Constructing Next-Generation Multiplexed CRISPRi Libraries for Comprehensive Pathway Discovery
This article provides a comprehensive guide for researchers and drug development professionals on the construction and application of multiplexed CRISPR interference (CRISPRi) libraries for systematic pathway discovery.
Constructing Next-Generation Multiplexed CRISPRi Libraries for Comprehensive Pathway Discovery
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on the construction and application of multiplexed CRISPR interference (CRISPRi) libraries for systematic pathway discovery. It covers the foundational principles of dCas9-based gene repression, detailing the latest advancements in highly active, compact library design, including dual-sgRNA strategies and optimized effectors like Zim3-dCas9. The content explores methodological workflows from gRNA selection and vector design to delivery and screening in diverse cell models. It further addresses critical troubleshooting for off-target effects and cytotoxicity, and concludes with rigorous validation frameworks and comparative analyses against CRISPRn and CRISPRa, establishing multiplexed CRISPRi as a premier tool for functional genomics and therapeutic target identification.
The Core Principles of Multiplexed CRISPRi for Systematic Gene Perturbation
Clustered Regularly Interspaced Short Palindromic Reference interference (CRISPRi) has emerged as a transformative technology for functional genomics, offering distinct advantages over traditional CRISPR nuclease approaches for pathway-level analysis. This technical guide examines the core mechanisms of CRISPRi, its application in multiplexed library screening, and its growing importance in drug discovery and microbial engineering. We present quantitative comparisons, detailed methodologies, and specialized visualization to equip researchers with the framework necessary to implement CRISPRi for comprehensive pathway discovery.
Traditional CRISPR-Cas9 nuclease editing permanently disrupts gene function through double-strand breaks and subsequent non-homologous end joining (NHEJ) repair, which often introduces insertion/deletion (indel) mutations [1] [2]. While effective for complete gene knockouts, this approach presents limitations for pathway-level interrogation: essential gene knockouts are lethal, preventing study of their function, and the binary nature of knockouts poorly mimics the partial inhibition characteristic of most pharmaceutical treatments [3] [4].
CRISPRi addresses these limitations by enabling reversible, tunable gene repression without permanent DNA alteration. By utilizing a catalytically dead Cas9 (dCas9) fused to repressive domains, CRISPRi blocks transcription through steric hindrance of RNA polymerase [3]. This technology has become particularly valuable for studying essential genes, complex genetic networks, and for creating more accurate models of therapeutic intervention in drug discovery pipelines [5] [3].
Core Mechanisms: CRISPR Nuclease vs. CRISPR Interference
Fundamental Operational Differences
The fundamental distinction between CRISPR nuclease and CRISPRi lies in the Cas9 protein and its operational outcome. CRISPR nuclease utilizes an active Cas9 that creates double-strand breaks in DNA, leading to permanent gene knockout via error-prone NHEJ repair. In contrast, CRISPRi employs a catalytically dead Cas9 (dCas9) with inactivated nuclease domains that binds DNA without cutting it, thereby physically obstructing transcription machinery [3] [2].
Table 1: Core Functional Differences Between CRISPR Nuclease and CRISPRi
| Feature | CRISPR Nuclease | CRISPRi |
|---|---|---|
| Cas9 Type | Wild-type (active) | dCas9 (catalytically dead) |
| DNA Cleavage | Yes (double-strand breaks) | No |
| Genetic Outcome | Permanent knockout (indels) | Reversible knockdown |
| Mechanism | NHEJ/HDR repair | Steric hindrance of RNA polymerase |
| Effect on Essential Genes | Lethal | Enables study of dosage effects |
| Therapeutic Mimicry | Poor (complete elimination) | Good (partial inhibition) |
CRISPRi Molecular Architecture
In prokaryotes, dCas9 alone can achieve effective repression by blocking RNA polymerase binding or progression. In mammalian systems, enhanced repression is achieved by fusing dCas9 to repressor domains such as the Kruppel associated box (KRAB), which recruits additional chromatin-modifying complexes to silence gene expression in an inducible, reversible, and non-toxic manner [3]. Guide RNAs are typically designed to target the template strand within -50 to +300 bp relative to the transcription start site (TSS), though optimal positioning varies by organism [6].
Key Advantages for Pathway-Level Interrogation
Studying Essential Genes and Dosage Sensitivity
CRISPRi enables the systematic discovery of haploinsufficient and dosage-sensitive genes that cannot be studied with conventional knockouts. By partially repressing essential genes rather than eliminating them entirely, researchers can investigate the effects of gene dosage on cellular fitness and pathway function [6]. This approach has been successfully employed in yeast to identify haploinsufficient genes through simple outgrowth experiments, demonstrating how graded reduction of gene expression can reveal fitness defects not observable with all-or-nothing knockout approaches [6].
Superior Modeling of Pharmacological Interventions
Most therapeutic drugs achieve partial rather than complete inhibition of their targets. CRISPRi better mimics this clinical reality by generating hypomorphic alleles that titrate gene expression levels, creating more biologically relevant models for drug discovery [3] [2]. This partial repression capability allows researchers to model drug action more accurately than complete knockouts and identify genes that confer resistance or sensitivity to chemical compounds when partially inhibited [3].
Reduced Off-Target Effects Compared to Alternative Technologies
CRISPRi demonstrates higher specificity than RNA interference (RNAi), the previous gold standard for gene knockdown. While RNAi suffers from both sequence-independent and sequence-dependent off-target effects that can trigger interferon responses and silence unintended targets, CRISPRi exhibits far fewer off-target effects due to more precise guide RNA design and validation tools [2]. This enhanced specificity reduces phenotypic noise in pathway screens, increasing confidence in genotype-phenotype relationships.
Tunable and Reversible Modulation
Unlike permanent knockouts, CRISPRi enables reversible modulation of gene expression through inducible systems. The tetracycline-regulated expression system allows controlled timing and duration of repression [6]. This tunability extends beyond simple on/off states – recent advances enable precise control of repression levels through engineered sgRNA variants that modulate binding affinity against dCas9, achieving transcriptional regulation at various levels between fully-repressing and non-repressing states with greater than 45-fold dynamic range [7].
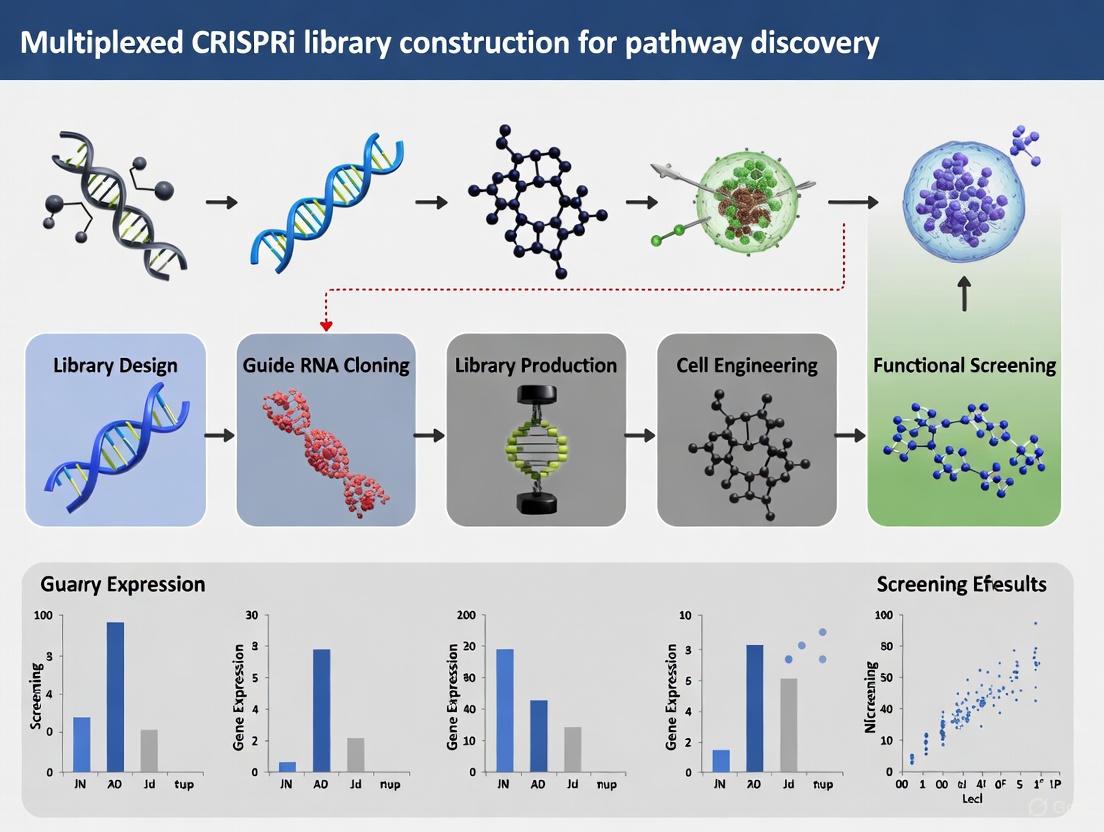
Multiplexed CRISPRi Library Construction for Pathway Discovery
Library Design Considerations
Effective CRISPRi library design requires organism-specific parameter optimization. In Saccharomyces cerevisiae, the optimal targeting window is a 200 bp region immediately upstream of the transcription start site, with particular efficacy in the region between TSS and 125 bp upstream [6]. Design must account for nucleosome occupancy, nucleotide features, and potential for cross-talk between adjacent genes sharing intergenic regions [6].
Essential quality control metrics include:
- Specificity Validation: Filtering sgRNAs with potential off-target effects using strict thresholds (e.g., 20% of on-target score for exonic regions)
- Efficiency Prediction: Calculating "on-target" scores using cutting frequency determination (CFD) and discarding sgRNAs scoring below 0.8
- Coverage Requirements: Designing 6-12 sgRNAs per gene to ensure adequate targeting [6] [8]
Advanced Multiplexing Strategies
Randomized Array Assembly (MuRCiS)
The Multiplexed, Randomized CRISPRi Sequencing (MuRCiS) approach enables comprehensive interrogation of genetic combinations by self-assembling CRISPR arrays from synthetic oligonucleotide pairs. This method uses "R-S-R" (repeat-spacer-repeat) building blocks composed of a 24 bp spacer flanked by partial repeat sequences with complementary overhangs that ligate randomly, creating diverse arrays capable of targeting multiple gene combinations simultaneously [9] [10]. This randomized assembly is particularly valuable for identifying synthetic lethal combinations among redundant gene families that escape detection when targeting predetermined gene sets [9].
Multi-Targeted Library Design
For organisms with extensive genetic redundancy, such as plants where 64.5% of genes belong to paralogous families, multi-targeted CRISPR libraries can overcome functional redundancy. Using algorithms like CRISPys, sgRNAs are designed to target conserved sequences across multiple genes within families, enabling simultaneous editing of several functionally related genes. This approach has been successfully implemented in tomato with 15,804 unique sgRNAs targeting 10,036 genes, with approximately 95% of sgRNAs targeting groups of 2-3 genes [8].
Table 2: CRISPRi Library Implementation Across Model Organisms
| Organism | Library Scale | Targeting Specificity | Key Applications |
|---|---|---|---|
| S. cerevisiae | >51,000 sgRNAs | -50 to +300 bp from TSS | Haploinsufficiency screening, metabolic engineering |
| S. oneidensis | 30,804 sgRNAs | 7 sgRNAs/coding gene | Extracellular electron transfer, bioremediation |
| Tomato | 15,804 sgRNAs | Conserved family domains | Fruit development, pathogen response, nutrient uptake |
| L. pneumophila | 44 gene combinations | Randomized multiplexing | Virulence factor identification |
Experimental Workflow and Validation
The standard workflow for genome-wide CRISPRi screening involves:
- Library Cloning: Pooled oligonucleotide amplification and Gibson Assembly into plasmid backbones containing dCas9-repressor fusions
- Transformation: Library introduction into host cells (e.g., via LiAc/PEG method for yeast) with >100x coverage per sgRNA
- Selection: Growth under selective pressure in semisolid 3D media to minimize competition bias
- Screening: Next-generation sequencing of sgRNA barcodes from pre- and post-selection populations
- Hit Identification: Statistical analysis of enriched/depleted sgRNAs to identify genes affecting fitness [6]
Validation should include qRT-PCR confirmation of repression efficiency (typically achieving 10-fold repression over 24 hours) and correlation analysis between biological replicates to ensure library representation and reproducibility [6].
Applications in Pathway Discovery and Functional Genomics
Metabolic Pathway Engineering
CRISPRi enables precise redistribution of metabolic flux without accumulating toxic intermediates. By titrating expression of multiple pathway enzymes simultaneously, researchers can optimize production of valuable compounds. This approach has successfully increased violacein derivatives and lycopene production in engineered strains, demonstrating how multiplexed CRISPRi can predictably manipulate complex metabolic networks [7]. The tunable nature of CRISPRi is particularly valuable for balancing growth and production pathways where complete gene knockout would be detrimental.
Genetic Interaction Mapping
CRISPRi facilitates systematic genetic interaction screening by enabling knockdown of gene pairs to identify synthetic sick/lethal relationships. This approach has revealed novel redundant virulence factors in Legionella pneumophila, where simultaneous repression of lpg2888 and lpg3000 impaired macrophage infection despite single knockdowns showing no phenotype [9] [10]. Such interactions illuminate functional redundancies in biological systems and identify compensatory pathways.
Essential Gene Identification
Genome-wide CRISPRi screens can systematically identify condition-specific essential genes. In Shewanella oneidensis, CRISPRi libraries have defined essential gene sets under both aerobic and anaerobic conditions, revealing how environmental factors reshape genetic requirements [4]. Unlike knockout approaches, CRISPRi allows quantification of fitness defects across a continuum of expression levels, providing more nuanced understanding of gene essentiality.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for CRISPRi Library Construction and Screening
| Reagent/Resource | Function | Implementation Example |
|---|---|---|
| dCas9-KRAB Fusion | Transcriptional repressor | Mammalian gene repression via chromatin modification |
| tetO-modified RPR1 Promoter | Inducible sgRNA expression | Anhydrotetracycline (ATc)-regulated expression in yeast [6] |
| R-S-R Oligonucleotides | Randomized array building blocks | MuRCiS assembly for combinatorial screening [9] |
| Gibson Assembly Master Mix | Multiplex cloning | Library construction from pooled oligonucleotides |
| PacBio Long-Read Sequencing | Array sequence resolution | Identifying complex sgRNA combinations in multiplex screens [9] |
| CRISPys Algorithm | Multi-target sgRNA design | Designing sgRNAs targeting conserved gene family domains [8] |
Visualization of Workflows and Molecular Relationships
Comparative Mechanism of Action
Multiplexed CRISPRi Library Screening Workflow
CRISPRi technology represents a paradigm shift in functional genomics, offering nuanced control over gene expression that enables pathway-level interrogation impossible with traditional nuclease approaches. The development of multiplexed CRISPRi libraries, combined with advanced screening methodologies, has empowered researchers to systematically dissect complex genetic networks, identify synthetic lethal interactions, and model disease states with unprecedented precision. As library design strategies continue to evolve toward greater scalability and organism-specific optimization, CRISPRi promises to accelerate both basic biological discovery and applied biotechnology across diverse species and biological contexts.
Why Multiplex? Addressing Genetic Redundancy and Polygeneic Networks
The Core Imperative for Multiplexing
In functional genomics, the ability to interrogate one gene at a time has often proven insufficient for unraveling complex biological systems. Multiplexed CRISPR technologies, particularly those based on CRISPR interference (CRISPRi), have emerged as a transformative platform for pathway discovery by enabling the simultaneous regulation of multiple genetic targets within a single cell [11] [12]. This capability is crucial for addressing two fundamental biological challenges: genetic redundancy and polygenic networks.
Genetic redundancy, where multiple genes perform overlapping functions, is pervasive across biology. In plants, for example, gene duplications and gene families are widespread, making the functional dissection of causal genes difficult without simultaneously targeting several paralogs [13]. Similarly, in bacteria, understanding complex processes like biofilm formation and antibiotic resistance requires perturbing multiple genes within a pathway concurrently [12]. Traditional single-gene knockout methods often fail to produce observable phenotypes under these conditions, as the function of one gene can be compensated by its redundant counterparts [13] [14].
Polygenic traits—those controlled by multiple genes and their complex interactions—represent another frontier where multiplexing excels. The simplicity of programming CRISPR-Cas systems by simply swapping guide RNAs (gRNAs) makes them ideally suited for multiplexed applications [15] [11]. Unlike earlier genome editing technologies like ZFNs and TALENs that required re-engineering proteins for each new target, CRISPR systems can target multiple genomic regions by expressing corresponding gRNAs [15]. This programmability has facilitated the development of sophisticated pooled screening approaches that can map complex genetic networks and identify combinatorial gene functions at an unprecedented scale [16] [17].
Technical Implementation of Multiplexed CRISPR Systems
Strategies for Multiplexed Guide RNA Expression
Implementing multiplexed CRISPR systems requires the coordinated expression of multiple gRNAs. Three primary genetic architectures have been established for this purpose, each with distinct advantages [11].
Table 1: Comparison of Multiplexed gRNA Expression Systems
| System Type | Key Feature | Mechanism of gRNA Processing | Typical Applications |
|---|---|---|---|
| Multi-cassette (Monocistronic) | Each gRNA has its own promoter and terminator [11] [14] | Direct transcription of individual gRNAs | Simplex multiplexing (2-4 gRNAs); when individual gRNA validation is needed [14] |
| Single-cassette (Polycistronic) | Multiple gRNAs encoded in a single transcript [11] [14] | Endogenous tRNA-processing system (RNases P and Z) [11] | High-level multiplexing (5-10+ gRNAs); species-agnostic applications [11] [14] |
| CRISPR Array-Based | Artificial CRISPR arrays mimicking native systems [11] [13] | Cas12a self-processing or co-expressed processing enzymes (e.g., Csy4) [11] | Large-scale screening libraries; when using Cas12a systems [11] |
The polycistronic tRNA-gRNA (PTG) system has emerged as particularly powerful for high-level multiplexing. This approach exploits the evolutionary conservation of tRNA processing enzymes, which cleave flanking tRNAs to release individual gRNAs from a single transcript [11] [14]. A significant advantage of the PTG system is its compatibility with RNA polymerase II (Pol II) promoters, enabling tissue-specific or inducible gRNA expression—a capability not easily achieved with the Pol III promoters typically used for gRNA expression [14].
CRISPRi for Pathway Discovery
CRISPR interference (CRISPRi) has become the preferred platform for multiplexed functional genomics in pathway discovery research [16] [12]. By using a catalytically dead Cas9 (dCas9) that binds DNA without cutting it, CRISPRi enables reversible, programmable gene repression without introducing DNA damage [16] [12]. This is particularly valuable for studying essential genes, where complete knockout would be lethal [12].
The effectiveness of CRISPRi depends critically on proper gRNA design. In prokaryotes, tiling screens have revealed that the most effective sgRNAs for gene repression target the non-template strand within the first 5% of the coding region proximal to the start codon [16]. For robust hit-calling in pooled screens, a minimum of 10 sgRNAs per gene is recommended, though this number can be optimized based on the specific application and screening parameters [16].
Table 2: CRISPRi Performance Characteristics in Functional Genomics
| Parameter | Performance Characteristic | Experimental Validation |
|---|---|---|
| Optimal sgRNA positioning | Within first 5% of ORF (non-template strand) [16] | Tiling screen with 2,281 sgRNAs targeting 44 E. coli genes [16] |
| Minimal sgRNAs per gene | 10 for reliable hit calling [16] | Computational sampling of sgRNA subsets [16] |
| Specificity | Superior to Tn-seq for short genes and ncRNAs [16] | Essential gene identification in E. coli [16] |
| Multiplexing capacity | Efficient silencing of multiple genes simultaneously [12] | Biofilm formation genes in E. faecalis [12] |
Experimental Framework for Multiplexed CRISPRi Screening
Library Design and Construction
A robust multiplexed CRISPRi screening workflow begins with comprehensive library design. For genome-scale screens, sgRNA libraries can be designed to target every protein-coding gene, non-coding RNA, or custom subsets of the genome [16] [18]. The library is typically synthesized as an oligonucleotide pool via microarray oligonucleotide synthesis, then amplified and cloned into an appropriate expression vector [16].
For pathway-specific discovery, a focused library targeting genes within a biological process of interest offers deeper coverage at lower cost. Essential design considerations include:
- Specificity: Ensure minimal off-target effects by rigorous bioinformatic screening [16]
- Efficiency: Prioritize sgRNAs with proven on-target activity [16] [18]
- Control elements: Include non-targeting sgRNAs and targeting essential genes as positive controls [16]
Library complexity typically ranges from thousands to hundreds of thousands of sgRNAs, with current human genome-wide libraries containing approximately 60,000 sgRNAs targeting around 20,000 genes [18].
Delivery and Screening Workflow
The following diagram illustrates a generalized workflow for a pooled CRISPRi screen:
The screening process involves transducing cells at a low multiplicity of infection (MOI ~0.3) to ensure most cells receive only one sgRNA, maintaining population heterogeneity [16]. After selection with antibiotics (e.g., puromycin), cells are subjected to phenotypic challenges such as drug treatment, nutrient limitation, or other biological stresses [16] [17]. The abundance of each sgRNA before and after selection is quantified by next-generation sequencing, with sgRNA depletion or enrichment indicating genes affecting fitness under the screening condition [16].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of multiplexed CRISPRi screening requires several key reagents and tools:
Table 3: Essential Research Reagents for Multiplexed CRISPRi Screening
| Reagent Category | Specific Examples | Function and Importance |
|---|---|---|
| CRISPRi Vectors | pMSP3545-dCas9, pGCP123-sgRNA [12] | Dual-vector system for inducible dCas9 and sgRNA expression; enables tunable repression [12] |
| Pooled Libraries | Human CRISPR Knockout Libraries (Jacquere) [18] | Genome-wide sgRNA collections with optimized on-target activity; available from repositories like Addgene [18] |
| Selection Markers | Puromycin, Blasticidin resistance genes [18] | Enable selection of successfully transduced cells; critical for maintaining library representation [16] |
| Induction Systems | Nisin-inducible promoter [12] | Allows temporal control of CRISPRi activity; essential for studying essential genes [12] |
| Analysis Tools | MAGeCK, PinAPL-Py [16] | Bioinformatics pipelines for quantifying sgRNA abundance and statistical analysis of screen results [16] |
Applications in Addressing Biological Complexity
Overcoming Genetic Redundancy
Multiplexed CRISPRi has proven particularly powerful for dissecting the functions of redundant gene families. In plants, for example, durable resistance to powdery mildew requires simultaneous knockout of multiple MLO genes—Atmlo2, Atmlo6, and Atmlo12 in Arabidopsis and Csmlo1, Csmlo8, and Csmlo11 in cucumber [13]. Single-gene knockouts in these systems typically fail to confer full resistance, demonstrating how multiplexing can reveal biological functions obscured by genetic redundancy [13].
