for investigating bacterial growth, metabolism, and pathogenicity mechanisms.
Metabolic Engineering: CRISPRi enables precise control of metabolic pathways in industrial microorganisms. For example, tunable CRISPRi has been used to repress multiple genes simultaneously to increase n-butanol yield and productivity in recombinant Escherichia coli [6].
Drug Target Validation: The reversibility and titratability of CRISPRi make it ideal for validating potential antibiotic targets by mimicking drug effects through targeted gene repression [6] [11].
The dCas9-sgRNA steric hindrance mechanism continues to evolve with engineering of improved repressor domains and system optimizations, further solidifying CRISPRi as an indispensable tool for transcriptional regulation in bacterial research and therapeutic development [9].
The repurposing of the CRISPR-Cas9 system into a programmable transcriptional regulator, known as CRISPR interference (CRISPRi), represents a groundbreaking advancement for functional genomics in bacterial research. This technical guide provides an in-depth examination of the three core components that form the foundation of CRISPRi technology: the catalytically dead Cas9 (dCas9), the single guide RNA (sgRNA), and the protospacer adjacent motif (PAM) sequence. We explore the structure-function relationships, molecular mechanisms, and optimization strategies for each component, with a specific focus on achieving efficient gene repression in bacterial systems. By synthesizing recent research findings and experimental data, this whitepaper serves as a comprehensive resource for researchers aiming to design and implement robust CRISPRi experiments for precise transcriptional control.
CRISPR interference (CRISPRi) has emerged as a powerful tool for sequence-specific gene repression without altering the underlying DNA sequence. Derived from the adaptive immune system of bacteria and archaea, CRISPRi represents a repurposed molecular machinery that allows researchers to probe gene function with unprecedented specificity [1]. The system functions as a programmable transcriptional regulator that can be targeted to specific genetic loci to suppress gene expression, making it particularly valuable for functional genomics studies, genetic circuit engineering, and drug target validation [9].
At its core, the CRISPRi system for transcriptional repression consists of two principal components: a catalytically dead Cas9 (dCas9) protein that retains DNA-binding capability but lacks cleavage activity, and a single guide RNA (sgRNA) that directs dCas9 to specific DNA sequences [1]. The binding of the dCas9-sgRNA complex to a target gene results in transcriptional interference by blocking RNA polymerase binding or elongation, effectively repressing gene expression at the DNA level [1]. This mechanism differs fundamentally from RNA interference (RNAi), which operates at the post-transcriptional level by degrading mRNA molecules [1].
The efficiency of CRISPRi is heavily dependent on the proper assembly and optimization of its core components, each of which presents unique considerations for experimental design. This whitepaper provides a comprehensive technical examination of these key system components—dCas9, sgRNA, and the PAM requirement—within the context of bacterial research, offering researchers a detailed framework for implementing effective CRISPRi-mediated transcriptional repression.
dCas9: The Engine of CRISPRi
Structural Basis and Functional Mechanism
The catalytically dead Cas9 (dCas9) protein serves as the central effector molecule in CRISPRi systems. dCas9 is derived from the native Cas9 nuclease through targeted point mutations (D10A and H840A in Streptococcus pyogenes Cas9) that inactivate the RuvC and HNH nuclease domains while preserving DNA-binding functionality [12]. This engineered protein retains the molecular architecture of Cas9, comprising a recognition lobe (REC) and a nuclease lobe (NUC), but functions as a programmable DNA-binding protein rather than a DNA-cleaving enzyme [12].
The REC lobe facilitates binding between the guide RNA and target DNA through its bridge helix and REC domains, while the NUC lobe contains the inactivated nuclease domains and the PAM-interacting domain (PI) that recognizes specific DNA sequences adjacent to the target site [12]. When dCas9 is directed to a target DNA sequence by sgRNA, it binds without introducing double-strand breaks, thereby physically obstructing transcriptional initiation or elongation by RNA polymerase [1]. This steric hindrance mechanism forms the basis for CRISPRi-mediated gene repression in bacterial systems.
Advanced dCas9 Repressor Fusions
To enhance repression efficiency beyond mere steric hindrance, dCas9 is often fused to transcriptional repressor domains. The Krüppel-associated box (KRAB) domain from the human KOX1 protein was among the first repressor domains used in CRISPRi systems [9]. Recent advancements have identified more potent repressor combinations, such as dCas9-ZIM3(KRAB)-MeCP2(t), which demonstrates significantly improved gene repression of endogenous targets at both transcript and protein levels [9]. These engineered repressors show reduced dependence on guide RNA sequences and more consistent performance across different gene targets and cell lines.
Table 1: Evolution of dCas9 Repressor Architectures
| Repressor Architecture | Key Components | Repression Mechanism | Performance Notes |
|---|---|---|---|
| dCas9-only | dCas9 | Steric hindrance of RNA polymerase | Basic repression, variable efficiency |
| dCas9-KOX1(KRAB) | dCas9 + KOX1 KRAB domain | Recruitment of heterochromatin-inducing factors | Improved repression over dCas9 alone |
| dCas9-ZIM3(KRAB) | dCas9 + ZIM3 KRAB domain | Enhanced recruitment of repressive complexes | Superior to KOX1-based repressors |
| dCas9-KOX1(KRAB)-MeCP2 | dCas9 + KRAB + MeCP2 | Combined repression mechanisms | "Gold standard" until recent improvements |
| dCas9-ZIM3(KRAB)-MeCP2(t) | dCas9 + ZIM3 KRAB + truncated MeCP2 | Synergistic repression pathways | Next-generation platform with highest efficiency |
sgRNA: The Guidance System
Structural Components and Design Principles
The single guide RNA (sgRNA) serves as the targeting module of the CRISPRi system, providing the sequence specificity that directs dCas9 to particular genomic loci. sgRNA is a synthetic fusion of two natural RNA components: the CRISPR RNA (crRNA), which contains a 17-20 nucleotide spacer sequence complementary to the target DNA, and the trans-activating crRNA (tracrRNA), which serves as a binding scaffold for dCas9 [13]. This chimeric RNA molecule simplifies the system from three components (crRNA, tracrRNA, and Cas9) to two (sgRNA and dCas9), facilitating experimental implementation [13].
Proper sgRNA design is critical for CRISPRi efficiency and specificity. Key considerations include:
- Target Sequence Length: Typically 17-23 nucleotides for S. pyogenes Cas9-derived systems [13]
- GC Content: Optimal range of 40-60% for stability and binding efficiency [12]
- Specificity: Minimization of off-target potential through careful sequence selection
- Absence of Self-Complementarity: Prevention of secondary structure formation that could interfere with dCas9 binding [13]
Optimized sgRNA Architectures
Recent research has revealed that modifications to the canonical sgRNA structure can significantly enhance CRISPRi performance. Systematic investigation has demonstrated that extending the duplex region by approximately 5 base pairs and mutating the fourth thymine in the continuous thymine sequence (often a polymerase III termination signal) to cytosine or guanine markedly improves gene repression efficiency [14]. These structural optimizations enhance sgRNA stability and transcription efficiency, leading to more consistent and potent target repression.
Table 2: sgRNA Format Comparison for CRISPRi Experiments
| sgRNA Format | Production Method | Key Advantages | Limitations | Typical Editing Efficiency |
|---|---|---|---|---|
| Plasmid-expressed | Cloning into expression vectors | Cost-effective for large-scale experiments | Prolonged expression may increase off-target effects; requires 1-2 weeks for cloning | Variable; can be prone to off-target effects |
| In vitro transcribed (IVT) | Transcription from DNA templates | No cloning required | Labor-intensive; lower quality RNA may require additional purification | Moderate; depends on purification quality |
| Synthetic sgRNA | Chemical synthesis | Highest purity and consistency; rapid availability | Higher cost for large-scale experiments | Highest efficiency and reproducibility |
Position-Dependent Efficiency in CRISPRi
The genomic targeting position of sgRNA significantly influences CRISPRi efficiency. Research demonstrates that sgRNAs targeting regions from -50 to +300 base pairs relative to the transcription start site (TSS) show optimal repression efficiency [15]. Furthermore, the precise identification of the TSS using specialized databases like FANTOM5/CAGE significantly improves sgRNA performance prediction [15]. Chromatin accessibility also plays a crucial role, with efficient sgRNAs preferentially targeting open chromatin regions [15].
PAM Requirement: The Recognition Signal
Biological Function and Recognition Mechanisms
The protospacer adjacent motif (PAM) is a short, specific DNA sequence (typically 2-6 base pairs) adjacent to the target DNA region that is essential for Cas9 activation and DNA binding [16]. From an evolutionary perspective, the PAM serves as a critical self versus non-self discrimination mechanism in bacterial adaptive immunity, preventing the CRISPR system from targeting the bacterium's own genome [17]. The PAM sequence is not part of the sgRNA target but must be present in the genomic DNA immediately downstream of the target site for successful dCas9 binding [18].
The molecular mechanism of PAM recognition involves the PAM-interacting domain in the Cas9 protein, which scans DNA for the appropriate motif [12]. Upon PAM identification, Cas9 undergoes conformational changes that facilitate DNA unwinding and subsequent RNA-DNA hybridization [12]. For the most commonly used Cas9 from Streptococcus pyogenes (SpCas9), the PAM sequence is 5'-NGG-3', where "N" can be any nucleotide base [16].
PAM Diversity and Engineering Solutions
Different Cas proteins from various bacterial species recognize distinct PAM sequences, providing researchers with a toolkit for targeting diverse genomic loci. The PAM requirement fundamentally constrains targetable sites in the genome, driving the exploration of natural Cas protein variants and engineered mutants with altered PAM specificities [19].
Table 3: PAM Sequences for Diverse CRISPR-Cas Effectors
| Cas Nuclease | Source Organism | PAM Sequence (5' to 3') | Notable Features |
|---|---|---|---|
| SpCas9 | Streptococcus pyogenes | NGG | Most widely characterized; broad application |
| SaCas9 | Staphylococcus aureus | NNGRRT or NNGRRN | Smaller size for constrained delivery systems |
| NmeCas9 | Neisseria meningitidis | NNNNGATT | Longer PAM for enhanced specificity |
| CjCas9 | Campylobacter jejuni | NNNNRYAC | Intermediate size and specificity |
| Cas12a (Cpf1) | Acidaminococcus sp. | TTTV | Creates staggered cuts; different mechanism |
| hfCas12Max | Engineered from Cas12i | TN and/or TTTN | Engineered for expanded targeting range |
| Cas12f | Various | NTTR | Ultra-small size for therapeutic applications |
Engineering approaches have generated Cas9 variants with altered PAM specificities, significantly expanding the targeting range of CRISPRi systems. For instance, xCas9 and SpCas9-NG variants recognize NG PAMs instead of NGG, while Cas12a Ultra nucleases recognize TTTN PAM sites rather than the wild-type TTTV [19]. These engineered variants maintain high on-target activity while substantially increasing the number of targetable sites in bacterial genomes.
Integrated Experimental Framework for CRISPRi in Bacteria
Component Assembly and Delivery Strategies
Implementing an effective CRISPRi system in bacteria requires careful consideration of component assembly and delivery methods. The dCas9 repressor fusion and sgRNA must be co-expressed within the bacterial cell, typically achieved through plasmid-based systems. Common approaches include:
- Dual-Plasmid Systems: Separate plasmids for dCas9-repressor fusions and sgRNA expression
- Single-Plasmid Systems: Combined expression cassettes for simplified transformation and maintenance
- Integrated Systems: Chromosomal integration of dCas9 with plasmid-based sgRNA expression
Delivery methods for CRISPRi components into bacterial cells include:
- Electroporation: Application of electric fields to create temporary pores in cell membranes
- Chemical Transformation: Use of calcium chloride or other salts to facilitate DNA uptake
- Conjugation: Transfer of genetic material between bacterial cells through direct contact
The choice of delivery method depends on the bacterial species, with electroporation being widely applicable for many laboratory strains.
Protocol for CRISPRi Knockdown Efficiency Evaluation
To assess CRISPRi-mediated gene repression, researchers can implement the following experimental protocol:
sgRNA Design and Cloning:
- Identify target gene transcription start site using appropriate annotation databases
- Design sgRNAs targeting regions from -50 to +300 bp relative to TSS
- Clone sgRNA sequences into appropriate expression vectors
dCas9-Repressor Transformation:
- Introduce dCas9-repressor fusion construct into target bacterial strain
- Select for successful transformants using appropriate antibiotics
CRISPRi Induction:
- Introduce sgRNA construct into dCas9-expressing bacteria
- Induce sgRNA expression with appropriate inducers if necessary
Efficiency Assessment:
- Measure transcript levels using RT-qPCR 24-72 hours post-induction
- Compare to non-targeting sgRNA controls
- Evaluate protein levels if antibodies are available
- Assess phenotypic consequences where applicable
This protocol enables systematic evaluation of CRISPRi efficiency and provides benchmarks for optimization.
Troubleshooting Common Challenges
Several challenges commonly arise in CRISPRi implementation:
- Incomplete Knockdown: Optimize sgRNA position relative to TSS; test alternative repressor domains; evaluate delivery efficiency
- Variable Performance Across Targets: Design multiple sgRNAs per target; verify TSS annotations; assess chromatin accessibility
- Off-Target Effects: Improve sgRNA specificity using computational tools; consider high-fidelity Cas9 variants; validate off-target transcription
- Toxicity or Growth Defects: Modulate expression levels; use inducible systems; verify target essentiality
Visualization of CRISPRi Mechanism and Workflow
Diagram 1: CRISPRi Molecular Mechanism. The dCas9-repressor fusion protein is guided by sgRNA to specific genomic loci, where PAM recognition enables target binding and transcriptional repression.
Research Reagent Solutions
Table 4: Essential Research Reagents for CRISPRi Experiments
| Reagent Category | Specific Examples | Function | Implementation Notes |
|---|---|---|---|
| dCas9 Repressors | dCas9-ZIM3(KRAB)-MeCP2(t), dCas9-KOX1(KRAB) | Programmable DNA binding and transcriptional repression | Select based on required repression strength; newer fusions show improved performance |
| sgRNA Formats | Synthetic sgRNA, Plasmid-expressed sgRNA, IVT sgRNA | Target recognition and dCas9 guidance | Synthetic sgRNA offers highest efficiency; plasmid-based is cost-effective for screening |
| Delivery Vectors | Lentiviral vectors, Plasmid systems, Integrated cassettes | Component delivery into bacterial cells | Consider stability and expression level requirements |
| PAM-Specific Cas Variants | SpCas9 (NGG), SaCas9 (NNGRRT), hfCas12Max (TN/TTTN) | Expanded targeting range | Choose based on genomic target sequence constraints |
| Design Tools | CHOPCHOP, Synthego Design Tool, Cas-Offinder | sgRNA design and off-target prediction | Essential for optimizing specificity and efficiency |
| Efficiency Assays | RT-qPCR primers, Antibodies for target proteins, Phenotypic assays | Knockdown validation | Implement multiple validation methods for robust results |
The CRISPRi system, comprising dCas9, sgRNA, and the PAM requirement, provides researchers with a powerful and specific tool for transcriptional repression in bacterial systems. Continued optimization of each component—from engineered dCas9 repressors with enhanced efficiency to structurally optimized sgRNAs and expanded PAM compatibility—has progressively overcome initial limitations of the technology.
Future developments in CRISPRi will likely focus on further expanding targeting capabilities through continued PAM engineering, enhancing repression efficiency with novel repressor domains, and improving system specificity to minimize off-target effects. Additionally, the integration of CRISPRi with other regulatory modalities and the development of more sophisticated delivery systems will expand the application scope of this technology in bacterial research.
As CRISPRi systems become increasingly refined and accessible, they promise to accelerate functional genomics studies, enable precise genetic circuit engineering, and facilitate drug target validation in bacterial systems. By understanding and strategically implementing the core components detailed in this technical guide, researchers can harness the full potential of CRISPRi for advanced genetic manipulation and transcriptional control.
CRISPR interference (CRISPRi) has emerged as a premier technology for programmable gene silencing in bacteria, enabling unprecedented functional genomic studies and synthetic biology applications. Derived from the adaptive immune systems of prokaryotes, CRISPRi repurposes a nuclease-deficient Cas protein (dCas9) and a single-guide RNA (sgRNA) to achieve highly specific transcriptional repression [20] [6]. This technical guide details the core properties of programmability, high efficiency, and specificity that make CRISPRi an indispensable tool in bacterial research, providing a comprehensive resource for scientists and drug development professionals. The content is framed within the broader context of utilizing CRISPRi as a mechanistic tool for investigating transcriptional regulation and gene function in bacterial systems.
The Core Mechanism of CRISPRi-Mediated Transcriptional Repression
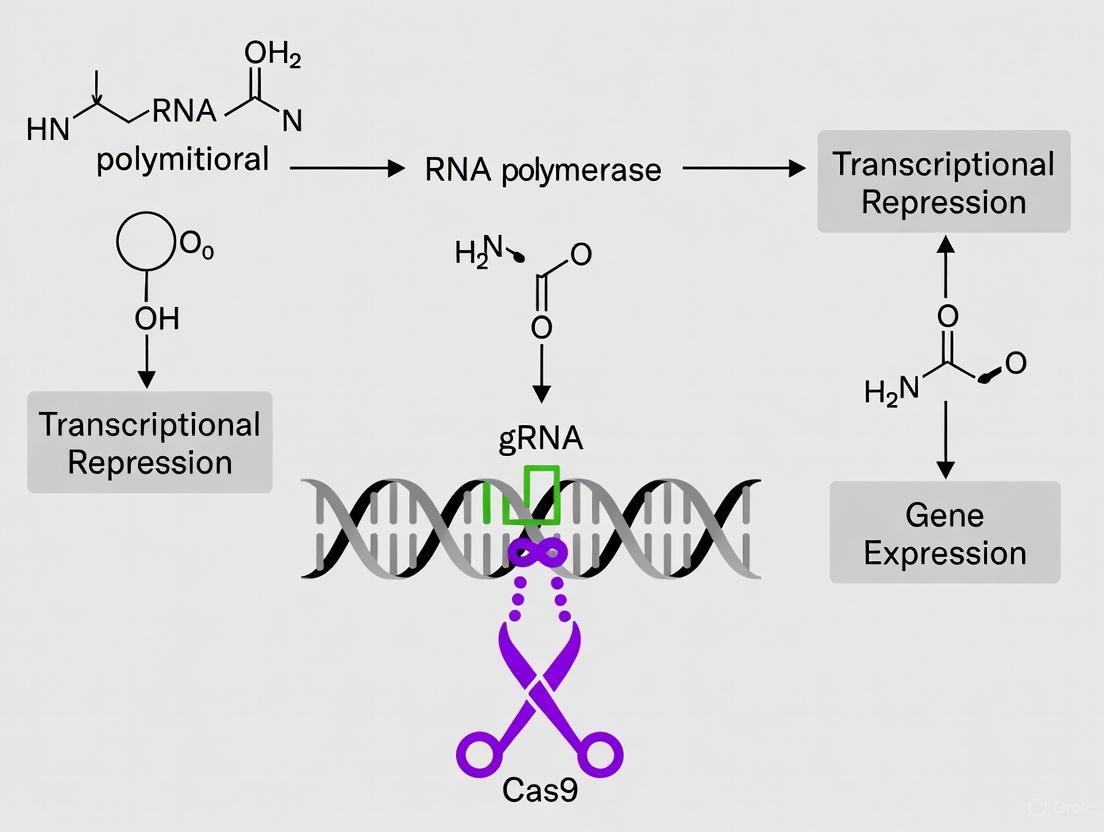
The fundamental CRISPRi system consists of two components: a catalytically dead Cas9 (dCas9) and a sequence-specific single-guide RNA (sgRNA) [6]. dCas9 is generated through point mutations (e.g., D10A and H840A in S. pyogenes Cas9) that inactivate the RuvC and HNH nuclease domains, rendering the protein incapable of DNA cleavage while preserving its DNA-binding capability [6]. The sgRNA is a chimeric noncoding RNA, typically 102 nucleotides in length, comprising a 20-nucleotide base-pairing region that defines target specificity, a 42-nucleotide Cas9-binding "handle," and a 40-nucleotide transcription terminator [10].
Upon formation of the dCas9-sgRNA complex, the system is directed to genomic target sites via Watson-Crick base pairing between the sgRNA's 20-nucleotide guide sequence and the complementary DNA strand. Successful binding requires the presence of a protospacer adjacent motif (PAM)—for the commonly used S. pyogenes dCas9, this is NGG (where N is any nucleotide)—immediately adjacent to the target sequence [10] [6]. Once bound to DNA, the dCas9-sgRNA complex functions as a physical roadblock to the transcribing RNA polymerase, thereby inhibiting either transcription initiation (when targeted to promoter regions) or transcription elongation (when targeted within coding sequences) [6]. This mechanism allows for robust, sequence-specific gene repression without altering the underlying DNA sequence.
Diagram 1: CRISPRi Mechanism of Action. The dCas9-sgRNA complex binds to target DNA adjacent to a PAM sequence, physically blocking RNA polymerase and inhibiting transcription.
Fundamental Properties of Bacterial CRISPRi Systems
Programmability
The programmability of CRISPRi is its most defining characteristic, allowing researchers to target virtually any gene of interest by simply redesigning the 20-nucleotide guide sequence within the sgRNA. This programmability enables several advanced applications:
- Multiplexed Gene Regulation: CRISPRi can simultaneously regulate multiple genes by expressing several sgRNAs targeting different genomic loci, enabling the study of complex genetic networks and metabolic pathways without cross-talk [6]. Studies have successfully used multiplexed CRISPRi to manipulate essential genes in biosynthetic pathways, such as those involved in 4HB synthesis for regulating P(3HB-co-4HB) composition [6].
- Titratable and Reversible Repression: The system allows for fine control over the level and timing of gene repression. By placing dCas9 or sgRNA expression under inducible promoters (e.g., anhydrotetracycline-inducible promoters), repression can be precisely titrated by varying inducer concentration or completely reversed by removing the inducer [6]. This provides a dynamic range of over two orders of magnitude in repression efficiency [6]. Partial repression can also be achieved by introducing mismatches between the sgRNA and its target DNA [6].
High Efficiency
CRISPRi achieves highly efficient gene silencing, with repression levels reaching up to 300-fold reduction in gene expression in E. coli [10]. Several factors influence this efficiency:
- Target Site Selection: The highest repression efficiency is achieved when the sgRNA targets the non-template DNA strand within the coding region, with positioning closer to the 5' end of the gene generally resulting in stronger repression [10]. Targeting the template strand typically yields only mild repression (~50%) [10].
- Gene-Specific Factors: Recent machine learning approaches analyzing genome-wide CRISPRi screens have revealed that target gene expression levels significantly impact guide depletion, with highly expressed genes showing greater susceptibility to CRISPRi-mediated silencing [21].
- sgRNA Design Considerations: While gene-specific factors play a major role, guide-specific features such as distance to the transcriptional start site also contribute to efficiency, though to a lesser extent [21].
Table 1: Factors Influencing CRISPRi Guide Efficiency
| Factor Category | Specific Feature | Impact on Efficiency | Optimization Strategy |
|---|---|---|---|
| Target Position | DNA strand targeted | ~300-fold repression on non-template strand vs. ~50% on template strand [10] | Target non-template strand |
| Distance to transcriptional start site | Moderate effect [21] | Position closer to 5' end of gene | |
| Genomic Context | Gene expression level | Major impact; higher expression associated with greater depletion [21] | Consider native expression levels during experimental design |
| Presence of downstream essential genes | Strong effect due to polar effects [21] | Target first gene in operon or use CRISPRi-ART to avoid polar effects [22] | |
| Sequence Features | PAM proximity | Essential for binding [10] | Ensure NGG PAM immediately adjacent to target |
| GC content | Moderate effect [21] | Consider during guide design |
Specificity
CRISPRi maintains high specificity with minimal off-target effects when properly designed [10]. Key aspects of its specificity include:
- Sequence-Specific Recognition: The requirement for a 20-nucleotide match between the sgRNA and target DNA, combined with the necessity of an adjacent PAM sequence, ensures highly specific binding. Mismatches, particularly in the PAM-adjacent 12-nucleotide "seed" region, can reduce off-target binding by an order of magnitude or more [10].
- Avoidance of Polar Effects with CRISPRi-ART: A significant advancement in specificity comes with CRISPRi-ART (CRISPR Interference through Antisense RNA-Targeting), which utilizes RNA-targeting dCas13d instead of DNA-targeting dCas9. This approach allows for targeted inhibition of protein translation without causing polar effects on downstream genes in operons, a common confounding factor in traditional CRISPRi essentiality screens [22]. In contrast, DNA-targeting dCas12a-based CRISPRi has been shown to misclassify non-essential genes as essential due to these polar effects [22].
Experimental Protocol for Implementing CRISPRi in Bacteria
sgRNA Design and Cloning
sgRNA Design Considerations [10]:
- Target Selection: Identify a 20-nucleotide target sequence 5' to an NGG PAM on the non-template DNA strand within the coding region of your gene of interest. Prefer regions closer to the 5' end.