In bacteria, multiplexed CRISPRi has enabled the study of stage-specific gene requirements in biofilm formation. By simultaneously targeting multiple pilus genes (ebpA, ebpB, ebpC) in Enterococcus faecalis, researchers demonstrated their collective importance in biofilm initiation, maturation, and maintenance [12]. This approach could target genes throughout the operon without the polar effects common in traditional mutagenesis [12].
Mapping Polygenic Networks and Genetic Interactions
Beyond redundancy studies, multiplexed CRISPRi enables systematic mapping of genetic interactions—where the combined effect of perturbing multiple genes differs from expected based on single perturbations [15] [16]. These synthetic lethal or synthetic sick interactions reveal functional relationships between genes and can identify potential combination drug targets [16].
Combinatorial CRISPR screening approaches have been developed specifically for this purpose. The CDKO (CRISPR-based double-knockout) library designed by the Bassik group, for example, uses paired gRNAs to target gene combinations, enabling genome-scale mapping of genetic interactions [15]. Such screens have identified synthetic lethal combinations that could inform cancer therapeutic development [15] [17].
The following diagram illustrates how multiplexed CRISPRi reveals genetic interactions in polygenic networks:
Future Directions and Concluding Perspectives
The field of multiplexed CRISPRi continues to evolve rapidly. Emerging applications include large-scale chromosomal engineering [15] [13], combinatorial epigenetic editing [15], and increasingly sophisticated single-cell CRISPR screening methodologies that combine perturbation with transcriptomic readouts [19]. The integration of artificial intelligence and machine learning into gRNA design and outcome prediction promises to further enhance the efficiency and specificity of these approaches [13].
For pathway discovery researchers, multiplexed CRISPRi offers an unparalleled toolkit for dissecting complex genetic networks. By enabling the systematic interrogation of genetic redundancy and polygenic traits, this technology moves beyond the one-gene-at-a-time paradigm to provide a more holistic understanding of biological systems. As toolkits continue to evolve—with improvements in vector design, delivery efficiency, and analytical methods—multiplexed CRISPRi is poised to become a foundational technology for functional genomics and drug target discovery [13] [17].
The demonstrated success of these approaches in identifying synthetic lethal interactions [15], mapping complex genetic networks [16], and revealing functionally redundant pathways [13] [12] underscores their transformative potential for both basic research and therapeutic development. For scientists grappling with the complexities of genetic redundancy and polygenic inheritance, multiplexing has evolved from a technical option to a methodological imperative.
CRISPR interference (CRISPRi) has emerged as a powerful technology for programmable, reversible, and titratable repression of gene expression in mammalian cells, establishing itself as an indispensable tool for pathway discovery research [20]. At its core, the CRISPRi system consists of two fundamental components: (1) an effector protein, typically a catalytically dead Cas9 (dCas9), which is fused to one or more transcription repressor domains; and (2) a single guide RNA (sgRNA), which directs the effector protein to a specific target DNA sequence [20]. Unlike nuclease-active Cas9, which creates double-strand breaks, dCas9 lacks endonuclease activity, thereby repurposing the system for targeted gene regulation without altering the underlying DNA sequence [21]. This technical guide delves into the key components of the CRISPRi system, detailing their structure, function, and optimization for the construction of multiplexed CRISPRi libraries aimed at unraveling complex biological pathways.
dCas9 Effectors: The Engine of Targeted Repression
The dCas9 protein serves as the programmable DNA-binding backbone of the CRISPRi system. It is engineered from the wild-type Streptococcus pyogenes Cas9 nuclease by introducing two point mutations (D10A and H840A) that inactivate the RuvC and HNH nuclease domains, respectively [21]. This renders the protein catalytically dead while retaining its ability to bind DNA in a guide RNA-directed manner.
Comparative Performance of dCas9 Effector Fusions
The repressive efficacy of dCas9 is significantly enhanced by fusion to transcriptional repressor domains. Recent systematic comparisons of CRISPRi effectors have been critical for establishing best practices.
Table 1: Comparison of dCas9-Repressor Domain Effectors
| Effector Protein | Key Repressor Domain(s) | On-Target Knockdown Efficacy | Non-Specific Effects on Cell Growth/Transcriptome | Recommended Use Case |
|---|---|---|---|---|
| dCas9-KRAB (Kox1) | Krüppel-associated box (KRAB) | Strong | Moderate | General purpose screening [20] |
| Zim3-dCas9 | Engineered repressor domain (Zim3) | Strong | Minimal | Recommended best practice for optimal balance of efficacy and specificity [20] |
| dCas9 alone | None | Variable/Modest | Low | Basal repression, but insufficient for most screening applications [20] [21] |
As evidenced from empirical data aggregated from 126 screens, the Zim3-dCas9 effector provides a superior balance, delivering robust on-target knockdown without introducing significant non-specific effects on cell viability or the global transcriptome, making it the current effector of choice for generating high-quality CRISPRi cell models [20].
Repressor Domains: Mediators of Transcriptional Silencing
Repressor domains are protein modules fused to dCas9 that recruit endogenous chromatin-modifying complexes to the target genomic locus, leading to epigenetic silencing and transcriptional repression.
- KRAB (Krüppel-associated box): This is a well-characterized repressor domain that recruits factors such as KAP1, which in turn facilitates histone methylation (H3K9me3) and the formation of heterochromatin, effectively shutting down transcription initiation and elongation [20] [21].
- Engineered Domains (e.g., Zim3): Recent advances have led to the development of novel, engineered repressor domains like Zim3. These are designed to maximize repression potency and minimize cellular toxicity, which is a crucial consideration for long-term or genome-wide genetic screens [20].
The fusion of dCas9 to these repressor domains creates a powerful "active repressor" complex that sterically blocks RNA polymerase while simultaneously establishing a repressive chromatin environment at the promoter region of the target gene [20] [21].
sgRNA Architecture: Design Principles for Efficacy and Specificity
The sgRNA is a chimeric RNA molecule that combines the functions of the native crRNA and tracrRNA. Its architecture is critical for determining the efficiency and specificity of DNA binding [21]. The 20-nucleotide base-pairing region at the 5' end of the sgRNA is responsible for target recognition via Watson-Crick base pairing with the complementary DNA strand [21].
Key Design Parameters for Optimal sgRNA Activity:
- Target Site Selection: The sgRNA must be designed to target a genomic region with an adjacent Protospacer Adjacent Motif (PAM), which for S. pyogenes dCas9 is the sequence NGG [21]. The optimal target sites are typically located within the promoter or enhancer regions or at the beginning of the coding sequence (5' end) of the target gene [21].
- Seed Sequence: The "seed" region, comprising the PAM-proximal 7-12 nucleotides, is critical for binding specificity. Mismatches in this region can severely compromise sgRNA activity [22].
- Off-Target Assessment: The specificity of the designed 20-nt sgRNA sequence must be verified using tools like BLAST or SeqMap to ensure it has minimal homology to other genomic sites, particularly in the seed region, to avoid unintended repression [21].
- sgRNA Scaffold: The secondary structure of the sgRNA itself (the dCas9 handle and terminator) can influence its stability and efficiency. Tools like RNAfold can be used to predict and avoid unfavorable structures [21].
Multiplexed sgRNA Strategies for Enhanced Knockdown and Library Design
A pivotal innovation in CRISPRi library design is the move from single-sgRNA to dual- or multi-sgRNA constructs. This strategy substantially improves the robustness and consistency of gene knockdown [20].
Dual-sgRNA Cassettes
This approach involves targeting a single gene with two distinct, highly active sgRNAs expressed from a tandem cassette within a single lentiviral vector. Empirical data from genome-wide growth screens demonstrate that dual-sgRNA libraries produce significantly stronger growth phenotypes for essential genes compared to single-sgRNA libraries (mean 29% decrease in growth rate vs. 20%), indicating more potent and reliable gene depletion [20]. This allows for the creation of ultra-compact, highly active genome-wide libraries where each gene is targeted by only one or two library elements, reducing library size and enabling more complex screens in settings with limited cell numbers [20].
Implementing Multiplexed sgRNA Expression
Several molecular strategies have been developed to express multiple sgRNAs from a single vector, which is essential for dual-targeting and combinatorial screening:
- Tandem Expression Cassettes: This is the most common approach, where each sgRNA is expressed from its own RNA Polymerase III promoter (e.g., human U6, mouse U6). Using different promoters for each sgRNA can help prevent homologous recombination [15] [23].
- Artificial Multi-sgRNA Precursors: A single long transcript encoding multiple sgRNAs can be expressed and subsequently processed into individual, functional sgRNAs by incorporating self-cleaving ribozymes or Csy4 endoribonuclease recognition sites [23]. This strategy is advantageous for using inducible or tissue-specific Pol II promoters.
Experimental Protocol: Implementing a CRISPRi Screen for Pathway Discovery
The following protocol outlines the key steps for executing a genome-wide CRISPRi screen using a dual-sgRNA library, from initial design to functional validation.
Library Design and Cloning
- Select Target Genes: Define the gene set relevant to your pathway of interest (e.g., whole genome, kinase family).
- sgRNA Design: For each gene, select two highly active sgRNAs based on empirical design rules, targeting the promoter region near the transcription start site [20] [21].
- Library Synthesis: Clone the pooled sgRNA sequences into a lentiviral vector containing the dual-sgRNA expression cassette. Use methods like Golden Gate assembly for efficient and high-fidelity cloning of large libraries [15] [23].
Cell Line Engineering
- Generate Stable dCas9 Effector Cell Line: Introduce a construct for stable, inducible expression of the chosen dCas9 repressor (e.g., Zim3-dCas9) into your target cell line (e.g., K562, RPE1, Jurkat). Select and validate clones for consistent dCas9 expression and minimal impact on baseline growth [20].
- Transduce with sgRNA Library: Transduce the engineered dCas9 cell line with the pooled lentiviral sgRNA library at a low Multiplicity of Infection (MOI ~0.3) to ensure most cells receive only one viral construct. Use puromycin selection to enrich for successfully transduced cells [20] [19].
Biological Selection and Screening
- Apply Selective Pressure: Culture the transduced cell pool under the biological challenge of interest (e.g., drug treatment, metabolic stress, viral infection) for an extended period (e.g., 2-3 weeks) [20] [19].
- Harvest Samples for Sequencing: Collect genomic DNA from cells at baseline (T0) and at the endpoint of the selection (Tfinal).
Sequencing and Data Analysis
- Amplify and Sequence sgRNAs: Amplify the integrated sgRNA cassettes from genomic DNA by PCR and subject them to next-generation sequencing [20].
- Quantify Enrichment/Depletion: For each sgRNA, calculate its fold-change in abundance (Tfinal vs. T0) using specialized analysis pipelines (e.g., MAGeCK). Statistically significant depletion of sgRNAs targeting a specific gene indicates that its knockdown confers a fitness defect under the selection pressure, implicating it in the pathway [19].
The Scientist's Toolkit: Essential Reagents for CRISPRi Screening
Table 2: Key Research Reagent Solutions for Multiplexed CRISPRi
| Reagent / Resource | Function and Description | Example/Source |
|---|---|---|
| Zim3-dCas9 Expression Construct | Plasmid for stable expression of the optimized dCas9-effector fusion. | Available from Jost & Weissman labs (e.g., Addgene) [20]. |
| Ultra-Compact Dual-sgRNA Library | Pooled lentiviral library targeting each gene with a dual-sgRNA cassette. | Jost & Weissman lab "next-generation" library [20]. |
| Lentiviral Packaging System | Plasmids (psPAX2, pMD2.G) to produce lentiviral particles for sgRNA delivery. | Common commercial and nonprofit repositories (e.g., Addgene). |
| Stable dCas9 Cell Lines | Pre-engineered mammalian cell lines with robust Zim3-dCas9 expression. | K562, RPE1, Jurkat, HT29 lines from published studies [20]. |
| sgRNA Amplification Primers | Custom oligonucleotides for PCR amplification of sgRNA cassettes from genomic DNA prior to sequencing. | Designed per library specification [20]. |
| Bioinformatic Analysis Pipeline | Software for quantifying sgRNA abundance and identifying hit genes from sequencing data. | Tools like MAGeCK [19]. |
The concerted optimization of dCas9 effectors, repressor domains, and sgRNA architecture has propelled CRISPRi into a premier technology for high-throughput functional genomics. The adoption of high-performance effectors like Zim3-dCas9 and compact, highly active dual-sgRNA libraries provides a robust framework for constructing multiplexed CRISPRi screening platforms. This powerful combination enables researchers to systematically dissect complex genetic pathways, identify key regulatory nodes, and uncover novel therapeutic targets with unprecedented precision and efficiency, thereby accelerating discovery in basic biology and drug development.
Theoretical Foundations of Programmable Transcriptional Repression
Programmable transcriptional repression, primarily facilitated by CRISPR interference (CRISPRi), represents a cornerstone technology in modern functional genomics. This technique leverages a catalytically dead Cas9 (dCas9) protein, which acts as a programmable DNA-binding entity guided by a short RNA sequence to specific genomic loci without introducing double-strand breaks. Upon binding, the dCas9 complex sterically obstructs RNA polymerase, leading to targeted gene silencing [24]. Within the context of a broader thesis on multiplexed CRISPRi library construction, this technology transitions from a tool for studying single genes to a powerful platform for systematic pathway discovery and genetic interaction mapping. This guide details the core mechanisms, optimization strategies, and practical implementation of CRISPRi, providing a foundation for its application in large-scale, multiplexed screening for pathway discovery research.
Core Mechanism and System Components
The fundamental CRISPRi system consists of two core components: the dCas9 protein and a single-guide RNA (sgRNA). The dCas9 is directed by the sgRNA to a specific DNA sequence adjacent to a Protospacer Adjacent Motif (PAM), where it binds and functions as a physical roadblock to the transcription machinery [24]. This blockade can inhibit either transcription initiation, when targeted to a promoter region, or transcription elongation, when targeted within the coding sequence [25] [24].
Repression efficiency is highly dependent on the genomic context of the target site. In bacteria, dCas9 binding alone is often sufficient for effective repression. However, in eukaryotic systems, dCas9 is typically fused to transcriptional repressor domains, such as the Krüppel-associated box (KRAB), to recruit chromatin-modifying complexes and enhance silencing [26]. The system is highly programmable, inducible, and reversible, allowing for precise temporal control of gene expression, which is critical for studying essential genes and dynamic biological processes [24].
Diagram: Core Mechanism of CRISPRi-Based Transcriptional Repression
Optimization of Repression Efficiency
Selection of Cas9 Orthologs
The choice of Cas9 ortholog is a critical determinant of CRISPRi efficacy. While the initial CRISPRi systems utilized Streptococcus pyogenes dCas9 (dCas9Spy), its performance in non-model bacteria like mycobacteria was suboptimal, yielding only ~3-4 fold repression and exhibiting significant proteotoxicity [25]. A systematic screen of 11 diverse Cas9 orthologs identified Streptococcus thermophilus CRISPR1 Cas9 (dCas9Sth1) as particularly effective in mycobacteria. This ortholog typically achieves 20–100 fold knockdown of endogenous gene expression with minimal toxicity [25]. Notably, dCas9Sth1 maintains robust repression even when targeted far from the transcription start site, providing greater flexibility for interrogating gene function within operons [25].
Table 1: Performance Comparison of Selected dCas9 Orthologs
| dCas9 Ortholog | Origin | PAM Sequence | Repression Efficiency | Key Characteristics | Ideal Application Context |
|---|---|---|---|---|---|
| dCas9Sth1 | Streptococcus thermophilus | NGGNG | 20-100 fold knockdown [25] | Minimal proteotoxicity; effective at long-range | Mycobacterial studies; operon dissection |
| dCas9Spy | Streptococcus pyogenes | NGG | ~3-4 fold (1st gen.); improved in 2nd gen. [25] | Well-characterized; but can cause proteotoxicity | Model organisms (E. coli, B. subtilis) |
| Type IIC Cas9s | Various | Varies | <5 fold knockdown [25] | Limited double-stranded DNA helicase activity | Not recommended for robust CRISPRi |
Engineering Enhanced Repressor Domains
In mammalian cells, repression efficiency can be significantly enhanced by fusing dCas9 to potent repressor domains. Recent engineering efforts have screened over 100 bipartite and tripartite repressor fusions, leading to the development of next-generation effectors such as dCas9-ZIM3(KRAB)-MeCP2(t) [26]. This optimized repressor demonstrates ~20–30% better knockdown than the previously established gold-standard dCas9-ZIM3(KRAB) and shows reduced performance variability across different sgRNA sequences and cell lines [26]. This consistency is paramount for ensuring the reliability of genome-wide CRISPRi screens.
Experimental Protocol for a Basic CRISPRi Experiment
The following protocol outlines the key steps for implementing a CRISPRi system to repress a target gene of interest, forming the basis for more complex multiplexed library construction.
System Selection and Vector Construction
- Choose CRISPRi Machinery: Select an appropriate dCas9 ortholog (e.g., dCas9Sth1 for mycobacteria [25] or dCas9-KRAB fusions for mammalian cells [26]) and a matching sgRNA backbone.
- Clone the dCas9 Component: Clone the codon-optimized
dcas9gene, often with a fused repressor domain (e.g., KRAB, MeCP2(t)), into an expression vector under the control of an inducible promoter (e.g., anhydrotetracycline (ATc)-induciblePtet). Use a low-copy plasmid if proteotoxicity is a concern [25]. - Clone the sgRNA Component: Design and synthesize an oligonucleotide corresponding to the 20-nucleotide spacer sequence that is complementary to the target genomic site. Clone this into an sgRNA expression vector under a constitutive promoter (e.g., U6 for mammalian Pol III or a strong synthetic promoter for bacteria). For high-throughput library construction, this step is automated using pooled oligonucleotide synthesis and advanced cloning techniques like Golden Gate assembly [15] [27].
Delivery and Induction
- Deliver Plasmids: Introduce the dCas9 and sgRNA plasmids into the target cells via transformation (bacteria) or transfection (mammalian cells). For stable expression, the
dcas9gene can be integrated into a neutral genomic site [24]. - Induce Repression: Add the inducer molecule (e.g., ATc) to the culture medium to trigger expression of dCas9 and/or the sgRNA. The timing and concentration of the inducer can be used to tune the level of repression [24].
Validation and Phenotyping
- Measure Knockdown Efficiency: After a suitable induction period (e.g., 24-48 hours), harvest cells and quantify transcript levels of the target gene using quantitative PCR (qPCR). Protein levels can be assessed by western blotting or flow cytometry if a fluorescent reporter is used [28] [26].
- Assess Functional Phenotype: Monitor for expected phenotypic consequences, such as growth defects for essential genes [25] or sensitivity to specific chemical compounds [28].
Application in Multiplexed Library Construction for Pathway Discovery
The true power of CRISPRi is realized in its multiplexed, library-scale format, which enables the systematic interrogation of gene function and genetic pathways across the genome.
Library Design and Workflow
Multiplexed CRISPRi libraries consist of pools of thousands to hundreds of thousands of unique sgRNAs, each designed to repress a specific gene. For pathway discovery, libraries can be designed to target entire genomes or curated gene sets (e.g., all kinases or transcription factors). A key advancement is the multi-targeted sgRNA library, where a single sgRNA is designed to target conserved sequences across multiple genes within a family, effectively overcoming functional redundancy [27].
Diagram: Workflow for Multiplexed CRISPRi Library Screening
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for CRISPRi Library Construction and Screening
| Reagent / Tool | Function | Example & Notes |
|---|---|---|
| dCas9-Repressor Vector | Constitutively expresses the dCas9-repressor fusion protein. | dCas9-ZIM3(KRAB)-MeCP2(t) for high-efficacy repression in mammalian cells [26]. |
| sgRNA Library Pool | A pooled collection of sgRNA constructs for high-throughput screening. | A genome-wide library targeting 10,036 tomato genes with 15,804 unique sgRNAs [27]. |
| Inducible Expression System | Allows temporal control over sgRNA and/or dCas9 expression. | Anhydrotetracycline (ATc)-inducible promoter (Ptet) for titratable repression [28] [24]. |
| Barcode Tagging System | Enables tracking of individual sgRNAs in a pooled screen. | CRISPR-GuideMap, a double-barcode system for mapping sgRNAs in plant lines [27]. |
| Next-Generation Sequencing (NGS) | Quantifies sgRNA abundance before and after screening to identify hits. | Used to determine which sgRNAs are enriched/depleted under selective pressure [28] [27]. |
The theoretical and practical foundations of programmable transcriptional repression establish CRISPRi as an exceptionally powerful and versatile tool for functional genomics. The ongoing optimization of Cas9 orthologs and repressor domains continues to enhance the efficiency, specificity, and reliability of this technology. By moving into a multiplexed library format, researchers can leverage CRISPRi to systematically dissect complex genetic pathways and interactions on a genome-wide scale. This approach provides an unparalleled methodology for pathway discovery research, with profound implications for understanding disease mechanisms and identifying novel therapeutic targets.
A Step-by-Step Guide to Designing and Building Optimized CRISPRi Libraries
The construction of multiplexed CRISPR interference (CRISPRi) libraries represents a powerful approach for pathway discovery and functional genomics. At the core of these screens lies the choice of the effector protein—a catalytically dead Cas9 (dCas9) fused to repressive domains that determine the efficiency and reliability of gene knockdown. This technical guide provides an in-depth comparison of dCas9 variants, with a focus on the advanced ZIM3-dCas9 system, and details experimental protocols for their use in large-scale genetic screens. We present quantitative data on the performance of next-generation effectors, including the novel dCas9-ZIM3(KRAB)-MeCP2(t) fusion, which demonstrates significantly enhanced repression across diverse cell lines and targets. Structured for the practicing scientist, this whitepaper integrates performance metrics, optimized protocols, and reagent solutions to inform the design of robust CRISPRi libraries for pathway discovery research.
CRISPR interference (CRISPRi) utilizes a catalytically dead Cas9 (dCas9) fused to transcriptional repressor domains to achieve programmable gene silencing without altering DNA sequence. This technology is particularly valuable for functional genomics and pathway discovery in mammalian cells, as it enables reversible gene knockdown, avoids confounders associated with DNA damage, and is well-suited for multiplexed screening [29]. The efficacy of a CRISPRi system is largely dictated by the repressor domain(s) fused to dCas9. The Krüppel-associated box (KRAB) domain is the most widely used repressor, which recruits co-factors to establish heterochromatin and silence gene expression [30]. However, traditional CRISPRi platforms can suffer from incomplete knockdown and performance variability across different cell lines and gene targets [29]. This variability poses a significant challenge for large-scale library screens, where consistent and potent repression is required to confidently identify genetic interactions and pathway members. Consequently, selecting an optimal dCas9 effector is a critical first step in constructing a reliable multiplexed CRISPRi library. Recent efforts have focused on engineering enhanced dCas9 effectors through the screening of novel repressor domains and the construction of multi-domain fusion proteins to overcome these limitations [30] [29].
Comparative Analysis of Key dCas9 Variants
The landscape of dCas9 effectors has evolved significantly from the initial dCas9-KOX1(KRAB) system. The following table summarizes the key performance characteristics of major dCas9 variants, providing a basis for effector selection.