- Genomic Specificity: Perform BLAST analysis of the designed sgRNA sequence against the host genome to ensure no off-target sites with perfect or near-perfect matches, especially in the seed region.
- Folding Quality (Optional): Predict the secondary structure of the full sgRNA using algorithms like ViennaRNA to ensure the dCas9-binding handle remains accessible.
Single-sgRNA Cloning via Inverse PCR [10]:
- Materials: E. coli sgRNA expression plasmid (e.g., Addgene ID 44251), designed forward sgRNA primers (5'-N~20~ GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGC-3'), universal reverse primer, Phusion PCR Master Mix, DpnI, Quick Ligase Kit, competent E. coli cells.
- Procedure:
- Perform inverse PCR using the sgRNA expression plasmid as template with primers containing the new 20-nt guide sequence.
- Digest template plasmid with DpnI.
- Purify PCR product and self-ligate using Quick Ligase.
- Transform into competent E. coli and select on ampicillin-containing plates.
- Verify constructs by sequencing.
CRISPRi Repression Assay
Materials [10]:
- E. coli test strain (e.g., K12-strain MG1655)
- dCas9 expression plasmid with chloramphenicol resistance (e.g., Addgene ID 44249)
- Constructed sgRNA plasmid
- Anhydrotetracycline for induction
- RNA purification kit (e.g., RNeasy), DNA-free Kit, cDNA synthesis system, qPCR reagents
Procedure [10]:
- Co-transform dCas9 and sgRNA plasmids into the target bacterial strain.
- Grow colonies in appropriate antibiotics and induce dCas9 expression with anhydrotetracycline.
- Harvest cells during mid-log phase for RNA extraction.
- Purify RNA and treat with DNase to remove genomic DNA contamination.
- Synthesize cDNA using reverse transcriptase with random hexamers.
- Perform qPCR with gene-specific primers to quantify repression levels.
- Calculate fold-repression compared to non-targeting sgRNA control.
Diagram 2: CRISPRi Experimental Workflow. Key steps from sgRNA design to repression efficiency assessment.
Advanced Applications and Recent Developments
High-Throughput Functional Genomics
CRISPRi has revolutionized functional genetic screening in bacteria, enabling genome-wide interrogation of gene function [20] [6]. Unlike transposon mutagenesis, CRISPRi can directly target specific genes of interest without requiring large mutant libraries to achieve gene saturation [21]. The programmability of CRISPRi allows for the construction of high-saturation sgRNA libraries targeting entire genomes, facilitating the identification of essential genes and genes involved in specific pathways or stress responses [6].
CRISPRi-ART for Phage Functional Genomics
A recent breakthrough, CRISPRi-ART (CRISPR Interference through Antisense RNA-Targeting), leverages RNA-targeting dCas13d to selectively interfere with phage protein translation [22]. This approach enables transcriptome-wide measurement of phage gene fitness and has been successfully applied across diverse phage phylogeny, including ssRNA, ssDNA, and dsDNA phages [22]. Key advantages include:
- Broad Applicability: Effective against phages with diverse genomic contents and modifications that evade DNA-targeting tools [22].
- Avoidance of Polar Effects: Unlike DNA-targeting CRISPRi, CRISPRi-ART does not cause false positive essentiality assignments due to operon polarity [22].
- Synergistic Multiplexing: Combining multiple crRNAs targeting different essential genes can nearly completely eliminate phage infectivity [22].
Table 2: Research Reagent Solutions for CRISPRi Implementation
| Reagent Type | Specific Examples | Function | Key Features |
|---|---|---|---|
| dCas9 Plasmids | Addgene ID 44249 [10] | Expresses dCas9 protein | Chloramphenicol resistance; anhydrotetracycline-inducible promoter |
| sgRNA Plasmids | Addgene ID 44251 [10] | Expresses sequence-specific sgRNA | Ampicillin resistance; strong constitutive promoter J23119 |
| sgRNA Cloning Primers | Forward: 5'-N~20~GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGC-3'Reverse: 5'-ACTAGTATTATACCTAGGACTGAGCTAGC-3' [10] | Inverse PCR cloning | Contains homology to vector and 20-nt guide sequence |
| Inducers | Anhydrotetracycline [10] | Induces dCas9 expression | Enables titratable and reversible repression |
| Competent Cells | One Shot TOP10 chemically competent E. coli [10] | Plasmid propagation and testing | High transformation efficiency |
| Analysis Tools | CrisprVi software [23] | CRISPR sequence visualization and analysis | Graphic user interface for analyzing DRs and spacers |
Predictive Modeling of Guide Efficiency
Recent advances in machine learning have led to improved prediction of CRISPRi guide efficiency. Mixed-effect random forest models that incorporate both guide-specific and gene-specific features provide better estimates of guide performance [21]. These models reveal that:
- Gene-specific features (expression levels, GC content, operon position) explain most of the variation in guide depletion [21].
- Data fusion from multiple CRISPRi screens significantly improves prediction accuracy [21].
- Features that can be manipulated during guide design (e.g., distance to transcriptional start site) have smaller effects than fixed gene-specific features [21].
CRISPRi technology represents a powerful and versatile platform for precise gene regulation in bacterial systems. Its fundamental properties of programmability, high efficiency, and specificity make it ideal for a wide range of applications, from targeted gene silencing to genome-wide functional screens. Recent developments, including CRISPRi-ART for phage studies and machine learning approaches for guide efficiency prediction, continue to expand its capabilities and applications. As CRISPRi technology evolves, it will undoubtedly remain a cornerstone tool for bacterial genetics, metabolic engineering, and antibacterial drug development, providing researchers with unprecedented control over gene expression for both basic science and applied biotechnology.
Implementing CRISPRi: From Single-Gene Knockdown to Genome-Scale Functional Screens
Clustered Regularly Interspaced Short Palindromic Repeats interference (CRISPRi) has emerged as a powerful technology for programmable transcriptional repression in bacteria, enabling functional genomics studies and metabolic engineering without permanent DNA alteration. This system utilizes a nuclease-deficient Cas9 (dCas9) protein that binds to DNA targets under the guidance of a sequence-specific single-guide RNA (sgRNA), forming a physical barrier that blocks RNA polymerase and aborts transcription [6]. The efficacy of CRISPRi is profoundly influenced by the method chosen for delivering the dCas9 component into bacterial cells, with plasmid-based and chromosomal integration representing the two primary strategies [24] [6]. The selection between these approaches involves critical trade-offs between expression stability, control over leakiness, and experimental throughput, making the delivery strategy a fundamental consideration in experimental design. This whitepaper provides a comprehensive technical comparison of these delivery platforms, offering detailed protocols and quantitative analyses to guide researchers in selecting and implementing the optimal system for their specific bacterial applications.
Core System Components and Repression Mechanism
Molecular Components of the CRISPRi System
The CRISPRi system requires two core molecular components for targeted gene repression. First, the dCas9 protein serves as the DNA-binding effector, engineered through point mutations (typically D10A and H840A for SpCas9) to eliminate endonuclease activity while retaining DNA-binding capability [6]. Second, the sgRNA functions as the targeting module, a chimeric RNA molecule comprising a 20-nucleotide base-pairing region that defines genomic specificity, a Cas9-binding handle, and a transcriptional terminator [10]. The dCas9 and sgRNA form a ribonucleoprotein complex that specifically binds to DNA sequences complementary to the sgRNA's base-pairing region, provided these targets are adjacent to a protospacer adjacent motif (PAM), typically 5'-NGG-3' for the commonly used Streptococcus pyogenes Cas9 [10] [25].
Mechanism of Transcriptional Repression
CRISPRi mediates gene silencing through steric occlusion of transcriptional machinery without cleaving DNA. When the dCas9-sgRNA complex binds within a promoter region, it prevents transcription initiation by blocking RNA polymerase binding. When bound within the coding sequence, it impedes transcription elongation by physically obstructing the progressing RNA polymerase [6]. This mechanism is reversible and tunable, allowing for precise control over gene expression levels. Repression efficiency depends on several factors, including the target location (with sites near the transcription start site and on the non-template strand typically performing best), sgRNA design, and intracellular concentrations of dCas9 and sgRNA [10].
Figure 1: CRISPRi System Workflow and Delivery Pathways. The diagram illustrates the two primary delivery strategies for dCas9 systems and their common pathway to transcriptional repression. Plasmid-based systems (yellow) typically yield higher dCas9 expression but may suffer from leakiness, while chromosomally integrated systems (green) offer tighter regulation through single-copy genomic integration.
Comparative Analysis of Delivery Strategies
Performance Characteristics
The choice between plasmid-based and chromosomally integrated dCas9 delivery involves significant trade-offs that impact experimental outcomes. The table below summarizes the key performance characteristics of each system:
Table 1: Quantitative Comparison of Plasmid-Based vs. Chromosomally Integrated dCas9 Systems
| Parameter | Plasmid-Based System | Chromosomally Integrated System |
|---|---|---|
| dCas9 Expression Level | High (approximately 20-fold higher than chromosomal) [24] | Low (approximately 5% of plasmid expression) [24] |
| Basal Expression (Leakiness) | Significant; mutant phenotypes observed without induction [24] | Minimal; tight regulation with no phenotype without induction [24] |
| Repression Efficiency | High (74% luciferase repression observed) [24] | Sufficient for essential gene silencing (ybeY repression) [24] |
| Multiplexing Capacity | Moderate (limited by plasmid size and compatibility) [6] | High (stable dCas9 base, sgRNA library compatibility) [6] |
| Experimental Timeline | Rapid implementation (days) [6] | Extended setup (weeks including integration) [24] |
| Genetic Stability | Variable (plasmid loss possible without selection) [6] | High (stable inheritance) [24] |
| Tunability | Moderate (inducer concentration titration) [6] | High (inducer concentration and sgRNA design) [6] |
Applications and Suitability
The distinct characteristics of each delivery system make them suitable for different research applications. Plasmid-based systems excel in proof-of-concept experiments and rapid screening due to their ease of implementation and high expression levels [6]. Their modular nature allows for quick testing of multiple sgRNAs and targets. However, leaky expression can obscure phenotypes or cause artificial toxicity, particularly when studying essential genes [24]. Chromosomally integrated systems are preferable for long-term functional studies, essential gene analysis, and high-throughput screening where tight regulation and genetic stability are paramount [24] [6]. The significantly reduced basal expression prevents confounding phenotypes and enables studies of genes where even low-level dCas9 activity might be detrimental.
Experimental Protocols and Implementation
Implementing a Chromosomally Integrated dCas9 System
Strain Construction Protocol:
- Integration Site Selection: Identify a transcriptionally silent genomic locus (e.g., pseudo29 in L. lactis) to minimize disruption to cellular physiology [24].
- Vector Construction: Clone the dCas9 gene (optionally fused to sfGFP for monitoring) under control of an inducible promoter (e.g., PnisA for nisin induction) into an integration vector containing flanking homology arms for the target locus.
- Chromosomal Integration: Introduce the construct into the target strain and select for integration via double-crossover homologous recombination using appropriate selection markers.
- Verification: Confirm correct chromosomal integration via PCR amplification across integration junctions and sequence verification.
- sgRNA Co-transformation: Introduce sgRNA expression plasmids (constitutively expressed from Pusp45) targeting genes of interest [24].
Induction and Repression Assay:
- Inoculate single colonies of the integrated strain carrying sgRNA plasmids into appropriate medium with selective antibiotics.
- Grow cultures to mid-exponential phase (OD600 ≈ 0.3-0.5).
- Induce dCas9 expression with optimal inducer concentration (e.g., 5-10 ng/mL nisin for PnisA in L. lactis).
- Monitor repression efficiency via:
Plasmid-Based dCas9 System Implementation
Dual-Plasmid System Construction:
- dCas9 Plasmid: Clone dCas9 under inducible control (e.g., PLTetO-1 for anhydrotetracycline induction) in a medium-copy vector with appropriate resistance marker [10].
- sgRNA Plasmid: Clone sgRNA expression cassette with target-specific 20-nt sequence under constitutive promoter (e.g., J23119) in a compatible vector with different resistance marker [10].
- Co-transformation: Introduce both plasmids into the target bacterial strain and select with both antibiotics.
- Induction and Validation: Follow similar induction and validation protocols as for chromosomal systems, noting that induction parameters may require optimization for specific plasmid systems.
Figure 2: Experimental Workflow for CRISPRi System Implementation. The decision pathway guides researchers through delivery strategy selection based on experimental goals, followed by streamlined protocols for system construction and validation. Chromosomal integration requires significantly more time but offers superior stability for long-term studies.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for CRISPRi Implementation in Bacterial Systems
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| dCas9 Expression Systems | pNZ-PnisA-dcas9-sfgfp (plasmid) [24], pseudo29::PnisA-dcas9-sfgfp (chromosomal) [24] | Provides regulated dCas9 expression; fluorescent fusions enable expression monitoring |
| sgRNA Cloning Vectors | pTLR-based backbones with Pusp45 promoter [24], Addgene #44251 [10] | Enables sgRNA expression with constitutive promoters |
| Induction Compounds | Nisin (for PnisA) [24], Anhydrotetracycline (for PLTetO-1) [10] | Controls dCas9 expression in inducible systems |
| Cloning Reagents | Phusion PCR Master Mix, Quick Ligase Kit, DpnI [10] | Facilitates molecular cloning of sgRNA and dCas9 constructs |
| Competent Cells | One Shot TOP10 E. coli [10], NZ9000 L. lactis [24] | Strain-specific transformation hosts |
| Validation Reagents | qPCR primers, RNA purification kits, SYBR Green Master Mix [10] | Enables quantification of repression efficiency |
| Selection Antibiotics | Ampicillin, Chloramphenicol [10] | Maintains plasmid selection pressure |
Technical Considerations and Optimization Strategies
sgRNA Design Principles
Effective sgRNA design is critical for successful CRISPRi implementation. Key considerations include:
- Strand Selection: Target the non-template DNA strand for significantly higher repression efficiency [10]
- PAM Proximity: Ensure the target site is immediately 5' to an NGG PAM sequence [10]
- Genomic Specificity: Perform BLAST analysis to ensure minimal off-target binding, especially in the PAM-adjacent 12-nt "seed" region [10]
- Target Position: For maximal repression, target sites near the 5' end of the coding sequence or within promoter regions [10]
- Folding Considerations: Verify that the sgRNA secondary structure does not disrupt the dCas9 binding handle using prediction tools like ViennaRNA [10]
Troubleshooting Common Challenges
Addressing Leaky Expression:
- For plasmid systems: Reduce copy number, optimize inducer concentration, or use tighter regulatory systems
- For chromosomal systems: Verify integration integrity and promoter functionality [24]
Improving Repression Efficiency:
- Screen multiple sgRNAs targeting different regions of the same gene
- Optimize dCas9 and sgRNA expression levels through promoter engineering
- For essential genes, titrate induction levels to achieve partial repression compatible with viability [24] [6]
Ensuring Genetic Stability:
- For plasmid systems: Maintain consistent antibiotic selection
- For chromosomal systems: Verify integration stability through serial passage without selection
- Implement regular stock culture renewal to prevent accumulation of suppressor mutations
The selection between plasmid-based and chromosomally integrated dCas9 delivery strategies represents a fundamental experimental design decision in bacterial CRISPRi applications. Plasmid-based systems offer rapid implementation and high expression levels suitable for preliminary screening and proof-of-concept studies, while chromosomally integrated systems provide tight regulation and genetic stability essential for investigating essential genes and long-term functional studies. The quantitative data and standardized protocols presented in this technical guide enable researchers to make informed decisions and successfully implement the optimal delivery strategy for their specific research objectives. As CRISPRi technology continues to evolve, advancements in delivery system engineering will further enhance the precision and applicability of this powerful transcriptional control platform in bacterial research and biotechnology.
CRISPR interference (CRISPRi) has emerged as a powerful tool for precise transcriptional repression in bacteria, enabling functional genomic studies and metabolic engineering. This technology utilizes a catalytically dead Cas9 (dCas9) protein that binds to DNA targets under the guidance of a single-guide RNA (sgRNA) without cleaving the DNA backbone. The resulting dCas9-sgRNA complex functions as a steric barrier, physically obstructing RNA polymerase (RNAP) and thereby inhibiting transcription [6]. The efficacy of this repression is highly dependent on the strategic design of the sgRNA, particularly the choice between targeting the promoter region or the coding sequence (CDS) of a gene. This guide synthesizes current research to establish evidence-based principles for designing sgRNAs that achieve strong, specific transcriptional repression in bacterial systems, a critical consideration for research and drug development professionals working with bacterial models.
Fundamental Mechanisms: How sgRNA Targeting Location Affects Repression
The mechanism of transcriptional repression differs significantly depending on whether the dCas9-sgRNA complex binds to a promoter or within the coding sequence of a gene. Understanding these distinct mechanisms is fundamental to rational sgRNA design.
Figure 1: Mechanisms of CRISPRi Repression. Targeting the promoter region prevents transcription initiation by physically blocking RNA polymerase binding. Targeting the coding sequence allows initiation but blocks elongating RNA polymerase, resulting in truncated mRNA transcripts.
When the dCas9-sgRNA complex binds to a promoter region, it physically blocks the binding of RNA polymerase, thereby preventing transcription initiation. In contrast, when the complex binds within the coding sequence, it allows transcription initiation but blocks the progression of the elongating RNA polymerase, leading to abortive transcription [6] [26]. This mechanistic distinction has profound implications for repression efficiency. In bacteria, targeting the coding sequence, particularly the non-template strand, often results in more effective knockdown because it guarantees interference with an elongating RNA polymerase and does not require precise knowledge of promoter architecture [27].
Quantitative Comparison of Targeting Strategies
The choice between promoter and CDS targeting involves balancing multiple factors including repression efficiency, design flexibility, and practical implementation. The following table summarizes key performance characteristics and design considerations for both strategies, synthesized from current bacterial CRISPRi research.
Table 1: Performance Comparison of Promoter vs. CDS Targeting Strategies in Bacteria
| Design Parameter | Promoter Targeting | Coding Sequence (CDS) Targeting |
|---|---|---|
| Optimal Targeting Window | ~100 bp window upstream of Transcription Start Site (TSS) [28] | Within the 5' region of the coding sequence [27] |
| Repression Mechanism | Blocks transcription initiation | Blocks transcription elongation |
| Typical Repression Efficiency | Variable; often lower in bacteria [26] | High; often more effective in prokaryotes [27] |
| Strand Preference | Not well-specified | Non-template strand strongly preferred [27] |
| Knowledge Requirements | Requires accurate TSS and promoter annotation | Requires only gene annotation |
| Design Flexibility | Limited to narrow upstream region | Flexible across gene length |
| Advantages | Prevents transcription at source | More reliable, less dependent on promoter mapping |
| Disadvantages | Sensitive to imperfect TSS annotation | May not completely eliminate initiated transcripts |
Beyond the basic targeting location, several sequence-specific factors significantly influence sgRNA efficacy. Research demonstrates that thermodynamic features describing sgRNA:target interactions, particularly the minimum free energy of gRNA:DNA hybridization, are critical predictors of repression efficiency [21]. Additionally, gene-specific features such as target gene expression levels, GC content, and operon context can substantially impact the observed repression, sometimes more than guide sequence features themselves [21].
Advanced Design Principles for Enhanced Repression
Multi-gRNA Strategies for Synergistic Repression
Employing multiple sgRNAs against a single gene can dramatically enhance repression efficacy through synergistic effects. A key design consideration is whether to use heterogeneous target sites (each gRNA has a unique sequence) or identical target sites (the same gRNA sequence is repeated). Simulation-based analysis in plant systems suggests that identical gRNA target sites yield far more effective transcriptional repression than heterogeneous sites, as they reduce competition between gRNA species and may allow dCas9 to occupy multiple sites through lateral diffusion along DNA [29]. While this principle was demonstrated in plants, it likely applies to bacterial systems where dCas9 binding kinetics are similar.
Tunable Repression through Mismatch Engineering
For applications requiring partial rather than complete gene repression, introducing strategic single-nucleotide mismatches between the sgRNA and DNA target provides a method to titrate knockdown efficacy [27] [6]. This approach is particularly valuable when studying essential genes, where complete silencing would be lethal, or for mimicking the partial inhibition achieved by pharmacological agents. Computational pipelines have been developed to predict the knockdown efficacies of mismatch-containing sgRNAs, enabling the design of graded repression libraries [27].
Experimental Protocol for sgRNA Design and Validation
Computational sgRNA Design Workflow
The following diagram outlines a comprehensive workflow for designing and filtering effective sgRNAs for bacterial CRISPRi experiments, incorporating both promoter and CDS targeting strategies.
Figure 2: sgRNA Design and Selection Workflow. A systematic computational pipeline for identifying and filtering potential sgRNAs for CRISPRi experiments in bacteria.
Step-by-Step Design Methodology
Genome Sequence Acquisition: Download the complete genome sequence of your bacterial strain in GenBank format from NCBI or other databases. Strain-specific sequences are crucial for accurate design [27].
Target Region Definition:
- For promoter targeting: Identify the transcription start site (TSS) using validated databases where possible. Define a target window from approximately 100 nucleotides upstream of the TSS to the TSS itself [28].
- For CDS targeting: Identify the protein-coding sequence of your target gene. Prioritize the 5' region of the CDS (from start codon to approximately 20-30% of gene length) to maximize early termination of transcription. Verify targeting to the non-template strand for enhanced efficacy [27].
sgRNA Identification: Scan the target region for NGG protospacer adjacent motif (PAM) sequences. Extract the 20 nucleotides immediately upstream of each PAM as the potential sgRNA spacer sequence [27].
sgRNA Filtering:
- Calculate GC content for each sgRNA spacer; optimal range is typically 40-60%.
- Perform off-target analysis by searching for identical or nearly identical sequences elsewhere in the genome, focusing particularly on the "seed" region proximal to the PAM [27] [28].
- Utilize available prediction algorithms (e.g., those incorporating thermodynamic features of gRNA:target interactions) to rank sgRNAs by predicted efficiency [21].
Final Selection: Select 2-3 top-ranked sgRNAs per target gene to enable experimental validation and control for potential variability in individual sgRNA performance.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Bacterial CRISPRi Experiments
| Reagent / Resource | Function | Implementation Notes |
|---|---|---|
| dCas9 Expression System | Catalytically dead Cas9 effector | Use a single-plasmid or chromosomally integrated system with inducible promoter for controlled expression [6] |
| sgRNA Expression Vector | Guides dCas9 to specific DNA targets | May use single or dual plasmid systems; consider library vectors for high-throughput screening [6] |
| Computational Design Pipeline | Identifies and filters potential sgRNAs | Tools like generate_sgrnas.py script [27] or models from [21] can predict sgRNA efficacy |
| Genome-Scale sgRNA Libraries | Enables high-throughput genetic screens | Available for essential gene identification and functional genomics [21] |
| Flow Cytometry Calibration Beads | Standardizes fluorescence measurements | Enables quantification of repression efficiency in reporter assays (e.g., MEFL units) [29] |
Effective sgRNA design for CRISPRi in bacteria requires careful consideration of targeting strategy, with coding sequence targeting typically providing more robust repression than promoter targeting. The principles outlined in this guide—including strand preference, multi-guide strategies, and tunable repression through mismatch engineering—provide a framework for designing effective sgRNAs for transcriptional repression. As machine learning approaches continue to improve predictions of guide efficiency [21], and as new Cas variants with altered PAM specificities become available [30], the flexibility and efficacy of CRISPRi in bacterial systems will continue to expand. For research and drug development applications, these design principles enable more precise genetic manipulation, facilitating both basic research into gene function and the development of engineered bacterial strains for therapeutic and industrial applications.
In bacterial research, the ability to precisely control gene expression is fundamental to dissecting complex biological systems. While CRISPR interference (CRISPRi) has emerged as a powerful tool for programmable gene repression in bacteria, traditional approaches often generate binary on/off states, limiting their utility for investigating dosage-sensitive genetic interactions and essential genes. The repurposing of the CRISPR system for transcriptional control uses a catalytically dead Cas9 (dCas9) that binds DNA without cutting it, sterically blocking RNA polymerase elongation in bacteria [2]. This technical whitepaper explores two sophisticated methods—inducible promoters and mismatched sgRNAs—that enable titratable repression and fine-tuned knockdown, advancing beyond all-or-nothing silencing to facilitate precise functional genomics in bacterial systems.
The fundamental mechanism of bacterial CRISPRi differs from eukaryotic systems in its simplicity: dCas9, guided by sgRNA, binds within coding sequences to physically obstruct transcription elongation by bacterial RNA polymerase [2]. This mechanistic understanding provides the foundation for developing titration strategies that modulate the efficiency of this blocking process, allowing researchers to stage bacterial cells along a continuum of gene expression levels to probe sensitive genetic networks and pathway dynamics.
Methodological Foundations for Titratable Repression
Mismatched sgRNAs: Programmable Titration Through Guide Engineering
The strategic introduction of mismatches between the sgRNA and its DNA target site represents a powerful approach to fine-tune CRISPRi efficacy. Single mismatches in the sgRNA base-pairing region can generate the full spectrum of repression, from no efficacy to complete silencing equivalent to perfectly matched sgRNAs [31]. This method leverages predictable reductions in dCas9 binding affinity to create defined knockdown levels without altering the underlying genetic machinery.
The position and type of mismatch significantly influence the resulting repression level. Research in both E. coli and B. subtilis demonstrates that mismatches closer to the PAM (protospacer adjacent motif) sequence, particularly in the seed region 10-12 bases upstream of the PAM, cause more substantial reductions in CRISPRi activity [31]. Different base substitutions (e.g., rG:dT mismatches) exhibit varying effects on sgRNA activity, enabling researchers to select specific mismatches that yield desired repression levels [32].
Table 1: Effects of Mismatch Position on sgRNA Activity in Bacterial CRISPRi
| Mismatch Position Relative to PAM | Relative sgRNA Activity | Recommended Application |
|---|---|---|
| Distal region (positions -18 to -20) | High (70-100%) | Moderate knockdown |
| Intermediate region (positions -9 to -17) | Variable (10-90%) | Tunable repression |
| Seed region (positions -1 to -8) | Low (0-30%) | Mild repression or essential genes |
Inducible Promoters: External Control of CRISPRi Components
Inducible promoter systems provide temporal and dosage control over CRISPRi components, enabling researchers to precisely regulate both the timing and intensity of gene repression. By placing dCas9 expression under the control of promoters that respond to specific chemical inducers, researchers can initiate repression at defined experimental timepoints and modulate repression strength by varying inducer concentration [33].
In Clostridium species, native bacterial microcompartment (BMC) promoters induced by choline (Pcholine1) or 1,2-propanediol (P1,2-PD) have been successfully employed to control CRISPR/Cas9 systems [33]. These systems demonstrate how native bacterial regulatory elements can be repurposed for genetic control, with the P1,2-PD promoter enabling highly efficient gene editing when used to drive Cas9 expression [33]. Similarly, tetracycline-inducible promoters have proven effective for controlling dCas9 expression in Clostridium autoethanogenum, achieving editing efficiencies exceeding 50% while minimizing the toxicity associated with constitutive Cas9 expression [33].
Experimental Implementation and Workflows
Mismatched sgRNA Library Design and Screening
Implementing a mismatched sgRNA approach requires systematic library design and robust phenotypic screening. The following workflow outlines the key steps for creating and testing mismatched sgRNA libraries in bacterial systems:
Table 2: Key Research Reagent Solutions for Titratable CRISPRi
| Reagent Type | Specific Examples | Function in Experimental System |
|---|---|---|
| dCas9 Effectors | dCas9 alone; dCas9-repressor fusions | Provides DNA-binding backbone for CRISPRi |
| sgRNA Scaffolds | Native sgRNA; modified constant regions | Directs targeting and modulates activity |
| Inducible Systems | P1,2-PD; Pcholine1; tetracycline-inducible | Controls timing and level of dCas9 expression |
| Reporter Systems | GFP; RFP; antibiotic resistance markers | Quantifies repression efficiency |
| Delivery Vectors | Lentiviral; plasmid; integrative vectors | Introduces CRISPRi components into cells |
Step 1: Target Selection and sgRNA Design
- Identify target genes and design fully complementary sgRNA sequences using standard tools
- Select target sites within the open reading frame for optimal bacterial CRISPRi activity [31]
- Consider GC content and avoid homopolymeric sequences that may impair sgRNA function [34]
Step 2: Mismatch Introduction
- Generate single mismatch variants for each position in the sgRNA targeting region
- Include multiple mismatch types (e.g., rG:dT, rA:dC) to cover a range of potential activities
- For comprehensive coverage, design libraries containing all possible single mismatches and a subset of double mismatches [31]
Step 3: Library Construction
- Clone sgRNA variants into appropriate expression vectors
- For bacterial systems, ensure proper promoter selection (e.g., J23119 for E. coli) and terminator sequences
- Include barcodes for pooled screening approaches when applicable
Step 4: Phenotypic Screening
- Transform library into bacterial strains expressing dCas9
- Conduct pooled growth screens under selective conditions
- Monitor sgRNA abundance over time using next-generation sequencing [31]
- Calculate fitness scores based on sgRNA depletion/enrichment patterns
Step 5: Model Building and Validation
- Analyze the relationship between mismatch characteristics and repression efficacy
- Build predictive models using parameters including mismatch position, type, and local sequence context [31]
- Validate model predictions for selected sgRNAs using individual growth assays and transcriptional measurements
Figure 1: Experimental workflow for mismatched sgRNA library screening in bacterial CRISPRi.
Inducible CRISPRi System Optimization
Implementing inducible CRISPRi systems requires careful optimization of several parameters to balance efficiency with minimal toxicity:
Component Selection and Vector Design
- Select inducible promoters based on induction kinetics, dynamic range, and compatibility with host organism
- For tight control of dCas9 expression, consider integrating the dCas9 gene under inducible control into the genome [33]
- Design sgRNA expression cassettes with constitutive promoters optimized for the host bacterium
Toxicity Mitigation
- Use inducible systems to minimize basal dCas9 expression, which can cause cellular toxicity in bacteria [33]
- Consider two-plasmid systems separating dCas9 and sgRNA expression to reduce vector size and improve transformation efficiency [33]
- For essential gene targeting, titrate inducer concentration to achieve partial knockdown compatible with viability
Efficiency Optimization
- Measure knockdown efficiency across a range of inducer concentrations
- Determine optimal induction timing relative to experimental readouts
- For genome-wide applications, verify consistent performance across multiple genomic loci
Quantitative Analysis of Titration Efficiency
Performance Metrics for Titratable CRISPRi Systems
The efficacy of titratable CRISPRi systems can be evaluated using multiple quantitative metrics. For mismatched sgRNAs, the correlation between predicted and measured activity provides a key quality indicator. In foundational work, a linear model using mismatch position, base substitution type, and spacer GC content accurately predicted the relative efficacy of mismatched sgRNAs in both E. coli and B. subtilis (R² = 0.65-0.71 between predicted and measured activities) [31].
For inducible systems, critical performance parameters include:
- Induction fold-change: Ratio of maximal to basal repression activity
- Dynamic range: Span of achievable repression levels
- Kinetics: Time required for full induction and reversal
- Titration linearity: Relationship between inducer concentration and repression strength
Table 3: Comparison of Titration Methods for Bacterial CRISPRi
| Parameter | Mismatched sgRNAs | Inducible Promoters |
|---|---|---|
| Titration Mechanism | Alters sgRNA:DNA binding affinity | Controls dCas9 expression level |
| Dynamic Range | Full spectrum (0-100% activity) [31] | Dependent on promoter strength and induction |
| Temporal Control | Fixed upon sgRNA expression | Tunable via inducer timing |
| Library Complexity | High (requires multiple sgRNAs per target) | Lower (single construct per target) |
| Experimental Throughput | High (pooled screening compatible) | Moderate (individual induction required) |
| Best Applications | Essential gene profiling, expression-fitness mapping | Temporal studies, toxic gene targeting |
Applications in Bacterial Functional Genomics
Titratable CRISPRi systems have enabled sophisticated functional genomics applications in bacteria:
Expression-Fitness Relationship Mapping
- Mismatched sgRNA libraries targeting essential genes in E. coli and B. subtilis have revealed diverse expression-fitness relationships, from linear to biphasic responses [31]
- These relationships are largely conserved between homologs despite ~2 billion years of evolutionary separation, suggesting shared constraints on essential gene expression levels [31]
Pathway-Specific Genetic Interactions
- Titratable repression allows identification of dosage-sensitive genetic interactions where partial knockdown of one gene sensitizes cells to knockdown of another
- This approach can reveal buffering relationships and redundant pathway functions
Essential Gene Characterization
- Traditional knockout approaches cannot study essential genes, but titratable CRISPRi enables systematic analysis of how reduced expression affects cellular fitness
- This has revealed gene-specific expression thresholds below which fitness defects emerge [31]
Figure 2: Research applications enabled by titratable CRISPRi methods in bacterial systems.
Technical Considerations and Optimization Strategies
Enhancing Titration Precision and Dynamic Range
Several strategies can improve the performance and reliability of titratable CRISPRi systems:
Multiparameter sgRNA Engineering
- Combine mismatch introduction with constant region modifications to fine-tune sgRNA activity [32]
- Explore truncated sgRNAs with varying lengths to modulate binding affinity [34]
- Implement dual-sgRNA approaches where two sgRNAs targeting the same gene are co-expressed to enhance repression and provide more titration points [35]
Promoter System Engineering
- Combine multiple inducible systems for tighter control and broader dynamic range
- Engineer promoter variants with different induction thresholds to create a spectrum of expression levels
- Incorporate regulatory elements such as riboswitches for translational control in addition to transcriptional control [33]
System Integration and Validation
- For mismatched sgRNAs, validate predicted activities for a subset of targets before full-library implementation
- For inducible systems, carefully characterize kinetics and dose-response relationships for each new bacterial strain
- Implement orthogonal reporter systems to monitor repression efficiency in real-time without affecting the target gene
Troubleshooting Common Implementation Challenges
Addressing Variable Knockdown Efficiency
- If certain targets show inconsistent repression, verify sgRNA target accessibility and avoid regions with secondary structure
- For inducible systems, optimize dCas9 expression levels to balance efficacy and toxicity
- Consider using multiple sgRNAs targeting different regions of the same gene to overcome position-dependent effects
Minimizing Off-Target Effects
- Design sgRNAs with minimal similarity to off-target genomic sites, particularly in the PAM-proximal seed region
- Use modified high-fidelity dCas9 variants when available to reduce non-specific binding
- Include appropriate controls to distinguish specific from nonspecific phenotypic effects
Optimizing Delivery and Stability
- For library screens, ensure high-efficiency transformation to maintain library representation
- Include barcodes to track individual sgRNAs in pooled formats
- For inducible systems, minimize basal expression through promoter engineering and genetic insulation
Titratable repression methods represent a significant advancement in bacterial functional genomics, enabling researchers to move beyond binary knockouts to explore the quantitative relationship between gene expression and phenotype. Both mismatched sgRNAs and inducible promoters provide powerful, complementary approaches for achieving precise control over gene expression levels in diverse bacterial species.
As these technologies continue to evolve, we anticipate several exciting developments. The integration of machine learning approaches with expanded empirical data will enhance prediction of sgRNA activity for both perfectly matched and mismatched guides [32]. Additionally, the discovery and engineering of novel CRISPR-Cas systems with different PAM requirements will expand the targetable genomic space. Finally, the combination of titratable repression with single-cell readouts will enable high-resolution mapping of expression-phenotype relationships in heterogeneous bacterial populations.
These methodological advances in titratable CRISPRi are transforming our ability to probe bacterial gene function with unprecedented precision, opening new avenues for understanding essential genes, genetic interactions, and the fundamental principles of microbial life.
CRISPR interference (CRISPRi) is a powerful genetic perturbation technique derived from the adaptive immune system of bacteria that allows for sequence-specific repression of gene expression. The system requires only two components: a catalytically inactive Cas9 (dCas9) protein, which lacks endonuclease activity but retains DNA-binding capability, and a customizable single guide RNA (sgRNA) that directs dCas9 to specific genomic loci through Watson-Crick base pairing [3] [2]. When deployed in bacterial systems, this dCas9-sgRNA complex binds to target DNA and creates a steric block that halts transcript elongation by RNA polymerase, resulting in potent repression of the target gene [3].
The targeting specificity of the CRISPRi system is jointly determined by the sgRNA-DNA base pairing and the presence of a short protospacer adjacent motif (PAM) sequence adjacent to the target site [3]. For the most commonly used Streptococcus pyogenes Cas9, the PAM sequence is 5'-NGG-3', which limits potential target sites but provides sufficient specificity for most bacterial genomes [3] [2]. CRISPRi operates at the transcriptional level, distinguishing it from RNA interference (RNAi) which functions at the mRNA level, making CRISPRi particularly valuable for probing essential genes and engineering metabolic pathways in bacteria [2].
CRISPRi Mechanism for Targeted Gene Repression
Fundamental Molecular Mechanism
The CRISPRi system exerts its repressive function through two primary mechanisms depending on the targeted genomic location. When the dCas9-sgRNA complex binds to the protein-coding region of a gene, particularly to the nontemplate DNA strand, it creates a physical roadblock that prevents the elongating RNA polymerase from progressing, thereby causing aborted transcription [3]. Research has demonstrated that repression is stronger when the sgRNA is complementary to the nontemplate strand, likely due to the activity of helicase which unwinds RNA:DNA heteroduplexes when sgRNA binds to the template strand [2].
Alternatively, when targeting promoter regions, including RNA polymerase-binding sites (e.g., -35 or -10 boxes in bacterial promoters) or transcription factor binding sites, the dCas9-sgRNA complex functions as a steric hindrance that prevents the association of key cis-acting DNA motifs with their cognate trans-acting transcription factors, leading to inhibition of transcription initiation [3]. The repression efficiency in bacteria can reach up to 99.9%, making it an exceptionally powerful tool for genetic studies [3] [2].
Visualizing the CRISPRi Mechanism
The following diagram illustrates the core mechanistic principles of CRISPRi-mediated transcriptional repression in bacteria:
Advantages of CRISPRi for Bacterial Genetics
CRISPRi offers several distinct advantages over traditional genetic engineering approaches for bacterial research:
High Specificity and Efficiency: CRISPRi enables highly specific gene repression with minimal off-target effects, achieving up to 99.9% repression in bacterial systems [3] [2]. The specificity is determined by a 14-nt recognition sequence (12 nt of sgRNA and 2 nt of PAM), which provides sufficient uniqueness for most bacterial genomes [3].
Reversible and Tunable Control: Unlike permanent gene knockouts, CRISPRi-mediated repression is fully reversible, allowing for transient gene silencing [3] [36]. repression levels can be finely tuned by introducing mismatches in the sgRNA base-pairing region or by targeting multiple sites within the same gene, enabling the creation of hypomorphs with varying expression levels [3] [2].
Multiplexing Capability: Multiple sgRNAs can be co-expressed to simultaneously repress several genes, enabling the study of genetic interactions, synthetic lethality, and pathway-level regulation [2] [36]. This capability is particularly valuable for metabolic engineering where coordinated regulation of multiple genes is often required [37] [38].
Application to Essential Genes: CRISPRi allows partial repression of essential genes that would be lethal if completely knocked out, facilitating the study of gene function and essential cellular processes [37] [36]. This reversible knockdown approach is especially useful for investigating genes involved in fundamental bacterial physiology and antibiotic mode of action.
Interrogating Essential Genes with CRISPRi
Theoretical Framework for Essential Gene Studies
Essential genes are those required for fundamental cellular processes such as replication, transcription, translation, and cell division. Traditional knockout approaches cannot be applied to these genes as their complete deletion is lethal to the cell. CRISPRi provides an ideal solution for studying these genes through partial and reversible repression, allowing researchers to investigate gene function without causing cell death [37] [36].
The ability to create tunable knockdowns enables the generation of hypomorphs with varying expression levels, effectively creating an allelic series that can be used to probe gene function in a dose-dependent manner [2]. This approach has been successfully applied in mycobacterial species, including Mycobacterium tuberculosis, where essential genes represent promising targets for novel antibacterial therapies [36].
Methodological Approach for Essential Gene Interrogation
The following protocol outlines the systematic process for interrogating essential genes using CRISPRi in bacteria:
Step 1: Target Selection and Validation
- Identify essential genes through existing databases or comparative genomics
- Select target sites within the coding region, preferably complementary to the nontemplate strand for stronger repression [3] [2]
- Avoid regions with secondary structure or potential off-target binding sites
- Verify the presence of an appropriate PAM sequence (NGG for S. pyogenes Cas9) adjacent to the target site [3]
Step 2: sgRNA Design and Optimization
- Design sgRNAs with 20-nt complementary regions specific to the target essential gene
- For tunable repression, design multiple sgRNAs with varying complementarity or target different regions of the same gene [3]
- Computational tools should be used to predict potential off-target effects by searching for the 14-nt specificity region (12-nt seed + 2-nt PAM) throughout the genome [3]
Step 3: Vector Construction and Cloning
- Clone the customized sgRNA sequence into an appropriate expression vector containing the dCas9 gene
- For mycobacterial species, specialized vectors such as the pJL series that incorporate fluorescent markers for selection and visualization are recommended [36]
- For essential gene studies, use inducible promoters (e.g., anhydrotetracycline-inducible) to control the timing and duration of repression [37] [36]
Step 4: Bacterial Transformation and Selection
- Introduce the CRISPRi construct into the target bacterial strain using appropriate transformation methods
- Select for successful transformants using antibiotic resistance markers or fluorescent markers for rapid identification [36]
- For slow-growing mycobacteria, utilize fluorescence-based selection to efficiently identify recombinant clones [36]
Step 5: Induction of Gene Repression
- Activate dCas9 and sgRNA expression using the appropriate inducer (e.g., IPTG, aTc) at varying concentrations to achieve different levels of repression [37]
- Include uninduced controls to account for basal expression effects
- Monitor bacterial growth throughout induction to assess viability and potential lethality
Step 6: Phenotypic Characterization and Validation
- Quantify repression efficiency using qRT-PCR to measure transcript levels [37]
- Assess functional consequences through growth assays, morphological analysis, or specific functional assays relevant to the target gene
- For essential genes, expect to observe growth defects or morphological changes proportional to the repression level [36]
- Use complementary approaches such as protein quantification or metabolite profiling to validate the phenotypic effects
Applications in Mycobacterial Research
CRISPRi has proven particularly valuable for studying essential genes in mycobacterial species, including Mycobacterium tuberculosis. Recent advances have incorporated fluorescence-based CRISPRi systems that enable both genetic repression and live single-cell imaging, providing powerful tools for investigating bacterial physiology and gene function [36]. These systems have been successfully used to target essential genes such as rpoB (encoding the RNA polymerase beta subunit) and mmpL3 (involved in mycolic acid transport), demonstrating the utility of CRISPRi for probing essential cellular processes in pathogenic bacteria [36].
High-Throughput CRISPRi Screening for Pathway Engineering
Design Principles for CRISPRi Libraries
High-throughput CRISPRi screening enables systematic interrogation of gene function and genetic interactions on a genome-wide scale. The design of effective CRISPRi libraries involves several key considerations:
Library Coverage and Specificity: For comprehensive coverage, design multiple sgRNAs per gene (typically 3-10) targeting different regions to account for variations in repression efficiency [37] [39]. Each sgRNA should be computationally screened for potential off-target effects by identifying similar sequences elsewhere in the genome [3].
Target Site Selection: For metabolic pathway engineering, target both structural genes encoding enzymes and regulatory genes controlling pathway flux [37]. Strategic targeting of promoter regions can modulate transcription initiation, while targeting coding regions can block transcript elongation [3].
Multiplexing Capacity: Design sgRNA arrays that enable simultaneous repression of multiple genes within the same pathway. Recent advances such as Extra-Long sgRNA Arrays (ELSAs) allow direct synthesis of 12-sgRNA arrays that can be integrated into the bacterial genome [2].
Implementation of Genome-Wide CRISPRi Screens
Large-scale CRISPRi screens have been successfully implemented in various bacterial systems, including E. coli and Corynebacterium glutamicum. A notable example is the construction of a CRISPRi library targeting 74 genes in C. glutamicum, including genes encoding enzymes of glycolysis, the pentose phosphate pathway, tricarboxylic acid cycle, and specialized metabolic pathways such as the methylerythritol phosphate and carotenoid biosynthesis pathways [37].
The experimental workflow for high-throughput CRISPRi screening involves:
- Library Construction: Synthesis of pooled sgRNAs targeting multiple genes or pathways
- Transformation and Selection: Introduction of the library into the bacterial population expressing dCas9
- Phenotypic Screening: Application of selective pressure or screening for desired traits
- Sequencing and Hit Identification: Quantification of sgRNA abundance before and after selection to identify genes affecting the phenotype of interest
Case Study: Metabolic Engineering of Carotenoid Biosynthesis
A compelling application of high-throughput CRISPRi screening is the optimization of decaprenoxanthin biosynthesis in C. glutamicum [37]. The screening identified 14 genes that significantly affected carotenoid production when repressed, with 11 genes showing decreased and 3 genes showing increased decaprenoxanthin levels upon repression [37]. Follow-up deletion studies confirmed that deletion of pgi (phosphoglucose isomerase) and gapA (glyceraldehyde-3-phosphate dehydrogenase) improved decaprenoxanthin production by 43-fold and 9-fold, respectively [37]. This demonstrates how CRISPRi screening can rapidly identify metabolic engineering targets for enhanced product synthesis.
Quantitative Analysis of CRISPRi Performance
Efficiency Metrics Across Bacterial Systems
Table 1: CRISPRi Repression Efficiency in Various Bacterial Systems
| Bacterial Species | Repression Efficiency | Key Applications | Notable Features |
|---|---|---|---|
| Escherichia coli | Up to 99.9% repression [3] | Metabolic engineering, essential gene studies | Rapid one-step oligo recombineering [2] |
| Corynebacterium glutamicum | Significant pathway modulation [37] | Amino acid and carotenoid production | Identification of 14 genes affecting decaprenoxanthin biosynthesis [37] |
| Mycobacterium spp. | Effective essential gene knockdown [36] | Antibiotic target validation, pathogenesis | Fluorescence-enabled selection and live single-cell imaging [36] |
| Streptococcus thermophilus | Multiplex pathway optimization [38] | Exopolysaccharide biosynthesis | Systematic optimization of UDP-glucose metabolism [38] |
Comparison of Genetic Perturbation Methods
Table 2: Comparison of CRISPRi with Alternative Genetic Perturbation Methods
| Parameter | CRISPRi | RNAi | Traditional Knockout | Zinc Finger/TALE |
|---|---|---|---|---|
| Mechanism of Action | Transcriptional repression [3] [2] | mRNA degradation [3] | Gene deletion | Transcriptional regulation [3] |
| Reversibility | Fully reversible [3] [36] | Reversible | Irreversible | Reversible |
| Applicability to Essential Genes | Excellent (partial repression) [37] [36] | Good | Poor (lethal) | Good |
| Multiplexing Capacity | High (multiple sgRNAs) [2] | Moderate | Low | Low (complex design) [3] |
| Development Time | Fast (1-2 weeks) [3] | Moderate | Slow | Slow (complex protein engineering) [3] |
| Off-Target Effects | Low with proper design [3] | High [3] | Minimal | Moderate |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for CRISPRi Experiments in Bacterial Systems
| Reagent | Function | Examples/Specifications |
|---|---|---|
| dCas9 Expression Vector | Catalytically dead Cas9 for targeted DNA binding | dCas9 with D10A and H840A mutations [3] [2] |
| sgRNA Expression System | Target-specific guide RNA delivery | Customizable 20-nt complementary region with Cas9-binding hairpin [3] |
| Inducible Promoter Systems | Controlled temporal expression of CRISPRi components | IPTG-inducible, anhydrotetracycline-inducible systems [37] |
| Fluorescent Markers | Selection and tracking of recombinant clones | mCherry, GFP integrated into CRISPRi vectors [36] [40] |
| Library Cloning Platforms | High-throughput sgRNA library construction | ELSAs for 12-sgRNA arrays [2] |
| Specialized Bacterial Vectors | Species-specific delivery of CRISPRi components | pJL series for mycobacteria [36] |
Advanced Applications and Future Directions
Multiplexed Pathway Engineering
The true power of CRISPRi for metabolic engineering emerges when multiple pathway genes are coordinately regulated. In Streptococcus thermophilus, CRISPRi has been successfully applied for multiplex repression of genes in the uridine diphosphate glucose sugar metabolism pathway to optimize exopolysaccharide biosynthesis [38]. This approach enables fine-tuning of metabolic flux without the need for permanent genetic modifications, allowing dynamic control of pathway intermediates and end products.
Integration with Single-Cell Analysis Technologies
Recent advances combine CRISPRi with single-cell analysis platforms such as droplet-based sequencing, enabling high-resolution functional genomics at the single-cell level [40]. This integration allows researchers to quantify mutational co-occurrences, zygosity status, and the occurrence of Cas9 edits with single-cell resolution, providing unprecedented insight into cellular heterogeneity and genetic interactions [40].
Next-Generation CRISPRi Systems
Ongoing research focuses on enhancing CRISPRi efficiency through engineering novel repressor domains. Recent work has identified improved CRISPRi platforms such as dCas9-ZIM3(KRAB)-MeCP2(t), which shows enhanced gene repression of endogenous targets with reduced variability across gene targets and cell lines [9]. These next-generation systems address limitations of earlier platforms, particularly the inconsistent performance dependent on guide RNA sequence and local chromatin context.