Table 1: Performance Comparison of Key dCas9 Effector Variants
| dCas9 Variant | Key Components | Reported Repression Efficiency | Key Advantages | Primary Applications |
|---|---|---|---|---|
| dCas9-KOX1(KRAB) | dCas9 + KOX1 KRAB domain | Baseline | Pioneer system; well-characterized | General gene repression; early screens [29] |
| dCas9-ZIM3(KRAB) | dCas9 + ZIM3 KRAB domain | ~40-50% stronger than KOX1 [31] | Exceptional potency; compact design | High-demand repression; genome-wide screens [30] [29] |
| dCas9-KOX1-MeCP2 | dCas9 + KOX1 KRAB + MeCP2 (283aa) | Superior to single-domain fusions [29] | Synergistic repression; strong knockdown | Transcriptional regulation studies [29] [30] |
| dCas9-ZIM3-MeCP2(t) | dCas9 + ZIM3 KRAB + truncated MeCP2 (80aa) | ~20-30% better than dCas9-ZIM3 [29] | Highest potency; reduced variability; consistent cross-cell line performance | Multiplexed library screens; pathway discovery; challenging targets [29] |
The progression from traditional to next-generation effectors is characterized by the strategic combination of potent repressor domains. The discovery of the ZIM3 KRAB domain marked a significant advance, identified from a screen of 57 human KRAB domains as an exceptionally potent repressor that silences gene expression more efficiently than the canonical KOX1-based platform [30]. Further gains in repression efficacy have been achieved by creating multi-domain fusion proteins. For instance, fusing the MeCP2 repressor domain to dCas9-KOX1(KRAB) creates a synergistic repressor that outperforms either domain alone [29]. The most recent innovations, such as dCas9-ZIM3(KRAB)-MeCP2(t), combine the best-performing domains into a single effector. This variant incorporates the potent ZIM3 KRAB domain with a truncated, 80-amino-acid MeCP2 domain, resulting in a repressor that demonstrates significantly enhanced gene repression at the transcript and protein level across several cell lines, with reduced performance variability dependent on the sgRNA sequence used [29].
Figure 1: Workflow for Engineering and Selecting Optimal dCas9 Effectors. The process involves iterative discovery and benchmarking, culminating in the selection of a highly potent multi-domain fusion for library construction.
Experimental Protocols for Effector Evaluation and Library Construction
Protocol for Evaluating Novel dCas9 Effectors
Before deploying a dCas9 effector in a large-scale library, it is crucial to empirically validate its repression efficiency in your specific cellular context. The following protocol, adapted from recent studies, outlines a robust methodology for head-to-head comparison [29].
- Construct Cloning: Clone the coding sequences for the dCas9 effector variants (e.g., dCas9-ZIM3(KRAB), dCas9-ZIM3(KRAB)-MeCP2(t)) into a lentiviral expression vector under a strong, ubiquitous promoter (e.g., SFFV or EF1α).
- Reporter Cell Line Generation: Create a stable reporter cell line (e.g., in HEK293T or K562) harboring a stably integrated construct with an eGFP gene under the control of a constitutively active promoter (e.g., SV40). This provides a quantifiable readout for repression.
- sgRNA Transduction: For the reporter cell line, also express a sgRNA that targets the dCas9 effector to the promoter driving eGFP expression. A dual-targeting sgRNA can enhance repression.
- Flow Cytometry Analysis: After 96-120 hours of effector and sgRNA expression, analyze the cells using flow cytometry.
- Measurement: Measure the median fluorescence intensity (MFI) of eGFP in the transfected cell population.
- Controls: Compare against a non-targeting sgRNA control and a dCas9-only control to differentiate repressor-mediated knockdown from steric blockade.
- Data Analysis: Calculate the percentage knockdown as
(1 - (MFI_sample / MFI_control)) * 100. Perform experiments with at least 6 biological replicates to ensure statistical power.
- Endogenous Target Validation: Select 3-5 endogenous genes and design 3-5 sgRNAs per gene targeting their transcription start sites. Transduce the dCas9 effector and sgRNAs into the desired cell line and quantify knockdown efficiency via RT-qPCR or Western blot 5-7 days post-transduction.
Protocol for Multiplexed CRISPRi Library Construction
For pathway discovery, constructing a multiplexed library that can target gene families is essential to overcome functional redundancy. The MuRCiS (Multiplexed, Randomized CRISPRi Sequencing) approach provides a framework for this [9].
- sgRNA Library Design:
- Target Selection: Define the gene set or pathway of interest. For comprehensive coverage, include all known members of redundant gene families.
- Multi-Target sgRNA Design: Use algorithms like CRISPys to design sgRNAs that target conserved sequences across multiple genes within a family. This allows a single sgRNA to knock down several paralogs simultaneously [8].
- Specificity Filtering: Apply computational tools to score sgRNAs for on-target efficiency (e.g., CFD score > 0.8) and filter out guides with potential off-target effects, applying stricter thresholds for exonic regions [8].
- Randomized Array Assembly (for MuRCiS):
- Oligo Design: Synthesize DNA oligonucleotides (R-S-Rs: repeat-spacer-repeat) where each spacer (targeting sequence) is flanked by partial repeat sequences with 4bp Golden Gate overhangs (e.g., TGAA) [9].
- Golden Gate Assembly: Use a Golden Gate reaction to mix the pooled R-S-R oligonucleotides, which self-assemble randomly into extended CRISPR arrays via their complementary overhangs. This creates a highly diverse library of arrays with varying spacer combinations and orders [9].
- Cloning and Sequencing: Clone the assembled arrays into a lentiviral vector backbone containing the optimized dCas9 effector (e.g., dCas9-ZIM3-MeCP2(t)). Use PacBio long-read sequencing to map the specific spacer combinations within the library [9].
- Library Production and Screening:
- Virus Production: Package the lentiviral library in HEK293T cells and titrate the virus to achieve a low multiplicity of infection (MOI ~0.3-0.4) to ensure most cells receive only one sgRNA array.
- Cell Transduction and Selection: Transduce the target cell population at a high coverage (e.g., 500x representation of the library) and select with antibiotics if needed.
- Phenotypic Screening: Challenge the cells with the relevant selective pressure (e.g., pathogen infection, nutrient stress, drug treatment). After a suitable period, harvest genomic DNA from the surviving population and the initial library pool.
- Sequencing and Analysis: Amplify and sequence the sgRNA regions from both populations. Compare sgRNA abundance using specialized analysis tools (e.g., MAGeCK) to identify guides (and thus gene combinations) that are enriched or depleted under the selection [30].
Successful execution of a multiplexed CRISPRi screen requires a suite of well-characterized reagents. The table below lists key resources for building and deploying an optimized CRISPRi system.
Table 2: Essential Research Reagents for Advanced CRISPRi Screening
| Reagent / Resource | Function / Description | Example Identifiers / Notes |
|---|---|---|
| Optimized Effector Plasmids | Lentiviral vectors for high-efficacy dCas9 repressors. | Addgene: 154472 (pLX303-ZIM3-KRAB-dCas9) [30]; dCas9-ZIM3(KRAB)-MeCP2(t) [29] |
| sgRNA Backbone | Vector for cloning and expressing sgRNA libraries. | Compatible with chosen dCas9 effector (e.g., for S. pyogenes dCas9) |
| Library Cloning System | Method for scalable, multiplexed sgRNA array assembly. | Randomized R-S-R oligo assembly (MuRCiS) [9] |
| Analysis Software | Computational tool for analyzing screen sequencing data. | MAGeCK [30], BAGEL [30] |
| Validated Control sgRNAs | Pre-tested non-targeting and positive control sgRNAs. | Essential for assay normalization and quality control |
| Reporter Cell Line | Cell line with fluorescent reporter for effector validation. | e.g., HEK293T-SV40-eGFP [29] |
Figure 2: Multiplexed CRISPRi Library Workflow. The process from computational design of multi-targeting sgRNAs to bioinformatic analysis of screening results, highlighting the key step of randomized array assembly.
The selection of the dCas9 effector is a foundational decision that dictates the success and reproducibility of multiplexed CRISPRi libraries for pathway discovery. The evidence clearly indicates that next-generation effectors, particularly the dCas9-ZIM3(KRAB)-MeCP2(t) fusion, offer a superior combination of potent repression, consistency across diverse cell lines and gene targets, and reduced sgRNA-dependent variability. Integrating this optimized protein backbone with sophisticated library design strategies—such as multi-targeting sgRNAs and randomized array assembly—enables researchers to systematically dissect complex biological processes, even in the face of significant genetic redundancy. As the field progresses, future efforts will likely focus on further refining effector specificity, developing orthogonal systems for simultaneous manipulation of multiple pathways, and creating more sophisticated computational models to predict and account for context-specific performance. By adopting these advanced tools and protocols, scientists can construct more reliable and informative CRISPRi libraries, accelerating the pace of discovery in functional genomics and therapeutic development.
For pathway discovery research using multiplexed CRISPR interference (CRISPRi) libraries, effective sgRNA design is paramount. Such libraries enable the systematic repression of genes across biological pathways to decode their function. Success hinges on the reliable and efficient knockdown of every targeted gene, which is directly determined by the application of robust sgRNA design rules. Research has demonstrated that highly active sgRNAs are not random; they are defined by the integration of three critical feature sets: the chromatin environment of the target site, its sequence composition, and its position relative to the transcription start site (TSS). This guide synthesizes the key design principles from foundational and recent studies, providing a technical framework for constructing optimized multiplexed CRISPRi libraries for pathway discovery.
Foundational Design Parameters
Positional Features: Targeting the Promoter
The position of the sgRNA relative to the TSS is one of the most critical determinants of CRISPRi efficacy. Optimal positioning ensures that dCas9-effector complexes effectively block the binding or progression of RNA polymerase.
- TSS Annotation is Key: The choice of TSS annotation database significantly impacts performance. Evidence indicates that the FANTOM5/CAGE promoter atlas represents the most reliable source for TSS annotations, as it maps robust, empirically defined TSSs, leading to superior prediction accuracy [32] [33].
- Optimal Targeting Window: The highest efficacy for CRISPRi repression is achieved within a specific window downstream of the TSS. The most effective sgRNAs typically target the region from -50 to +300 bp relative to the annotated TSS, with the nucleosome-deprived region immediately downstream of the TSS often yielding the strongest activity [32] [33].
Table 1: Influence of sgRNA Position on CRISPRi Efficacy
| Positional Feature | Optimal Design Rule | Biological Rationale |
|---|---|---|
| TSS Annotation | Use FANTOM5/CAGE-defined TSSs | Captures empirically used start sites, improving prediction accuracy |
| Distance from TSS | Target region from -50 to +300 bp | Enables dCas9 to block transcriptional initiation and early elongation |
| Nucleosome Phasing | Avoid nucleosome-occupied regions; favor nucleosome-depleted regions | Nucleosomes directly block dCas9 access to DNA [32] |
Sequence Features: Optimizing sgRNA-Target Interaction
Beyond position, the nucleotide sequence of the sgRNA and its target DNA dictates the stability of the binding interaction and its efficiency.
- PAM-Adjacent Nucleotides: A guanine (G) nucleotide directly downstream of the PAM sequence (which is 5'-NGG-3' for S. pyogenes Cas9) is disfavored and is associated with reduced activity [32].
- sgRNA Secondary Structure: The internal structure of the sgRNA itself can impact its function. Guides with stable secondary structures, particularly in the seed region, should be avoided, as they can hinder the formation of the sgRNA-DNA complex. Tools like ViennaRNA can be used to predict and avoid such structures [32].
- Homopolymeric Sequences: sgRNAs containing continuous runs of identical nucleotides, especially four or more consecutive thymines (T), should be avoided. Such "T-stretches" can act as premature termination signals for RNA Polymerase III, reducing sgRNA expression [34].
Chromatin Features: Navigating the Epigenetic Landscape
The local chromatin environment presents a physical barrier to dCas9 binding. Accounting for this is essential for predicting sgRNA activity.
- Nucleosome Occupancy: Nucleosomes are fundamental units of chromatin that directly and profoundly impede dCas9 access to DNA. Target sites in nucleosome-dense regions are consistently less effective. Therefore, sgRNAs should be designed to target nucleosome-depleted regions [32].
- Chromatin Accessibility: More broadly, sgRNAs targeting regions of open chromatin, as defined by assays like DNase-seq or ATAC-seq, show higher efficiency. The chromatin accessibility of a target site is a strong independent predictor of CRISPRi functionality [33].
Table 2: Chromatin and Sequence-Based Design Rules
| Feature Category | Parameter | Design Rule | Experimental Validation |
|---|---|---|---|
| Chromatin | Nucleosome Occupancy | Favor nucleosome-depleted regions | Machine learning models identified nucleosome positioning as a top feature [32] |
| Chromatin Accessibility | Prefer target sites in open chromatin domains | sgRNA efficiency correlates with DNase-hypersensitive sites [33] | |
| Sequence | PAM-Proximal Sequence | Avoid guanine (G) directly downstream of PAM | Recapitulated from multiple independent studies [32] |
| sgRNA Structure | Avoid stable secondary structures in seed region | sgRNA folding energy is a contributing parameter in predictive models [32] | |
| Homopolymers | Avoid >3 consecutive thymine (T) nucleotides | Prevents premature transcription termination [34] |
Integrated Design Workflow and Experimental Validation
A Machine-Learning-Informed Workflow
Modern, highly active sgRNA libraries are designed using comprehensive machine learning models that integrate the features described above. A general workflow, as employed for the design of the CRISPRi v2 library, involves:
- Training Data Curation: Aggregating data from dozens of prior CRISPRi screens to generate a large dataset of sgRNAs with normalized activity scores.
- Feature Engineering: Encoding each sgRNA based on its position (relative to FANTOM5 TSSs), sequence features, and chromatin features (nucleosome positioning, accessibility).
- Model Training: Using regularized linear regression (e.g., elastic net) or other algorithms to learn the weighted contribution of each feature to the final sgRNA activity score.
- Library Design: Selecting the top-ranked sgRNAs for each gene based on the model's predictions, resulting in a library highly enriched for active guides [32].
The following diagram illustrates the logical flow of this integrated design and validation process.
Experimental Protocols for Library Validation
After library design and construction, it is crucial to validate its performance through a focused screen. A genome-wide growth screen for essential genes serves as a robust benchmark.
Protocol: Essential Gene Growth Screen for CRISPRi Library Validation
Cell Line Engineering:
- Generate a clonal cell line (e.g., K562, RPE1) that stably expresses the dCas9-effector protein (e.g., dCas9-KRAB, Zim3-dCas9) via lentiviral transduction and selection.
- Validate effector expression and function by testing knockdown of a control gene.
Library Transduction:
- Transduce the sgRNA library into the effector-expressing cells at a low Multiplicity of Infection (MOI ~0.3) to ensure most cells receive only one sgRNA. Include a representation of at least 200-500 cells per sgRNA to maintain library complexity.
- Use puromycin selection (e.g., 2 µg/ml for 3-7 days) to eliminate untransduced cells.
Phenotypic Selection:
- Harvest a reference sample of cells immediately after puromycin selection (T0).
- Culture the remaining cells for approximately 14-20 population doublings to allow depletion of cells targeting essential genes.
- Harvest the final population (Tfinal).
Sequencing and Analysis:
- Extract genomic DNA from T0 and Tfinal samples.
- Amplify the integrated sgRNA cassettes from the genomic DNA using PCR with primers containing Illumina adapters and sample barcodes.
- Pool the PCR products and perform next-generation sequencing.
- Map sequencing reads to the sgRNA library and count the abundance of each guide in T0 and Tfinal samples.
- Calculate a phenotype (e.g., growth score γ) for each sgRNA and gene using statistical models (e.g., MAGeCK, PinAPL-Py). Essential genes will be significantly depleted in the Tfinal sample [32] [35].
Advanced Strategies for Multiplexed Library Construction
Overcoming Functional Redundancy with Multi-Targeted sgRNAs
A significant challenge in pathway discovery is functional redundancy among genes in the same family. A powerful solution is the design of multi-targeted sgRNA libraries, where a single sgRNA is designed to target conserved sequences across multiple paralogous genes.
- Design Principle: Using algorithms like CRISPys, phylogenetic trees of gene families are reconstructed. sgRNAs are then designed to target homologous sequences shared among closely related subgroup members, ideally within the first two-thirds of the coding sequence [8].
- Application: This approach has been successfully implemented to create genome-scale libraries in plants and can be directly adapted for human pathway discovery. It allows for the simultaneous knockout of multiple redundant genes with a single sgRNA, unmasking phenotypes that would be buffered in a single-gene knockout [8].
Enhancing Efficacy with Dual-sgRNA and Optimized Effectors
To maximize knockdown efficacy and enable the use of more compact libraries, two advanced strategies have emerged:
- Dual-sgRNA Libraries: Instead of using a single sgRNA per gene, library elements can encode a tandem cassette expressing the two most active sgRNAs for a target gene. This approach has been shown to produce significantly stronger phenotypic effects (e.g., a 29% greater decrease in growth rate for essential genes) compared to single-sgRNA designs, enabling the creation of highly compact and effective libraries [35].
- Optimized Effector Proteins: The choice of the repressor domain fused to dCas9 impacts both on-target efficacy and non-specific toxicity. Comparative studies have shown that the Zim3-dCas9 effector provides an excellent balance, offering strong on-target knockdown with minimal non-specific effects on cell growth or the transcriptome [35].
Table 3: Key Research Reagents for Multiplexed CRISPRi Library Construction
| Reagent / Resource | Function / Description | Example / Source |
|---|---|---|
| dCas9-Effector Plasmids | Catalytically dead Cas9 fused to a repressor domain for CRISPRi. | Zim3-dCas9 (Addgene #167165), dCas9-KRAB (Addgene #109028) [35] |
| sgRNA Backbone | Vector for cloning and expressing sgRNAs, often with a U6 promoter. | pU6-sgRNA-EF1α-puro-T2A-BFP (Addgene #60955) [33] |
| Validated sgRNA Libraries | Pre-designed, sequence-verified libraries for genome-scale screening. | hCRISPRi-v2 (Addgene #113869), Dolcetto (Compact dual-sgRNA) [32] [35] |
| TSS Annotations | Reference data for accurate transcription start site locations. | FANTOM5/CAGE Robust Promoter Set [32] [33] |
| Chromatin Data | Reference data on nucleosome positioning and open chromatin. | ENCODE DNase-seq, ATAC-seq, and MNase-seq datasets [33] |
| Design Algorithms | Computational tools for predicting highly active sgRNAs. | Machine learning model from Horlbeck et al. 2016; CRISPys for multi-targeting [32] [8] |
| Analysis Software | Tools for quantifying sgRNA abundance and calculating gene phenotypes from screen data. | MAGeCK, PinAPL-Py [32] |
The following diagram outlines the key steps in the experimental workflow for conducting a CRISPRi screen, from cell line preparation to hit identification.
The construction of CRISPR-based genetic libraries represents a cornerstone of modern functional genomics. As research expands into more complex biological models and pathways, the need for highly efficient and compact libraries has intensified. This technical guide examines the strategic choice between single-sgRNA and dual-sgRNA library architectures, focusing on their application in multiplexed CRISPRi library construction for pathway discovery research. We present quantitative comparisons of knockdown efficacy, library size compression, and functional performance across diverse screening contexts. The implementation of dual-sgRNA libraries enables significant library miniaturization while maintaining or enhancing screening sensitivity, offering researchers powerful tools for dissecting complex genetic networks and identifying synthetic lethal interactions in disease-relevant pathways.
CRISPR interference (CRISPRi) technology utilizes a catalytically dead Cas9 (dCas9) fused to transcriptional repressor domains to achieve programmable gene knockdown without DNA cleavage [36]. This approach provides reversible, titratable repression of gene expression, making it particularly valuable for studying essential genes and complex genetic pathways [35]. The design of sgRNA libraries is a critical determinant of screening success, balancing the competing priorities of high on-target efficacy, minimal off-target effects, and practical library size. Single-sgRNA libraries, which typically employ multiple guides per gene (commonly 3-10), have been the conventional workhorse for genome-wide screens [32]. More recently, dual-sgRNA libraries, where two highly active guides are combined in a single construct to target the same gene, have emerged as a strategy to enhance knockdown efficacy and enable substantial library compression [35]. For researchers focused on pathway discovery, the choice between these architectures influences not only screening sensitivity and cost but also the ability to interrogate genetic interactions and redundant biological systems.
Performance Comparison: Quantitative Analysis
Direct comparisons between single and dual-sgRNA libraries reveal distinct performance advantages and considerations for each approach. The tables below summarize key quantitative findings from recent benchmark studies.
Table 1: Performance Metrics of Single versus Dual-sgRNA Libraries in Essentiality Screens
| Library Type | Average Guides per Gene | Essential Gene Depletion (γ) | Non-essential Gene Enrichment | Key Observations |
|---|---|---|---|---|
| Standard Single (Yusa v3) | 6 | Moderate | Standard | Baseline performance |
| Optimized Single (Top3-VBC) | 3 | Strong | Standard | Performance comparable or superior to larger libraries |
| Dual-sgRNA | 2 (as pairs) | Strongest | Weaker | Enhanced essential gene depletion; potential fitness cost |
Data derived from benchmark studies demonstrate that libraries with fewer but better-designed guides can perform as well as or better than larger libraries [37]. In growth-based essentiality screens, a dual-sgRNA library targeting essential genes produced significantly stronger growth phenotypes (mean growth rate γ = -0.26) compared to a single-sgRNA library (mean γ = -0.20) [35]. This suggests that dual targeting confers stronger depletion of target genes, potentially through more potent knockdown.
Table 2: Library Size and Efficiency Comparison
| Parameter | Conventional Single Library | Minimized Single Library | Dual-sgRNA Library |
|---|---|---|---|
| Typical Guides Per Gene | 5-10 | 2-3 | 1-2 elements (2 guides) |
| Relative Library Size | 100% | 30-50% | 20-40% |
| Screening Cost | High | Reduced | Significantly Reduced |
| Feasibility in Complex Models | Limited | Improved | Greatly Improved |
Compact libraries enable more cost-effective screens with reduced reagent and sequencing costs, increased throughput, and improved feasibility for applications with limited material, such as organoids or in vivo models [37]. The dual-sgRNA approach has been successfully implemented to create an "ultra-compact" genome-wide CRISPRi library where each gene is targeted by a single library element encoding a dual-sgRNA cassette [35].
Mechanisms of Enhanced Knockdown
Dual-sgRNA Synergy
The enhanced efficacy observed with dual-sgRNA libraries stems from several synergistic mechanisms. When two sgRNAs are directed against the same gene, the repressor complexes can bind to multiple sites within the promoter region, potentially leading to more potent transcriptional repression by occluding a larger region of the DNA from transcriptional machinery [35]. This multi-site blocking is particularly effective in the nucleosome-depleted region immediately downstream of the transcription start site (TSS), which is crucial for transcriptional initiation [32]. Furthermore, the presence of multiple dCas9-repressor complexes may facilitate cooperative interactions, leading to more stable and prolonged repression.