CRISPRi technology has revolutionized the study of essential genes and metabolic pathway engineering in bacterial systems. Its capacity for specific, reversible, and tunable gene repression enables researchers to interrogate gene function with unprecedented precision, while its compatibility with high-throughput screening approaches allows systematic exploration of genetic interactions and pathway optimization. As CRISPRi platforms continue to evolve with enhanced repressor domains, improved delivery systems, and integration with single-cell technologies, their impact on bacterial genetics, metabolic engineering, and therapeutic development will undoubtedly expand, offering new avenues for fundamental discovery and applied biotechnology.
Overcoming Challenges: A Guide to Optimizing CRISPRi Efficiency and Specificity
Identifying and Mitigating Off-Target Effects and sgRNA Toxicity
The precision of CRISPR interference (CRISPRi) has established it as a powerful tool for programmable transcriptional repression in bacterial research and beyond. This technology, which utilizes a catalytically dead Cas9 (dCas9) fused to repressor domains to silence gene expression without altering the DNA sequence, is invaluable for functional genomics and metabolic engineering [9]. However, its application is constrained by two significant technical challenges: off-target effects and sgRNA toxicity. Off-target effects occur when the dCas9-sgRNA complex binds to unintended genomic loci, leading to spurious gene repression, while sgRNA toxicity can manifest as reduced cell viability due to unintended interactions with the host machinery [41] [42]. For researchers employing CRISPRi in microbial cell factories, these issues can confound experimental results and reduce the efficiency of engineering endeavors [43]. This guide provides an in-depth analysis of the mechanisms underlying these challenges and outlines state-of-the-art, practical strategies for their identification and mitigation, framed within the context of bacterial research.
Mechanisms of Off-Target Effects and sgRNA Toxicity
A comprehensive understanding of the mechanisms behind off-target effects and sgRNA toxicity is fundamental to developing effective countermeasures.
Mechanisms of Off-Target Interactions
Off-target activity in CRISPRi systems is primarily influenced by the promiscuity of the sgRNA-DNA interaction. The following factors have been identified as key contributors:
- Mismatch Tolerance: The RNA-DNA hybrid can tolerate a certain number of base-pair mismatches, particularly if they are located in the distal region from the Protospacer Adjacent Motif (PAM) and are compensated by strong sequence matches elsewhere in the guide [41] [42].
- DNA Context and Chromatin Accessibility: The local DNA environment, including chromatin structure and accessibility, significantly influences off-target binding. Open chromatin regions are more prone to off-target interactions [44].
- sgRNA Secondary Structure: The intrinsic secondary structure of the sgRNA itself can affect its binding specificity. Guides with stable secondary structures may have reduced on-target efficiency and altered off-target profiles [41] [42].
- Energetics of RNA-DNA Hybrid: The overall binding energy of the RNA-DNA duplex plays a critical role; off-target sites with favorable binding energetics, even with mismatches, are more likely to be bound [42].
- Cellular Enzyme Concentration: High intracellular concentrations of the dCas9-repressor fusion protein can exacerbate off-target effects by increasing the likelihood of binding to lower-affinity, off-target sites [41].
Mechanisms of sgRNA Toxicity
sgRNA toxicity, often observed as impaired cell growth or viability, can arise from several mechanisms:
- High-Efficiency On-Target Repression: The repression of essential genes, even when targeted intentionally, naturally leads to growth defects. However, toxicity can occur even when targeting non-essential genes if the repression is exceptionally efficient, potentially by disrupting finely balanced metabolic or regulatory networks.
- Immunogenic Response: In some host systems, the bacterial components of the CRISPR system or the expressed RNAs can trigger host immune or stress responses, leading to cellular toxicity [45].
- Off-Target Repression of Essential Genes: As a specific case of off-target effects, the unintended binding and repression of genes essential for cell growth or metabolism is a direct cause of sgRNA toxicity. This underscores the critical link between accurate targeting and cellular health.
The diagram below illustrates the core mechanism of CRISPRi and the primary origins of its off-target effects and associated toxicity.
Quantitative Assessment of Contributing Factors
The propensity for off-target effects is not random; it is governed by quantifiable factors. Understanding these allows for predictive modeling and proactive guide design. The table below summarizes the key factors and their impact based on empirical data.
Table 1: Factors Influencing CRISPRi Off-Target Effects and Their Characterization
| Factor | Impact on Off-Target Risk | Experimental Evidence |
|---|---|---|
| Mismatch Position | Mismatches in the PAM-distal seed region (nucleotides 10-20) are more tolerated and pose a higher risk than PAM-proximal mismatches [41] [42]. | High-throughput screening with sgRNA variant libraries [41]. |
| sgRNA Length | Truncated sgRNAs (shorter than 20 nt) can increase specificity but may reduce on-target efficiency. | Specificity profiling in bacterial and human cells [43]. |
| GC Content | Very high or very low GC content in the sgRNA spacer can influence binding stability and specificity. | Analysis of editing outcomes from thousands of sgRNAs [44]. |
| Chromatin Accessibility | Off-target binding is significantly more likely in regions of open chromatin [44]. | CRISPRi screens coupled with ATAC-seq or DNase-seq data. |
| dCas9 Concentration | High intracellular dCas9 concentration linearly correlates with increased off-target binding events [41]. | Titration experiments measuring repression at known off-target sites. |
| TSS Annotation Accuracy | Use of inaccurate Transcription Start Site (TSS) data is a major source of functional "off-target" effects (i.e., poor on-target repression) [44]. | Comparison of CRISPRi efficiency using different TSS annotations (e.g., FANTOM5/CAGE). |
Methodologies for Identification and Detection
Accurately identifying off-target sites is a critical step in profiling and mitigating their effects. The following protocols detail both computational and empirical methods.
Computational Prediction and Guide RNA Design
Purpose: To in silico predict potential off-target sites for a given sgRNA prior to experimental use. Principle: Bioinformatics algorithms scan the genome for sequences with significant homology to the sgRNA spacer, allowing for mismatches, bulges, and varying PAM sequences.
Experimental Protocol:
- sgRNA Sequence Input: Obtain the 20-nt spacer sequence of your sgRNA.
- Algorithm Selection: Utilize established off-target prediction tools. Recent studies have shown that machine learning models, particularly RNN-GRU and 5-layer feedforward neural networks, coupled with cosine similarity metrics for transfer learning, provide superior prediction accuracy [46].
- Parameter Setting:
- Maximum Mismatches: Typically set to 3-5.
- PAM Flexibility: For SpCas9, the canonical PAM is "NGG". Include non-canonical PAMs (e.g., "NAG", "NGA") with lower weighting.
- Seed Region Weighting: Assign higher penalty scores to mismatches within the PAM-distal seed region (e.g., nucleotides 10-20).
- Genome Scanning: Execute the tool against the reference genome of your bacterial strain.
- Output Analysis: Generate a ranked list of potential off-target sites based on a composite score (e.g., MIT specificity score). Sites with high scores should be prioritized for empirical validation, especially if they lie within coding regions or regulatory elements.
Empirical Detection of Off-Target Binding
Purpose: To experimentally identify the genome-wide binding profile of the dCas9-sgRNA complex, including all off-target sites. Principle: Methods like DISCOVER-Seq and its derivatives leverage the recruitment of DNA repair factors to dCas9-bound sites (even without cleavage) to pull down and sequence these loci.
Experimental Protocol (Adapted from DISCOVER-Seq and AutoDISCO):
- Cell Transformation: Introduce the dCas9-repressor and sgRNA constructs into your bacterial cells.
- Cross-Linking: Fix cells with formaldehyde to cross-link DNA-protein complexes.
- Cell Lysis and Chromatin Shearing: Lyse cells and fragment the chromatin via sonication to an average size of 200-500 bp.
- Immunoprecipitation: Use an antibody specific to a relevant DNA-binding protein (e.g., MRE11, a key repair factor recruited to Cas9-bound DNA) to pull down the protein-DNA complexes. AutoDISCO refines this step for scalability and minimal tissue input, making it suitable for varied workflows [46].
- Washing and Elution: Wash away non-specifically bound material and elute the immunoprecipitated DNA.
- Cross-Link Reversal and Purification: Reverse the cross-links and purify the DNA.
- Library Preparation and Sequencing: Prepare a next-generation sequencing library from the purified DNA and perform high-throughput sequencing.
- Bioinformatic Analysis: Map the sequencing reads to the reference genome. Peaks of enriched reads indicate binding sites of the dCas9-sgRNA complex. Compare samples with and without sgRNA to distinguish specific binding.
The workflow for a comprehensive off-target assessment, integrating both prediction and empirical discovery, is visualized below.
Strategies for Mitigation
Once off-target sites are identified, several robust strategies can be employed to mitigate their effects and associated toxicity.
sgRNA and System Optimization
- Precise TSS Mapping: The efficiency of CRISPRi is highly dependent on sgRNA binding near the true Transcription Start Site (TSS). Using inaccurate annotations is a primary source of failure. Rely on experimentally defined TSS atlases, such as FANTOM5/CAGE, for the organism of interest to guide sgRNA design [44].
- Truncated sgRNAs (tru-gRNAs): Using sgRNAs with a shorter spacer sequence (16-18 nt instead of 20 nt) can reduce mismatch tolerance and improve specificity, though this must be balanced against potential reductions in on-target potency [43].
- Moderate dCas9 Expression: Since off-target binding is concentration-dependent, using a weaker, inducible promoter to control dCas9-repressor expression can significantly reduce off-target effects while maintaining sufficient on-target activity [41].
- High-Fidelity dCas9 Variants: While more common for nuclease-active Cas9, engineering efforts have produced dCas9 variants with mutations that tighten the binding specificity, reducing affinity for mismatched targets.
Advanced Repressor Engineering
A powerful approach to mitigate indirect toxicity from excessive on-target repression is to use highly efficient repressor domains. This allows for effective silencing at lower complex concentrations, thereby reducing off-target risk. Recent protein engineering has created novel repressor fusions that outperform traditional ones.
Table 2: Engineered CRISPRi Repressor Domains for Enhanced Efficiency
| Repressor Domain/Fusion | Key Feature | Reported Performance Gain | Proposed Mechanism |
|---|---|---|---|
| dCas9-ZIM3(KRAB) | Uses a KRAB domain from the ZIM3 protein, identified as more potent than the historical KOX1(KRAB) [9] [47]. | Superior gene silencing compared to dCas9-KOX1(KRAB) [9]. | More effective recruitment of endogenous transcriptional repressive machinery. |
| dCas9-KOX1(KRAB)-MeCP2 | A bipartite repressor combining KRAB with a truncation of MeCP2 [9]. | Established "gold standard" for high-efficacy knockdown [9]. | Synergistic repression via multiple chromatin-modifying pathways. |
| dCas9-ZIM3(KRAB)-MeCP2(t) | A next-generation fusion combining the best-in-class ZIM3(KRAB) with a truncated MeCP2 repressor [9]. | ~20-30% better knockdown than dCas9-ZIM3(KRAB) alone; lower variability across gene targets [9]. | Maximized recruitment efficiency and stability of repressive complexes. |
| dCas9-ZIM3-NID-MXD1-NLS | An optimized tripartite repressor featuring an ultra-compact NCoR/SMRT interaction domain (NID) from MeCP2 and an optimized nuclear localization signal (NLS) [47]. | Gene knockdown performance enhanced by ~40% (from NID) and a further ~50% (from NLS optimization) over canonical repressors [47]. | Optimized domain function and enhanced nuclear import. |
Empirical Screening for sgRNA Toxicity
Purpose: To rapidly identify sgRNAs that cause growth defects due to off-target repression of essential genes or overwhelming on-target repression. Principle: A pooled library of sgRNAs is transformed into a population of cells expressing dCas9-repressor. sgRNAs that target essential genes or have high toxicity will be depleted from the population over time, which can be measured by sequencing.
Experimental Protocol:
- Library Design: Synthesize a pooled oligonucleotide library containing hundreds to thousands of sgRNA sequences.
- Cloning and Delivery: Clone the library into an appropriate dCas9-compatible vector and transform it into your bacterial host at a high coverage (e.g., >500x per sgRNA).
- Growth Passage: Grow the transformed culture for multiple generations, ensuring sufficient passaging to allow for the depletion of slow-growing clones.
- Sample Collection: Collect genomic DNA at the initial time point (T0) and after several generations of growth (T-final).
- Amplification and Sequencing: Amplify the sgRNA cassette from the genomic DNA and perform high-throughput sequencing.
- Analysis of Depletion: Calculate the fold-depletion of each sgRNA from T0 to T-final. sgRNAs with significant depletion scores are considered toxic and should be avoided.
Table 3: Key Research Reagents and Tools for Off-Target and Toxicity Analysis
| Tool / Reagent | Function | Example / Note |
|---|---|---|
| Off-Target Prediction Software | In silico identification of potential off-target sites. | CRISPRon/off [43], tools with RNN-GRU or MLP architectures [46]. |
| Empirical Validation Kits | Experimental detection of genome-wide dCas9 binding. | AutoDISCO reagents [46] (for low-input, scalable discovery). |
| High-Fidelity Repressor Plasmids | Provides optimized, high-efficiency dCas9-repressor fusions. | Plasmids encoding dCas9-ZIM3(KRAB)-MeCP2(t) [9] or dCas9-ZIM3-NID-MXD1-NLS [47]. |
| TSS Annotation Database | Provides accurate TSS locations for effective sgRNA design. | FANTOM5/CAGE promoter atlas [44]. |
| Pooled sgRNA Library | Enables large-scale functional screening for on- and off-target effects. | Custom-designed libraries targeting pathways of interest; available from various commercial suppliers. |
| Inducible Promoter Systems | Allows for precise temporal control of dCas9-repressor expression. | anhydrotetracycline (aTc)- or arabinose-inducible promoters for fine-tuning expression levels. |
The challenges of off-target effects and sgRNA toxicity are significant but manageable hurdles in the application of CRISPRi for bacterial research. A multi-faceted approach is essential for success. This begins with rigorous in silico sgRNA design informed by accurate TSS data and predictive algorithms, and is followed by empirical validation of binding specificity using modern, sensitive methods like AutoDISCO. Mitigation hinges on system optimization, including the use of high-efficiency repressor domains like dCas9-ZIM3(KRAB)-MeCP2(t) to achieve robust silencing at lower, safer expression levels, and the implementation of toxicity screens to filter problematic guides. By systematically integrating these strategies—computational prediction, empirical verification, and protein engineering—researchers can harness the full potential of CRISPRi with heightened confidence in its specificity and minimal toxicity, thereby advancing more reliable and reproducible genetic research in microbes and beyond.
Addressing Transcriptional Polarity in Operons and Reverse Polarity Effects
In bacterial genetics, the presence of genes within multi-gene operons presents a fundamental challenge for precise genetic manipulation. Transcriptional polarity refers to the phenomenon where disruption of an upstream gene in an operon negatively affects the expression of downstream genes. This occurs because traditional gene disruption methods, including transposon insertions and even CRISPR-mediated transcriptional repression, often create polar effects that silence not only the targeted gene but also other genes in the same transcriptional unit [48] [49]. In Escherichia coli, approximately 68% of all genes are organized into multi-gene operons, with some operons containing as many as 14 genes [49]. This prevalent genomic organization means that polar effects pose a significant challenge for researchers attempting to study individual gene function or engineer bacterial metabolism.
The core issue stems from the fundamental structure of bacterial operons, where multiple genes are transcribed as a single polycistronic mRNA molecule. When a disruption occurs upstream—whether through insertional mutagenesis, transcriptional termination, or steric blockade of RNA polymerase—it can prevent adequate transcription of downstream genes. This effect complicates functional genomics studies, as phenotypes observed from targeting one gene may actually result from silencing multiple genes. Furthermore, for metabolic engineers seeking to optimize biosynthetic pathways, polarity effects prevent fine-tuned regulation of individual enzymatic steps within operon-encoded pathways [49]. Addressing these challenges requires innovative approaches that can achieve more precise, gene-specific regulation within operonic contexts.
CRISPRi Mechanisms and Polar Effects
dCas9-Based Transcriptional Repression
The advent of CRISPR interference (CRISPRi) technology promised more precise genetic control, but conventional dCas9-based systems still produce significant polar effects. When catalytically dead Cas9 (dCas9) is targeted to a gene's transcription start site or coding region, it forms a steric blockade that prevents transcription elongation by RNA polymerase [9] [50]. While effective for gene repression, this approach suffers from the same polarity issues as traditional methods when applied to operons. As dCas9 binding disrupts the entire transcription process, genes downstream of the target site in the same operon are inevitably silenced alongside the intended target [49].
The limitations of dCas9-mediated repression have driven the development of more sophisticated CRISPRi platforms. Recent advances include engineered repressor domains fused to dCas9 to enhance repression efficiency. For instance, the dCas9-ZIM3(KRAB)-MeCP2(t) repressor demonstrates significantly improved gene silencing compared to earlier dCas9-KRAB fusions [9]. However, while these enhanced repressors improve knockdown efficiency, they do not fundamentally solve the polarity problem when multiple genes share a single transcript.
dCas13-Based Translational Repression
A promising solution to transcriptional polarity emerges with RNA-targeting CRISPR systems. Unlike DNA-targeting dCas9, catalytically inactive Cas13 (dCas13) operates at the translational level by binding to mRNA transcripts and blocking ribosome access or progression [49]. This mechanism allows for gene-specific regulation within operons because it targets individual mRNAs after transcription has occurred, leaving the overall transcript intact.
Research directly comparing dCas9 and dCas13 systems demonstrates that dCas13-mediated repression exhibits up to 6-fold lower polar effects compared to dCas9 [49]. By targeting the translation process rather than transcription, dCas13 enables more precise control of individual genes within multi-gene operons, overcoming a fundamental limitation of DNA-targeting approaches. This translational CRISPRi (tlCRISPRi) approach represents a significant advancement for bacterial genetics and metabolic engineering applications.
Table 1: Comparison of CRISPR Approaches for Addressing Transcriptional Polarity
| Feature | dCas9 (Transcriptional CRISPRi) | dCas13 (Translational CRISPRi) |
|---|---|---|
| Target | DNA | mRNA |
| Mechanism | Steric blockade of RNA polymerase | Blockade of ribosome access/translation |
| Polar Effects | High (affects entire operon) | Low (6-fold lower than dCas9) |
| Operon Application | Represses entire operon | Can target individual genes within operon |
| Tunability | Moderate (via guide RNA design) | High (via guide RNA design and expression levels) |
| Implementation | Well-established in bacteria | Emerging, with demonstrated efficacy in E. coli |
Experimental Approaches and Methodologies
Implementing dCas13 for Reduced-Polarity Repression
The application of dCas13 for translational repression requires careful experimental design. Below is a detailed protocol for implementing dCas13-mediated CRISPRi in E. coli, based on established methodologies [49]:
Plasmid Design and Construction:
- Express dCas13 from a medium-strength constitutive promoter (e.g., J23107) to balance repression efficiency and cellular burden.
- Clone guide RNAs (crRNAs) under a strong constitutive promoter (e.g., J23119) in the same or compatible plasmid.
- Design crRNAs with 28-nt spacer sequences for optimal activity, targeting the ribosome binding site or early coding region of the target mRNA.
- Include a 23-nt direct repeat sequence in the crRNA scaffold, specific to the Cas13 ortholog used.
Experimental Workflow:
- Transform the dCas13 expression plasmid and crRNA plasmid(s) into the target E. coli strain.
- For multiplexed repression, clone multiple crRNA expression cassettes in a single array.
- Culture transformed cells in appropriate selective media and measure repression efficiency via fluorescence reporters or quantitative PCR.
- For temporal control, consider inducible promoter systems (e.g., tetracycline-inducible) to initiate repression at specific growth phases.
Control Experiments:
- Include non-targeting crRNAs to account for potential off-target effects.
- Compare phenotypes with dCas9-based repression targeting the same gene.
- Measure expression of both targeted and non-targeted genes in the same operon to quantify polar effects.
Table 2: Key Research Reagents for dCas13-Mediated Repression
| Reagent | Function | Example/Details |
|---|---|---|
| dCas13 Expression Vector | Expresses catalytically inactive Cas13 | p15A origin, medium-strength promoter (J23107) |
| crRNA Expression Cassette | Guides dCas13 to target mRNA | Strong promoter (J23119), direct repeat sequence |
| Fluorescent Reporters | Quantify repression efficiency | mRFP1, sfGFP, mTagBFP2 |
| Selective Antibiotics | Maintain plasmid selection | Carbenicillin, Chloramphenicol, Kanamycin |
| Minimal Promoter Reporters | Measure CRISPRi dynamic range | BBaJ23110 or BBaJ23117 promoters |
Combinatorial Transcriptional and Translational Control
For advanced applications requiring both operon-wide and gene-specific regulation, researchers can combine dCas9 and dCas13 systems [49]. This approach enables sophisticated control strategies where:
- Transcriptional activation of an entire operon is achieved using dCas9-based CRISPR activation (CRISPRa) systems.
- Translational repression of specific genes within the activated operon is implemented using dCas13.
This combinatorial method was successfully demonstrated for optimizing biosynthesis of human milk oligosaccharides (HMOs) in E. coli, where it improved production yields compared to transcriptional control alone [49]. The protocol for this approach involves:
System Configuration:
- Express dCas9 from a genome-integrated copy to reduce plasmid burden.
- Use a separate plasmid for dCas13 expression to maintain compatibility.
- Design guide RNAs for dCas9 to target activation domains (e.g., MCP-SoxS) to operon promoters.
- Design crRNAs for dCas13 to target specific genes within the operon that require down-regulation.
Implementation Steps:
- Engineer the production host to contain genome-integrated dCas9 with an appropriate activation domain.
- Introduce the dCas13 expression plasmid and relevant crRNA plasmids.
- Optimize the expression levels of both CRISPR systems to balance activation and repression.
- Monitor metabolic output and adjust guide RNA combinations iteratively.
Research Applications and Case Studies
Essential Gene Analysis in Pseudomonas aeruginosa
CRISPRi has been powerfully applied for genome-wide essentiality studies in pathogenic bacteria. Researchers developed a titratable tetracycline-inducible CRISPRi system for Pseudomonas aeruginosa that enables precise control of gene repression timing and magnitude [50]. This system addresses several limitations of traditional transposon mutagenesis approaches:
- Temporal control allows analysis of essential genes whose complete repression would be lethal.
- Titratable repression enables assessment of gene vulnerability (how much repression impairs fitness) and responsiveness (how quickly fitness declines after repression).
- Reduced polar effects compared to transposon insertions, especially when carefully targeting non-operonic regions.
The system was used to perform genome-wide CRISPRi-seq screens, identifying essential genes and their characteristics under different growth conditions [50]. This approach revealed FprB as a synergistic target for gallium therapy in P. aeruginosa, demonstrating how CRISPRi screens can identify potential drug targets that enhance existing antimicrobial therapies.
Metabolic Pathway Optimization
The combination of transcriptional and translational CRISPR tools enables unprecedented precision in metabolic engineering. In one application, researchers optimized the production of lacto-N-tetraose (LNT), a human milk oligosaccharide with therapeutic potential [49]:
- The entire LNT biosynthetic operon was transcriptionally activated using dCas9-based CRISPRa.
- Specific genes within the operon were translationally repressed using dCas13 to rebalance metabolic flux.
- This combinatorial approach increased LNT production compared to transcriptional activation alone.
This case study demonstrates how addressing transcriptional polarity enables fine-tuning of complex metabolic pathways, moving beyond simple gene knockout or universal activation strategies toward precisely balanced expression of multi-gene pathways.
Visualization of Experimental Approaches
Comparative Mechanisms of dCas9 and dCas13
Combinatorial CRISPR Approach Workflow
Transcriptional polarity presents a persistent challenge in bacterial genetics and metabolic engineering, particularly given the prevalence of multi-gene operons in species like E. coli and P. aeruginosa. While traditional CRISPRi approaches using dCas9 have improved precision compared to transposon mutagenesis, they still produce significant polar effects that complicate the interpretation of genetic experiments and limit metabolic engineering applications.
The emergence of dCas13-based translational repression offers a promising solution, demonstrating substantially reduced polarity effects—up to 6-fold lower than dCas9-based approaches [49]. Furthermore, the strategic combination of transcriptional and translational CRISPR tools enables sophisticated control schemes that can activate entire operons while selectively repressing specific genes within them. These advances are already proving valuable for essential gene identification, drug target discovery, and metabolic pathway optimization.
As CRISPR technologies continue to evolve, the ability to precisely control individual genes within operonic structures will remain crucial for advancing both basic bacterial genetics and applied biotechnology. The methodologies and case studies presented here provide a framework for researchers to address transcriptional polarity challenges in their own work, enabling more accurate functional analyses and more efficient engineering of bacterial systems for therapeutic and industrial applications.
Managing dCas9 Expression to Balance Efficacy and Cellular Toxicity
In bacterial CRISPR interference (CRISPRi) research, achieving optimal silencing efficacy while minimizing cellular toxicity is a fundamental challenge. The core of this balance lies in the precise management of catalytically dead Cas9 (dCas9) expression. CRISPRi, a powerful genetic perturbation technique, uses dCas9 in conjunction with a sequence-specific guide RNA (sgRNA) to repress transcription without cleaving DNA [6] [2]. While this system enables programmable and highly efficient gene knockdown, excessive dCas9 expression or improper sgRNA design can lead to significant fitness costs for the bacterial host, including growth defects and off-target effects [6]. This guide details the sources of dCas9-induced toxicity and provides actionable, quantitative strategies for researchers to fine-tune dCas9 expression, ensuring robust experimental outcomes in functional genomics and metabolic engineering applications.
Core Mechanisms of dCas9-Induced Toxicity
Understanding the sources of dCas9 toxicity is prerequisite to managing it. The primary mechanisms include off-target binding, resource sequestration, and protein overaccumulation.
- Off-Target Binding and Genomic Integrity: Although dCas9 lacks nuclease activity, its binding is not perfectly specific. The dCas9-sgRNA complex can bind to genomic sites with partial complementarity to the sgRNA, particularly if the seed sequence (the PAM-adjacent 10-12 nucleotides) is perfectly matched [2]. This promiscuous binding can disrupt transcription by physically blocking RNA polymerase (RNAP), leading to unintended gene silencing and potential disruption of essential cellular processes [6]. Figure 1 illustrates how both on-target and off-target binding can impede transcription.
Figure 1: Mechanisms of dCas9-Mediated Transcriptional Repression and Toxicity. The dCas9-sgRNA complex can bind to both intended target sites and off-target sites with sufficient sequence similarity, blocking RNA polymerase and leading to intended gene repression or unintended toxic effects.
- Cellular Resource Sequestration: High-level, constitutive expression of dCas9 places a significant burden on the host cell's transcriptional and translational machinery. This can deplete nucleotides, amino acids, and energy (ATP), redirecting resources away from essential growth functions and ultimately reducing the bacterial growth rate and culture density [6].
- Protein Overaccumulation and Aggregation: The dCas9 protein is relatively large (~160 kDa). When expressed at high levels, it can exceed the solubility capacity of the bacterial cytoplasm, leading to protein aggregation and inclusion body formation [51]. These aggregates can be toxic, disrupt proteostasis, and sequester other vital cellular proteins.
Quantitative Comparison of dCas9 Expression Control Systems
Selecting the appropriate expression system is the most critical step in managing dCas9 levels. The following table summarizes the key inducible systems validated for dCas9 expression in bacteria, along with their performance characteristics.
Table 1: Inducible Promoter Systems for Tunable dCas9 Expression in Bacteria
| Promoter System | Inducer | Dynamic Range | Leakiness | Key Features & Applications |
|---|---|---|---|---|
| PBAD (Arabinose) [10] [6] | L-Arabinose | >100-fold [6] | Low | Fine-tunable with arabinose concentration; well-characterized in E. coli. |
| PLTetO-1 (Anhydrotetracycline) [10] | aTc | High (Not specified) | Very Low | Tightly repressed; excellent for strong, uniform induction. |
| Ptet (Tetracycline) [6] | aTc/Tetracycline | Broad range [6] | Low | Titratable repression; suitable for precise knockdown studies. |
| PrhaBAD (Rhamnose) [52] | L-Rhamnose | High (Not specified) | Low | Tight regulation; used in recent optimized CRISPRa/i systems [52]. |
Beyond the promoter, the copy number and origin of replication (ori) of the plasmid harboring the dCas9 gene are crucial. A high-copy plasmid will produce significantly more dCas9 than a low-copy or single-copy plasmid, even under the same promoter. For long-term or genome-wide studies, integrating the dCas9 gene into a neutral site on the bacterial chromosome can be advantageous. This single-copy, stable configuration eliminates variability from plasmid loss and reduces the metabolic burden associated with plasmid maintenance [6].
Experimental Protocols for Optimization
Protocol: Titrating dCas9 Expression Using an Inducible System
This protocol outlines the steps to establish a dose-response relationship between inducer concentration and repression efficacy, enabling identification of the minimal effective dCas9 expression level.
- Strain and Plasmid Preparation: Transform your bacterial strain with two plasmids: one expressing dCas9 under the control of an inducible promoter (e.g., pLtetO-1-dCas9 [10]) and another expressing an sgRNA targeting a reporter gene (e.g., GFP or mRFP).
- Inducer Dilution Series: Prepare a culture of the transformed strain and aliquot it into separate flasks. Induce with a gradient of inducer concentration (e.g., 0, 10, 50, 100, 500 ng/mL aTc). Include an uninduced control (0 ng/mL) and a non-targeting sgRNA control [10].
- Culture Growth and Sampling: Grow cultures under optimal conditions (e.g., 37°C with shaking). Monitor the optical density at 600 nm (OD600) to track growth. Sample cells at mid-log phase (OD600 ~0.5-0.6) and at stationary phase.
- Efficacy and Toxicity Assessment:
- Repression Efficacy: For a fluorescent reporter, measure fluorescence intensity via flow cytometry or a plate reader. Calculate repression as (1 - (Fluorescenceinduced / Fluorescenceuninduced)) * 100%. For endogenous genes, quantify mRNA levels using RT-qPCR [10].
- Cellular Toxicity: Use the growth curve data. Compare the growth rate (doubling time) and final culture density (maximum OD600) of induced cultures against the uninduced control. A significant reduction indicates toxicity.
- Data Analysis: Plot repression efficacy and relative growth rate against the inducer concentration. The optimal inducer level is the point that achieves >90% repression with a growth rate reduction of less than 20%.
Protocol: Assessing and Mitigating Off-Target Effects
- sgRNA Design for Specificity: Use established algorithms to design sgRNAs with a 20-nt spacer region. Perform a BLAST search of the spacer sequence against the host genome to exclude sgRNAs with significant off-target potential, especially in the seed region [10] [2].
- Whole-Genome Expression Profiling: For critical sgRNAs, perform RNA-seq on the knockdown strain (induced) versus a non-targeting sgRNA control. This unbiased approach identifies all genes and pathways differentially expressed due to dCas9 binding.
- Validation: Confirm putative off-target effects by designing new sgRNAs specific to those genomic regions. If repression is observed with the new sgRNAs, it validates the off-target site. Redesign the original sgRNA to avoid these sites.
Figure 2 below provides a workflow integrating these optimization protocols.
Figure 2: Experimental Workflow for Balancing dCas9 Efficacy and Toxicity. A step-by-step guide for empirically determining the optimal dCas9 expression conditions, with troubleshooting pathways.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of titratable CRISPRi requires specific genetic tools and reagents. The table below lists key solutions for constructing and testing a tunable dCas9 system.
Table 2: Research Reagent Solutions for Bacterial CRISPRi
| Reagent / Tool | Function | Example & Source |
|---|---|---|
| Inducible dCas9 Plasmid | Expresses dCas9 protein under tunable control. | Addgene #44249 (dCas9 under pLtetO-1 in E. coli) [10]. |
| sgRNA Expression Plasmid | Expresses the custom guide RNA. | Addgene #44251 (sgRNA under J23119 promoter) [10]. |
| sgRNA Design Algorithm | Identifies specific target sites with minimal off-target potential. | BLAST for the host genome; Vienna suite for RNA folding analysis [10]. |
| Fluorescent Reporter Plasmid | Provides a quantitative readout for repression efficacy. | Plasmid expressing GFP or mRFP under a constitutive promoter [52] [10]. |
| Golden Gate Assembly Kit | Enables modular cloning of multiple sgRNAs for multiplexing. | Commercial kits with BsaI enzyme for efficient assembly [52] [10]. |
Precise management of dCas9 expression is not merely a technical optimization but a fundamental requirement for robust and reliable bacterial CRISPRi research. By leveraging titratable expression systems, employing rigorous sgRNA design, and systematically quantifying the trade-off between silencing strength and cellular fitness, researchers can mitigate the inherent toxicity of dCas9. The strategies and protocols outlined in this guide provide a clear roadmap for achieving this balance, thereby enhancing the quality of functional genomic screens and the efficiency of metabolic engineering efforts in bacterial systems.
CRISPR interference (CRISPRi) has become an indispensable technique for programmable gene repression in bacteria. The core system utilizes a catalytically dead Cas protein (dCas) that binds to DNA in a guide RNA-programmed manner without cleaving it, thereby physically blocking transcription. While the initial CRISPRi systems provided a foundational technology, two critical areas for enhancement have emerged: the development of more effective repressor domains and the engineering of Cas variants with relaxed Protospacer Adjacent Motif (PAM) requirements. PAM restrictions significantly limit the targeting scope of CRISPR systems, confining them to genomic regions flanked by specific nucleotide sequences. This technical guide examines recent advances in both novel repressor domains and PAM-flexible Cas variants, providing a comprehensive resource for researchers aiming to implement these enhanced CRISPRi systems in bacterial hosts. Through structured data comparison, detailed protocols, and practical implementation guidelines, we frame these technological developments within the broader context of optimizing CRISPRi mechanisms for transcriptional repression in bacterial research.
Novel Repressor Domains for Enhanced CRISPRi Efficacy
Engineering the cAMP Receptor Protein (CRP) as a Versatile Effector
The native dCas9 protein provides baseline repression through steric hindrance, but its efficacy can be substantially enhanced through fusion to optimized repressor domains. While domains like KRAB are well-established in eukaryotic systems, bacterial CRISPRi has benefited from recent engineering of prokaryotic transcriptional regulators. A significant advancement comes from the engineering of the Escherichia coli cAMP receptor protein (CRP), a global transcriptional regulator that naturally controls hundreds of genes involved in carbon metabolism and energy homeostasis [52].
Through systematic optimization of CRP domains and linkers, researchers have developed a versatile effector capable of precise gene expression control when combined with an evolved PAM-flexible dxCas9. This engineered dxCas9-CRP system demonstrates robust repression of coding sequences while also enabling targeted activation of upstream regulatory regions, creating a dual-mode CRISPRa/i system [52]. When applied to metabolic engineering in E. coli, this system enabled genome-scale activation and repression to increase violacein production by successfully identifying key regulatory targets [52].
Table 1: Performance Comparison of CRISPRi Repressor Domains in Bacteria
| Repressor Domain | Origin | Mechanism of Action | Key Features | Validated Applications |
|---|---|---|---|---|
| Native dCas9 | S. pyogenes | Steric hindrance of RNA polymerase | Baseline repression, no fusion required | Essential gene knockdowns [21] |
| KRAB | Eukaryotic | Chromatin modification | Strong repression in eukaryotes; limited efficacy in bacteria | Primarily eukaryotic systems [53] |
| Engineered CRP | E. coli | Modulation of RNA polymerase interaction | Dual-mode activation/repression, native bacterial compatibility | Metabolic engineering, genome-scale screening [52] |
| ω-subunit (RpoZ) | Bacterial | Direct binding to RNA polymerase | Prokaryotic-specific mechanism | Limited testing in CRISPRi context [52] |
Implementation Considerations for Novel Repressor Domains
When implementing novel repressor domains like engineered CRP in bacterial CRISPRi systems, several technical factors critically influence performance:
Linker Optimization: The flexible peptide linker connecting dCas9 to the repressor domain requires careful optimization to balance spatial flexibility and structural stability. Systematic testing of linker length and composition was essential for the CRP fusion functionality [52].
Cellular Context: Repressor domain efficacy can vary significantly across bacterial species due to differences in transcriptional machinery. The dxCas9-CRP system has demonstrated functionality in both E. coli and Pseudomonas putida, suggesting broad applicability [52].
Expression Balancing: The repressor domain fusion can impact dCas9 expression, stability, and DNA binding efficiency. Inducible promoters like the rhamnose-inducible PrhaBAD allow controlled expression to balance efficacy and cellular toxicity [52].
PAM-Flexible Cas Variants for Expanded Targeting Scope
Engineering Strategies for PAM Relaxation
The stringent PAM requirement of native SpCas9 (5'-NGG-3') represents a fundamental limitation for comprehensive genomic targeting. Recent protein engineering efforts have produced several Cas variants with relaxed PAM requirements, dramatically expanding the targetable genome space. Two primary approaches have driven this advancement: structure-guided engineering and directed evolution [54] [53].
Structure-guided engineering focuses on modifying residues in the PAM-interacting (PI) domain responsible for recognizing specific DNA sequences. In SpCas9, residues R1333 and R1335 form specific contacts with the guanines in the NGG PAM through bidentate hydrogen bonds [54]. Rational mutagenesis of these residues has yielded variants with altered PAM specificities:
- SpCas9-NG: Contains mutations R1335V/L1111R/D1135V/G1218R/E1219F/A1322R/T1337R that enable recognition of NG PAMs [54] [53].
- SpdNG-LWQT: A derivative with additional mutations (R1333Q/V1335T) that further expands PAM recognition to preferentially accommodate NRN PAMs, with enhanced compatibility for the 5'-CAT-3' PAM found at start codons [54].
- xCas9: Developed through phage-assisted continuous evolution, recognizes NG PAMs with varying efficiency across genomic contexts [53].
Table 2: Comparison of PAM-Flexible Cas9 Variants for Bacterial CRISPRi
| Variant | Mutations | PAM Recognition | Relative Efficiency* | Key Applications |
|---|---|---|---|---|
| Wild-type SpCas9 | None | 5'-NGG-3' | Reference (100%) | Baseline CRISPRi [53] [21] |
| xCas9 | D10A/E480K/E543D/E1219V/A262T/S409I/M694I/H840A | 5'-NG-3' | ~43% of WT Cas9 | Transcriptional activation [53] |
| Cas9-NG | D10A/H840A/R1335V/L1111R/D1135V/G1218R/E1219F/A1322R/T1337R | 5'-NG-3' | ~64% of WT Cas9 | Start codon targeting [54] [53] |
| SpdNG-LWQT | Includes Cas9-NG + R1333Q/V1335T | 5'-NRN-3' (preferential) | ~52% repression at 5'-CAT-3' PAM | Universal gene repression [54] |
| xCas9-NG | Combines xCas9 and Cas9-NG mutations | 5'-NG-3' | Improved over both parents | Hybrid nuclease and CRISPRi applications [53] |
Relative nuclease activity compared to WT SpCas9 at NGG PAMs based on pooled competition screens [53]
Performance Trade-offs in PAM-Flexible Variants
High-throughput comparative analyses reveal that PAM flexibility comes with measurable efficiency costs. Pooled competition screens comparing Cas9 variants across thousands of genomic loci demonstrate that wild-type Cas9 consistently outperforms PAM-flexible variants at canonical NGG PAMs across nuclease, activation, and repression modalities [53].
For NGH PAMs (H = A, C, or T), Cas9-NG universally outperforms xCas9, though with 2- to 4-fold lower activity than wild-type Cas9 at NGG PAMs [53]. This performance trade-off necessitates careful experimental design when implementing these variants, potentially requiring the testing of multiple guides to achieve sufficient repression efficiency.
Experimental Framework for CRISPRi Implementation
Protocol for CRISPRi System Assembly and Validation
Implementing a novel CRISPRi system with engineered repressor domains and PAM-flexible Cas variants requires a structured experimental approach:
A. Plasmid Construction and Transformation
- Vector Selection: Utilize a modular plasmid system such as pACCRi as the backbone for CRISPRi components [52].
- dCas9-Repressor Fusion: Clone the PAM-flexible dCas9 variant (e.g., dxCas9 or SpdNG-LWQT) fused to the engineered repressor domain (e.g., CRP) under control of an inducible promoter (e.g., PrhaBAD) [52].
- Guide RNA Expression: Express sgRNAs from a constitutive promoter (e.g., BBa_J23119) on a separate plasmid or in an array format [52].
- Transformation: Introduce constructs into the target bacterial strain using appropriate transformation methods, selecting with compatible antibiotics.
B. Guide RNA Design and Validation
- Target Selection: For repression, design guides targeting the template strand of coding sequences near the start codon [54] [21].
- PAM Consideration: Identify available PAM sites based on the variant's specificity (NG for Cas9-NG/xCas9, NRN for SpdNG-LWQT) [54] [53].
- Efficiency Prediction: Apply machine learning tools that incorporate sequence features, genomic context, and gene expression data to predict guide efficiency [21].
- Experimental Validation: Test multiple guides per target using a reporter system or direct measurement of target gene expression.
C. Functional Validation
- Repression Efficiency Quantification: Measure knockdown efficiency using qRT-PCR or fluorescent reporters 4-24 hours after dCas9 induction [52] [21].
- Growth Phenotyping: Assess fitness costs of dCas9 and repressor domain expression through growth curve analysis [52].
- Specificity Verification: RNA-seq to assess off-target effects, particularly important for engineered repressor domains with broader transcriptional effects [55].
Figure 1: CRISPRi System Implementation Workflow. This diagram outlines the key stages in implementing a novel CRISPRi system, from initial design to functional validation.
Protocol for Genome-Scale CRISPRi Screening
For comprehensive gene function discovery or metabolic engineering applications, genome-scale CRISPRi screening provides a powerful approach:
Library Design: Design a genome-wide gRNA library targeting all non-essential genes with 3-5 guides per gene, focusing on regions near transcription start sites with available PAM sequences for your chosen Cas variant [52] [21].
Library Construction: Synthesize pooled oligonucleotide libraries and clone into appropriate gRNA expression vectors using high-efficiency Golden Gate assembly [52].
Screen Execution: Transform the gRNA library into strains expressing the dCas9-repressor fusion at a representation of >500x coverage. Include appropriate controls (non-targeting gRNAs, essential gene targets) [52] [21].
Phenotypic Selection: Apply relevant selective pressure (e.g., substrate limitation, toxin exposure) over multiple generations to enrich for gRNAs conferring fitness advantages or disadvantages [21].
Sequencing and Analysis: Harvest genomic DNA at multiple timepoints, amplify gRNA regions, and sequence to quantify gRNA abundance changes using specialized analysis pipelines [21].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Advanced CRISPRi Research
| Reagent Category | Specific Examples | Function | Implementation Notes |
|---|---|---|---|
| Cas Variants | dxCas9-3.7, SpCas9-NG, SpdNG-LWQT | DNA binding with flexible PAM recognition | Select based on required PAM flexibility and efficiency trade-offs [52] [54] |
| Repressor Domains | Engineered CRP, KRAB, RpoZ | Transcriptional repression | CRP optimized for bacterial systems [52] |
| Expression Plasmids | pACCRi, psgRNA | Modular component expression | Use different antibiotic resistance for compatible co-expression [52] |
| Inducible Promoters | PrhaBAD, PLlacO1 | Controlled dCas9 expression | Balance repression efficiency with cellular toxicity [52] [54] |
| Guide RNA Libraries | Genome-wide, pathway-specific | Targeted gene repression | Include non-targeting controls and essential gene targets [52] [21] |
| Machine Learning Tools | Mixed-effect random forest models | gRNA efficiency prediction | Incorporates guide and gene-specific features [21] |
Implementation Considerations and Future Directions
Practical Guidelines for System Selection
Choosing the appropriate CRISPRi system requires balancing multiple experimental factors:
Targeting Scope vs. Efficiency: When comprehensive targeting is prioritized over maximum repression, PAM-flexible variants like Cas9-NG or SpdNG-LWQT are preferable despite their reduced efficiency. For applications requiring strong repression at specific loci, wild-type SpCas9 may remain optimal if NGG PAMs are available [54] [53].
Multiplexing Capability: Newer vector systems enable coordinated repression of multiple targets, which is particularly valuable for metabolic engineering applications. The compact nature of some PAM-flexible variants facilitates more complex multiplexing arrangements [56] [55].
Species Compatibility: While most CRISPRi tools are developed in E. coli, many show functionality in related Gram-negative bacteria. Verification of expression system compatibility and gRNA specificity is essential when working with non-model organisms [52].
Emerging Technologies and Future Developments
The CRISPRi landscape continues to evolve with several promising developments on the horizon:
AI-Designed Editors: Protein language models trained on diverse CRISPR-Cas sequences are now generating novel Cas proteins with optimized properties, including potentially improved PAM flexibility and DNA binding affinity [57].
Enhanced Prediction Algorithms: Mixed-effect machine learning models that incorporate both guide-specific and gene-specific features are improving the accuracy of gRNA efficiency predictions, reducing experimental optimization time [21].
Expanded Delivery Methods: While plasmid-based delivery remains standard for bacterial systems, new conjugation and transduction methods may facilitate CRISPRi implementation in less genetically tractable species [58].
Integration with Multi-omics: Combining CRISPRi with transcriptomics, metabolomics, and 13C-metabolic flux analysis provides systems-level understanding of repression effects, guiding more effective strain engineering strategies [55].
Figure 2: CRISPRi Repression Mechanism. This diagram illustrates the core mechanism of CRISPRi, showing how the dCas9-repressor fusion binds DNA through guide RNA programming and PAM recognition to block RNA polymerase and repress transcription.
Through strategic implementation of novel repressor domains and PAM-flexible Cas variants, researchers can achieve unprecedented control over bacterial gene expression. The continued refinement of these tools promises to accelerate both fundamental research and applied biotechnology in prokaryotic systems.
CRISPR interference (CRISPRi) has emerged as a powerful tool for programmable transcriptional repression in bacterial research, enabling targeted gene knockdown without altering the underlying DNA sequence. This guide provides an in-depth technical resource for researchers, scientists, and drug development professionals working with CRISPRi systems in bacterial contexts. Within the broader thesis of CRISPRi mechanisms for transcriptional repression, we focus specifically on troubleshooting common experimental pitfalls and optimizing data interpretation strategies to enhance reproducibility and reliability in your research.
Core Principles of CRISPRi in Bacteria
CRISPRi functions through a two-component system: a catalytically dead Cas protein (dCas9, dCas12a, or other variants) fused to transcriptional repressor domains, and a guide RNA (gRNA) that directs this complex to specific DNA sequences through base-pair complementarity. Unlike nuclease-active CRISPR-Cas9 systems that create double-strand breaks, CRISPRi achieves reversible gene expression control without inducing DNA damage, which can confound screening results through activation of DNA repair pathways and cellular stress responses [9].
In bacteria, CRISPRi binding to a target gene's promoter region or coding sequence can block transcription initiation or elongation, effectively knocking down gene expression. The system's precision allows for the investigation of essential genes, metabolic pathway mapping, and identification of genetic vulnerabilities without permanent genetic alterations [52] [9].
Common Technical Pitfalls and Solutions
Inefficient Gene Repression
Problem: Incomplete gene knockdown leads to subtle phenotypic effects and inconclusive results.
Root Causes:
- Suboptimal gRNA binding position relative to the transcription start site (TSS)
- Weak repressor domain efficacy
- Insufficient expression of CRISPRi components
- Target site inaccessibility due to chromatin structure or DNA supercoiling
Solutions:
- gRNA Positioning: For dCas9 systems, design gRNAs to target the non-template strand between -35 and +1 relative to the TSS, as this region shows highest repression efficiency. For dCas12a systems, optimal positioning may vary [59].
- Enhanced Repressor Domains: Utilize improved repressor fusion proteins. Recent research has identified novel combinations that significantly improve knockdown:
CRISPRi Repressor Domain Enhancement Workflow
- Expression Optimization: Ensure strong, inducible promoters drive dCas9-repressor fusions, and use moderate-copy number plasmids to maintain consistent expression levels without burdening the host [52] [59].
Variable Performance Across gRNAs
Problem: Significant differences in repression efficiency between gRNAs targeting the same gene.
Root Causes:
- Sequence-dependent gRNA binding affinity
- Local DNA accessibility variations
- gRNA secondary structure interference
- Off-target binding effects
Solutions:
- Multiplexed gRNA Design: Always design 3-5 gRNAs per target gene with diverse genomic positions to account for efficiency variability.
- gRNA Efficiency Prediction: Utilize computational tools that consider local GC content, absence of self-complementarity, and position relative to TSS.
- Empirical Validation: Conduct small-scale pilot experiments to quantify repression efficiency of each gRNA before proceeding to large-scale screens [9].
Cell Line-Specific Variability
Problem: CRISPRi system performance varies significantly between different bacterial strains.
Root Causes:
- Differences in endogenous transcription machinery
- Variable expression of co-factors required for repressor function
- Strain-specific genetic backgrounds affecting dCas9 expression or gRNA processing
Solutions:
- System Customization: Optimize induction conditions and expression levels for each bacterial strain.
- Repressor Domain Screening: Test multiple repressor domain combinations to identify the most effective configuration for your specific bacterial host.
- Control Validation: Include strong positive and negative controls specific to each strain to establish baseline performance metrics [9].