Algorithmic Guide RNA Design
Advanced computational algorithms have been developed to optimize sgRNA selection, incorporating chromatin accessibility, nucleosome positioning, sequence features, and secondary structure predictions. Machine learning models trained on empirical screening data can accurately predict highly effective sgRNAs by integrating multiple parameters [32]. The position relative to the TSS, particularly within the nucleosome-deprived region immediately downstream, is a major determinant of CRISPRi activity [32]. These algorithmic advances enable the selection of a minimal number of highly effective guides for both single and dual-library architectures, maximizing the efficacy of compressed libraries.
Experimental Protocols and Workflows
Dual-sgRNA Library Construction
The construction of a dual-sgRNA library requires careful planning and execution. The following protocol outlines the key steps for generating a lentiviral dual-sgRNA library for CRISPRi screening:
- sgRNA Selection and Oligonucleotide Design: Select the top two predicted sgRNAs per gene using established design algorithms (e.g., VBC scores, Rule Set 3) [37]. Design oligonucleotides encoding each sgRNA with appropriate flanking sequences for cloning, including unique 4-bp overhangs (e.g., TGAA) for Golden Gate assembly [9].
- Vector Preparation: Select a lentiviral backbone containing two distinct RNA polymerase III promoters (e.g., human U6 and mouse U6) to drive expression of the two sgRNAs and minimize recombination [15]. Digest the vector with appropriate restriction enzymes to create compatible ends.
- Golden Gate Assembly: Perform a one-pot Golden Gate assembly reaction using the synthesized oligonucleotide pool and prepared vector backbone. This method utilizes a type IIS restriction enzyme (e.g., BsaI) that cleaves outside its recognition site, enabling seamless assembly of multiple DNA fragments [15].
- Library Transformation and Amplification: Transform the assembled library into a highly competent E. coli strain (e.g., Endura Electrocompetent Cells) to ensure maximum library diversity. Harvest the plasmid library from the bacterial culture using a maxiprep kit.
- Quality Control: Validate library representation by next-generation sequencing (NGS) to ensure even distribution of all sgRNA pairs. Check for contamination via PCR and gel electrophoresis [38].
- Lentiviral Production: Package the sgRNA library into lentiviral particles by co-transfecting the library plasmid with packaging plasmids (psPAX2 and pMD2.G) into HEK293T cells. Harvest the virus-containing supernatant, concentrate if necessary, and titer the lentiviral stock.
Screening Implementation and Hit Identification
For the functional screen, transduce target cells expressing the appropriate dCas9-effector (e.g., dCas9-KRAB or Zim3-dCas9) with the lentiviral dual-sgRNA library at a low multiplicity of infection (MOI ~0.3) to ensure most cells receive only one sgRNA pair. Apply selection pressure (e.g., puromycin) to eliminate untransduced cells. At this point (T0), harvest a reference sample of cells for genomic DNA extraction. Culture the remaining cells under the selective condition of interest for an appropriate duration (e.g., 14-21 days for a negative selection screen), then harvest the final population (Tfinal). Extract genomic DNA from both timepoints, amplify the integrated sgRNA cassettes by PCR, and quantify sgRNA abundance by high-throughput sequencing. Analyze the data by comparing the fold-change of each sgRNA pair from T0 to Tfinal using specialized algorithms (e.g., MAGeCK or Chronos) to identify hits [37].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Dual-sgRNA CRISPRi Library Construction and Screening
| Reagent / Tool | Function | Examples / Specifications |
|---|---|---|
| dCas9 Effector | Transcriptional repressor fusion | dCas9-KRAB, Zim3-dCas9, dCas9-SALL1-SDS3 [35] [36] |
| Algorithmically Designed Guides | Target-specific sgRNA selection | VBC scores, Rule Set 3; designed for regions 0-300 bp downstream of TSS [37] [32] |
| Dual-Promoter Vector | Simultaneous expression of two sgRNAs | Lentiviral backbone with human U6 and mouse U6 promoters [15] |
| Golden Gate Assembly System | Modular cloning of sgRNA pairs | Type IIS restriction enzymes (e.g., BsaI) with unique 4-bp overhangs [9] [15] |
| NGS Platform | Library quality control and hit deconvolution | Illumina sequencing for sgRNA abundance quantification |
| Analysis Software | Statistical analysis of screen data | MAGeCK, Chronos algorithm for time-series data [37] |
Applications in Pathway Discovery and Addressing Redundancy
Multiplexed CRISPRi libraries, particularly dual-sgRNA formats, are uniquely powerful for dissecting biological processes characterized by functional redundancy, such as signaling pathways with parallel arms or metabolic routes with isozymes. In such cases, silencing a single gene often fails to produce a phenotype due to compensation by related genes, making traditional screening approaches ineffective. The MuRCiS (Multiplexed, Randomized CRISPRi Sequencing) approach addresses this by enabling the comprehensive interrogation of all pairwise combinations of genes from a targeted set to identify synthetic lethal interactions [9]. This method was successfully applied to Legionella pneumophila, where it identified previously unknown synthetic lethal pairs among virulence genes that were missed when silencing predetermined groups of ten genes [9]. For pathway discovery in mammalian systems, dual-sgRNA libraries can be designed to target multiple components of hypothesized pathways simultaneously, revealing functional connections and compensatory mechanisms that define network architecture and robustness. This approach is invaluable for identifying therapeutic targets in complex diseases like cancer, where pathway redundancy often underlies treatment resistance.
Considerations and Future Directions
While dual-sgRNA libraries offer significant advantages, they also present unique considerations. Some studies have reported a modest fitness cost associated with dual targeting, even for non-essential genes, potentially due to an heightened DNA damage response from multiple dCas9 binding events [37]. This observation warrants caution in certain screening contexts. The choice of CRISPRi effector also impacts performance; recent comparisons indicate that the Zim3-dCas9 effector provides an excellent balance between strong on-target knockdown and minimal non-specific effects on cell growth or the transcriptome [35].
Future research directions will likely focus on further library optimization, including the exploration of 2-guide single libraries and the refinement of dual-sgRNA pairing strategies. The integration of dual-sgRNA libraries with single-cell readouts (e.g., Perturb-seq) represents a powerful frontier for unraveling complex transcriptional responses to combinatorial gene knockdown [15]. Additionally, the development of inducible and tissue-specific systems will enhance the application of these compact libraries in vivo and in complex disease models.
The strategic implementation of dual-sgRNA libraries represents a significant advancement in CRISPRi technology, offering enhanced knockdown efficacy and substantial library compression. For pathway discovery research, this architecture enables more cost-effective and feasible screens in complex models, while providing a powerful tool for probing genetic redundancy and identifying synthetic lethal interactions. As algorithmic design improves and our understanding of the potential cellular responses to multiplexed knockdown deepens, dual-sgRNA and other compact library formats are poised to become indispensable tools for systematically mapping genetic networks and accelerating therapeutic discovery.
Vector Systems and Cloning Strategies for Multiplexed gRNA Expression
The capacity to express multiple guide RNAs (gRNAs) from a single construct represents a pivotal advancement in CRISPR-based research, enabling sophisticated experimental applications that were previously challenging or impossible. Multiplexed CRISPR systems fundamentally enhance the scope of pathway discovery research by allowing simultaneous perturbation of multiple genetic targets within a single cell [15] [11]. This capability is particularly valuable for addressing functional redundancy in gene families, modeling polygenic diseases, and conducting comprehensive genetic screens that more accurately reflect the complexity of biological systems [8] [13]. For researchers constructing CRISPRi libraries for pathway discovery, multiplexing provides a powerful framework for deciphering complex genetic interactions and identifying synthetic lethal relationships that remain invisible in single-gene perturbation experiments [39] [40].
The technological foundations for multiplexed gRNA expression stem from natural CRISPR systems, where organisms inherently process multiple CRISPR RNAs from a single transcriptional unit [11]. Synthetic biology has harnessed and refined these natural processing mechanisms to develop the arrayed systems in use today. This technical guide provides a comprehensive overview of current vector systems, cloning methodologies, and experimental considerations for implementing multiplexed gRNA strategies in pathway discovery research, with a specific focus on CRISPRi library construction.
Vector Architectures for Multiplexed gRNA Expression
The design of the vector backbone directly influences the efficiency, stability, and functional output of multiplexed CRISPR systems. Three primary architectural strategies have emerged for expressing multiple gRNAs, each with distinct advantages and considerations for library construction.
Multi-Promoter Systems
The most straightforward approach involves assembling multiple individual promoter-gRNA-terminator cassettes within a single vector. This architecture provides independent transcriptional control for each gRNA and typically utilizes RNA polymerase III promoters (such as U6, H1, or 7SK) due to their precise transcription initiation and termination characteristics [41] [11]. For mammalian systems, the pX333 vector exemplifies this approach, containing humanized wild-type Cas9 and two different U6 promoters (human and mouse) to minimize recombination between identical promoter sequences [41]. The Gersbach lab multiplexing system extends this concept to 2-4 gRNAs using four different promoters (7SK, human U6, mouse U6, and human H1) with destination vectors that offer fluorescent selection markers to identify cells with high Cas9 and gRNA expression [41].
A significant challenge with this approach is the potential for homologous recombination between identical promoter sequences, which can lead to vector instability during bacterial amplification and eventual gRNA cassette loss [41] [11]. Strategies to mitigate this include using heterologous promoters with varying sequences but similar transcriptional activity or implementing recombination-deficient bacterial strains during plasmid propagation.
Polycistronic Transcript Systems
Polycistronic systems consolidate multiple gRNA sequences into a single transcriptional unit that is subsequently processed into individual functional gRNAs. This approach offers advantages in vector size and delivery efficiency, particularly critical for viral vector systems with limited packaging capacity [41] [11]. Several processing mechanisms enable this strategy:
- Csy4 Processing: The Csy4 RNA nuclease from Pseudomonas aeruginosa efficiently cleaves gRNAs flanked by 28-base recognition sequences. The pSQT1313 system from the Joung lab implements this approach, enabling temporal and/or spatial control of gRNA processing based on Csy4 expression [41].
- tRNA Processing: Eukaryotic RNases P and Z recognize and cleave tRNA sequences, enabling the processing of tRNA-gRNA arrays. The polycistronic tRNA-gRNA (PTG) system developed by the Yang lab can express up to 8 gRNAs simultaneously and has been successfully implemented in plants, with vectors available for both transient expression and Agrobacterium-mediated transformation [41] [8].
- Ribozyme Processing: Self-cleaving ribozymes (such as Hammerhead and hepatitis delta virus ribozymes) flank each gRNA sequence, enabling precise excision without requiring additional protein co-factors. This system functions with both Pol II and Pol III promoters and has been demonstrated in multiple organisms [11] [42].
Endogenous CRISPR Array Processing
Some CRISPR systems inherently process multiple gRNAs from a single transcript. Most notably, Cas12a (Cpf1) possesses intrinsic RNase activity that cleaves pre-crRNA arrays, making it particularly suitable for multiplexed applications without requiring additional processing components [11]. This approach has been successfully implemented in human cells, plants, yeast, and bacteria, with demonstrations of up to five simultaneous genetic cleavages and ten concurrent transcriptional regulations [11].
Table 1: Comparative Analysis of Multiplexed gRNA Vector Architectures
| Architecture Type | Maximum gRNAs Demonstrated | Key Advantages | Key Limitations | Ideal Applications |
|---|---|---|---|---|
| Multi-Promoter Systems | 7 [41] | Independent transcriptional control; Predictable expression levels | Homologous recombination between identical promoters; Large vector size | Focused libraries; Dual targeting for large deletions |
| Polycistronic Transcript (Csy4) | 12 [11] | Compact design; Titratable control with Csy4 expression | Requires Csy4 co-expression; Potential cytotoxicity at high levels | Viral delivery; Inducible systems |
| Polycistronic Transcript (tRNA) | 8 [41] [8] | Endogenous processing; No additional components needed | Variable processing efficiency; Strain-dependent activity | Plant systems; Large-scale libraries |
| Polycistronic Transcript (Ribozyme) | 4 [42] | Self-processing; No protein cofactors required | Complex construct design; Larger sequence footprint | Eukaryotic systems; Stable cell lines |
| Endogenous Processing (Cas12a) | 10+ [11] | Native processing; Simplified design | Limited orthogonality; PAM sequence constraints | Bacterial systems; Transcriptional regulation |
Cloning Strategies for gRNA Array Assembly
The assembly of multiple gRNA expression cassettes presents technical challenges due to sequence repetition and the need for high-fidelity construction. Several cloning methodologies have been specifically adapted or developed to address these challenges.
Golden Gate Assembly
Golden Gate assembly utilizes Type IIS restriction enzymes (such as BsaI and BsmBI) that cleave outside their recognition sequences, creating unique overhangs that enable ordered, directional assembly of multiple fragments [41] [11]. This method is particularly well-suited for constructing gRNA arrays because it allows precise modular assembly without introducing scar sequences. The Yamamoto Lab Multiplex CRISPR/Cas9 Assembly Kit employs Golden Gate assembly to express up to 7 gRNAs, with custom destination vectors optimized for different gRNA numbers [41]. Similarly, systems developed for plants by the Qi-Jun Chen Lab and Liu Lab enable assembly of 2-4 and up to 8 gRNAs, respectively, using Golden Gate or Gibson Assembly methods [41].
STAgR (String Assembly gRNA Cloning)
The STAgR method represents an innovative approach that uses the gRNA targeting sequences (N20) themselves as homologies for Gibson assembly [43]. This system employs a universal "String" DNA fragment containing the gRNA scaffold and transcriptional terminator, which is amplified with primers containing the specific targeting sequences. The resulting PCR products assemble seamlessly in a single reaction, offering exceptional customization flexibility for gRNA numbers, combinations, and vector backbones [43]. STAgR has demonstrated functionality for both genome and transcriptome editing applications in vitro and in vivo, providing a cost-effective and efficient alternative to traditional methods.
Gateway Assembly
The Multiple Lentiviral Expression (MuLE) system from the Frew lab adapts Gateway recombination technology for multiplexed gRNA vector construction [41]. This system uses entry vectors containing U6 promoters and gRNA scaffolds compatible with type IIS enzyme BfuAI. Gateway cloning then combines multiple gRNAs and Cas9 into a single plasmid, enabling construction of lentiviral vectors expressing wild-type humanized Cas9 and up to three gRNAs [41]. While offering high efficiency and reliability, this system may have limitations in scalability compared to Golden Gate-based approaches.
BioBrick Assembly
For yeast systems, a sequential BioBrick assembly approach has proven effective for constructing gRNA arrays [42]. This method involves assembling individually expressed gRNAs as standardized genetic parts, enabling the construction of arrays with up to five gRNAs demonstrated in Saccharomyces cerevisiae [42]. The system utilizes SNR52 promoters to avoid strain-dependent activity issues associated with tRNA promoters and provides reliable performance without requiring additional processing components like ribozymes or Csy4 endonuclease [42].
Table 2: Cloning Method Comparison for Multiplexed gRNA Vector Construction
| Cloning Method | Principle | Key Enzymes/Reagents | Typical Throughput | Technical Considerations |
|---|---|---|---|---|
| Golden Gate Assembly | Type IIS restriction enzymes creating unique overhangs | BsaI, BsmBI | 2-10 gRNAs | Most widely adopted; Modular and scalable |
| STAgR | Gibson assembly using N20 as homologies | Gibson assembly mix | Highly customizable (demonstrated with high numbers) | Single-step reaction; Highly flexible |
| Gateway Assembly | Site-specific recombination | BP/LR Clonase | Up to 3 gRNAs | High efficiency; Limited by entry vector availability |
| BioBrick Assembly | Standardized part assembly | Restriction enzymes (EcoRI, XbaI, SpeI, PstI) | Up to 5 gRNAs (demonstrated) | Standardized parts; Sequential cloning required |
Experimental Design and Workflow
Implementing a robust multiplexed CRISPR system requires careful planning and execution across multiple stages. The following workflow outlines key considerations for successful implementation in pathway discovery research.
gRNA Selection and Design
Effective multiplexed editing begins with strategic gRNA design. For CRISPRi library construction targeting biological pathways, gRNAs should be designed to target early exons or promoter regions of genes within the pathway of interest [39] [40]. Computational tools such as CRISPys enable the design of sgRNAs that target conserved sequences across multiple gene family members, simultaneously editing several genes to overcome functional redundancy [8]. When designing libraries, calculate on-target scores using established algorithms like Cutting Frequency Determination (CFD) and apply strict thresholds (e.g., >0.8) to ensure high cleavage efficacy [8]. Specificity must be verified through comprehensive off-target scanning with stricter thresholds for exonic regions (e.g., 20% of on-target score) compared to other genomic regions (e.g., 50% of on-target score) [8].
Vector Assembly and Validation
Following the selection of an appropriate cloning strategy, careful attention to vector assembly is critical. For Golden Gate assembly, prepare high-quality plasmid DNA for all modular parts and optimize enzyme-to-DNA ratios to ensure complete digestion and efficient ligation [41]. When using systems requiring protein co-expression (e.g., Csy4), ensure balanced expression of all components to minimize potential cytotoxicity [41] [11]. Following assembly, validate constructs through restriction digest patterns and Sanger sequencing of junction regions, with particular attention to repetitive elements that may be prone to recombination [42]. For large-scale library construction, implement barcode tagging systems like CRISPR-GuideMap, which uses double barcodes to enable efficient sgRNA tracking in generated cell lines or organisms [8].
Delivery and Selection
Lentiviral delivery remains the most efficient method for introducing multiplexed CRISPR libraries into mammalian cells [39] [40]. For CRISPRi applications, utilize doxycycline-inducible dCas9 systems inserted at safe harbor loci (such as AAVS1) to enable controlled gene repression [40]. When transducing cells, maintain a low multiplicity of infection (MOI < 0.3) to ensure most cells receive only one gRNA construct and implement appropriate selection markers (e.g., puromycin, blasticidin) to enrich for successfully transduced cells [39] [40]. For arrayed library formats, as demonstrated in the SNAP (Streptococcus mutans Arrayed CRISPRi) library, array individual CRISPRi strains in microtiter plates for high-throughput phenotypic screening [39].
Functional Validation
Comprehensive validation is essential to confirm system functionality. Assess editing efficiency through T7 endonuclease assays or tracking of indels by decomposition (TIDE) for knockout approaches [42]. For CRISPRi applications, measure target gene knockdown using RT-qPCR and confirm phenotypic outcomes through pathway-specific functional assays [39] [40]. In arrayed library screens, calculate metrics such as area under the curve (AUC) for growth curves to quantify growth defects in essential gene repression strains compared to controls [39]. For in vivo applications, such as the zebrafish system developed by the Wenbiao Chen lab, include visible markers (e.g., tyr gRNA causing hypopigmentation) to identify successfully modified cells [41].
Visualization of Experimental Workflow
The following diagram illustrates the comprehensive workflow for constructing and implementing multiplexed gRNA systems for pathway discovery research:
Diagram 1: Comprehensive Workflow for Multiplexed gRNA System Implementation. This workflow outlines the key decision points and processes for constructing and implementing multiplexed CRISPR systems, from initial design to data analysis.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of multiplexed gRNA systems requires access to specialized reagents and resources. The following table catalogues essential research tools referenced in this guide:
Table 3: Essential Research Reagents for Multiplexed gRNA Systems
| Reagent/Resource | Function/Application | Key Features | Source/Reference |
|---|---|---|---|
| pX333 Vector | Two-gRNA expression in mammalian systems | Contains hCas9 + two different U6 promoters | Ventura Lab [41] |
| Yamamoto Lab Multiplex Kit | Golden Gate assembly for up to 7 gRNAs | Custom destination vectors for different gRNA numbers | Yamamoto Lab [41] |
| Gersbach Lab Multiplex System | 2-4 gRNA expression with fluorescent selection | Four different promoters to prevent recombination | Gersbach Lab [41] |
| STAgR Cloning System | One-step gRNA multiplexing using Gibson assembly | Highly customizable; Uses N20 as homologies | Stricker Lab [43] |
| Multiple Lentiviral Expression (MuLE) | Gateway-based multiplexing for up to 3 gRNAs | Lentiviral delivery; Compatible with various Cas9 variants | Frew Lab [41] |
| Csy4 Processing System | Polycistronic gRNA processing | Titratable control with Csy4 expression; Compact design | Joung Lab (pSQT1313) [41] |
| tRNA-gRNA (PTG) System | Polycistronic processing using endogenous RNases | No additional components needed; Up to 8 gRNAs | Yang Lab [41] [8] |
| CRISPys Algorithm | Computational sgRNA design for gene families | Designs sgRNAs targeting multiple gene family members | [8] |
| CRISPR-GuideMap | Double barcode system for sgRNA tracking | Enables large-scale sgRNA identification in plants | [8] |
Applications in Pathway Discovery Research
Multiplexed gRNA systems offer particular advantages for pathway discovery research, where biological responses often emerge from complex genetic interactions rather than single gene effects.
Functional Genomics Screening
Arrayed CRISPRi libraries enable systematic interrogation of gene function within biological pathways. The SNAP library in Streptococcus mutans exemplifies this approach, with arrayed strains targeting >250 essential and growth-supporting genes enabling high-throughput characterization of gene function in pathways relevant to virulence and biofilm formation [39]. Similarly, in human cells, inducible CRISPRi screens in hiPS cells and differentiated lineages have revealed cell-type-specific dependencies on mRNA translation machinery, identifying context-specific essential genes that would be missed in traditional cell line models [40].
Overcoming Genetic Redundancy
Many signaling pathways contain paralogous genes with overlapping functions, creating buffering capacity that confounds traditional genetic approaches. Multiplexed CRISPR systems directly address this challenge by enabling simultaneous targeting of multiple family members. The tomato multi-targeted CRISPR library applies this strategy at genome scale, with 15,804 unique sgRNAs designed to target multiple genes within the same families, successfully generating mutants with distinct phenotypes in fruit development, flavor, nutrient uptake, and pathogen response [8]. Similar approaches have been applied to study powdery mildew resistance in plants, where multiplex knockouts of three clade V MLO genes (Csmlo1 Csmlo8 Csmlo11) were necessary to achieve full resistance [13].
Polygenic Trait Engineering
Many agriculturally and clinically relevant traits are polygenic, controlled by multiple genes acting in concert. Multiplexed editing enables trait stacking through simultaneous modification of multiple genetic loci. The BREEDIT tool in maize demonstrates this application, facilitating multiplexed genome editing of several gene families to improve complex traits such as yield and drought tolerance [8]. In biomedical research, multiplexed CRISPRi enables the study of combinatorial gene repression in disease-relevant pathways, modeling the polygenic nature of complex diseases more accurately than single-gene approaches [40] [17].
Technical Considerations and Troubleshooting
Successful implementation of multiplexed gRNA systems requires attention to several technical considerations that can impact experimental outcomes.
Optimization Strategies
- Promoter Selection: Balance promoter strength with the potential for recombination. When using multiple identical promoters, consider incorporating point mutations in non-essential regions to reduce homology while maintaining function [11].