Essential Research Reagent Solutions
Table 1: Key Reagents for CRISPRi Experiments in Bacteria
| Reagent Category | Specific Examples | Function & Application Notes |
|---|---|---|
| dCas9 Variants | dCas9(3.7), dxCas9 (PAM-flexible) | DNA binding moiety; PAM-flexible variants expand targeting range [52] |
| Repressor Domains | KOX1(KRAB), ZIM3(KRAB), MeCP2(t), KRBOX1(KRAB) | Transcriptional repression modules; combination domains show enhanced efficacy [9] |
| Bacterial Activation Domains | SoxS (R93A variant), CRP, RpoZ | For CRISPRa systems; enable both upregulation and repression studies [52] [59] |
| gRNA Expression Systems | BBa_J23119 promoter, psgRNA plasmid | Constitutive gRNA expression; ensure compatibility with bacterial host [52] |
| Inducible Systems | PrhaBAD (rhamnose), other inducible promoters | Controlled dCas9-repressor expression; minimize fitness burden [52] [59] |
Data Interpretation Framework
Quality Control Metrics
Before interpreting screening results, establish these quality control benchmarks:
- Essential Gene Enrichment: In genome-wide screens, essential genes should show significant depletion (p < 0.05, log2 fold change ≤ -2) in the negative selection.
- gRNA Consistency: At least 2/3 gRNAs targeting the same gene should produce concordant phenotypic effects.
- Control Performance: Non-targeting gRNAs should cluster separately from essential gene-targeting gRNAs in principal component analysis [60].
Normalization Strategies
Table 2: Data Normalization Methods for CRISPRi Screens
| Method | Application Context | Implementation | Advantages/Limitations |
|---|---|---|---|
| Median Normalization | Initial data scaling | Subtract median of all gRNAs from each individual gRNA | Simple implementation; assumes most genes have no effect |
| Control-Based Normalization | Screens with non-targeting controls | Use non-targeting gRNAs as reference distribution | Removes technical artifacts; requires sufficient controls |
| Quantile Normalization | Multi-replicate experiments | Force distribution equality across replicates | Improves comparability; may remove biological signals |
| RLE (Relative Log Expression) | Large-scale screens | Normalize using the geometric mean of all gRNAs | Robust to outliers; standard in RNA-seq analysis |
Statistical Analysis for Hit Identification
- Fold Change Calculations: Compute log2(fold change) for each gRNA using normalized read counts (late timepoint/early timepoint).
- Gene-Level Scoring: Apply robust statistical aggregation (e.g., α-RRA) to combine gRNA-level effects into gene-level scores.
- Multiple Testing Correction: Use Benjamini-Hochberg procedure to control false discovery rate (FDR < 0.1-0.25 depending on screen size) [61] [60].
Advanced Methodologies
Experimental Protocol: Genome-Wide CRISPRi Screen in Bacteria
Stage 1: Library Design and Construction
- gRNA Library Design: Design 5-10 gRNAs per gene targeting the template strand near the TSS (-50 to +100). Include at least 100 non-targeting control gRNAs with no matches in the host genome.
- Oligonucleotide Pool Synthesis: Synthesize oligonucleotide library containing gRNA sequences with appropriate adapter sequences for cloning.
- Vector Construction: Clone the gRNA library into the appropriate expression vector using Golden Gate assembly or similar high-efficiency methods [52].
- Library Validation: Sequence the pooled plasmid library to confirm representation and uniformity.
Stage 2: Screen Implementation
- Transformation: Transform the gRNA library into your bacterial strain expressing the dCas9-repressor fusion at appropriate coverage (>500x per gRNA).
- Selection and Expansion: Plate transformed cells on selective media and harvest a portion as the T0 reference timepoint.
- Experimental Conditions: Apply the specific selective pressure (antibiotic treatment, nutrient limitation, etc.) or growth condition of interest for an appropriate number of generations.
- Endpoint Sampling: Harvest cells at the endpoint (T1) for genomic DNA extraction [52] [60].
Stage 3: Sequencing and Analysis
- gRNA Amplification: Amplify gRNA sequences from genomic DNA using PCR with barcoded primers for multiplexing.
- High-Throughput Sequencing: Sequence amplified fragments with sufficient depth (>100 reads per gRNA minimum).
- Read Alignment and Counting: Align sequences to the gRNA library reference and count reads for each gRNA in T0 and T1 samples.
- Statistical Analysis: Normalize counts, calculate fold changes, and identify significantly enriched/depleted gRNAs using appropriate statistical methods [60].
CRISPRi Screening Workflow
Multi-Omics Integration for Enhanced Interpretation
Integrating CRISPRi screening data with other omics datasets provides a more comprehensive understanding of gene function and network relationships:
- Transcriptomic Correlation: Compare CRISPRi hit genes with differentially expressed genes from RNA-seq under similar conditions.
- Proteomic Validation: Verify protein-level changes for top hits using targeted proteomics.
- Metabolomic Context: Interpret gene essentiality within the context of metabolic pathway usage through integration with metabolomics data [62].
Validation Strategies
Secondary Assay Design
- Individual Knockdown Validation: Clone individual gRNAs targeting hit genes and quantify repression efficiency using qRT-PCR.
- Phenotypic Confirmation: Assess growth defects or relevant phenotypes in individual knockout strains.
- Complementation Testing: Express a repression-resistant version of the target gene to confirm phenotype specificity [60].
Orthogonal Validation Approaches
- Chemical Inhibition: Where possible, use specific small-molecule inhibitors of hit gene products to confirm phenotypes.
- Traditional Genetic Approaches: Compare CRISPRi results with transposon mutagenesis or gene deletion studies.
- Cross-Species Validation: Leverage conservation of essential genes across related bacterial species [61].
Effective troubleshooting of CRISPRi screens in bacterial systems requires careful attention to system design, implementation quality control, and appropriate data interpretation frameworks. By addressing common pitfalls in gRNA design, repressor selection, and experimental execution, researchers can significantly enhance the reliability and reproducibility of their CRISPRi screening outcomes. The continued development of optimized repressor domains, improved computational tools, and multi-omics integration approaches will further strengthen the utility of CRISPRi technology for bacterial research and therapeutic development.
CRISPRi vs. Other Technologies: Validation, Advantages, and Strategic Selection
{*}
Benchmarking Repression Efficiency: CRISPRi vs. RNA Interference (RNAi) and TALENs
Within bacterial research, the precise repression of gene expression is a cornerstone for functional genomics and metabolic engineering. The development of robust transcriptional repression tools has been vital for probing essential genes, understanding pathogenesis, and optimizing industrial strains. This whitepaper benchmarks three primary technologies for gene repression: CRISPR interference (CRISPRi), RNA interference (RNAi), and Transcription Activator-Like Effector Nucleases (TALENs). While RNAi has been a traditional method for gene knockdown and TALENs can be repurposed for repression, the newer CRISPRi system offers a distinct mechanism and several advantages in a prokaryotic context [63] [6]. It is critical to note that RNAi machinery is predominantly active in eukaryotes and is largely ineffective in most bacteria, limiting its direct application in bacterial studies without significant modification [63] [6]. This analysis focuses on the mechanistic and practical attributes of these tools to guide researchers in selecting the optimal system for their investigations in bacterial systems.
Mechanisms of Action
Understanding the fundamental mechanisms of each technology is key to appreciating their applications and limitations. The following diagram illustrates the core mechanistic pathways for CRISPRi, RNAi, and TALEN-based repression.
CRISPR Interference (CRISPRi)
CRISPRi is an emerging technology that exploits a catalytically inactive Cas9 (dCas9) protein and a single-guide RNA (sgRNA) to repress sequence-specific genes in bacteria [6]. The dCas9 protein, carrying mutations (e.g., D10A and H840A in SpCas9) that abolish its nuclease activity, retains its ability to bind DNA based on sgRNA complementarity [6]. The dCas9-sgRNA complex binds to the target DNA at the promoter or the coding sequence and acts as a physical roadblock to the elongating RNA polymerase (RNAP), thereby aborting transcription initiation or elongation [6]. The system is programmable, highly efficient, and specific. Furthermore, by fusing dCas9 to potent transcriptional repressor domains like Krüppel-associated box (KRAB), the repression efficiency can be significantly enhanced, as these domains recruit chromatin-modifying complexes to induce a repressive state, even in the absence of steric occlusion [9]. Recent advances have led to novel repressor fusions such as dCas9-ZIM3(KRAB)-MeCP2(t), which show improved gene repression across various targets and cell types [9].
RNA Interference (RNAi)
RNAi is an evolutionarily conserved endogenous pathway in eukaryotic cells that regulates gene expression post-transcriptionally [63] [64]. The process can be hijacked by introducing synthetic small RNAs, such as short-interfering RNAs (siRNAs) or short-hairpin RNAs (shRNAs) [63]. These exogenous RNAs are loaded into the RNA-induced silencing complex (RISC), which then promotes the degradation of perfectly complementary target mRNA or blocks its translation [63] [64]. A crucial limitation for bacterial researchers is that the RNAi machinery is not naturally present in most prokaryotes, confining its conventional application to eukaryotic systems [6]. While the primary function of RNAi is to regulate gene expression, it can also confer resistance to viral infections in eukaryotes [64].
TALEN Repressors
TALENs are artificial proteins that consist of a Transcription Activator-Like Effector (TALE) DNA-binding domain fused to a FokI nuclease domain [63] [65]. For gene repression (rather than cleavage), the nuclease domain can be omitted, and the TALE DNA-binding domain can be fused directly to a transcriptional repressor domain like KRAB [63]. This creates a TALE-repressor that binds to the transcription start site (TSS) and prevents transcription in the nucleus [63]. The TALE domain is composed of 33-35 amino acid repeats, with two amino acids (Repeat-Variable Diresidue, or RVD) in each repeat determining nucleotide specificity (e.g., NG for T, NI for A, HD for C, and NN for G) [63] [66]. This modularity allows for the design of proteins that can target specific DNA sequences. However, for each new target site, a new, relatively large (~500-700 amino acids) protein must be designed and cloned, which can be a lengthy process [63].
Quantitative Benchmarking of Repression Technologies
The following table summarizes the key performance characteristics of CRISPRi, RNAi, and TALEN-based repression systems, with a focus on data relevant to bacterial and mammalian cell applications.
Table 1: Performance Benchmarking of Gene Repression Technologies
| Feature | CRISPRi | RNAi | TALEN Repressor |
|---|---|---|---|
| Repression Level | Knockdown (Tunable) [67] [6] | Knockdown (Hypomorphic) [63] [65] | Knockdown (Hypomorphic) [63] |
| Max Repression Efficiency | Up to 45-fold (tunable systems) [67] & >90% (novel repressors) [9] | Varies; often incomplete [63] | High, but less efficient than CRISPRi [63] |
| Mechanism of Action | Transcriptional repression via dCas9 steric hindrance and/or repressor domains [6] [9] | Post-transcriptional mRNA degradation or translational inhibition [63] [64] | Transcriptional repression via recruiter repressor domains [63] |
| Target | DNA [6] | mRNA (Cytoplasmic) [63] | DNA [63] |
| Efficiency in Bacteria | High (Endogenous system repurposed) [6] | Not applicable (Machinery absent) [6] | Possible, but delivery is challenging [68] |
| Off-Target Effects | Moderate to Low (Sequence-specific) [6] [64] | High (Sequence-dependent and independent) [63] [64] | Low (High specificity DNA-binding protein) [63] [68] |
| Key Advantage | Programmable, tunable, reversible, and highly efficient in bacteria [67] [6] | Simple transient knockdown in eukaryotes; rapid setup [65] [64] | High specificity; lower off-targets than CRISPR [68] |
| Key Disadvantage | PAM sequence requirement; potential for off-target binding [66] [6] | High off-target effects; ineffective for nuclear transcripts and bacteria [63] [6] | Complex, time-consuming protein design and cloning [63] [68] |
Experimental Workflows
A successful repression experiment requires careful planning and execution. The workflows for implementing CRISPRi, RNAi, and TALENs differ significantly in their complexity and requirements, as outlined below.
Detailed CRISPRi Protocol for Bacterial Research
The following protocol provides a step-by-step methodology for implementing a CRISPRi system in E. coli, as supported by recent literature [67] [6].
Strain and Plasmid Preparation
- Bacterial Strains: Commonly used laboratory strains like E. coli Mach1-T1R or NEB 5-alpha can be used for cloning and screening. For final experiments, use the target strain of interest [67].
- Plasmid System: A dual-plasmid system is often preferred for simplicity and stability. One plasmid carries the gene for the dCas9-repressor fusion (e.g., dCas9-KOX1(KRAB) or more advanced variants like dCas9-ZIM3(KRAB)-MeCP2(t)), under the control of an inducible promoter (e.g., anhydrotetracycline (aTc)-inducible promoter). The second, smaller plasmid carries the sgRNA expression cassette [6] [9]. Alternatively, a single plasmid system or chromosomal integration of dCas9 can be employed [6].
sgRNA Design and Library Cloning
- Target Selection: Identify a target site within the promoter or the 5' coding sequence (CDS) of the gene of interest. The target must be adjacent to a Protospacer Adjacent Motif (PAM). For the commonly used SpCas9, the PAM is 5'-NGG-3' [67] [6].
- sgRNA Design: Design the sgRNA spacer sequence (typically 20 nucleotides) to be complementary to the target DNA. To achieve tunable repression, a library of sgRNAs with mutations in the tetraloop and flanking regions can be constructed to modulate binding affinity to dCas9, resulting in varying levels of repression efficiency [67].
- Library Construction: Synthesize the sgRNA library oligos and clone them into the sgRNA expression plasmid using high-fidelity DNA polymerase (e.g., Q5 High-Fidelity) and appropriate restriction enzymes or Gibson assembly [67].
Transformation and Culture
- Transformation: Co-transform the dCas9-repressor plasmid and the sgRNA plasmid into the target bacterial strain. If using an inducible system, include the appropriate antibiotic in the culture media for plasmid selection [67] [6].
- Culture Conditions: Inoculate overnight cultures and refresh the media the next day to an OD600 of approximately 0.05. Incubate until the OD600 reaches 0.6-0.8. To induce repression, add the inducer (e.g., 20 ng/ml aTc) to the culture and continue incubation for a set period (e.g., 4-6 hours) [67].
Validation and Analysis
- Transcript Quantification: Extract total RNA using a commercial kit (e.g., RNeasy Mini Kit) with on-column DNA digestion. Perform RT-qPCR using a one-step kit (e.g., Luna Universal One-Step RT-qPCR Kit). Use a stable reference gene (e.g., cysG in E. coli) for relative quantification via the delta-delta-Ct method [67].
- Phenotypic Assays: Measure the resulting phenotype, such as the production of a metabolite (e.g., violacein or lycopene) or growth in a particular condition, to correlate gene repression with functional output [67].
- High-Throughput Screening: For genome-wide screens, use fluorescence-activated cell sorting (FACS) to sort cells based on reporter fluorescence (e.g., GFP) linked to repression efficiency. Sequence the sgRNAs from sorted populations to identify hits [67] [9].
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs the key reagents and their functions required for establishing a CRISPRi system in a bacterial context.
Table 2: Essential Reagents for Bacterial CRISPRi Research
| Reagent / Solution | Function / Description | Example Products / Components |
|---|---|---|
| dCas9-Repressor Plasmid | Expresses the nuclease-dead Cas9 fused to transcriptional repressor domain(s). The backbone of the innovation in modern CRISPRi. | dCas9-ZIM3(KRAB)-MeCP2(t) [9]; dCas9-KOX1(KRAB) [6] |
| sgRNA Expression Plasmid | A small plasmid expressing the single-guide RNA that targets the dCas9-repressor complex to specific DNA loci. | High-copy plasmid with a constitutive promoter [6] |
| sgRNA Library | A pooled collection of sgRNA constructs targeting multiple genes or designed for tunable repression. | Synthesized oligo library cloned into the sgRNA plasmid backbone [67] |
| Inducer Molecule | A small molecule that triggers the expression of dCas9 or sgRNA from inducible promoters, allowing temporal control. | Anhydrotetracycline (aTc) for Tet promoters [67] [6] |
| High-Fidelity Polymerase | Used for error-free amplification of DNA fragments during plasmid construction. | Q5 High-Fidelity DNA Polymerase [67] |
| RNA Extraction Kit | For purifying high-quality total RNA from bacterial cells for downstream transcriptomic analysis. | RNeasy Mini Kit (Qiagen) [67] |
| One-Step RT-qPCR Kit | Enables quantitative analysis of mRNA transcript levels in a single tube, crucial for validation. | Luna Universal One-Step RT-qPCR Kit [67] |
Applications in Bacterial Research and Future Perspectives
CRISPRi has rapidly become an indispensable tool for functional genomics and metabolic engineering in bacteria. Its primary application lies in high-throughput genetic screening to identify essential genes and map genetic interactions [6]. By using genome-scale sgRNA libraries, researchers can systematically repress every gene in a bacterium and identify those critical for growth under various conditions or during infection. Furthermore, CRISPRi is exceptionally powerful in metabolic engineering for redistributing cellular flux. For example, it has been used to tune the expression of multiple genes in a pathway to optimize the production of valuable chemicals like n-butanol, lycopene, and violacein derivatives without accumulating toxic intermediates [67] [6]. The ability to partially repress essential genes also allows for the study of their functions without being lethal to the cell [6].
The future of CRISPRi is bright, with ongoing innovations focusing on enhancing repression efficiency and specificity. The development of novel repressor domains, as demonstrated with the dCas9-ZIM3(KRAB)-MeCP2(t) fusion, points toward a trend of engineering more potent and consistent effectors [9]. The compatibility of these systems with delivery technologies like lipid nanoparticles (LNPs), as shown in mammalian studies, hints at future therapeutic applications, including targeting bacterial infections or modulating the microbiome [58]. As the toolset expands, CRISPRi is poised to remain at the forefront of bacterial genetic research, offering an unprecedented level of control over gene expression.
Comparative Analysis of Specificity and Off-Target Profiles
The advent of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology has revolutionized genetic engineering, with CRISPR interference (CRISPRi) emerging as a particularly powerful tool for precise transcriptional repression in bacteria. Unlike nuclease-active CRISPR-Cas9 systems that introduce double-strand breaks (DSBs), CRISPRi utilizes a catalytically dead Cas9 (dCas9) that binds target DNA without causing cleavage, thereby repressing transcription through steric hindrance of RNA polymerase (RNAP) [6]. This mechanism offers significant advantages for functional genomics studies, especially for investigating essential genes where knockout would be lethal. However, the specificity of CRISPRi and its potential for off-target effects remain critical considerations for research and therapeutic development. This technical analysis examines the specificity parameters of CRISPRi within the broader landscape of CRISPR-based technologies, providing a comprehensive framework for assessing and mitigating off-target effects in bacterial systems.
Mechanisms of CRISPRi and Specificity Determinants
Fundamental Mechanism of Transcriptional Repression
CRISPRi functions through a programmable ribonucleoprotein complex consisting of dCas9 and a single-guide RNA (sgRNA). The sgRNA directs dCas9 to specific DNA sequences via Watson-Crick base pairing, where it binds without cleaving the DNA backbone. This binding physically blocks the progression of RNAP along the template, effectively repressing transcription initiation or elongation [6]. The system requires the presence of a protospacer adjacent motif (PAM) adjacent to the target site; for the commonly used Streptococcus pyogenes Cas9, this PAM is 5'-NGG-3' [10]. The specificity of CRISPRi is inherently high because it relies on two simultaneous recognition events: sgRNA-DNA base pairing and dCas9-PAM interaction.
Diagram: CRISPRi Repression Mechanism in Bacteria
Figure 1: CRISPRi transcriptional repression mechanism. The dCas9-sgRNA complex binds target DNA via complementary base pairing, creating a physical barrier that blocks RNA polymerase progression and leads to gene repression.
Key Factors Governing CRISPRi Specificity
The precision of CRISPRi is governed by multiple interdependent factors. The PAM recognition requirement serves as the initial specificity gatekeeper, restricting potential binding sites to sequences adjacent to appropriate motifs [10]. The sgRNA-DNA complementarity, particularly in the 10-12 nucleotide "seed region" proximal to the PAM, is crucial for stable binding; mismatches in this region significantly reduce binding affinity [69]. Cellular context including chromatin structure, DNA accessibility, and repair pathway activity further influences binding specificity and functional outcomes [70]. Unlike nuclease-active CRISPR systems that induce complex DNA damage responses including non-homologous end joining (NHEJ) and homology-directed repair (HDR), CRISPRi minimizes these confounding genotoxic stresses, potentially reducing indirect off-target effects associated with DNA repair processes [71] [6].
Comparative Analysis of Off-Target Effects Across CRISPR Systems
Spectrum of Genomic Alterations in Nuclease-Active CRISPR vs. CRISPRi
Nuclease-active CRISPR systems introduce double-strand breaks that can lead to diverse unintended genomic consequences beyond simple point mutations. Recent studies reveal that these systems can generate large structural variations (SVs) including kilobase- to megabase-scale deletions, chromosomal translocations, and chromothripsis [71]. Particularly concerning is the finding that strategies to enhance homology-directed repair (HDR), such as DNA-PKcs inhibitors, can exacerbate these aberrations, with one study reporting a thousand-fold increase in translocation frequency [71]. These SVs raise substantial safety concerns for clinical applications, as they can disrupt tumor suppressor genes or activate oncogenes. In contrast, CRISPRi significantly reduces this risk profile by eliminating the induction of DSBs altogether, instead creating reversible, titratable gene repression without permanent DNA alterations [6].
Quantitative Comparison of Off-Target Frequencies
Table 1: Comparative Analysis of Off-Target Profiles Across CRISPR Technologies
| Technology | Primary Mechanism | Common Off-Target Effects | Reported Off-Target Frequencies | Key Influencing Factors |
|---|---|---|---|---|
| CRISPRi (dCas9) | Steric blockage of transcription | Non-specific binding; partial repression at homologous sites | Minimal observed off-target repression in E. coli [10] | sgRNA design; PAM specificity; cellular delivery efficiency |
| Nuclease-Active Cas9 | DNA double-strand break induction | Small indels; large deletions (>1kb); chromosomal translocations | Off-target sites with up to 6 mismatches reported [70] [69]; Megabase-scale deletions in 5-20% of alleles [71] | DNA repair pathway modulation; sgRNA specificity; chromatin state |
| High-Fidelity Cas9 Variants | Engineered DNA cleavage with reduced off-target activity | Reduced but not eliminated off-target cleavage | ~10-100 fold reduction in off-target activity while maintaining on-target efficiency [72] | Protein engineering strategy; PAM restrictions; guide specificity |
| Base Editors | Chemical conversion of specific nucleotides without DSBs | Off-target DNA/RNA editing; bystander edits | Varies by editor; can be limited with optimization [70] | Editor architecture; delivery method; cellular context |
Methodologies for Off-Target Detection and Validation
Experimental Approaches for Comprehensive Off-Target Assessment
Accurate assessment of off-target activity requires complementary methodological approaches. Biochemical methods including DIGENOME-seq, CIRCLE-seq, and CHANGE-seq utilize purified genomic DNA incubated with Cas9-sgRNA complexes to map potential cleavage sites in vitro, offering high sensitivity but lacking cellular context [69] [73]. Cellular methods such as GUIDE-seq, DISCOVER-seq, and HTGTS detect editing events within living cells, capturing the influences of chromatin structure and DNA repair pathways but requiring efficient delivery systems [70] [72] [73]. In silico prediction tools including Cas-OFFinder, CCTop, and CFD scoring provide initial off-target risk assessment based on sequence homology but may miss biologically relevant sites affected by cellular context [70] [69].
Table 2: Experimental Methods for Off-Target Detection in CRISPR Studies
| Method | Principle | Sensitivity | Advantages | Limitations |
|---|---|---|---|---|
| GUIDE-seq | Captures double-stranded oligodeoxynucleotides into DSB sites | High sensitivity; low false positive rate | Detects off-targets in native chromatin context | Requires efficient dsODN delivery; potential cellular toxicity |
| DIGENOME-seq | Whole-genome sequencing of Cas9-digested genomic DNA | Moderate; requires high sequencing depth | Works with any reference genome; no special reagents | Cell-free system may overestimate cleavage; lacks biological context |
| CIRCLE-seq | Circularization of sheared DNA followed by Cas9 digestion and sequencing | High sensitivity; lower sequencing depth needed | Enhanced detection of rare off-target sites | In vitro conditions may not reflect cellular environment |
| DISCOVER-seq | Relies on MRE11 recruitment to DSBs via ChIP-seq | High precision in cells | Identifies biologically relevant off-target edits; works in primary cells | May have false positives; requires specific antibodies |
| BLESS/BLISS | Direct in situ labeling of DSB ends | Moderate; limited by labeling efficiency | Preserves nuclear architecture; captures breaks in situ | Technically complex; lower throughput |
Experimental Protocol for CRISPRi Off-Target Assessment in Bacteria
For researchers implementing CRISPRi in bacterial systems, the following protocol provides a comprehensive approach to assess specificity:
Stage 1: sgRNA Design and Specificity Validation
- Identify target sequences following established design rules: 20-nt base pairing region, NGG PAM, and positioning within the non-template DNA strand of the coding region for optimal repression efficiency [10]
- Conduct BLAST analysis of the sgRNA base pairing region against the host genome to identify potential off-target sites with high sequence similarity
- Evaluate predicted RNA secondary structure using tools like ViennaRNA to ensure the dCas9 binding handle remains accessible [10]
Stage 2: Plasmid Construction and Transformation
- Employ a dual-plasmid system with dCas9 and sgRNA on separate vectors to simplify cloning and improve stability [6]
- For single-sgRNA cloning, utilize inverse PCR with target-specific primers containing the 20-nt base pairing region
- For multiplexed repression, apply Golden Gate Cloning to assemble multiple sgRNA expression cassettes into a single vector [10]
- Transform constructs into the target bacterial strain (e.g., E. coli K12-strain MG1655) with appropriate antibiotic selection
Stage 3: Specificity Validation and Off-Target Assessment
- Induce dCas9 expression with anhydrotetracycline and quantify repression of the target gene via RT-qPCR
- Assess potential off-target effects by measuring expression at the top 5-10 computationally predicted off-target loci
- For genome-wide screening, employ RNA-seq to transcriptome profile edited cells versus controls
- Validate putative off-target hits through targeted sequencing of affected regions
Diagram: CRISPRi Off-Target Assessment Workflow
Figure 2: Comprehensive workflow for assessing CRISPRi specificity and off-target effects in bacterial systems, integrating computational prediction with experimental validation.