- gRNA Stoichiometry: For polycistronic systems, gRNA position within the array can influence expression levels. Place critical gRNAs in 5' positions if using systems with potential processing inefficiencies [41] [11].
- Delivery Optimization: For lentiviral delivery, carefully titrate viral particles to achieve optimal transduction efficiency without multiple integrations. Include fluorescence markers or antibiotic resistance genes to enrich for successfully transduced cells [39] [40].
Troubleshooting Common Issues
- Low Editing Efficiency: Verify Cas9/dCas9 expression and nuclear localization. For CRISPRi, confirm efficient target gene repression through RT-qPCR before phenotypic assessment [39] [40].
- Vector Instability: Use recombination-deficient bacterial strains (e.g., Stbl3) for plasmid propagation and minimize the number of serial amplifications. For systems with high sequence repetition, consider direct synthesis of repetitive regions [11] [42].
- Inconsistent gRNA Processing: For polycistronic systems, verify processing enzyme expression (e.g., Csy4) and consider adding nuclear localization signals to ensure proper cellular compartmentalization [41] [11].
Future Perspectives
The field of multiplexed genome engineering continues to evolve rapidly, with several emerging trends particularly relevant to pathway discovery research. Long-read sequencing technologies are improving the resolution of complex editing outcomes, including structural variations often missed by standard genotyping when targeting repetitive or tandemly spaced loci [13]. There is growing demand for user-friendly computational workflows that integrate gRNA design, construct assembly, and mutation analysis in a seamless pipeline [13]. Additionally, inducible and tissue-specific systems will enable more precise temporal and spatial control of multiplexed editing, allowing researchers to probe pathway function at specific developmental stages or in particular cell types [13]. As these tools mature, multiplexed CRISPR editing is poised to become a foundational technology for next-generation functional genomics and pathway discovery research.
Pooled CRISPR interference (CRISPRi) screening represents a powerful functional genomics platform that enables systematic, targeted gene silencing at scale. This technology utilizes a catalytically dead Cas9 (dCas9) fused to transcriptional repressors, which is guided by single guide RNAs (sgRNAs) to silence specific genomic loci without introducing DNA double-strand breaks. Unlike CRISPR knockout screens that permanently disrupt gene function, CRISPRi produces reversible knockdown effects, making it particularly valuable for studying essential genes and complex genetic interactions [44] [45]. The programmability of CRISPRi allows researchers to construct comprehensive libraries targeting thousands of genes, facilitating unbiased discovery of gene functions and pathways in diverse biological contexts.
The application of pooled CRISPRi screening has transformed multiple research domains, from basic biology to drug discovery. In primary human T cells, CRISPRi screening has identified regulators of T cell receptor signaling and genes that negatively tune proliferation following stimulation [46]. In bacterial systems, multiplexed CRISPRi approaches have helped overcome functional redundancy to identify critical virulence factor combinations [47]. As a scalable and cost-effective method, pooled CRISPRi screening continues to evolve with advanced readouts including single-cell RNA sequencing and high-content imaging, further enhancing its resolution and application space [19] [45].
CRISPRi Library Design and Construction
Fundamental Principles of Library Design
Effective CRISPRi library design requires careful consideration of multiple parameters to ensure optimal silencing efficiency and minimal off-target effects. CRISPRi libraries typically include multiple sgRNAs per gene (usually 4-10) to account for variable guide efficiency and provide robust coverage [44]. The libraries can range from focused sets targeting specific gene families to genome-wide collections encompassing all protein-coding genes. For CRISPRi applications, sgRNAs are typically designed to target the 5' region of genes, specifically from the transcriptional start site (TSS) to approximately 50-500 bp downstream, as this positioning maximizes transcriptional repression efficiency [47].
Several optimized CRISPRi library designs have been publicly available, including the Dolcetto library for human genome-wide CRISPRi containing five guides per gene [45]. The specificity of sgRNAs is paramount, and computational tools incorporating cutting frequency determination (CFD) scoring help identify guides with high on-target activity while minimizing off-target effects [8]. Modern design algorithms apply strict thresholds for off-target potential, with more stringent parameters for exonic regions compared to other genomic areas to maintain robustness in genome-scale applications [8].
Advanced and Specialized Library Designs
Multiplexed and Combinatorial Libraries: To address biological redundancy and identify genetic interactions, researchers have developed multiplexed CRISPRi systems capable of simultaneously silencing multiple genes. The MuRCiS (Multiplexed, Randomized CRISPR interference Sequencing) approach exemplifies this advancement, utilizing randomized self-assembly of CRISPR arrays from synthetic oligonucleotide pairs to comprehensively interrogate pairwise gene combinations [47]. This method is particularly valuable for investigating synergistic processes typically carried out by groups of redundant proteins with similar functions.
Multi-Targeted Libraries: For organisms with significant genetic redundancy, such as plants where approximately 64.5% of genes belong to paralogous gene families, specialized library designs have emerged. Multi-targeted CRISPR libraries incorporate sgRNAs that target conserved sequences across multiple genes within the same family, enabling simultaneous editing of several functionally redundant genes [8]. This approach effectively overcomes phenotypic buffering that often limits traditional genetic screens.
Barcoded Library Systems: Recent advancements incorporate barcoding strategies to enhance sgRNA tracking and quantification. Double-barcode tagging systems, such as CRISPR-GuideMap, significantly improve the scalability and precision of sgRNA identification in complex pooled screens [8]. Similarly, the Perturb-seq vector incorporates guide barcodes (GBCs) on synthetic polyadenylated transcripts that are captured during single-cell RNA sequencing, enabling precise mapping of perturbations to transcriptional profiles [48].
Table 1: CRISPR Library Types and Their Applications
| Library Type | Key Features | Target Capacity | Primary Applications |
|---|---|---|---|
| Genome-wide | 4-10 sgRNAs per gene | ~20,000 genes | Unbiased discovery, essential gene identification [44] [45] |
| Multiplexed/Combinatorial | Multiple sgRNAs per vector | 10+ genes simultaneously | Genetic interactions, synthetic lethality [47] |
| Multi-targeted | sgRNAs targeting conserved family regions | 2-8 genes per sgRNA | Overcoming genetic redundancy [8] |
| Focused | High coverage of specific gene families | 100-1,500 genes | Pathway-specific screening [45] |
| CRISPRa | dCas9 fused to transcriptional activators | ~20,000 genes | Gain-of-function studies [44] [45] |
Delivery Methods Across Cell Types
Lentiviral Delivery Systems
Lentiviral transduction remains the gold standard for delivering sgRNA libraries into diverse cell types due to its high efficiency and stable genomic integration. The process involves transducing cells at low multiplicity of infection (MOI typically 0.3-0.5) to ensure most infected cells receive only one sgRNA construct, maintaining the crucial single-perturbation-per-cell principle [44]. Following transduction, antibiotic selection or fluorescence-based methods eliminate uninfected cells, generating a mutagenized population where each cell carries a defined genetic perturbation [45].
For successful library representation, genome-wide screens require maintaining 500-1,000 cells per sgRNA throughout the experiment. This demands starting populations of 50-100 million cells for a 100,000 sgRNA library to prevent stochastic guide loss from random sampling effects [44]. The lentiviral vectors used in these systems often incorporate advanced features, such as the Perturb-seq vector which contains both an RNA polymerase II-driven "guide barcode expression cassette" and an RNA polymerase III-driven "sgRNA expression cassette" to enable simultaneous perturbation and tracking [48].
Specialized Delivery Methods for Challenging Systems
Primary Human T Cells: Delivery of CRISPR components to primary human immune cells presents unique challenges due to their limited ex vivo lifespan and resistance to standard transfection methods. The SLICE (sgRNA lentiviral infection with Cas9 protein electroporation) platform addresses this by combining lentiviral sgRNA delivery with electroporation of Cas9 protein, enabling efficient genome editing in primary T cells [46]. This hybrid approach has successfully identified regulators of T cell stimulation responses and mediators of immunosuppression in genome-wide screens.
Insect Cells: The IntAC (integrase with anti-CRISPR) system was developed for Drosophila and mosquito cell lines where retroviral delivery is inefficient. This method utilizes site-specific recombination coupled with transient anti-CRISPR protein expression to suppress Cas9 activity until stable sgRNA integration occurs [49]. The IntAC approach dramatically improves genotype-phenotype mapping accuracy by preventing early CRISPR activity that can cause discrepancies between genome edits and integrated sgRNAs.
Bacterial Systems: In organisms like Legionella pneumophila, CRISPRi delivery utilizes chromosomal integration of dCas9 followed by plasmid-based sgRNA library delivery [47]. This system enables multiplexed gene silencing even in bacterial species lacking efficient DNA repair mechanisms for handling double-strand breaks.
Table 2: Delivery Methods for Different Cell Types
| Cell Type | Delivery Method | Key Features | Applications |
|---|---|---|---|
| Primary Human T Cells | SLICE: Lentiviral sgRNA + Cas9 protein electroporation | Overcomes low transduction efficiency; enables screening in short-lived primary cells [46] [50] | Immunotherapy target discovery; T cell activation regulation [46] |
| Standard Mammalian Cell Lines | Lentiviral transduction (stable dCas9 + sgRNAs) | High efficiency; stable integration; well-established protocols [44] [45] | Essential gene identification; drug mechanism of action [44] |
| Insect Cells | IntAC: Plasmid transfection with anti-CRISPR and integrase | Site-specific integration; temporal control of Cas9 activity [49] | Drosophila genetic screening; toxin resistance studies [49] |
| Bacterial Cells | Chromosomal dCas9 integration + plasmid sgRNAs | Enables CRISPRi in DNA repair-deficient strains [47] | Bacterial virulence factor identification; essential gene mapping [47] |
Screening Protocols and Workflows
Core Screening Methodologies
Negative Selection Screens: These screens identify genes essential for cell survival or proliferation under specific conditions. Cells transduced with sgRNA libraries are passaged continuously for 2-4 weeks while maintaining library representation. sgRNAs targeting essential genes become progressively depleted as cells containing these knockouts fail to proliferate or die. The comparison of sgRNA abundance at the final timepoint versus the initial population (T0) reveals genetic dependencies in the experimental conditions [44]. Negative selection screens have successfully identified cell-line-specific essential genes and context-dependent vulnerabilities across hundreds of cancer cell lines.
Positive Selection Screens: In these screens, selective pressures (e.g., drug treatment, toxin exposure) kill most cells, enriching for sgRNAs conferring survival advantage. Resistance screens treat library-infected cells with therapeutics at lethal concentrations, enriching for sgRNAs that disrupt drug targets, activate resistance pathways, or block cell death signaling [44]. Positive selection has identified both known and novel resistance mechanisms, providing insights into drug mechanisms of action and potential clinical resistance pathways.
FACS-Based Screens: Fluorescence-activated cell sorting enables screens for genes regulating any fluorescently measurable phenotype. Cells expressing fluorescent reporters under pathway control are sorted based on expression levels, with sgRNA abundance compared between populations [44]. This approach identifies both positive regulators (enriched when disrupted in low-expression populations) and negative regulators (enriched when disrupted in high-expression populations). Multiparameter FACS further enhances phenotypic discrimination by simultaneously measuring multiple markers.
Advanced Screening Modalities
Single-Cell RNA Sequencing Readouts: Technologies like Perturb-seq and CROP-seq combine pooled CRISPR screening with single-cell RNA sequencing, enabling high-content transcriptional profiling of genetic perturbations. These methods incorporate cell barcoding strategies that encode perturbation identity in expressed transcripts, which are captured during droplet-based single-cell RNAseq [48] [45]. This approach allows decomposition of complex cellular responses and identification of distinct transcriptional programs affected by genetic perturbations.
Image-Based Screening: Automated microscopy coupled with sophisticated image analysis enables screens for morphological phenotypes inaccessible to other methods. Unlike pooled screens, image-based approaches typically use arrayed formats (one or few guides per well), extracting hundreds of morphological features per cell [44]. Machine learning algorithms then classify phenotypes, identifying guides that produce characteristic morphological changes. Live-cell imaging extends this capability to track dynamic processes like cell division or migration over time.
Synthetic Lethality Screening: These screens identify genetic interactions where simultaneous perturbation of two genes is lethal while individual perturbations are viable. Combinatorial screens using dual-sgRNA libraries systematically test gene pairs, mapping genetic networks and identifying potential combination therapeutic strategies [44]. While technically challenging due to factorial increases in library complexity, focused combinatorial screens targeting druggable gene families reveal therapeutic combinations that could prevent resistance.
CRISPRi Screening Workflow: This diagram outlines the major steps in a pooled CRISPRi screen, from library design through hit validation, highlighting key decision points for phenotypic readout methods.
Pathway Discovery Through Multiplexed CRISPRi
Unraveling Biological Redundancy
Multiplexed CRISPRi screening has proven particularly valuable for dissecting biological processes characterized by functional redundancy. The MuRCiS approach demonstrates this capability in Legionella pneumophila, where it identified synthetic lethal combinations among 44 virulence genes encoding conserved transmembrane proteins [47]. This randomized array assembly method enabled near-comprehensive interrogation of all pairwise combinations, revealing that lpg2888 and lpg3000 function redundantly during human macrophage infection, while lpg3000 alone was essential for virulence in amoebae hosts. Such insights would remain inaccessible through conventional single-gene perturbation approaches due to pervasive redundancy in virulence factor arsenals.
In mammalian systems, Perturb-seq has enabled functional clustering of genes within complex pathways like the unfolded protein response (UPR). By combining single and combinatorial CRISPRi perturbations with single-cell RNA sequencing, researchers decoupled the three UPR branches (IRE1α, ATF6, and PERK) and revealed bifurcated activation patterns among cells subject to identical perturbations [48]. This high-resolution approach identified an isolated feedback loop between the translocon and IRE1α, demonstrating how specialized monitoring systems operate within broader stress response pathways.
Mapping Genetic Networks
Combinatorial CRISPRi screening facilitates systematic genetic interaction mapping, revealing synthetic lethal relationships and functional relationships between genes. The ability to simultaneously repress multiple genes with high efficacy using programmable CRISPR arrays has transformed our approach to genetic network analysis [48]. By systematically targeting gene pairs, researchers can construct comprehensive genetic interaction maps that reveal functional relationships and compensatory pathways, providing insights into cellular robustness mechanisms.
Advanced CRISPRi platforms now enable repression of up to three genes with high homogeneity across cell populations [48]. This capability is essential for precise genetic interaction studies, as variable repression efficiency can obscure true genetic interactions. The development of tandem sgRNA expression cassettes with different RNA polymerase III promoters minimizes intramolecular recombination during lentiviral transduction, ensuring maintained library diversity and representation [48].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for CRISPRi Screening
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| dCas9-KRAB Fusion | Catalytically dead Cas9 fused to KRAB repressor domain; enables transcriptional repression without DNA cleavage [48] [45] | Core component for CRISPRi; provides consistent repression across genomic contexts |
| Optimized sgRNA Libraries | Collection of guide RNAs targeting genes of interest; designed for minimal off-target effects [44] [45] | Available as genome-wide (e.g., Dolcetto) or focused libraries; include multiple guides per gene for robustness |
| Lentiviral Packaging System | Produces viral particles for efficient sgRNA library delivery [44] [45] | Enables stable genomic integration; requires biosafety level 2 containment |
| Perturb-seq Vectors | Specialized lentiviral vectors with guide barcode expression cassettes [48] | Enables single-cell RNA sequencing readouts; incorporates unique barcodes for perturbation tracking |
| Anti-CRISPR Proteins | Temporarily inhibits Cas9 activity to control editing timing [49] | Used in IntAC system to improve genotype-phenotype linkage; enables stronger sgRNA promoters |
| Barcoded Guide Libraries | sgRNA libraries with molecular barcodes for enhanced tracking [8] [45] | Improves quantification accuracy; enables complex screening paradigms |
| Stable Cell Lines | Cells with integrated dCas9 repressor systems [44] [45] | Provides uniform CRISPRi background; requires monitoring for silencing of integrated constructs |
The field of pooled CRISPRi screening continues to evolve with emerging technologies that enhance scale, resolution, and application scope. Base editor screens represent a promising frontier, introducing precise point mutations rather than complete gene knockout and enabling systematic mutagenesis of protein domains or regulatory elements [45]. Similarly, prime editing screens promise even greater precision for introducing or correcting specific mutations. These next-generation approaches will enable detailed dissection of protein structure-function relationships and disease-associated variants at unprecedented scale.
The integration of CRISPRi screening with advanced readouts is expanding the phenotypic space accessible to functional genomics. Methods combining optical phenotyping with padlock-based rolling circle amplification enable guide identification directly on microscopy slides, preserving spatial information while maintaining pooled screening efficiency [45]. As these technologies mature, they will provide increasingly multidimensional views of gene function across diverse biological contexts.
In conclusion, pooled CRISPRi screening has established itself as an indispensable tool for pathway discovery and functional genomics. The continuous refinement of delivery methods, library designs, and readout technologies has expanded its application from standard model cell lines to challenging primary cells and diverse organisms. As screening platforms become increasingly sophisticated, integrating multiple perturbation types with high-content phenotyping, they will continue to drive fundamental biological insights and accelerate therapeutic development across disease areas.
Solving Common Challenges in CRISPRi Library Construction and Deployment
CRISPR off-target editing refers to the non-specific activity of the Cas nuclease at sites other than the intended target, causing undesirable or unexpected effects on the genome [51]. This phenomenon occurs because wild-type CRISPR systems maintain a reasonable level of tolerance for mismatches between their target sequence and guide RNA (gRNA) – for instance, the wild-type Cas9 from Streptococcus pyogenes (SpCas9) can tolerate between three and five base pair mismatches, enabling potential double-stranded breaks at multiple genomic sites with similarity to the intended target and correct PAM sequence [51]. In the context of multiplexed CRISPRi library construction for pathway discovery, where dozens to hundreds of guide RNAs are deployed simultaneously, the risk of off-target effects multiplies, potentially confounding experimental results and leading to erroneous conclusions about gene function and pathway architecture.
The implications of off-target effects are particularly significant in functional genomics applications. When determining gene function via CRISPR knockout, off-target activity can make it difficult to ascertain whether observed phenotypes result from the intended edit or off-target effects [51]. For drug development professionals relying on these datasets to identify therapeutic targets, such ambiguity introduces unacceptable risk in the target validation pipeline. This technical guide outlines comprehensive computational and experimental strategies to identify, quantify, and minimize off-target effects, with specific consideration for the unique challenges presented by multiplexed CRISPRi screening approaches.
Computational Prediction and gRNA Design Strategies
Computational methods for off-target prediction leverage algorithmic models to identify potential unintended genomic sites associated with CRISPR/Cas9 editing [52]. These methods typically begin by comparing the target sgRNA sequence against the entire reference genome to locate potential off-target sites, evaluating factors such as sequence similarity, thermodynamic stability of regions adjacent to the PAM, and chromatin accessibility [52]. For multiplexed CRISPRi library construction, incorporating robust computational prediction at the design phase represents the most effective first line of defense against off-target effects.
Table 1: Computational Tools for Off-Target Prediction and gRNA Design
| Tool/Method | Primary Function | Key Features | Considerations for Multiplex Screening |
|---|---|---|---|
| CRISPOR | Guide RNA selection | Specialized algorithms for CRISPR off-target prediction; provides off-target scores [51] | Batch processing of multiple gRNAs essential for library design |
| CRISPys | Multi-targeted gRNA design | Designs sgRNAs targeting multiple genes within gene families; uses phylogenetic trees [8] | Particularly valuable for addressing functional redundancy in pathways |
| Cutting Frequency Determination (CFD) Scoring | Off-target potential quantification | Calculates on-target scores; typically filters sgRNAs with scores <0.8 [8] | Standardized thresholding enables consistent library-wide quality control |
| COSMID | Identification and validation of off-target sites | Web-based tool for identifying and validating CRISPR/Cas off-target sites [52] | Complementary validation for pre-selected gRNAs |
Advanced gRNA Design Principles for Multiplex Libraries
When designing gRNAs for multiplexed libraries, several sequence-specific factors significantly influence off-target potential. The seed region – the PAM-proximal 10–12 nucleotide region of the sgRNA – is particularly crucial for specific recognition and cleavage of target DNA [52]. Mismatches in this region are less tolerated than those in the distal region, informing design algorithms to prioritize perfect seed sequence complementarity [52].
For pathway discovery research, where targeting multiple genes within the same family is common, the multi-targeted CRISPR library approach offers strategic advantages. This method designs sgRNAs that target conserved sequences across multiple genes, allowing simultaneous editing of several gene family members while minimizing the total number of guides required [8]. In practice, one study designed 15,804 unique sgRNAs, each targeting multiple genes within the same families, with approximately 95% of sgRNAs targeting groups of two or three genes [8]. This strategy reduces overall library size and potential off-target sites while effectively addressing functional redundancy in biological pathways.
Additional gRNA optimization strategies include careful attention to GC content (optimal between 40-60%), which stabilizes the DNA:RNA duplex and increases on-target editing efficiency [51] [53]. Guide length also influences off-target likelihood, with shorter gRNAs of 20 nucleotides or less generally presenting lower risk [51]. Chemical modifications such as 2'-O-methyl analogs (2'-O-Me) and 3' phosphorothioate bond (PS) modifications can further reduce off-target edits while increasing editing efficiency at the target site [51].
Experimental Detection and Validation Methods
After computational prediction and gRNA design, experimental validation of off-target effects is essential, particularly for multiplexed CRISPRi libraries where complex interactions may occur. Detection methods generally fall into three categories: in vitro assays, in vivo/cellular assays, and post-screening validation techniques, each with distinct advantages and limitations for different experimental contexts.
Table 2: Experimental Methods for Detecting Off-Target Effects
| Method | Type | Key Principle | Detection Capability | Considerations for Multiplex Screening |
|---|---|---|---|---|
| Digenome-seq | In vitro | In vitro digestion of genomic DNA using Cas9/sgRNA complexes; sequencing of cleavage sites [52] | Genome-wide; sensitive | Requires high sequencing depth; suitable for pre-validation of library gRNAs |
| GUIDE-seq | Cellular | Uses oligonucleotide tags integrated at DSB sites; NGS identification [51] | Genome-wide; high sensitivity | More complex experimentally; may be challenging for pooled formats |
| CIRCLE-seq | In vitro | Circularization of genomic DNA and in vitro cleavage; high-sensitivity sequencing [51] | Highly sensitive; genome-wide | Cell-free system; can pre-screen library gRNAs before cellular experiments |
| BLESS | Cellular | Direct in situ breaks labelling, streptavidin enrichment and NGS [52] | Genome-wide; captures DSBs in fixed cells | Snapshots of DSBs at specific timepoints; requires optimization for cell type |
| Whole Genome Sequencing | Cellular | Comprehensive sequencing of entire genome [51] | Most comprehensive; detects chromosomal aberrations | Expensive; computational burden; typically used for final therapeutic validation |
Specialized Detection Strategies for Multiplexed CRISPRi Screening
For multiplexed CRISPRi library construction, traditional off-target detection methods may require adaptation to address the simultaneous presence of multiple gRNAs. Emerging approaches specifically designed for these applications include:
Multiplexed, single-cell CRISPR screening platforms combine droplet-based single-cell RNA-seq with barcoding strategies for CRISPR-mediated perturbations [54] [48]. This allows complex pools of cells with multiple perturbations to be interrogated in parallel, with single-cell resolution enabling detection of heterogeneous responses that might be masked in bulk measurements. In one implementation, researchers developed a robust cell barcoding strategy that encodes CRISPR perturbation identity in an expressed transcript captured during single-cell RNA-seq, enabling high-confidence assignment of gRNAs to individual cells [48].