Table 3: Key Research Reagents for CRISPRi Studies in Bacteria
| Reagent/Resource | Function | Example Sources/Identifiers |
|---|---|---|
| dCas9 Expression Plasmid | Catalytically inactive Cas9 for transcriptional repression | Addgene ID no. 44249 (aTc-inducible) [10] |
| sgRNA Expression Vector | Guide RNA delivery system | Addgene ID no. 44251 (constitutive promoter J23119) [10] |
| MCP-SoxS Fusion Protein | Transcriptional activator for CRISPRa applications | Custom construction required [26] |
| Anhydrotetracycline | Inducer for PLTetO-1 promoter controlling dCas9 | Commercially available (e.g., Clontech) [10] |
| Bacterial sgRNA Library | Pooled guides for high-throughput screening | Custom design and synthesis [6] |
| CAST-Seq/LAM-HTGTS Kits | Detection of structural variations and translocations | Commercial kits available [71] |
CRISPRi represents a significant advancement in bacterial genetic engineering, offering superior specificity compared to nuclease-active CRISPR systems while enabling reversible, titratable control of gene expression. The absence of double-strand breaks eliminates the risk of large structural variations and chromosomal rearrangements that pose substantial safety concerns in therapeutic applications. However, comprehensive off-target assessment remains essential, particularly as CRISPR-based technologies advance toward clinical applications. Future developments will likely focus on enhancing specificity through improved sgRNA design algorithms, engineered dCas9 variants with refined PAM requirements, and more sensitive detection methodologies that capture the full spectrum of potential off-target effects. For the bacterial research community, the continued refinement of CRISPRi specificity will enable more precise functional genomics studies and accelerate the development of synthetic biology applications in industrial and therapeutic contexts.
CRISPR interference (CRISPRi) has emerged as a powerful, programmable tool for transcriptional repression in bacterial functional genomics. This technology utilizes a catalytically inactive Cas9 (dCas9) that binds to target DNA sequences specified by a single-guide RNA (sgRNA) without introducing double-strand breaks. The bound dCas9 functions as a steric block to RNA polymerase, resulting in targeted and reversible repression of gene expression [74]. Within bacterial research, particularly for studying commensal and probiotic species, CRISPRi overcomes long-standing barriers to genetic manipulation, including restrictive transformation efficiencies and the presence of diverse restriction-modification systems that traditionally hindered functional studies [74]. This guide details the application of CRISPRi for mapping gene essentiality and genetic interactions, providing a framework for mechanistic studies in a wide range of bacterial species.
Core Mechanism and System Components
Molecular Basis of Transcriptional Repression
The CRISPRi system functions through the formation of a ribonucleoprotein complex where the dCas9 protein is directed to a specific genomic locus by its associated sgRNA. When the sgRNA-dCas9 complex binds to the non-template strand of a target gene, it creates a physical roadblock that prevents transcription elongation by the RNA polymerase [74]. The repression efficiency is influenced by several factors, including the binding location within the promoter or open reading frame, the concentration of the dCas9-sgRNA complex, and the accessibility of the target DNA. This mechanism allows for fine-tuned, sequence-specific knockdown of gene expression rather than complete knockout, enabling the study of essential genes that would be lethal if completely inactivated.
Essential Research Reagents
Successful implementation of CRISPRi requires several core components, each with a specific function as outlined in the table below.
Table 1: Essential Research Reagents for CRISPRi Experiments
| Reagent | Function | Examples & Specifications |
|---|---|---|
| dCas9 Ortholog | Catalytic core providing DNA-binding function; lacks nuclease activity. | Commonly used: dCas9 from Streptococcus thermophilus [74]. |
| Single-Guide RNA (sgRNA) | Specifies genomic target via 20-nucleotide spacer sequence complementary to target DNA. | Designed with 20nt guide sequence complementary to the non-template strand of the gene of interest [74]. |
| Expression System | Delivers genetic components into the bacterial cell. | Single-plasmid systems are preferred for ease of transformation [74]. |
| Selection Marker | Enables maintenance of the CRISPRi system in the bacterial population. | Antibiotic resistance genes (e.g., erythromycin, chloramphenicol) [74]. |
Experimental Workflows and Protocols
Implementing a CRISPRi System in Bacteria
The following diagram illustrates the key steps for establishing a functional CRISPRi system in a bacterial strain, from vector design to phenotypic analysis.
Diagram 1: CRISPRi System Workflow
To establish a functional CRISPRi system in a target bacterium, researchers must first clone the dCas9 gene and an sgRNA expression cassette into an appropriate expression vector. A single-plasmid system is often preferred for simplicity [74]. The dCas9 should be codon-optimized for the host species, and the sgRNA should be designed to target the non-template strand of the gene of interest. The plasmid is then introduced into the target bacterium via transformation. For bifidobacteria and other anaerobes, this requires specialized protocols, including growth in anaerobic chambers and the use of electroporation with cell wall weakening pre-treatments [74]. Following transformation, successful dCas9 expression and sgRNA function must be validated. This is typically confirmed through reverse transcription quantitative PCR (RT-qPCR) to measure transcript levels of the target gene and a control gene. Repression is then induced, often via a chemical inducer like anhydrotetracycline, depending on the promoter system. Finally, the functional consequence of gene repression is measured through growth assays, microscopy, or other phenotypic readouts.
Genome-Scale Essentiality Screening
For genome-wide mapping of essential genes, a pooled screening approach is highly effective. The process involves creating a comprehensive library of sgRNAs targeting nearly every gene in the genome. A high-density sgRNA library is crucial for high-resolution results, as it allows for multiple, independent sgRNAs per gene, reducing false positives and negatives from inefficient guides [75]. The following protocol outlines the key steps:
- Library Design and Cloning: A library of sgRNAs is designed to target a wide range of genes. For a high-resolution screen in E. coli, this can involve synthesizing a complex pool of oligos that are then cloned into a CRISPRi plasmid backbone [75].
- Library Transformation: The pooled sgRNA plasmid library is transformed into a bacterial population already expressing dCas9. The transformation must yield a high enough diversity to ensure representation of all sgRNAs in the final pool.
- Selection Under Stress: The transformed population is divided and grown under two conditions: a permissive control condition and a selective condition, such as in the presence of a sub-lethal concentration of an antibiotic [75].
- Sequencing and Analysis: Genomic DNA is harvested from populations before and after selection. The sgRNA inserts are amplified and sequenced via next-generation sequencing. sgRNA abundance is compared between conditions; sgRNAs that become depleted under selective pressure target genes essential for survival under that stress [75].
Table 2: CRISPRi Screening Parameters from Select Bacterial Studies
| Parameter | E. coli Antibiotic Resistance Screening [75] | Bifidobacterium Species Screening [74] |
|---|---|---|
| Primary Goal | Identify genes essential for antibiotic tolerance and resistance. | Repress genes in nucleotide and carbohydrate metabolism. |
| SgRNA Library Scale | Genome-scale, high-density. | Focused on specific pathways. |
| Phenotyping Method | Growth fitness under antibiotic stress. | Growth assays on different carbon sources. |
| Key Finding | Identified essential membrane proteins and transcriptional modulators in tolerance. | Achieved gene repression without extensive optimization across species. |
Delineating Genetic Interactions with Dual CRISPRi
Genetic interactions, such as synthetic lethality, can be systematically mapped using dual CRISPRi systems that express two sgRNAs simultaneously. This approach allows for the repression of two genes in the same cell, revealing non-additive effects on fitness [76]. The workflow, termed Dual CRISPRi-seq, involves the following steps:
- Dual sgRNA Library Design: A library of paired sgRNAs is generated, targeting combinations of genes hypothesized to have functional relationships.
- Library Delivery: This dual sgRNA library is introduced into a bacterial population expressing dCas9.
- Competitive Growth Assay: The population is grown competitively, and samples are taken at the beginning and end of the experiment.
- Sequencing and Genetic Interaction Score: The abundance of each dual-sgRNA construct is quantified by sequencing. A genetic interaction score is calculated for each gene pair by comparing the observed fitness effect to the expected effect based on the single repressions [76].
The following diagram visualizes the conceptual process of identifying a synthetic lethal genetic interaction using Dual CRISPRi.
Diagram 2: Synthetic Lethality Concept
Key Applications and Technical Considerations
Application 1: Investigating Antibiotic Resistance Mechanisms
CRISPRi screens have proven invaluable for dissecting complex bacterial responses to antibiotics. A genome-scale screen in E. coli exposed to various antibiotics successfully identified both known and previously unrecognized genes involved in resistance mechanisms [75]. This approach is particularly powerful because it can highlight the importance of transcriptional modulation of essential genes in antibiotic tolerance, revealing potential new targets for antimicrobial strategies [75]. The high-resolution nature of these screens allows for the rapid identification of key resistance genes, providing a valuable resource for understanding the genetic fitness landscape under antibiotic stress.
Application 2: Functional Genomics in Probiotic Species
CRISPRi has removed a significant bottleneck in the functional characterization of commercially important but genetically recalcitrant bacteria, such as bifidobacteria. A one-plasmid CRISPRi system based on a dCas9 from Streptococcus thermophilus has been shown to function across multiple Bifidobacterium species, including B. breve, B. animalis, and B. longum subsp. infantis [74]. This system enabled the repression of genes involved in carbohydrate metabolism and exopolysaccharide production, paving the way for mechanistic studies of probiotic functions. The key advantage is that this was achieved without the need for laborious optimization to bypass restriction-modification systems, which has traditionally been a major barrier in this genus [74].
Technical Considerations and Best Practices
- sgRNA Design: sgRNAs should be designed to target the non-template strand within the promoter or early in the coding sequence for optimal repression efficiency. It is critical to use multiple sgRNAs per gene to control for variation in guide efficiency.
- Control Experiments: Always include non-targeting sgRNAs as negative controls and sgRNAs targeting essential genes as positive controls for repression in your screen.
- dCas9 Expression Level: The level of dCas9 expression must be optimized. Too little may result in inefficient repression, while too much can lead to off-target effects and toxicity.
- Multi-sgRNA Systems: For dual CRISPRi, ensure the expression system reliably co-expresses both sgRNAs. The use of distinct promoters or a single promoter with a tRNA processing system can be effective [76].
CRISPRi technology provides a robust and flexible platform for functional validation in bacteria, enabling the systematic mapping of gene essentiality and genetic interactions. Its application ranges from fundamental investigations of bacterial physiology in model organisms like E. coli to unlocking the functional genomics of commercially significant but understudied probiotics like bifidobacteria. The continued development of streamlined systems, such as all-in-one plasmids, and sophisticated screening methods, like Dual CRISPRi-seq, will further empower researchers to decipher complex genetic networks and mechanisms underlying antibiotic resistance and probiotic functionality, accelerating both basic science and drug discovery.
CRISPR interference (CRISPRi) has emerged as a powerful and programmable technology for precise transcriptional repression in bacterial systems. This technology leverages a catalytically inactive Cas9 (dCas9) protein, which retains its DNA-binding capability but cannot cleave target DNA [6]. When guided to specific genomic loci by a single-guide RNA (sgRNA), the dCas9-sgRNA complex functions as a molecular roadblock, physically impeding the progression of RNA polymerase (RNAP) and thereby repressing transcription [7] [6]. The simplicity of this system—requiring only a single protein and a customizable guide RNA for targeted gene repression—has revolutionized functional genomics in bacteria [77].
In Escherichia coli, CRISPRi demonstrates remarkably high repression efficiency, achieving up to ~300-fold repression with minimal off-target effects [77]. This efficiency, combined with its inherent programmability, makes CRISPRi particularly valuable for probing gene function, essential gene analysis, and metabolic engineering. Unlike CRISPR nuclease systems that create double-strand breaks—which are typically lethal in bacteria due to inefficient non-homologous end joining (NHEJ) repair—CRISPRi offers a reversible and non-destructive means of gene perturbation [7]. This key distinction enables researchers to study essential genes whose complete disruption would be lethal to the bacterial cell, opening new avenues for investigating fundamental cellular processes and identifying novel antibiotic targets [7] [6].
Core Mechanisms and Key Advantages
The exceptional utility of CRISPRi in bacterial research stems from three fundamental advantages: its reversible and inducible nature, its capacity for titratable repression, and its unique ability to target non-coding genomic regions. These characteristics collectively provide researchers with an unprecedented level of precision in transcriptional control.
Reversibility and Inducibility
CRISPRi-mediated repression is inherently reversible, allowing for dynamic and temporal control of gene expression. This reversibility is primarily achieved by placing the expression of dCas9 and/or the sgRNA under the control of inducible promoters [7] [6].
- Mechanism of Reversibility: When the inducer is removed from the culture medium, the expression of the CRISPRi components ceases. Existing dCas9-sgRNA complexes are gradually diluted as cells divide, and transcription of the target gene resumes once the repression complex is no longer present at the target site [6]. This enables researchers to study the functional consequences of transient gene knockdown and monitor cellular recovery after repression.
- Experimental Implementation: Common inducible systems include the anhydrotetracycline (aTc)-inducible promoter and the arabinose-inducible PBAD promoter [7] [6]. For example, Li et al. (2016) achieved over two orders of magnitude dynamic range in repression by controlling dCas9 expression with the PBAD promoter [6]. This inducible control allows researchers to initiate repression at specific time points during bacterial growth, facilitating the study of essential genes required at different growth phases.
The reversible nature of CRISPRi makes it particularly valuable for studying essential bacterial processes where permanent gene knockout would be lethal, enabling functional analysis of genes involved in cell division, metabolism, and central dogma processes [7].
Titratable and Tunable Repression
A significant advantage of CRISPRi over complete gene knockout is the ability to achieve partial or graded repression, allowing researchers to study dose-dependent gene effects and mimic hypomorphic alleles.
- Titration Methods: CRISPRi repression levels can be precisely tuned through multiple strategies:
- Inducer Concentration: Using sub-saturating concentrations of inducer (e.g., varying aTc or arabinose levels) to modulate dCas9/sgRNA expression [7].
- Guide RNA Engineering: Employing truncated sgRNAs or introducing mismatches between the sgRNA and target DNA to reduce binding efficiency and create hypomorphic repression [7].
- Promoter Strength: Utilizing constitutive promoters of varying strengths to establish gradients of CRISPRi component expression [7].
- Practical Applications: Titratable repression is crucial for studying essential genes where complete repression would be lethal. It also allows for fine-tuning metabolic pathways in engineering applications, where balanced expression of multiple genes is required for optimal product yield [6]. For instance, Fontana et al. (2018) demonstrated a broad range of titration by controlling gRNA expression from a Ptet promoter in E. coli [6].
Table 1: Methods for Titrating CRISPRi Repression Levels in Bacteria
| Method | Mechanism | Dynamic Range | Applications |
|---|---|---|---|
| Inducer Titration | Varying concentration of inducer (e.g., aTc, arabinose) to control dCas9/sgRNA expression | >100-fold [6] | Temporal studies; essential gene analysis |
| sgRNA Engineering | Using truncated sgRNAs or mismatched guides to reduce binding efficiency | Variable depending on design [7] | Creating hypomorphic alleles; pathway tuning |
| Promoter Selection | Employing constitutive promoters of different strengths | Dependent on promoter collection [7] | Metabolic engineering; continuous cultures |
Targeting Non-Coding Genomic Regions
While much of traditional bacterial genetics has focused on protein-coding genes, CRISPRi provides a powerful tool for functionally characterizing the vast non-coding regions of bacterial genomes, including regulatory elements and non-coding RNAs.
- Promoter and Operator Targeting: By designing sgRNAs to target promoter regions or transcription factor binding sites, CRISPRi can competitively inhibit the binding of RNA polymerase or activators, effectively reducing transcription initiation [7] [6]. This approach allows researchers to dissect regulatory elements and study their contribution to gene expression control.
- Non-Coding RNA Studies: CRISPRi enables functional screening of non-coding RNAs, including small RNAs (sRNAs) and as-yet unannotated transcriptional units [78]. While most large-scale non-coding CRISPRi screens have been conducted in mammalian cells [39] [78], the same principles apply to bacterial systems. For example, the ability to target transcription start sites with high precision (within ~1 kb) allows specific perturbation of non-coding transcripts without affecting adjacent genes [78].
- Functional Genomics: CRISPRi can be scaled for high-throughput screening of non-coding elements, enabling systematic functional annotation of bacterial non-coding genomes [79]. This approach is particularly valuable for identifying regulatory elements that control virulence, antibiotic resistance, or metabolic adaptations in bacterial pathogens.
The combination of these three advantages—reversibility, titratability, and non-coding targeting capability—makes CRISPRi an exceptionally versatile tool for bacterial genetics, enabling research questions that were previously difficult or impossible to address with traditional knockout approaches.
Experimental Design and Implementation
CRISPRi System Configuration
Successful implementation of CRISPRi in bacteria requires careful consideration of system configuration and delivery. The two primary approaches are plasmid-based systems and chromosomal integration systems, each with distinct advantages.
Table 2: Comparison of CRISPRi Delivery Systems in Bacteria
| System Type | Components | Advantages | Limitations | Ideal Use Cases |
|---|---|---|---|---|
| Single-Plasmid System | dCas9 and sgRNA expression cassettes on a single vector | Simplified transformation; stable maintenance [6] | Large plasmid size may reduce cloning efficiency [6] | Single-gene repression; routine laboratory use |
| Dual-Plasmid System | dCas9 and sgRNA on separate plasmids | Simplified cloning; modular sgRNA replacement [6] | Potential plasmid incompatibility; requires multiple antibiotics [6] | High-throughput screening; multiplexed repression |
| Chromosomally Integrated System | dCas9 integrated at neutral site; sgRNA on plasmid | Reduced metabolic burden; stable dCas9 expression [6] | Requires genome engineering; less flexible for dCas9 variants [6] | Industrial applications; long-term studies |
For high-throughput screening applications, the chromosomally integrated dCas9 system combined with plasmid-based sgRNA libraries has become the gold standard, as it minimizes system loss and ensures consistent repression across a large population of cells [6].
Guide RNA Design and Optimization
The specificity and efficiency of CRISPRi repression are largely determined by guide RNA design. Optimal sgRNA design for bacterial CRISPRi follows several key principles:
- Target Site Selection: For transcriptional repression, sgRNAs should be designed to target the non-template strand within the promoter region or the early coding sequence (5' end) of the gene [7] [6]. Targeting the promoter region typically blocks transcription initiation, while targeting the coding sequence impedes transcription elongation.
- PAM Consideration: The Protospacer Adjacent Motif (PAM) requirement varies depending on the Cas9 ortholog used. The most commonly used Streptococcus pyogenes Cas9 (SpCas9) recognizes a 5'-NGG-3' PAM sequence [6]. The PAM must be present immediately adjacent to the target site for successful dCas9 binding.
- Specificity Verification: Potential off-target binding sites should be identified by searching for genomic sequences with significant similarity to the sgRNA spacer sequence, particularly in promoter regions of non-target genes. Mismatches in the seed region (PAM-proximal 8-12 nucleotides) are most critical for preventing off-target binding [6].
- Efficiency Prediction: While empirical testing remains the most reliable approach, computational tools can help predict sgRNA efficiency based on factors such as GC content, nucleotide composition, and genomic context [80].
For high-throughput applications, library design typically includes 3-10 sgRNAs per target to account for variable efficiency and ensure robust phenotypic detection [78].
Protocol for Inducible CRISPRi Repression in E. coli
The following protocol provides a detailed methodology for establishing inducible CRISPRi repression in Escherichia coli, adaptable to other bacterial species with appropriate modifications.
Day 1: Strain Preparation
- Transform competent cells expressing dCas9 (from a plasmid or chromosomal integration) with the sgRNA plasmid or library. Include appropriate antibiotic selection.
- Plate transformed cells on LB agar with appropriate antibiotics and incubate overnight at 37°C.
Day 2: Culture Inoculation
- Pick single colonies and inoculate 2-5 mL of LB medium with antibiotics.
- Incubate overnight at 37°C with shaking (200-250 rpm).
Day 3: Induction and Sampling
- Dilute overnight culture 1:100 in fresh medium with antibiotics.
- Divide culture into two flasks: one for induced repression and one as an uninduced control.
- Add inducer (e.g., aTc, arabinose, or IPTG) at predetermined concentration to the induced culture. Add equivalent volume of solvent to the control.
- Incubate with shaking at 37°C and monitor growth (OD600) periodically.
- Harvest samples at appropriate time points for downstream analysis (e.g., RNA extraction for qRT-PCR, protein extraction for Western blotting, or phenotypic assays).
Day 4: Assessment and Validation
- Analyze repression efficiency using qRT-PCR to measure transcript levels or other relevant phenotypic assays.
- For reversibility studies, wash cells to remove inducer and resuspend in fresh medium without inducer. Monitor recovery of gene expression over time.
This protocol can be adapted for high-throughput screening by performing these steps in multi-well plates and using pooled sgRNA libraries, with sgRNA abundance monitored by next-generation sequencing before and after selection [78].
Technical Considerations and Troubleshooting
While CRISPRi is a powerful tool, researchers should be aware of several technical considerations and potential pitfalls when implementing this technology in bacterial systems.
- Polar Effects: In bacteria, CRISPRi knockdown of upstream genes in an operon can cause polar effects on downstream genes [7]. Additionally, some systems exhibit "reverse polarity," where targeting downstream genes affects upstream gene expression, with severity varying by bacterial species [7].
- dCas9 and sgRNA Toxicity: High levels of dCas9 expression can be toxic in some bacterial strains [7] [6]. Similarly, certain sgRNA sequences can cause "bad seed" effects, leading to non-specific toxicity [7]. These issues can be mitigated by titrating expression levels and using multiple sgRNAs per target.
- Escape Mutants: When targeting essential genes, suppressor mutants may arise that inactivate the CRISPRi system [7]. Using freshly transformed strains and including appropriate controls can help identify and mitigate this issue.
- Variable Efficiency: Repression efficiency can vary based on chromosomal position, local chromatin state (in some bacteria), and transcription rate [6]. Testing multiple sgRNAs per target is recommended for critical applications.
Research Reagent Solutions
Table 3: Essential Research Reagents for Bacterial CRISPRi Studies
| Reagent / Tool | Function | Examples / Specifications | Key Considerations |
|---|---|---|---|
| dCas9 Variants | DNA-binding effector for repression | dCas9 (D10A, H840A mutations) [6]; dCas12a [81] | Orthologs with different PAM requirements expand targeting range |
| Guide RNA | Targets dCas9 to specific genomic loci | Synthetic sgRNA; plasmid-encoded sgRNA [80] | High-purity synthesis improves reproducibility [80] |
| Inducible Promoters | Controls timing and level of CRISPRi component expression | PBAD, Ptet, PaTc-inducible systems [7] [6] | Leaky expression should be characterized for each system |
| Delivery Vectors | Plasmid systems for CRISPRi component expression | Single-plasmid, dual-plasmid, or integrated systems [6] | Choice affects cloning efficiency and metabolic burden |
| Selection Markers | Maintains CRISPRi components in bacterial population | Antibiotic resistance genes (e.g., AmpR, KanR, CmR) | Should be compatible with host strain and experimental conditions |
| sgRNA Libraries | Pooled guides for high-throughput screening | Arrayed or pooled formats [78] | Library design should include controls and multiple guides/target |
Visualization of CRISPRi Workflows
Core CRISPRi Mechanism in Bacteria
Diagram 1: CRISPRi creates a roadblock that prevents transcription.
Inducible and Reversible CRISPRi System
Diagram 2: Inducible systems enable temporal control of repression.
Applications in Non-Coding Region Targeting
Diagram 3: CRISPRi enables functional screening of non-coding elements.