CRISPR-GuideMap, a double-barcode tagging system, enhances sgRNA tracking in generated plants or cells [8]. Originally developed for plant systems, this approach could be adapted for mammalian CRISPRi screens to maintain precise linkage between gRNAs and observed phenotypes, particularly valuable when investigating complex pathways with redundant components.
MuRCiS (Multiplexed, Randomized CRISPRi-Seq) represents another innovative approach, enabling randomized self-assembly of CRISPR arrays from synthetic oligonucleotide pairs [9]. When paired with long-read sequencing, this method allows near-comprehensive interrogation of all pairwise combinations of gene perturbations, identifying synthetic lethal relationships and functional redundancies within pathways while monitoring for unexpected phenotypes potentially indicative of off-target effects [9].
Diagram 1: Comprehensive workflow for off-target assessment in multiplexed CRISPRi screening, spanning design, detection, and validation phases.
Strategies to Minimize Off-Target Effects in Multiplexed Libraries
Selection of High-Fidelity Cas Variants
The choice of Cas nuclease fundamentally influences off-target potential. While SpCas9 remains widely used, numerous engineered variants offer improved specificity:
High-fidelity Cas9 variants such as SpCas9-HF1 and eSpCas9(1.1) were rationally designed to reduce non-specific interactions with DNA [51] [53]. These mutants retain on-target activity comparable to wild-type SpCas9 with >85% of sgRNAs tested in human cells while significantly reducing off-target effects [53]. The mechanism involves strategic mutations that create a "proofreading" mechanism, trapping the mutants in an inactive state when bound to mismatched targets [53].
Alternative Cas nucleases with different PAM requirements can also reduce off-target potential. For example, SaCas9 from Staphylococcus aureus recognizes a longer PAM sequence (5'-NNGRRT-3'), naturally restricting its targetable sites in the genome and reducing potential off-target loci [52] [53]. Similarly, Cas12a (Cpf1) systems offer distinct recognition and cleavage properties that may be advantageous for specific multiplex applications.
Cas9 nickase variants represent another strategic approach. By mutating one nuclease domain to create single-strand breaks rather than double-strand breaks, and using paired guides to generate staggered cuts, nickase systems significantly reduce off-target editing while maintaining on-target efficiency [53]. This approach is particularly valuable for therapeutic applications where maximal specificity is required.
Optimized Delivery Systems and Expression Control
The method and timing of CRISPR component delivery significantly impact off-target rates. Transient expression of editing components, rather than stable integration, limits the temporal window for off-target activity [51]. For multiplexed CRISPRi libraries, delivery as ribonucleoprotein (RNP) complexes rather than plasmid DNA can further reduce persistence time of active nucleases in cells [51].
In one optimized platform for multiplex screening, researchers used a piggyBac transposon-based gRNA expression vector (piggyFlex) for genomic integration and stable expression of gRNAs [54]. This system avoids issues that can arise due to recombination during viral packaging and enables both antibiotic and fluorescence-based selection for cells with higher numbers of gRNA integrants [54].
Chemical Modifications and gRNA Engineering
Strategic modifications to gRNA structure can significantly enhance specificity:
- Truncated gRNAs (tru-gRNAs) with 17-19 nucleotides instead of 20 show reduced off-target effects while maintaining on-target activity [52] [53].
- Chemically modified gRNAs with specific alterations to the ribose-phosphate backbone (e.g., 2'-O-methyl-3'-phosphonoacetate) can reduce off-target cleavage while maintaining on-target performance [53].
- The "GG20" technique involves replacing the GX19 sgRNAs at the 5' end with two guanines (creating ggX20 sgRNAs), which significantly reduces off-target effects while boosting specificity [53].
Table 3: Research Reagent Solutions for Minimizing Off-Target Effects
| Reagent Type | Specific Examples | Function in Off-Target Reduction | Application Context |
|---|---|---|---|
| High-Fidelity Cas Variants | SpCas9-HF1, eSpCas9(1.1) [53] | Engineered to reduce non-specific DNA binding | General CRISPR applications; therapeutic development |
| Alternative Cas Nucleases | SaCas9, Cas12a, NmCas9 [52] | Recognize longer or rarer PAM sequences | Applications requiring high specificity; complex genomes |
| CRISPRi/dCas9 Systems | dCas9-KRAB, dCas9 transcriptional inhibitors [55] [9] | Catalytically dead; no DNA cleavage | Gene regulation studies; essential gene analysis |
| Nickase Systems | Cas9n, paired nickase approaches [53] | Create single-strand breaks; require dual recognition | High-precision editing; therapeutic applications |
| Chemical Modification Kits | 2'-O-methyl analogs, 3' phosphorothioate bonds [51] | Enhance gRNA stability and specificity | Sensitive applications; long-term experiments |
| Specialized Delivery Vectors | piggyFlex, Perturb-seq vectors [54] [48] | Enable controlled expression and barcoding | Multiplexed screening; single-cell analysis |
Special Considerations for Multiplexed CRISPRi Library Construction
Multiplexed CRISPRi library construction for pathway discovery presents unique challenges for off-target control. Unlike single-gene editing, where off-target sites can be comprehensively mapped, multiplexed systems introduce complexity from multiple simultaneous perturbations, potential synergistic effects, and technical limitations in tracking numerous gRNAs.
Addressing Functional Redundancy Without Compromising Specificity
Pathway discovery research frequently encounters functional redundancy, where multiple genes perform overlapping functions, masking phenotypic effects when only single genes are perturbed. The multi-targeted CRISPR library approach addresses this by designing sgRNAs that target conserved sequences across multiple gene family members [8]. However, this strategy requires careful balancing between comprehensive target coverage and maintenance of specificity.
In one implementation for tomato genomics, researchers designed sgRNAs using the CRISPys algorithm, which reconstructs phylogenetic trees of gene families and designs sgRNAs that optimally target multiple members within subgroups [8]. Specificity was verified by scanning the rest of the genome for similar sequences, with stricter thresholds for exonic regions (20% of on-target score) compared to other genomic regions (50% of on-target score) [8]. This approach generated a library with 15,804 unique sgRNAs targeting 10,036 genes, with approximately 95% of sgRNAs targeting groups of two or three genes [8].
Advanced Screening Platforms for Complex Phenotypes
Traditional pooled screening approaches with simple phenotypic readouts may miss subtle off-target effects in multiplexed systems. Perturb-seq and related single-cell RNA-seq compatible platforms address this limitation by combining high-complexity pooled screening with rich transcriptional phenotyping at single-cell resolution [48].
In one representative study, researchers developed a robust barcoding strategy that encodes CRISPR perturbation identity in an expressed transcript, enabling pooled analysis of complex libraries while maintaining perturbation-transcriptome linkage [48]. This approach revealed bifurcated unfolded protein response activation among cells subject to the same perturbation – heterogeneity that would be masked in bulk measurements [48]. For pathway discovery research, such granularity enables detection of subtle off-target effects that might otherwise go unnoticed.
Diagram 2: Integrated approach to off-target management in multiplexed CRISPRi screening, highlighting the relationship between enhancement strategies, detection methods, and validation frameworks.
For pathway discovery research utilizing multiplexed CRISPRi libraries, minimizing off-target effects requires an integrated approach spanning computational design, experimental execution, and rigorous validation. The most effective strategy combines multiple complementary methods: conservative gRNA design using advanced algorithms; selection of high-fidelity editing systems; implementation of appropriate detection methods matched to screening scale; and systematic validation of screening hits through orthogonal approaches.
As CRISPR-based screening technologies continue to evolve toward higher complexity and greater analytical depth, maintaining rigor in off-target assessment becomes increasingly crucial for generating reliable pathway insights. By implementing the computational and experimental best practices outlined in this technical guide, researchers can maximize confidence in their findings and accelerate the translation of CRISPR screening results into meaningful biological discoveries and therapeutic opportunities.
The advent of CRISPR technologies has revolutionized genetic engineering, offering unprecedented tools for functional genomics and therapeutic development. However, the choice between different CRISPR modalities carries significant implications for experimental outcomes and cellular health. CRISPR nuclease editing (CRISPRn) and CRISPR interference (CRISPRi) represent two fundamentally distinct approaches to genetic manipulation. While CRISPRn utilizes nucleases like Cas9 to create double-strand breaks (DSBs) in DNA, CRISPRi employs a catalytically dead Cas9 (dCas9) fused to repressive domains to silence gene expression without altering the DNA sequence itself [56]. This technical distinction underlies a critical divergence in their safety profiles, particularly regarding cellular toxicity and genomic integrity. For researchers constructing multiplexed CRISPRi libraries for pathway discovery, understanding these differences is paramount to designing robust, interpretable experiments while maintaining cell viability and physiological relevance.
Mechanisms of Action and Associated Toxicity
CRISPR Nuclease Editing: Genomic Instability from DNA Breaks
CRISPRn functions by introducing targeted double-strand breaks in genomic DNA, relying on cellular repair mechanisms to generate genetic modifications. This process activates complex DNA damage response pathways and poses several substantial risks to genomic integrity:
On-Target Structural Variations: Beyond intended small insertions or deletions (indels), CRISPRn can induce large structural variations including kilobase- to megabase-scale deletions, chromosomal truncations, and translocations [57]. These aberrations are particularly exacerbated when using DNA-PKcs inhibitors to enhance homology-directed repair, with studies reporting a thousand-fold increase in translocation frequencies [57].
P53-Mediated Stress Responses: DSB-induced activation of the p53 pathway can trigger apoptosis, cell cycle arrest, or delayed proliferation across various cell types [57]. While transient p53 suppression can reduce chromosomal aberrations, it may also promote selective expansion of p53-deficient clones with potential oncogenic risk [57].
Analytical Challenges: Traditional short-read sequencing often fails to detect large deletions that eliminate primer-binding sites, leading to overestimated editing efficiency and underestimated collateral damage [57].
CRISPR Interference: A Non-Destructive Alternative
CRISPRi operates through a mechanistically distinct, non-destructive approach that significantly reduces genomic damage:
Catalytically Inactive Core: The dCas9 protein lacks nuclease activity, functioning instead as a targeted DNA-binding scaffold that recruits transcriptional repressive domains to gene promoters [56]. The most common configuration fuses dCas9 to the Krüppel-associated box (KRAB) domain, which silences transcription by promoting heterochromatin formation [56].
Reversible Suppression: Unlike permanent DNA mutations generated by CRISPRn, CRISPRi-mediated silencing is typically reversible, allowing transient phenotypic assessment without permanent genetic damage [56].
Reduced Genotoxic Stress: By avoiding DSBs entirely, CRISPRi circumvents the activation of DNA damage response pathways and associated p53-mediated toxicity, making it particularly suitable for studying essential genes and long-term experiments [56].
Table 1: Fundamental Mechanism Comparison Between CRISPRi and CRISPRn
| Feature | CRISPRi | CRISPR Nuclease Editing |
|---|---|---|
| Core enzyme | dCas9 (catalytically dead) | Cas9 nuclease |
| DNA cleavage | None | Double-strand breaks |
| Primary mechanism | Transcriptional repression | DNA repair-mediated mutagenesis |
| Genetic change | Reversible epigenetic effects | Permanent sequence alterations |
| P53 pathway activation | Minimal | Significant |
| Structural variation risk | Low | High (large deletions, translocations) |
| Reversibility | High (doxycycline-controlled) | None |
Diagram 1: Mechanisms of CRISPRi versus CRISPR Nuclease Editing
Quantitative Comparison of Toxicity and Efficiency
Direct comparative studies reveal substantial differences in performance and cellular impact between these technologies. A foundational study in human induced pluripotent stem cells (iPSCs) demonstrated that CRISPRi gene repression is more efficient and homogenous across cell populations compared to CRISPRn [56]. When targeting the pluripotency gene OCT4, CRISPRi achieved greater than 95% knockdown in bulk populations, while a significant fraction of CRISPRn cells (30-40%) remained OCT4-positive despite Cas9 expression [56]. This heterogeneity in CRISPRn outcomes stems from variable repair efficiencies and the stochastic nature of DSB repair.
Notably, long-term expression (3-4 weeks) of dCas9-KRAB showed no detectable cytotoxicity, decrease in proliferation, or morphological changes in iPSCs, whereas extended Cas9 nuclease expression frequently induces cellular stress responses [56]. RNA-seq analyses confirmed that dCas9-KRAB expression alone caused minimal transcriptomic perturbations, with virtually the entire transcriptome remaining unchanged upon induction [56].
Table 2: Experimental Performance Comparison in iPSCs
| Parameter | CRISPRi | CRISPR Nuclease Editing |
|---|---|---|
| Repression efficiency (OCT4) | >95% in bulk populations | 60-70% effective in bulk populations |
| Cellular heterogeneity | Low, uniform repression | High, mixed populations |
| Long-term cytotoxicity | None detected after 3 weeks | Variable, stress responses common |
| Transcriptomic impact | Minimal off-target effects | Broader transcriptomic disturbances |
| Phenotypic attribution | Clear genotype-phenotype links | Confounded by mixed populations |
Practical Implementation for Multiplexed Library Screening
CRISPRi Library Design and Construction
For pathway discovery research, multiplexed CRISPRi libraries enable systematic functional interrogation without the confounding toxicity of nuclease editing. Effective implementation requires strategic design and validation:
Barcoding Strategies: The Perturb-seq platform exemplifies robust barcoding, combining droplet-based single-cell RNA-seq with CRISPRi perturbation barcoding [48]. This system uses a lentiviral vector containing both a guide barcode (GBC) expression cassette and an sgRNA expression cassette, enabling pooled screening with single-cell transcriptomic readouts [48].
Combinatorial Targeting: Engineering vectors with tandem sgRNA expression cassettes using different RNA polymerase III promoters (e.g., U6, H1, 7SK) minimizes recombination and enables simultaneous repression of multiple pathway components [48]. Validation experiments demonstrate robust and homogeneous combinatorial repression across cell populations [48].
Specificity Optimization: Guide RNA design should prioritize high on-target scores (>0.8 using cutting frequency determination metrics) and minimize off-target potential through stringent mismatch thresholds (e.g., 20% of on-target score for exonic regions) [8]. Multi-targeted designs can address functional redundancy while maintaining specificity through careful consensus targeting [8].
Experimental Workflow for Toxicity-Free Screening
The following workflow outlines a comprehensive approach for implementing multiplexed CRISPRi screens while monitoring and minimizing cellular toxicity:
Diagram 2: Multiplexed CRISPRi Screening Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Multiplexed CRISPRi Library Construction and Screening
| Reagent/System | Function | Application Notes |
|---|---|---|
| dCas9-KRAB Expression System | Transcriptional repression engine | Use inducible (Tet-On) systems for temporal control; multiple clones available (Gen1, Gen2) [56] |
| Perturb-seq Vectors | Combined sgRNA and barcode delivery | Third-generation lentiviral vectors with GBC expression cassette and sgRNA expression cassette [48] |
| Multi-promoter sgRNA Arrays | Combinatorial gene targeting | Tandem U6, H1, 7SK promoters minimize recombination; enable 3+ gene repression [48] |
| Guide Barcode (GBC) System | Perturbation tracking in pooled screens | Polyadenylated transcript with unique barcode captured in scRNA-seq libraries [48] |
| Droplet-based scRNA-seq Platform | Single-cell transcriptional phenotyping | 10X Genomics Chromium system or similar; enables thousands of cells per run [48] |
Mitigation Strategies for Persistent Technical Challenges
Despite its favorable safety profile, CRISPRi implementation faces specific technical hurdles that require targeted mitigation:
Variable Repression Efficiency: Optimization of guide RNA positioning relative to transcription start sites (TSS) is critical. Empirical testing of multiple guides per gene with systematic TSS mapping significantly improves repression consistency [56].
Epigenetic Context Sensitivity: CRISPRi efficacy can be influenced by local chromatin architecture. Incorporating chromatin context metrics into guide design and utilizing dual sgRNA approaches for critical targets can overcome repression barriers in heterochromatic regions.
Partial Repression Limitations: Unlike complete knockout, CRISPRi typically achieves 80-95% repression. For essential pathway analysis, this partial suppression can actually be advantageous, revealing dose-sensitive relationships and avoiding complete viability loss [56].
Delivery Optimization: Lentiviral transduction efficiency must be carefully titrated to ensure single-copy integrations, with typical MOI ≤0.3 to prevent multiple guide incorporation. Chemical modifications (2'-O-methyl analogs, 3' phosphorothioate bonds) on synthetic gRNAs can enhance stability and performance [51].
CRISPRi represents a superior approach for multiplexed library screening in pathway discovery research, offering substantially reduced cellular toxicity while maintaining high efficacy and reproducibility. The avoidance of double-strand breaks and consequent structural variations enables more physiologically relevant phenotypic readouts without the confounding effects of DNA damage response activation and p53-mediated stress pathways. As CRISPR technology evolves, emerging approaches like Perturb-seq with combinatorial CRISPRi are paving the way for comprehensive genetic interaction mapping in complex biological systems [48]. For researchers embarking on large-scale functional genomics, prioritizing CRISPRi over nuclease-based approaches will yield more reliable, interpretable data while preserving cellular health throughout screening campaigns. The continued refinement of CRISPRi systems—including improved repressor domains, optimized delivery platforms, and enhanced specificity profiles—will further solidify its position as the method of choice for toxicity-aware genetic screening.
In the realm of functional genomics, the construction of multiplexed CRISPR interference (CRISPRi) libraries represents a powerful approach for elucidating complex genetic pathways. This technology, which employs a catalytically dead Cas9 (dCas9) fused to transcriptional repressor domains to silence gene expression, is particularly valuable for probing redundant biological processes and identifying synthetic lethal interactions [9] [58]. However, its utility in large-scale screens is often compromised by inconsistent performance—a challenge stemming from variable promoter strength, effector protein stability, and guide RNA (gRNA) efficacy. Inconsistencies in knockdown efficiency across different cell lines, gene targets, and single guide RNAs (sgRNAs) can hinder the reproducibility and interpretability of pathway discovery research [59] [29]. This technical guide synthesizes recent advances in CRISPRi platform optimization, providing a structured framework for researchers to maximize knockdown efficiency in the context of multiplexed library construction. We detail a multi-pronged engineering strategy encompassing novel repressor domains, combinatorial fusion architectures, nuclear localization signals (NLS), and rigorous gRNA validation protocols, all aimed at achieving potent, specific, and consistent transcriptional repression for uncovering novel biological insights.
Core Components of a High-Efficiency CRISPRi System
Optimizing the Repressor Effector: Beyond KRAB
The choice of repressor domain(s) fused to dCas9 is a primary determinant of knockdown efficiency. While the Krüppel-associated box (KRAB) domain has been a historical standard, recent protein engineering efforts have identified more potent alternatives and combinations.
- Novel KRAB Domains: Replacing the traditionally used KOX1(KRAB) domain with the KRAB domain from the ZIM3 protein (ZIM3(KRAB)) has been shown to significantly improve gene silencing capabilities [29] [36].
- The Power of Truncation: A key finding is that a compact, 80-amino acid truncation of the methyl-CpG binding protein 2 (MeCP2), known as the NCoR/SMRT interaction domain (NID), can enhance CRISPRi performance by an average of ~40% compared to canonical MeCP2 subdomains [59]. This MeCP2(t) or NID domain is highly effective in combinatorial fusions.
- Combinatorial Domain Fusion: The most potent repressors emerge from fusing multiple repressor domains in tandem to dCas9. Screening of bipartite and tripartite fusion libraries has yielded superior constructs, such as dCas9-ZIM3(KRAB)-MeCP2(t) and dCas9-ZIM3-NID-MXD1-NLS, which outperform previous gold-standard repressors across multiple cell lines and gene targets [59] [29]. The proprietary dCas9-SALL1-SDS3 fusion from Horizon Discovery also reports more potent repression compared to dCas9-KRAB variants [36].
The Critical Role of Nuclear Localization Signal (NLS) Configuration
The efficiency of the dCas9-repressor complex is contingent upon its localization to the nucleus. Optimization of the NLS configuration is not a mere detail but a powerful lever for enhancing performance. Studies systematically evaluating NLS placement found that affixing a single carboxy-terminal NLS to repressor fusions boosted gene knockdown efficiency by an average of ~50% [59]. This simple modification ensures robust nuclear import of the fully assembled CRISPRi machinery, a prerequisite for effective transcriptional repression.
Promoter Choice and gRNA Design for Reliable Targeting
The transcriptional regulatory elements driving gRNA expression are critical for minimizing recombination in multiplexed vectors and ensuring consistent gRNA abundance.
- Promoter Choice for Multiplexing: To avoid homologous recombination when cloning multiple gRNAs into a single lentiviral vector, it is essential to use heterogeneous promoters. A proven strategy involves combining the human U6 and mouse U6 promoters to express individual gRNAs within the same transcript [15]. This prevents recombination during library production and maintains library integrity.
- gRNA Design and Validation: For CRISPRi, gRNAs must be designed to target the region 0-300 base pairs downstream of the transcription start site (TSS) [36]. Computational tools that leverage machine learning, such as the CRISPRi v2.1 algorithm, can predict highly effective gRNA designs by incorporating TSS annotation, chromatin accessibility, and sequence data [36]. Furthermore, empirical validation is crucial. Pools of 3-4 sgRNAs per gene target often produce stronger and more reliable knockdown than individual guides [36].
Table 1: Optimized Repressor Effector Constructs for High-Efficiency CRISPRi
| Construct Name | Key Domains/Features | Reported Advantage | Primary Application Context |
|---|---|---|---|
| dCas9-ZIM3-NID-MXD1-NLS | ZIM3(KRAB), MeCP2 NID, MXD1, C-terminal NLS | Superior gene silencing; consistent performance across cell lines and sgRNAs [59] | Multiplexed genome-wide screening |
| dCas9-ZIM3(KRAB)-MeCP2(t) | ZIM3(KRAB), truncated MeCP2 | Improved repression of endogenous targets at transcript and protein level [29] | Endogenous gene knockdown; validation screens |
| dCas9-SALL1-SDS3 | Proprietary SALL1 and SDS3 repressor domains | More potent target gene repression compared to dCas9-KRAB; broad functionality [36] | Arrayed and pooled screening |
Experimental Protocols for Validation and Optimization
Protocol: Validating Novel Repressor Constructs
This protocol outlines a robust methodology for screening and validating the efficacy of novel dCas9-repressor fusions, adapted from high-impact studies [59] [29].