CRISPRi technology represents a transformative approach for bacterial genetics, offering unique capabilities that extend far beyond traditional gene knockout methods. The reversibility and inducibility of CRISPRi enable dynamic studies of gene function, while the ability to achieve titratable repression allows for fine dissection of dose-dependent phenotypes. Perhaps most significantly, the capacity to target non-coding genomic regions opens new frontiers in understanding bacterial regulatory networks.
These advantages make CRISPRi particularly valuable for functional genomics, essential gene analysis, metabolic engineering, and bacterial pathogenesis studies. As the field advances, ongoing optimization of CRISPRi systems—including the development of novel Cas orthologs with diverse PAM specificities, improved guide RNA designs, and enhanced delivery systems—will further expand its applications in bacterial research.
For researchers implementing CRISPRi, careful attention to system design, guide RNA selection, and appropriate controls will ensure robust and interpretable results. The protocols and considerations outlined in this technical guide provide a foundation for successful application of CRISPRi to diverse research questions in bacteriology.
Integrating CRISPRi with Other Omics Data for Robust Biological Validation
CRISPR interference (CRISPRi) has emerged as a powerful technology for programmable transcriptional repression in bacterial systems. This technical guide provides a comprehensive framework for integrating CRISPRi screening data with multi-omics datasets to achieve robust biological validation. We detail experimental protocols for implementing CRISPRi in bacteria, outline computational approaches for multi-omics integration, and present key case studies demonstrating how this synergistic approach can elucidate complex gene regulatory networks, validate gene function at scale, and accelerate therapeutic discovery. The methodologies described herein provide researchers with a standardized pipeline for maximizing the biological insights gained from CRISPRi experiments through systematic multi-omics validation.
CRISPRi technology repurposes components of the native bacterial Type II CRISPR-Cas immune system for targeted gene repression without altering DNA sequences. The system requires two fundamental components: a catalytically dead Cas9 (dCas9) protein that binds DNA without cleaving it, and a single guide RNA (sgRNA) that directs dCas9 to specific genomic loci through Watson-Crick base pairing [3]. In bacterial cells, the dCas9-sgRNA complex achieves transcriptional repression through two primary mechanisms:
- Transcription Elongation Block: When targeted to the protein-coding region of a gene, particularly the nontemplate DNA strand, the dCas9-sgRNA complex creates a steric barrier that physically blocks the progressing RNA polymerase, leading to aborted transcription [3].
- Transcription Initiation Inhibition: When targeted to promoter regions, including RNA polymerase binding sites (e.g., -35 or -10 boxes) or transcription factor binding sites, the complex prevents the assembly of essential transcription machinery, thereby inhibiting transcription initiation [3].
The targeting specificity of the CRISPRi system is jointly determined by the 20-nucleotide complementary region of the sgRNA and a short protospacer adjacent motif (PAM) sequence immediately following the target site. For the commonly used Streptococcus pyogenes Cas9, the PAM sequence is NGG (where N is any nucleotide), though some recognition of NAG PAMs has been reported [3]. This requirement places some constraint on targetable sites within bacterial genomes but maintains high specificity when properly designed.
Experimental Design and Implementation of CRISPRi in Bacteria
sgRNA Design Considerations for Bacterial Systems
Effective CRISPRi implementation begins with strategic sgRNA design. The following parameters must be considered for optimal repression efficiency:
- Target Strand Selection: For elongation blocking, sgRNAs must target the nontemplate DNA strand of the coding region. Targeting the template strand typically results in significantly weaker repression (~50% efficiency) [10].
- Target Position: For protein-coding genes, positioning the sgRNA target closer to the 5' end of the gene (just downstream of the transcription start site) generally yields stronger repression by blocking elongation early in transcription [3] [10].
- PAM Availability: The target must be immediately adjacent to an appropriate PAM sequence (NGG for S. pyogenes Cas9). The targetable sites are therefore restricted to 20-nt regions 5' to NGG in the genome [10].
- Genomic Specificity: The sgRNA base pairing region should be unique within the genome to avoid off-target effects. Bioinformatics tools (e.g., BLAST) should be used to ensure no identical 20-nt sequences with adjacent PAM sites exist elsewhere in the genome [3] [10].
- Folding Quality: The complete sgRNA sequence should be checked for secondary structure that might interfere with dCas9 binding. RNA folding algorithms (e.g., Vienna suite) can predict potential structural issues [10].
Table 1: Key Considerations for Bacterial CRISPRi sgRNA Design
| Design Parameter | Optimal Configuration | Rationale |
|---|---|---|
| Target Strand | Nontemplate strand | Effective blockage of RNA polymerase elongation |
| Target Region | Coding sequence near 5' end or promoter elements | Early transcription blockade or prevention of initiation complex formation |
| PAM Requirement | NGG adjacent to target site | Essential for dCas9 recognition and binding |
| Genomic Specificity | Unique 20-nt sequence with no off-target sites | Prevents unintended repression of non-target genes |
| Target Length | 20 nucleotides | Balance of specificity and binding efficiency |
Molecular Cloning and Delivery Protocols
The implementation of CRISPRi in bacteria typically involves plasmid-based expression of both dCas9 and sgRNA components. Below we outline a standardized protocol for E. coli, which can be adapted for other bacterial species:
Materials Required:
- dCas9 expression plasmid (e.g., Addgene ID #44249 with chloramphenicol resistance)
- sgRNA expression plasmid (e.g., Addgene ID #44251 with ampicillin resistance)
- Target bacterial strain (e.g., E. coli K12-strain MG1655)
- Primers for sgRNA cloning
- PCR purification and gel extraction kits
- Restriction enzymes and ligase
- Competent cells for transformation
- LB media with appropriate antibiotics
Protocol for Single-sgRNA Cloning Using Inverse PCR [10]:
sgRNA Insert Preparation:
- Design forward primer containing the 20-nt target-specific sequence (5'-N20 GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGC-3' for Addgene #44251)
- Use universal reverse primer (5'-ACTAGTATTATACCTAGGACTGAGCTAGC-3')
- Perform PCR amplification of sgRNA scaffold template
- Treat PCR product with DpnI to digest template DNA
- Purify PCR product using gel extraction kit
Plasmid Assembly:
- Phosphorylate and anneal oligonucleotides using T4 polynucleotide kinase
- Ligate into linearized sgRNA expression vector using Quick Ligase
- Transform into competent E. coli cells (e.g., TOP10 strain)
- Plate on LB agar with 100 μg/mL ampicillin
- Screen colonies for correct inserts by colony PCR or sequencing
Dual Plasmid Transformation:
- Co-transform validated sgRNA plasmid with dCas9 expression plasmid into target bacterial strain
- Select on LB agar containing both ampicillin (sgRNA plasmid) and chloramphenicol (dCas9 plasmid)
- Verify successful transformation by colony PCR and sequencing
Induction of CRISPRi:
- Culture transformed bacteria in appropriate media with antibiotics
- Induce dCas9 expression with anhydrotetracycline (if using inducible promoter)
- Monitor repression efficiency after 16-24 hours of induction
For multiplexed repression, multiple sgRNA expression cassettes can be assembled into a single vector using Golden Gate cloning with type IIS restriction enzymes (e.g., BsaI) [10].
Multi-Omics Integration Frameworks for CRISPRi Validation
Transcriptomic Integration Strategies
Integration of CRISPRi with transcriptomic profiling provides a direct readout of repression efficiency and secondary effects. Multiple approaches can be employed:
Bulk RNA Sequencing:
- Apply after CRISPRi-mediated repression (typically 24-48 hours post-induction in bacteria)
- Identify differentially expressed genes beyond the immediate target
- Assess specificity by evaluating off-target transcriptional effects
- Protocol: Extract RNA using commercial kits (e.g., RNeasy), prepare libraries with reverse transcription (e.g., Superscript III), and sequence on appropriate platform [10]
Single-Cell RNA Sequencing (scRNA-seq) with CRISPRi Perturbation:
- Enables resolution of heterogeneous responses to gene repression
- Recently adapted for bacterial systems through modified CROP-seq vectors
- Allows simultaneous capture of sgRNA identity and transcriptome in individual cells
- Protocol: Implement combinatorial indexing strategies (e.g., PerturbSci) that incorporate cell barcoding during reverse transcription [82]
Native Elongating Transcript Sequencing (NET-seq):
- Provides nucleotide-resolution mapping of RNA polymerase positions
- Directly validates transcription elongation blocks by CRISPRi
- Reveals precise locations of dCas9-induced transcription termination [3]
Table 2: Transcriptomic Methods for CRISPRi Validation
| Method | Resolution | Key Applications | Considerations |
|---|---|---|---|
| Bulk RNA-seq | Population average | Genome-wide differential expression, off-target effects | Masks cellular heterogeneity |
| scRNA-seq with Perturbation | Single-cell | Heterogeneous responses, complex regulatory networks | Technical noise, higher cost |
| NET-seq | Nucleotide | Transcription elongation dynamics, precise blockade mapping | Specialized protocol, lower throughput |
| 4sU Labeling (Nascent RNA) | Time-resolved | RNA kinetics, synthesis and degradation rates | Metabolic labeling, chemical conversion required |
Proteomic and Epigenomic Integration
Beyond transcriptomics, integrating proteomic and epigenomic data provides complementary validation of CRISPRi effects:
Proteomic Approaches:
- Mass spectrometry-based proteomics quantifies protein-level changes following CRISPRi
- Validates that transcriptional repression translates to functional protein reduction
- Reveals post-transcriptional compensation mechanisms
- Can be combined with pulsed stable isotope labeling with amino acids (pSILAC) for dynamic measurements
Epigenomic Profiling:
- Chromatin immunoprecipitation followed by sequencing (ChIP-seq) for histone modifications
- Assay for Transposase-Accessible Chromatin with sequencing (ATAC-seq) maps chromatin accessibility changes
- Validates indirect epigenetic consequences of targeted repression
- Identifies compensatory regulatory mechanisms activated upon perturbation
Multi-Omics Factor Analysis (MOFA+):
- Statistical framework for integrating multiple omics datasets
- Identifies latent factors that capture shared variation across data modalities
- Enables systems-level understanding of CRISPRi-induced perturbations [83]
Computational Analysis Pipelines for Integrated Data
Essential Bioinformatics Tools
Robust computational analysis is crucial for interpreting integrated CRISPRi-omics data. The following tools and approaches are recommended:
sgRNA Quantification and Quality Control:
- For bulk screens: MAGeCK for identifying essential genes and enriched/depleted sgRNAs [84]
- For single-cell screens: CROP-seq analysis pipeline for linking cellular transcriptomes to perturbations
- Quality metrics: sgRNA representation rate (>99% in initial library), read distribution, and correlation between replicates
Differential Expression Analysis:
- Standard tools: DESeq2, edgeR, or limma for bulk RNA-seq data
- Single-cell methods: Seurat, SCANPY, or Monocle3 for scRNA-seq data
- Specialized perturbation tools: Mixscape for distinguishing direct from indirect effects
Multi-Omics Integration Algorithms:
- MOFA+: Integrates multiple omics data types to identify latent factors [83]
- LIGER: Aligns datasets and identifies shared and dataset-specific patterns
- Schema: Specifically designed for CRISPR screen integration with transcriptomic data
Functional Enrichment Analysis:
- Gene set enrichment analysis (GSEA) for pathway-level interpretation
- Enrichr or clusterProfiler for ontology term enrichment
- Network visualization tools: Cytoscape for displaying regulatory networks
Data Visualization Approaches
Effective visualization is essential for communicating integrated CRISPRi-omics results:
- Uniform Manifold Approximation and Projection (UMAP): Visualizes single-cell transcriptomes colored by sgRNA identity [82]
- Volcano Plots: Highlights significant differentially expressed genes following repression
- Heatmaps: Displays expression patterns across multiple conditions and genes
- Genome Browser Tracks: Integrates CRISPRi targeting with epigenomic features and transcription data
- Network Graphs: Illustrates gene regulatory networks inferred from perturbation responses
Case Studies in Bacterial Systems
Metabolic Pathway Engineering
CRISPRi coupled with metabolomics has enabled precise optimization of bacterial metabolic pathways for industrial biosynthesis. In one application, researchers used multiplexed CRISPRi to systematically repress competing metabolic pathways in E. coli, while integrating LC-MS metabolomics to quantify pathway intermediates and products. This approach identified optimal repression patterns that increased target metabolite production by 3.7-fold compared to wild-type strains [26].
Bacterial Pathogenesis Studies
In studies of bacterial pathogenesis, CRISPRi has been integrated with dual RNA-seq (simultaneous sequencing of bacterial and host RNA) to dissect host-pathogen interactions. Repression of specific virulence factors combined with host transcriptome profiling revealed novel immune evasion mechanisms and identified host pathways targeted by bacterial effectors.
Genetic Network Mapping
Large-scale CRISPRi screens targeting transcription factors combined with RNA-seq have enabled comprehensive mapping of bacterial regulatory networks. By measuring the transcriptomic consequences of repressing individual regulators, researchers have reconstructed hierarchical regulatory networks and identified master regulators of bacterial stress responses, antibiotic resistance, and biofilm formation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for CRISPRi-Omics Integration in Bacteria
| Reagent/Resource | Function | Example Sources/IDs |
|---|---|---|
| dCas9 Expression Plasmid | Catalytically dead Cas9 for targeted binding | Addgene #44249 (E. coli) |
| sgRNA Expression Vector | Scaffold for target-specific sgRNAs | Addgene #44251 |
| sgRNA Library Cloning System | Multiplexed sgRNA expression | Golden Gate Assembly with BsaI [10] |
| RNA Purification Kit | High-quality RNA for transcriptomics | RNeasy Kit (Qiagen) |
| cDNA Synthesis Kit | Library preparation for RNA-seq | Superscript III System |
| Single-Cell Library Prep Kit | scRNA-seq with perturbation capture | PerturbSci protocol [82] |
| Analysis Software | CRISPR screen analysis | MAGeCK, CASA [84] [39] |
| Reference Genome | Alignment and annotation | NCBI RefSeq databases |
Workflow Visualization
The integration of CRISPRi with multi-omics technologies represents a powerful paradigm for biological validation in bacterial systems. This approach moves beyond simple gene repression to provide comprehensive insights into regulatory networks, compensation mechanisms, and system-wide responses to perturbation. As single-cell technologies advance and computational integration methods become more sophisticated, the resolution and scope of CRISPRi-omics will continue to expand. Emerging applications in bacterial consortia engineering, antibiotic development, and synthetic biology will further leverage these integrated approaches to address complex biological questions and develop novel biotechnological solutions.
Conclusion
CRISPRi has firmly established itself as a transformative technology for precise transcriptional control in bacteria. Its core mechanism of programmable steric blockade provides a reversible and tunable means to dissect gene function with high specificity, overcoming key limitations of older technologies like RNAi. The development of optimized systems, including novel repressor fusions and PAM-flexible Cas proteins, continues to enhance its robustness and applicability for genome-wide screens. As we look forward, the integration of CRISPRi with other technologies, such as CRISPRa for simultaneous activation, paves the way for complex metabolic engineering and the systematic deconvolution of intricate genetic networks. For biomedical and clinical research, the ability to perform high-throughput functional genomics in bacterial pathogens with CRISPRi promises to accelerate the discovery of new drug targets and innovative therapeutic strategies, solidifying its role as an indispensable tool in the modern molecular biology toolkit.
References
- 1. CRISPR/dCas9-Based Systems: Mechanisms and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8540305/]
- 2. CRISPR interference [https://en.wikipedia.org/wiki/CRISPR_interference]
- 3. CRISPR interference (CRISPRi) for sequence-specific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3922765/]
- 4. CRISPR Guide [https://www.addgene.org/guides/crispr/]
- 5. dCAS9 (Dead CAS system): Concept, Functions and ... [https://geneticeducation.co.in/dcas9-dead-cas-system-concept-functions-and-applications/]
- 6. Gene Silencing Through CRISPR Interference in Bacteria [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2021.635227/full]
- 7. CRISPRi functional genomics in bacteria and its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11332340/]
- 8. Metabolically-targeted dCas9 expression in bacteria [https://pubmed.ncbi.nlm.nih.gov/36629257/]
- 9. Engineering novel CRISPRi repressors for highly efficient ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03640-4]
- 10. Targeted Transcriptional Repression in Bacteria Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4845913/]
- 11. Genome-scale CRISPRi and base-editing libraries for ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925004123]
- 12. Cas9 Nuclease: Structure, Mechanism & Genome Editing [https://lifesciences.danaher.com/us/en/library/cas9-nuclease.html]
- 13. The Complete Guide to Understanding CRISPR sgRNA [https://www.synthego.com/guide/how-to-use-crispr/sgrna/]
- 14. Optimizing sgRNA structure to improve CRISPR-Cas9 ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0846-3]
- 15. Optimizing sgRNA position markedly improves the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5062975/]
- 16. Importance of the PAM Sequence in CRISPR Experiments [https://www.synthego.com/guide/how-to-use-crispr/pam-sequence/]
- 17. PAM identification by CRISPR-Cas effector complexes [https://pmc.ncbi.nlm.nih.gov/articles/PMC6546366/]
- 18. What Is the PAM Sequence for CRISPR and Where Is It? | IDT [https://www.idtdna.com/pages/support/faqs/what-is-a-pam-sequence-and-where-is-it-located]
- 19. Unlock CRISPR Gene Editing Potential: PAM sequences [https://www.idtdna.com/page/support-and-education/decoded-plus/crispr-cas-nucleases-understanding-pam-requirements-and-cas-diversification-strategies/]
- 20. Gene Silencing Through CRISPR Interference in Bacteria : Current ... [https://pubmed.ncbi.nlm.nih.gov/33868193/]
- 21. Improved prediction of bacterial guide CRISPRi from... efficiency [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03153-y]
- 22. CRISPRi-ART enables functional genomics of diverse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11879866/]
- 23. CrisprVi: a software for visualizing and analyzing CRISPR ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04716-9]
- 24. Chromosomal and Plasmid-Based CRISPRi Platforms for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12524933/]
- 25. CRISPR/Cas9 therapeutics: progress and prospects [https://www.nature.com/articles/s41392-023-01309-7]
- 26. Synthetic CRISPR-Cas gene activators for transcriptional ... [https://www.nature.com/articles/s41467-018-04901-6]
- 27. Computational pipeline for designing guide RNAs ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8121773/]
- 28. How to Design Your gRNA for CRISPR Genome Editing [https://blog.addgene.org/how-to-design-your-grna-for-crispr-genome-editing]
- 29. Simulations predict stronger CRISPRi transcriptional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12007490/]
- 30. An updated evolutionary classification of CRISPR–Cas ... [https://www.nature.com/articles/s41564-025-02180-8]
- 31. Mismatch-CRISPRi reveals the co-varying expression-fitness ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7704046/]
- 32. Titrating gene expression using libraries of systematically ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7065968/]
- 33. Inducible promoters of bacterial microcompartments ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12016505/]
- 34. Genome-Scale CRISPR-Mediated Control of Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4253859/]
- 35. Maximizing CRISPRi efficacy and accessibility with dual- ... [https://elifesciences.org/articles/81856]
- 36. Advancing Mycobacterial Research with CRISPR Technologies [https://scisimple.com/en/articles/2025-07-10-advancing-mycobacterial-research-with-crispr-technologies--a3ox4jo]
- 37. CRISPRi-Library-Guided Target Identification for Engineering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8064071/]
- 38. CRISPR/dCas9-based metabolic pathway engineering for ... [https://www.sciencedirect.com/science/article/pii/S002203022200337X]
- 39. Multicenter integrated analysis of noncoding CRISPRi ... [https://www.nature.com/articles/s41592-024-02216-7]
- 40. High throughput single-cell detection of multiplex CRISPR ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02174-1]
- 41. Off-target interactions in the CRISPR-Cas9 Machinery [https://pubmed.ncbi.nlm.nih.gov/40688512/]
- 42. Off-target interactions in the CRISPR-Cas9 Machinery [https://www.sciencedirect.com/science/article/pii/S2405580825002213]
- 43. Application of CRISPR-Cas9 in microbial cell factories [https://pubmed.ncbi.nlm.nih.gov/40259107/]
- 44. Optimizing sgRNA position markedly improves the ... [https://pubmed.ncbi.nlm.nih.gov/27353328/]
- 45. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 46. CMN Weekly (31 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-31-october-2025-your-weekly-crispr-medicine-news/]
- 47. A Next-Generation Platform for Highly Optimized CRISPR- ... [https://www.sciencedirect.com/science/article/abs/pii/S0168165625002391]
- 48. Quantitative essentiality in a reduced genome: a functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12494982/]
- 49. CRISPR-Cas tools for simultaneous transcription & ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11109947/]
- 50. CRISPRi screen identifies FprB as a synergistic target for ... [https://www.nature.com/articles/s41467-025-61208-z]
- 51. CRISPR 技术- 创新基因组学研究所(IGI) [https://innovativegenomics.org/zh-CN/%E6%B8%85%E8%84%86%E7%99%BE%E7%A7%91/%E4%BF%9D%E9%B2%9C%E6%8A%80%E6%9C%AF/]
- 52. Dual-mode CRISPRa/i for genome-scale metabolic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12370623/]
- 53. High-Throughput Screens of PAM-Flexible Cas9 Variants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7558435/]
- 54. Engineering a PAM-flexible SpdCas9 variant as a ... [https://www.nature.com/articles/s41467-021-27290-9]
- 55. Tuning a high performing multiplexed-CRISPRi ... [https://www.sciencedirect.com/science/article/pii/S2214030122000153]
- 56. Development of PAM-flexible multiplex genome editors for ... [https://www.sciencedirect.com/science/article/pii/S2214514125000571]
- 57. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 58. CMN Weekly (3 October 2025) [https://crisprmedicinenews.com/news/cmn-weekly-3-october-2025-your-weekly-crispr-medicine-news/]
- 59. Development of a CRISPR activation system for targeted ... [https://www.nature.com/articles/s42003-025-08164-y]
- 60. How to Accurately Interpret CRISPR Screening Data [https://www.cd-genomics.com/biomedical-ngs/resource/crispr-screening-data-interpretation-genomics.html]
- 61. Benchmarking genetic interaction scoring methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12464814/]
- 62. 2025 and Beyond: The Future of Genomic Data Analysis ... [https://blog.crownbio.com/2025-and-beyond-the-future-of-genomic-data-analysis-and-innovations-in-genomics-services]
- 63. Choosing the Right Tool for the Job: RNAi , TALEN or CRISPR - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4441801/]
- 64. RNAi vs. CRISPR: Guide to Selecting the Best Gene ... [https://www.synthego.com/blog/rnai-vs-crispr-guide/]
- 65. CRISPR Cas9 - Gene Silencing Methods: CRISPR vs . TALEN . vs RNAi [https://info.abmgood.com/crispr-cas9-talens-rnai-gene-silencing]
- 66. Precision gene editing: The power of CRISPR-Cas in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590234/]
- 67. CRISPRi-mediated tunable control of gene expression level ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10201414/]
- 68. Gene Editing Techniques: ZFNs, TALENs or CRISPR? [https://www.news-medical.net/life-sciences/Gene-Editing-Techniques-ZFNs-TALENs-or-CRISPR.aspx]
- 69. Methods for detecting off-target effects of CRISPR/Cas9 [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002368]
- 70. Off-target effects in CRISPR/Cas9 gene editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10034092/]
- 71. The hidden risks of CRISPR/Cas: structural variations and ... [https://www.nature.com/articles/s41467-025-62606-z]
- 72. Methods for Optimizing CRISPR-Cas9 Genome Editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4976696/]
- 73. BLOG: Selecting the Right Gene Editing Off-Target Assay [https://seqwell.com/guide-to-selecting-right-gene-editing-off-target-assay/]
- 74. A CRISPRi Gene Regulation System for Bifidobacteria - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12592239/]
- 75. Rapid identification of key antibiotic resistance genes in E. ... [https://pubmed.ncbi.nlm.nih.gov/40352728/]
- 76. Dual CRISPRi-seq for genome-wide genetic interaction ... [https://www.sciencedirect.com/science/article/pii/S2405471225002418]
- 77. Targeted Transcriptional in Repression Using CRISPR... Bacteria [https://pubmed.ncbi.nlm.nih.gov/25981485/]
- 78. CRISPRi-based genome-scale identification of functional long ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5394926/]
- 79. Decoding the noncoding genome via large-scale CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6252135/]
- 80. High Purity Guide RNA for CRISPR Screening Applications [https://www.technologynetworks.com/genomics/white-papers/reproducible-guide-rna-screening-for-crispr-applications-405171]
- 81. Modular and signal-responsive transcriptional regulation using... [https://jbioleng.biomedcentral.com/articles/10.1186/s13036-025-00526-8]
- 82. Dissecting key regulators of transcriptome kinetics through ... [https://www.nature.com/articles/s41587-023-01948-9]
- 83. A Single-Cell Transcriptomics CRISPR-Activation Screen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7383230/]
- 84. A multi-omics integrative analysis based on CRISPR ... [https://www.nature.com/articles/s42003-023-04700-w]