- Construct Assembly: Clone candidate repressor domain fusions (e.g., KRAB-NID, KRAB-MeCP2(t)) into a dCas9 expression backbone. Ensure the inclusion of an optimized NLS configuration (e.g., single C-terminal NLS).
- Reporter Cell Line Transduction: Generate a stable cell line (e.g., HEK293T) expressing a reporter construct, such as an SV40 promoter-driven enhanced Green Fluorescent Protein (eGFP) cassette.
- CRISPRi Transfection: Co-transfect the dCas9-repressor construct with a pool of sgRNAs designed to target the reporter gene's promoter (e.g., SV40) into the reporter cell line. Include controls:
- Positive Control: A previously validated repressor (e.g., dCas9-KOX1(KRAB)-MeCP2).
- Negative Control: dCas9 alone or with a non-targeting sgRNA.
- Flow Cytometry Analysis: Harvest cells 72-96 hours post-transfection. Analyze eGFP fluorescence intensity via flow cytometry. Quantify knockdown efficiency as the percentage reduction in median fluorescence intensity (MFI) compared to the negative control.
- Orthogonal Validation: Confirm hits on endogenous genes using RT-qPCR (for transcript level) and western blotting (for protein level) across multiple cell lines (e.g., RPE-1, K562, HeLa) to ensure broad functionality.
Protocol: gRNA Functional Validation and TSS Mapping
Accurate gRNA design and validation are paramount for successful CRISPRi experiments [60] [36].
- TSS Identification: Utilize curated databases like FANTOM and Ensembl to pinpoint the dominant TSS for your target gene. Be aware of alternative TSSs.
- gRNA Design and Synthesis: Using a specialized algorithm (e.g., CRISPRi v2.1), design 3-4 gRNAs targeting the region from 0 to 300 bp downstream of the TSS. Synthesize these as individual synthetic sgRNAs and as a pooled reagent.
- Transfection and Expression Analysis:
- Deliver the sgRNAs (individual and pooled) into a cell line stably expressing your optimized dCas9-repressor (e.g., dCas9-SALL1-SDS3).
- Use a transfection control (e.g., GFP mRNA) to monitor delivery efficiency [61].
- Harvest cells 72 hours post-transfection.
- Efficiency Quantification: Isolate total RNA, synthesize cDNA, and perform RT-qPCR. Calculate relative gene expression using the ∆∆Cq method, normalizing to a housekeeping gene (e.g., GAPDH, ACTB) and a non-targeting control sgRNA.
- Hit Selection: Select the individual sgRNA with the strongest knockdown or proceed with the pooled sgRNAs for maximal repression [36].
Diagram 1: gRNA validation workflow for optimal CRISPRi.
Protocol: Essential Controls for CRISPRi Experiments
Incorporating proper controls is non-negotiable for interpreting CRISPRi results and troubleshooting failures [61].
- Positive Editing Control: Use a validated sgRNA targeting a well-characterized, easily measurable gene (e.g., PPIB, HBP1) to confirm that your dCas9-repressor system is functional under your experimental conditions.
- Negative Editing Controls:
- Non-targeting sgRNA: A scramble sgRNA with no perfect genomic match, controlling for non-specific effects of dCas9-repressor binding.
- Targeting sgRNA without dCas9: Rules out off-target effects of the sgRNA itself.
- Transfection Control: Co-deliver a fluorescent reporter (e.g., GFP mRNA) to quantify delivery efficiency and distinguish between poor knockdown caused by inefficient transfection versus ineffective gRNA/repressor design.
- Mock Transfection: Cells subjected to the transfection reagent/protocol without any nucleic acids, controlling for cellular stress induced by the delivery process.
Application in Multiplexed Library Construction and Screening
The ultimate test of an optimized CRISPRi system is its performance in complex, multiplexed libraries designed for pathway discovery. The principles outlined above have been successfully implemented in large-scale efforts.
A landmark study demonstrated this by creating the "SPIDR" (Systematic Profiling of Interactions in DNA Repair) library, a combinatorial CRISPRi dual-guide library targeting 548 DNA damage response (DDR) genes [58]. This library, used in a growth-based screen, successfully mapped approximately 150,000 gene-level interactions, uncovering both known and novel synthetic lethal relationships. This work underscores the power of optimized multiplexed CRISPRi to systematically dissect complex biological networks with high precision. The library design utilized a dual-sgRNA lentiviral expression vector, mitigating recombination risks through careful design.
For researchers constructing custom multiplexed libraries, the following workflow is recommended:
- Select an Optimized Repressor: Base the library on a highly potent repressor like dCas9-ZIM3-NID-MXD1-NLS [59].
- Clone with Heterogeneous Promoters: Use a combination of human U6 and mouse U6 promoters to express individual gRNAs within the same vector backbone to maintain stability [15].
- Employ High-Quality gRNAs: For each gene target, include 3-4 gRNAs that have been pre-validated or designed using a state-of-the-art algorithm, and consider deploying them as pools for maximal robustness [36].
- Include Library-Specific Controls: Embed non-targeting control sgRNA pairs and target positive essential genes as internal controls for screen quality assessment [58].
Table 2: The Scientist's Toolkit: Essential Reagents for Optimized CRISPRi
| Reagent / Tool | Function | Example/Source |
|---|---|---|
| Optimized dCas9-Repressor | Core effector protein for targeted transcriptional repression | dCas9-ZIM3-NID-MXD1-NLS [59]; dCas9-SALL1-SDS3 [36] |
| Algorithmically-Designed gRNAs | Ensure high on-target efficiency and specificity for CRISPRi application | CRISPRi v2.1 algorithm [36] |
| Heterogeneous Promoters | Enable stable cloning of multiple gRNAs by preventing homologous recombination | Human U6 + Mouse U6 combination [15] |
| Synthetic sgRNA Pools | Fast, potent gene repression; ideal for validation and arrayed screens | Horizon Discovery synthetic sgRNA formats [36] |
| Validation Controls | Critical for experiment interpretation and troubleshooting | Positive control sgRNAs (e.g., for PPIB); non-targeting control sgRNAs [61] [36] |
Diagram 2: Multiplexed CRISPRi library construction and screening.
The journey toward maximizing knockdown efficiency in multiplexed CRISPRi libraries is a multi-parameter optimization problem. As detailed in this guide, solutions lie in the synergistic combination of next-generation repressor effectors like dCas9-ZIM3-NID-MXD1-NLS, strategic NLS configuration, careful promoter selection for library stability, and rigorous, algorithm-informed gRNA validation. By adopting this integrated framework, researchers can construct highly robust multiplexed CRISPRi libraries. This significantly enhances the reliability and depth of pathway discovery research, enabling the systematic deconvolution of complex genetic networks, the identification of novel therapeutic targets, and the uncovering of synthetic lethal interactions with high confidence and reproducibility.
Ensuring Uniform sgRNA Representation and Library Coverage
In multiplexed CRISPR interference (CRISPRi) screens for pathway discovery, the uniformity of single guide RNA (sgRNA) representation and the comprehensiveness of library coverage are not merely technical details—they are fundamental determinants of success. These factors directly impact statistical power, reduce false negatives, and enable the reliable identification of genetic interactions and subtle pathway regulators. Pooled CRISPR screens require distributing a complex library of lentivirally delivered sgRNAs across a population of cells; high variability in the abundance of individual guides necessitates immense cell numbers to ensure that guides with low initial representation remain detectable after a biological selection. This requirement poses a prohibitive logistical challenge for sophisticated screens in technically demanding models such as primary cells, differentiated cell lines, and adherent cultures [62]. Furthermore, incomplete library coverage can systematically miss biological insights. Recent analyses have revealed that confounding effects, such as the systematic under-calling of hits for genes targeted by low-specificity sgRNAs, can directly stem from inadequate library design [63]. This technical guide outlines established and emerging strategies to overcome these hurdles, providing a framework for constructing robust multiplexed CRISPRi libraries that yield biologically accurate and comprehensive data for pathway discovery research.
Core Principles: sgRNA Specificity and Library Uniformity
Quantifying Guide RNA Specificity with Advanced Tools
The problem of sgRNA off-target activity is a major source of false-positive and false-negative results in genetic screens. Low-specificity guides, which have numerous potential off-target binding sites in the genome, can produce strong confounding fitness effects. In CRISPR knockout (CRISPRko) screens, these guides can cause genotoxicity through excessive DNA cutting. In CRISPRi screens, a previously unobserved confounding effect has been identified: genes targeted by guides with low average specificity are systematically less likely to be identified as hits. This may occur because the dCas9 effector is diluted across an excessively large set of off-target sites, reducing its effective concentration at the primary target [63].
To mitigate this, the development of sophisticated gRNA design software is critical. GuideScan2 is a next-generation tool that addresses the limitations of its predecessor and other design tools. It uses a novel search algorithm based on the Burrows-Wheeler transform for indexing the genome, combined with simulated reverse-prefix trie traversals for exhaustive off-target enumeration. This approach offers a 50x improvement in memory efficiency (3.4 Gb for hg38 vs. 190 Gb for GuideScan) and a 65x acceleration in index construction time (30 minutes on a standard laptop), without compromising the accuracy of off-target identification [63]. Using such tools to filter out low-specificity guides during the library design phase is essential for minimizing off-target confounding effects.
The Impact of Library Bias on Screen Performance
Library uniformity refers to the evenness of representation of each sgRNA within the pooled library. A highly uniform library, where all guides are present at similar abundances, maximizes screening power and minimizes the number of cells required. The standard metric for quantifying this is the skew ratio, which compares the abundance of guides at different percentiles (e.g., 90th vs. 10th). A lower skew ratio indicates a more uniform library [62]. Early genome-wide libraries often suffered from high skew ratios and significant guide "dropouts" (guides that are completely missing from the pool), necessitating screening at 500-1000x cell coverage to reliably measure the effects of low-abundance guides. This translates to a requirement for tens to hundreds of millions of cells per experimental sample, which is often infeasible in biologically relevant but difficult-to-culture cell models [62].
Table 1: Quantitative Impact of Cloning Optimization on Library Uniformity
| Library Version | Description | 90/10 Skew Ratio | Key Improvement |
|---|---|---|---|
| Legacy Library | Library cloned with original protocol | >2 | Baseline |
| Optimized Library | Library cloned with full set of optimizations | <2 | ~2-fold improvement in uniformity, fewer dropouts [62] |
Experimental Protocols for Enhanced Library Construction
An Optimized Cloning Workflow for Maximal Uniformity
A systematically optimized cloning protocol can significantly reduce representation bias, allowing for screening at lower cell coverage. The following protocol, adapted from a 2024 study, details key modifications to the standard procedure for synthesizing and cloning sgRNA insert pools [62].
Procedure:
- Oligo Pool Synthesis: Order the single-stranded oligo templates in both forward and reverse complement orientations. Synthesis biases are sequence-specific; ordering both orientations ensures that dropouts in one orientation can be compensated by the other, resulting in a more complete final library [62].
- Polymerase Selection for Insert Amplification: Use a high-fidelity, thermostable polymerase such as NEB Q5 Ultra II for the PCR amplification of the double-stranded sgRNA insert from the oligo pool. Avoid mesophilic polymerases like Klenow, which can produce non-specific byproducts and lead to less uniform guide distributions [62].
- Minimize PCR Cycles: Use the minimum number of PCR cycles necessary to generate sufficient product for cloning. Over-amplification can lead to "bubble products" and biased dropout of certain sequences, harming uniformity. Optimize template concentration to minimize the required cycle number [62].
- Low-Temperature Gel Purification: After digesting the PCR product to create the final inserts, purify the DNA fragment by gel electrophoresis. Critically, perform the gel run and subsequent gel excision on ice to keep the apparatus cold. Elute the DNA from the gel slice at 4°C instead of room temperature or 37°C. This minimizes the Tm-dependent bias that causes under-representation of inserts with lower melting temperatures [62].
- Colony Validation: After ligation and transformation, perform colony PCR to verify that clones contain the correct single insert. The optimized protocol reduces the formation of hybrid clones with non-complementary inserts from 40% to ~1.2% [62].
Diagram: Optimized sgRNA Library Cloning Workflow. Key steps that enhance uniformity are highlighted in green, while critical quality control checkpoints are in red.
Alternative Strategies: Dual-sgRNA and Barcoding Approaches
Dual-sgRNA Library Design: For an ultra-compact and highly active library, consider a dual-sgRNA design. This approach targets each gene with a single lentiviral construct that expresses a tandem cassette of the two most effective sgRNAs. Empirical data from genome-wide growth screens demonstrate that dual-sgRNA libraries produce significantly stronger fitness phenotypes for essential genes compared to single-sgRNA libraries (mean growth rate γ = -0.26 vs. -0.20, p = 6 × 10⁻¹⁵), indicating more potent target gene depletion. This allows for a massive reduction in library size (e.g., 1-2 elements per gene instead of 5-10) while maintaining or even improving performance, enabling screens in contexts where cell numbers are limiting [35]. A challenge to note with this system is the potential for lentiviral template switching, which can create inaccurate hybrid vectors; however, optimized protocols for amplifying and sequencing these cassettes from genomic DNA are available [35].
Linear Amplification of Barcodes for Precise Quantification: To achieve more precise and robust measurements of guide abundance in pooled screens, implement a barcoding strategy with linear amplification. In this method, each guide RNA expression plasmid is linked to one of a few random nucleotide barcodes. When preparing sequencing libraries from pooled cell populations, subject the plasmid DNA to linear amplification by in vitro transcription (IVT), followed by reverse transcription and a limited PCR. This IVT-RT method demonstrates substantially lower technical noise compared to direct exponential PCR amplification, with replicate correlations improving from r=0.93 (PCR) to r=0.98 (IVT-RT). This enhanced precision increases statistical power to resolve subtle phenotypic differences in screens [64].
Table 2: Research Reagent Solutions for CRISPRi Library Construction
| Reagent / Resource | Function | Example & Notes |
|---|---|---|
| gRNA Design Software | Designs highly specific sgRNAs and enumerates off-targets. | GuideScan2 (command-line and web interface). Offers massive improvements in memory efficiency and speed [63]. |
| Optimized Polymerase | Amplifies sgRNA insert pool with high fidelity and uniformity. | NEB Q5 Ultra II. Outperforms mesophilic polymerases in producing uniform guide representation [62]. |
| Dual-sgRNA Library | Ultra-compact library for high-efficacy knockdown. | Enables strong phenotypes with 1-2 elements/gene. Publicly available sequences and cloning protocols [35]. |
| CRISPRi Effector | Catalytically dead Cas9 fused to repressor domains for gene knockdown. | Zim3-dCas9. Provides an excellent balance of strong on-target knockdown and minimal non-specific effects on cell growth/transcriptome [35]. |
| Barcoded Vector System | Allows precise quantification of guide abundance via linear amplification. | Plasmid system with embedded random barcodes. Use with IVT-RT library generation to reduce noise [64]. |
The construction of a multiplexed CRISPRi library is a foundational step in pathway discovery research, where the quality of the library directly dictates the quality of the biological insights. By integrating computational design for maximal sgRNA specificity with wet-lab protocols that maximize library uniformity, researchers can create powerful screening resources. The adoption of optimized cloning techniques, compact dual-sgRNA designs, and precise barcode-based quantification enables robust genetic screens in a wider range of biologically relevant cell models. Adhering to these best practices ensures that the resulting data is reliable, comprehensive, and capable of revealing the complex genetic architectures that underlie cellular pathways.
Benchmarking Performance and Validating Screening Outcomes
In pathway discovery research, the construction of a multiplexed CRISPR interference (CRISPRi) library represents only the initial step. The true determinant of experimental success lies in rigorous, multi-faceted assessment of both on-target knockdown efficacy and overall screen quality. Imperfect knockdown can lead to misinterpretation of gene function and failed pathway identification, while poor screen quality metrics can render entire datasets uninterpretable. Within the context of multiplexed CRISPRi library construction for pathway discovery, this technical guide establishes a comprehensive framework for validation, drawing on current methodologies and empirical data to ensure researchers can confidently extract biological insights from their screens. The integration of single-cell RNA sequencing (scRNA-seq) technologies with CRISPR screening, as exemplified by Perturb-seq, has revolutionized validation by enabling direct measurement of transcriptional consequences from genetic perturbations in thousands of individual cells simultaneously [48]. This guide details the quantitative metrics and experimental protocols essential for confirming that your library performs as intended, from individual gene targeting to genome-scale functional enrichment.
Core Metrics for Assessing Knockdown and Screen Performance
Quantitative Metrics for On-Target Efficacy
A multi-tiered approach to assessing on-target knockdown is essential. The table below summarizes the key metrics, their measurement methodologies, and target benchmarks for a successful screen.
Table 1: Key Metrics for Assessing On-Target Knockdown Efficacy
| Metric | Measurement Method | Target Benchmark | Key Considerations |
|---|---|---|---|
| Gene Expression Knockdown | RT-qPCR [65] or RNA-seq [48] | >75% mRNA reduction under optimal conditions [66] | Use multiple assays on the same transcript to control for artifacts [65]. |
| Phenotypic Effect Size | Growth-based lethality screens [67] [35] | Strong depletion of essential genes (e.g., γ ≈ -0.26 for dual-sgRNAs) [35] | Compare to gold-standard essential and non-essential gene sets [67]. |
| Single-Cell Resolution | Perturb-seq / single-cell RNA-seq [48] [54] | High-confidence guide barcode (GBC) assignment for >90% of cells [48] | Decouples heterogeneous responses and confirms perturbation identity at the single-cell level. |
| Library Element Efficacy | sgRNA abundance log-fold change (LFC) [37] | Strong negative LFC for essential gene targeting guides [37] | Dual-sgRNA constructs show significantly stronger phenotypes than single-sgRNAs [35]. |
Beyond individual gene knockdown, the overall quality of the screen must be validated using system-level metrics.
Table 2: Core Metrics for Assessing Overall Screen Quality
| Quality Metric | Description | Interpretation |
|---|---|---|
| Precision-Recall of Essential Genes | Ability to recall known essential genes with low false-positive rate [67] [37]. | AUC >0.90 is good; >0.98 is excellent [67] [35]. |
| Reagent-Level Reproducibility | Correlation of sgRNA or dual-sgRNA element phenotypes between replicates [67]. | High reproducibility (e.g., r > 0.8) indicates a robust screen [35]. |
| Guide-to-Cell Assignment Rate | Percentage of single-cell transcriptomes with a confidently assigned perturbation [48] [54]. | Rates of ~80% are achievable, enabling powerful paired phenotype analysis [54]. |
| Specificity & Off-Target Effects | Enrichment of non-essential genes or negative controls [37] [35]. | Minimal enrichment of non-targeting controls indicates low false-positive rate. |
Experimental Protocols for Key Validation Experiments
Protocol 1: Validating Knockdown Efficiency via RT-qPCR
For initial validation of CRISPRi library elements, RT-qPCR provides a rapid and quantitative measure of on-target mRNA knockdown.
- Assay Design: IDT recommends designing at least two PrimeTime qPCR assays spaced apart on the target transcript. This controls for potential artifacts, such as undiscovered gene isoforms not targeted by the knockdown reagent or blocking of reverse transcriptase enzyme [65].
- Execution: Transfert cells with your CRISPRi library elements or establish stable cell pools. Isolate RNA and synthesize cDNA. Perform qPCR using the designed assays and normalise data to appropriate housekeeping genes.
- Analysis: Calculate percentage knockdown relative to non-targeting control cells. Successful reagents should achieve robust knockdown, with premium reagents like ON-TARGETplus siRNA guaranteeing at least 75% silencing under optimal conditions [66]. While this benchmark is for siRNA, it provides a reasonable target for CRISPRi.
Protocol 2: Genome-Scale Fitness Screening for Library QC
This protocol assesses the functional performance of your entire library in a pooled growth screen.
- Library Transduction: Transduce your CRISPRi-ready cell line (e.g., K562s expressing Zim3-dCas9 [35]) with the lentiviral sgRNA library at a low MOI (~0.3) to ensure most cells receive a single library element. Include a puromycin selection marker and select for transduced cells.
- Time-Point Sampling: Harvest cell pellets at two time points: a baseline (T0) immediately after selection, and a final time point (Tfinal) after ~14-20 population doublings to allow for phenotypic depletion [35].
- Sequencing & Analysis: Extract genomic DNA from both pellets and amplify the integrated sgRNA cassettes for next-generation sequencing. Quantify the abundance of each sgRNA in T0 and Tfinal populations. Calculate a log-fold change (LFC) for each sgRNA. The screen quality is evidenced by strong depletion of sgRNAs targeting essential genes (e.g., gold-standard sets from DepMap [35]) and minimal change for non-targeting controls.
Protocol 3: Single-Cell Transcriptomic Validation (Perturb-Seq)
Perturb-seq provides the highest-content validation, linking each perturbation directly to its transcriptional outcome in thousands of single cells.
- Cell Preparation and Sequencing: Generate a single-cell suspension of your perturbed cells. Use a droplet-based single-cell RNA-seq platform (e.g., 10x Genomics) that captures both the cellular transcriptome and the expressed guide barcode (GBC) from the Perturb-seq vector [48] [54].
- Computational Analysis: A specialized computational pipeline is used to demultiplex the data. This assigns GBCs to individual cell transcriptomes, confidently identifying the perturbation in each cell [48]. Cells are then partitioned into test (has gRNA X) and control (lacks gRNA X) groups.
- Differential Expression Testing: For each targeted gene, perform differential expression analysis between test and control cells for all genes within a 1 Mb window of the target site [54]. This confirms not only the knockdown of the intended target but also identifies downstream transcriptional consequences, thereby validating the library's utility for pathway discovery.
The Scientist's Toolkit: Essential Research Reagents
The following reagents are critical for constructing and validating a high-performance multiplexed CRISPRi library.
Table 3: Key Reagent Solutions for CRISPRi Library Construction and Validation
| Reagent / Tool | Function | Example & Key Features |
|---|---|---|
| CRISPRi Effector | Engineered dCas9 fused to repressor domains for gene knockdown. | Zim3-dCas9: Provides optimal balance of strong on-target knockdown and minimal non-specific effects on cell growth/transcriptome [35]. |
| sgRNA Library Design | A collection of sgRNAs targeting genes of interest for pooled screening. | Ultra-Compact Dual-sgRNA Library: Targets each gene with a single element expressing two sgRNAs. Shows significantly stronger knockdown and phenotypic effects than single-sgRNA designs [35]. |
| Perturb-seq Vector | Lentiviral vector for delivering and tracking CRISPR perturbations in scRNA-seq. | Barcoded Lentivector: Contains both a Pol III-driven sgRNA cassette and a Pol II-driven Guide Barcode (GBC) expression cassette for unambiguous perturbation tracking in single cells [48]. |
| Validation Assays | Tools to quantitatively measure the success of gene knockdown. | RT-qPCR Assays: PrimeTime qPCR assays designed to multiple regions of the target transcript to control for artifacts and confirm knockdown [65]. |
| Positive Controls | Validated reagents that provide a known signal for assay optimization. | ON-TARGETplus Control siRNA/Pools: Non-targeting negative controls and silencing controls for housekeeping genes (e.g., Cyclophilin B, GAPD) [66]. |
The journey from a designed library to biological discovery hinges on rigorous validation. By systematically applying the metrics and protocols outlined in this guide—from RT-qPCR validation of individual targets to genome-scale fitness screens and ultimately high-content Perturb-seq—researchers can confidently determine the quality of their multiplexed CRISPRi screens. The adoption of advanced reagents, such as dual-sgRNA libraries and optimized effectors like Zim3-dCas9, further ensures that screens are powered by highly effective perturbations. A well-validated screen, where on-target efficacy is confirmed and quality metrics are met, provides a solid foundation for the subsequent and most critical phase: the unbiased discovery of novel functional pathways and genetic interactions that drive biological processes and disease states.
In the realm of functional genomics and pathway discovery research, CRISPR-based technologies have revolutionized our ability to interrogate gene function. While CRISPR knockout (CRISPRko) enables permanent gene disruption, and CRISPR activation (CRISPRa) facilitates gene upregulation, CRISPR interference (CRISPRi) occupies a unique niche with its reversible, tunable gene silencing capability. For researchers constructing multiplexed CRISPRi libraries to deconvolve complex biological pathways, understanding the comparative strengths and limitations of each approach is paramount. This technical analysis provides a comprehensive comparison of these three fundamental technologies, with particular emphasis on their application in multiplexed library screens for pathway discovery. Each technology offers distinct advantages—CRISPRko for complete gene disruption, CRISPRa for gain-of-function studies, and CRISPRi for reversible, titratable knockdown—enabling researchers to select the optimal approach based on their specific biological questions, whether investigating essential genes, non-coding elements, or complex genetic interactions.
Fundamental Mechanisms and Molecular Components
Core Machinery and Mechanism of Action
CRISPRko (Knockout) utilizes the wild-type Cas9 nuclease, which introduces double-strand breaks (DSBs) in target DNA guided by a single-guide RNA (sgRNA). These breaks are repaired by error-prone non-homologous end joining (NHEJ), often resulting in frameshift mutations and permanent gene disruption [24] [68].
CRISPRi (Interference) employs a catalytically dead Cas9 (dCas9) with inactivated nuclease domains (D10A and H840A for SpCas9) fused to repressor domains like the Krüppel-associated box (KRAB). The dCas9-KRAB complex binds to target DNA without cutting it, sterically hindering RNA polymerase and recruiting chromatin remodeling factors to suppress transcription [24] [3] [68].
CRISPRa (Activation) similarly uses dCas9 but fused to transcriptional activator domains such as VP64, p65, or Rta. More advanced systems like the Synergistic Activation Mediator (SAM) recruit multiple distinct activator domains to the target gene promoter, significantly enhancing transcription initiation [69] [68].
Table 1: Core Molecular Components of CRISPR Technologies
| Technology | Cas9 Variant | Effector Domains | Primary Molecular Action |
|---|---|---|---|
| CRISPRko | Wild-type Cas9 | Nuclease domains active | Creates double-strand breaks |
| CRISPRi | dCas9 (D10A, H840A) | KRAB repressor domain | Blocks transcription elongation |
| CRISPRa | dCas9 (D10A, H840A) | VP64, p65, Rta activators | Recruits transcriptional machinery |
Optimal Targeting Strategies and gRNA Design
The targeting strategies and gRNA design principles differ significantly between these approaches, directly impacting library construction considerations:
CRISPRko gRNAs typically target early exons in protein-coding regions to maximize the probability of generating frameshift mutations that disrupt functional protein domains [68].
CRISPRi gRNAs achieve optimal repression when targeting a window spanning from -50 to +300 base pairs relative to the transcriptional start site (TSS), with the most effective gRNAs binding within the first 100 bp downstream of the TSS [68].
CRISPRa gRNAs function most effectively when targeting regions between -400 to -50 bp upstream of the TSS, where they can best recruit transcriptional machinery to the promoter region [68].
For all technologies, gRNA efficacy is influenced by protospacer length (optimally ≤21 bp), absence of nucleotide homopolymers, and local chromatin accessibility [68]. In library-scale implementations, including 3-10 gRNAs per gene helps ensure target coverage despite variations in individual gRNA efficiency [68].
Comparative Performance Characteristics
Efficiency, Specificity, and Reversibility
Table 2: Performance Characteristics Comparison
| Parameter | CRISPRko | CRISPRi | CRISPRa |
|---|---|---|---|
| Gene Modulation Efficiency | High (complete knockout) | Moderate to high (60-99% knockdown) [3] [68] | Variable (up to 1,000-fold activation) [68] |
| Temporal Control | Permanent | Reversible, titratable [24] [68] | Reversible, titratable [68] |
| On-target Specificity | High, but DSB repair can cause indels | High, with minimal off-target repression [68] | High, with minimal off-target activation |
| Multiplexing Capacity | Moderate (toxicity concerns) | High (efficient 10+ gene knockdown) [9] | High |
| gRNA Design Complexity | Moderate | High (requires precise TSS annotation) [68] | High (requires precise TSS annotation) [68] |
Applications in Functional Genomics and Pathway Discovery
Each technology excels in distinct application spaces, particularly in the context of multiplexed library screening:
CRISPRko is ideal for complete loss-of-function studies, identifying essential genes, and validating gene function when full ablation is required. However, it is suboptimal for studying essential genes where complete knockout is lethal, or for investigating non-coding regions where small indels may not sufficiently alter function [68].
CRISPRi demonstrates particular strength in several key applications relevant to pathway discovery:
- Essential gene studies: Enables partial knockdown of essential genes where complete knockout would be lethal [24] [68]
- Non-coding gene perturbation: Effectively targets long non-coding RNAs and other non-coding elements [68]
- High-order multiplexing: Successfully applied to simultaneously target up to 10+ genes in pathways with redundant components [9]
- Genetic interaction mapping: CRISPRi–TnSeq enables systematic mapping of interactions between essential and non-essential genes [70]
- Titratable phenotypes: Allows fine-tuning of gene expression levels to model partial inhibition [22]
CRISPRa excels in gain-of-function studies, enabling:
- Endogenous gene activation: More physiological relevance compared to ORF overexpression [68]
- Non-coding region screening: Identification of enhancers and regulatory elements [69]
- Gene dosage studies: Investigating effects of moderate gene overexpression
Notably, CRISPRi outperforms RNA interference (RNAi) in large-scale screening applications by generating more robust phenotypes with fewer off-target effects, as CRISPRi components are entirely exogenous and avoid competition with endogenous RNA processing machinery [68].
Implementation in Multiplexed Library Screens
Experimental Workflows and Considerations
The implementation of each technology in multiplexed library screens follows similar overarching workflows but with critical technology-specific considerations:
Protocol for Multiplexed CRISPRi Library Screening
The following detailed protocol outlines a representative workflow for conducting a multiplexed CRISPRi screen, adaptable for various biological contexts:
1. Library Design and gRNA Selection
- Select 3-10 gRNAs per gene targeting the region from -50 to +300 bp relative to the TSS [68]
- Include non-targeting control gRNAs (typically 750-1000 sequences) for normalization [71]
- For enhanced repression, design gRNAs targeting the first 100 bp downstream of the TSS [68]
- Filter gRNAs with homopolymer sequences or potential off-target effects using algorithms like CFD scoring [8]
2. Stable Cell Line Generation
- Generate helper cell lines expressing dCas9-KRAB under inducible control (e.g., Tet-On system) [68] [71]
- Use lentiviral transduction followed by antibiotic selection to create stable pools
- Validate dCas9-KRAB expression and inducibility by Western blotting [71]
- Confirm minimal basal toxicity and normal growth kinetics in uninduced state
3. Library Delivery and Cell Selection
- Transduce dCas9-KRAB cells with the sgRNA library at low MOI (∼0.3) to ensure most cells receive single integrations
- Maintain >1000 cells per sgRNA throughout the screen to preserve library complexity [71]
- Apply selection (e.g., puromycin) for 3-7 days to eliminate untransduced cells
- Harvest baseline sample (T0) for reference before applying experimental perturbations
4. Induced Repression and Phenotypic Selection
- Induce dCas9-KRAB expression with doxycycline or other appropriate inducers
- Apply relevant phenotypic selection (e.g., drug treatment, growth factor withdrawal)
- Culture cells for 14-21 population doublings to allow phenotypic manifestation
- Maintain minimum 500x library coverage throughout selection phase
5. Sample Harvesting and Next-Generation Sequencing
- Harvest final cell population (T1) and extract genomic DNA
- Amplify integrated sgRNA cassettes with PCR using barcoded primers
- Sequence amplified products using Illumina platforms to determine sgRNA abundance
- Process raw sequencing data through alignment pipelines to count sgRNA reads
6. Data Analysis and Hit Identification
- Normalize sgRNA counts to total reads and compare T1 vs T0 abundances
- Calculate gene-level scores using robust ranking algorithms (e.g., MAGeCK, RSA)
- Identify significant hits based on comparison to negative control sgRNA distribution
- Validate top hits using individual sgRNAs in secondary assays
Advanced Applications: Bidirectional Epigenetic Editing
Recent advances enable more sophisticated screening approaches, such as CRISPRai, which applies activating and repressive perturbations to two loci simultaneously in the same cell. This bidirectional approach:
- Enables investigation of genetic interactions and epistasis [69]
- Identifies hierarchical relationships in gene regulatory networks [69]
- Maps enhancer-promoter interactions and regulatory landscapes [69]
- Uncovers compensatory relationships and redundant pathways [69]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Multiplexed CRISPR Screening
| Reagent Category | Specific Examples | Function & Importance |
|---|---|---|
| Cas9 Variants | Wild-type SpCas9 (CRISPRko), dCas9-KRAB (CRISPRi), dCas9-VPR (CRISPRa) [68] | Core effector proteins that determine technological approach |
| Delivery Systems | Lentiviral vectors, plasmid systems, barcoded libraries [8] [71] | Enable efficient, stable integration of CRISPR components |
| gRNA Libraries | Genome-scale libraries (3-10 gRNAs/gene), sub-libraries targeting specific gene families [8] [71] | Provide comprehensive gene coverage with built-in redundancy |
| Induction Systems | Doxycycline-inducible (Tet-On) promoters, chemical inducers [71] | Enable temporal control over CRISPR activity |
| Selection Markers | Puromycin, blasticidin, GFP/mCherry reporters [71] | Permit enrichment of successfully transduced cells |
| Sequencing Tools | Illumina platforms, barcoded PCR primers, sgRNA amplification kits [9] [71] | Facilitate quantification of sgRNA abundance changes |
CRISPRi represents a powerful addition to the functional genomics toolkit, particularly valuable for multiplexed library construction in pathway discovery research. Its capacity for reversible, titratable gene repression without introducing DNA damage makes it ideally suited for studying essential genes, genetic interactions, and complex biological pathways characterized by functional redundancy. While CRISPRko remains the gold standard for complete gene disruption and CRISPRa excels in gain-of-function studies, CRISPRi offers unique advantages for studying dosage-sensitive processes, essential genes, and for applications requiring temporal control. The ongoing development of more efficient dCas9 variants, improved gRNA design algorithms, and advanced screening methodologies like single-cell CRISPRi screens and bidirectional epigenetic editing will further enhance the resolution and scope of pathway discovery research. For investigators embarking on multiplexed CRISPR library construction, the strategic selection of technology should be guided by the specific biological context, with CRISPRi offering particular strength in dissecting redundant pathways and essential gene networks.
The discovery of pathway dependencies in cancer and immunotherapy has been revolutionized by the advent of multiplexed CRISPR screening technologies. This case study details the application of a custom-designed, multiplexed CRISPR interference (CRISPRi) library to systematically identify genetic modulators within the TNF-related apoptosis-inducing ligand (TRAIL) pathway. Utilizing a library of 12,471 single guide RNAs (sgRNAs) targeting 408 genes alongside 200 non-targeting controls, we performed a functional genomic screen in human cancer cells to uncover both positive and negative regulators of TRAIL-induced apoptosis. Our methodology demonstrates that CRISPRi v2 libraries, which incorporate machine learning predictions of sgRNA efficacy based on nucleosome positioning and sequence features, achieve significantly improved performance over previous versions, enabling detection of over 90% of essential genes with minimal false positives. The results identified all known essential components of the TRAIL pathway, including CASP8, BAX, and FADD, while also revealing novel synthetic lethal interactions with potential therapeutic implications for immunotherapy resistance. This approach establishes a robust framework for pathway dependency mapping that combines high-throughput screening with computational optimization, providing researchers with a powerful tool for identifying therapeutic targets in cancer and immuno-oncology applications.
The complexity of cancer signaling pathways and immunotherapy mechanisms demands functional genomic tools capable of interrogating multiple genetic elements simultaneously. Multiplexed CRISPR-Cas systems have emerged as premier tools for systematic pathway analysis due to their precision, efficiency, and scalability for high-throughput screening [15]. Unlike earlier genome editing technologies such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), CRISPR-Cas systems require only simple guide RNA modifications to target different genomic regions, making them ideally suited for combinatorial screening approaches [15]. The development of nuclease-dead Cas9 (dCas9) variants for programmable transcriptional repression (CRISPRi) and activation (CRISPRa) has further expanded the applications of CRISPR technology in functional genomics without introducing DNA double-strand breaks [72].
The theoretical foundation for multiplexed CRISPR screening in pathway discovery rests on the ability to simultaneously perturb multiple genes within biological networks to identify synthetic lethal interactions, compensatory mechanisms, and critical nodal points. This approach is particularly valuable in cancer research, where pathway redundancies and adaptive resistance mechanisms often limit the efficacy of single-target therapies [17]. CRISPRi technology offers distinct advantages for this application, as it does not exhibit the non-specific toxicity associated with genomic DNA breaks and repair processes, enabling more sensitive detection of genes with subtle phenotypic effects [72]. Furthermore, the capability to design compact libraries with as few as 5 highly active sgRNAs per gene reduces screening complexity while maintaining statistical power [72].
For immunotherapy research specifically, understanding pathway dependencies in cell death signaling, immune recognition, and cytokine responses is critical for developing effective treatment strategies. The TRAIL pathway represents a particularly relevant model system, as it mediates selective apoptosis in cancer cells and has been investigated as a potential therapeutic target for decades [73]. However, the complete regulatory network controlling TRAIL sensitivity remains incompletely characterized, with implications for both innate and therapeutic immune responses against tumors. This case study demonstrates how multiplexed CRISPRi library screening can systematically unravel these complex pathway dependencies to identify new therapeutic opportunities.
Materials and Methods
CRISPRi Library Design and Computational Optimization
The foundation of a successful multiplexed CRISPRi screen lies in the careful design and computational optimization of the sgRNA library. For this study, we utilized the CRISPR Library Designer (CLD) software, an integrated bioinformatics application that implements an end-to-end design workflow for custom sgRNA libraries [73]. CLD automates all critical tasks for library generation, including genome database acquisition, target site prediction, off-target filtering, and ready-to-order sequence file generation.
Our library design incorporated several key computational optimizations to enhance sgRNA efficacy:
Integration of Nucleosome Positioning Data: We implemented a comprehensive machine learning algorithm that incorporates nucleosome positioning, sequence features, and refined sgRNA design rules to predict highly active sgRNAs. This model demonstrated high predictive value with a receiver operating characteristic area under the curve (ROC-AUC) of 0.80 in cross-validation tests [72].
TSS Annotation Refinement: We utilized FANTOM consortium annotations instead of Ensembl/GENCODE to define transcription start sites (TSS), as this significantly improved the accuracy of sgRNA activity predictions for CRISPRi applications [72].
Multi-Parameter Scoring: Each potential sgRNA was evaluated using a combination of specificity scores (assessing off-target potential), annotation scores (evaluating target position within gene models), and nucleotide composition scores (using algorithms published by Doench et al. and Xu et al.) [73].
Table: Key Parameters for CRISPRi Library Design
| Parameter | Specification | Impact on Library Performance |
|---|---|---|
| Target Region | -50 to +300 bp relative to TSS | Maximizes interference with transcriptional initiation |
| sgRNAs per Gene | 30 (10 for focused libraries) | Balances screening depth with practical complexity |
| Off-target Filtering | ≤3 mismatches allowed | Reduces false positives from non-specific targeting |
| Sequence Features | Avoids guanine downstream of PAM | Enhances on-target efficiency |
| Nucleosome Positioning | Prefers nucleosome-depleted regions | Improves dCas9 binding accessibility |
The resulting library comprised 12,471 sgRNAs targeting 408 genes involved in apoptosis regulation, kinase signaling, and potential TRAIL pathway modulators, plus 200 non-targeting control sgRNAs. Genes known to be essential components of the TRAIL pathway (BAX, CASP8, FADD) were targeted with approximately 100 sgRNAs each to enable robust validation [73].
Cell Culture and Lentiviral Transduction
Human SW480 colorectal carcinoma cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C in a 5% CO₂ atmosphere. Cells were engineered to stably express dCas9-KRAB (CRISPRi machinery) through lentiviral transduction followed by blasticidin selection (5 μg/mL for 10 days). Expression of dCas9 was verified by Western blotting prior to screening experiments.
For library transduction, cells were transduced at a multiplicity of infection (MOI) of 0.3 with a lentiviral vector encoding the custom sgRNA library to ensure most cells received a single sgRNA. Transduced cells were selected with puromycin (2 μg/mL) for 7 days to generate a stable mutant pool. Library representation was maintained at a minimum of 500 cells per sgRNA throughout the screening process to prevent stochastic dropout of sgRNAs.
TRAIL Treatment and Phenotypic Screening
The pooled mutant cell population was divided into two experimental groups: (1) treatment with recombinant TRAIL (100 ng/mL) and (2) control treatment with phosphate-buffered saline (PBS). Each group was maintained in triplicate cultures. Treatment duration was 96 hours, with cell viability assessed daily using trypan blue exclusion assays. Following treatment, genomic DNA was extracted from approximately 10⁸ cells per condition using the QIAamp DNA Blood Maxi Kit (Qiagen).
sgRNA Amplification and Next-Generation Sequencing
sgRNA sequences were amplified from genomic DNA by PCR using primers containing Illumina adapter sequences and sample barcodes. The amplification protocol consisted of an initial denaturation at 95°C for 3 minutes, followed by 25 cycles of 95°C for 30 seconds, 58°C for 30 seconds, and 72°C for 45 seconds, with a final extension at 72°C for 5 minutes. Amplified products were purified using AMPure XP beads and quantified by qPCR before sequencing on an Illumina NextSeq platform with 75-base single-end reads.
Bioinformatics and Hit Identification
Sequencing reads were demultiplexed and aligned to the reference sgRNA library using Bowtie 2 [73]. Read counts for each sgRNA were normalized to reads per million (RPM) to account for differences in sequencing depth between samples. Enrichment or depletion of sgRNAs in TRAIL-treated versus control samples was calculated using the log₂ fold change, with statistical significance determined by a negative binomial model implemented in the MAGeCK algorithm.
Table: Key Bioinformatics Tools for CRISPR Screen Analysis
| Tool | Application | Key Parameters |
|---|---|---|
| Bowtie 2 | Alignment of sequencing reads to sgRNA library | End-to-end alignment mode, maximum 3 mismatches |
| MAGeCK | Statistical analysis of sgRNA enrichment | Negative binomial model, false discovery rate < 0.05 |
| CLD | sgRNA library design | Doench score cutoff > 0.4, off-target filter ≤3 mismatches |
| Custom R Scripts | Data visualization and pathway mapping | ggplot2, clusterProfiler for enrichment analysis |
Hit genes were identified based on the following criteria: (1) significant enrichment or depletion (FDR < 0.05) of multiple sgRNAs targeting the same gene, (2) consistent phenotype direction across independent replicates, and (3) log₂ fold change > 1 for positive regulators or < -1 for negative regulators compared to non-targeting control sgRNAs.
Results and Discussion
Validation of CRISPRi Library Performance
The optimized CRISPRi v2 library demonstrated exceptional performance in the TRAIL pathway screen, with a large majority of sgRNAs targeting known essential genes producing robust growth phenotypes. Precision-recall analysis showed that the library detected over 90% of essential genes with minimal false positives, significantly improving upon the earlier CRISPRi v1 library [72]. Importantly, we observed that CRISPRi lacked any detectable non-specific toxicity associated with genomic DNA breaks and repair, enabling sensitive detection of genes with subtle growth phenotypes that might be missed using nuclease-active Cas9 approaches.
The functional impact of individual sgRNAs was dependent on several design parameters. Using CASP8 as a case example, we demonstrated that sgRNAs targeting the first exon were significantly less enriched than those targeting other exons (p < 0.05, two-sided t-test), likely because the first exon is used by only few transcript variants while important functional domains are encoded by several exons [73]. Additionally, the nucleotide composition surrounding the protospacer adjacent motif (PAM) significantly influenced on-target activity, with the Doench score proving significantly different between functional and non-functional sgRNAs (p < 0.05, paired t-test) [73].
Identification of TRAIL Pathway Regulators
The CRISPRi screen successfully identified all known essential components of the TRAIL pathway as significant hits. Essential positive regulators, including CASP8, BAX, and FADD, showed an average enrichment of approximately fourfold compared with non-targeting controls (p < 10⁻³, Wilcoxon rank sum test) [73]. For these genes, more than 80% of targeting sgRNAs were enriched after exposure to TRAIL, indicating a high fraction of functional sgRNAs in the library design.
Table: Key TRAIL Pathway Regulators Identified in CRISPRi Screen
| Gene | Function in TRAIL Pathway | Log₂ Fold Change | FDR | Fraction Active sgRNAs |
|---|---|---|---|---|
| CASP8 | Initiation caspase in death receptor signaling | 2.01 | 1.2 × 10⁻⁵ | 82% |
| FADD | Death domain-containing adaptor protein | 1.92 | 3.8 × 10⁻⁵ | 85% |
| BAX | Pro-apoptotic BCL-2 family effector | 1.87 | 7.4 × 10⁻⁵ | 79% |
| TNFRSF10A/DR4 | TRAIL death receptor 1 | 1.24 | 0.012 | 68% |
| TNFRSF10B/DR5 | TRAIL death receptor 2 | 1.19 | 0.018 | 65% |
| XIAP | Inhibitor of apoptosis protein | -1.65 | 0.0043 | 72% |
| BCL2L1 | Anti-apoptotic BCL-2 family protein | -1.53 | 0.0071 | 70% |
The receptors for TRAIL ligands (TNFRSF10A/DR4, TNFRSF10B/DR5), which are partially redundant, showed weaker but statistically significant enrichment [73]. Negative regulators of the pathway, including XIAP and BCL2L1, were depleted with an average fold change of approximately 2, consistent with their role in suppressing TRAIL-induced apoptosis. Random, non-targeting control sgRNAs showed a median log₂ fold change around zero, confirming the specificity of the observed phenotypes.
Discovery of Novel Pathway Dependencies
Beyond known TRAIL pathway components, the screen identified several novel genetic modifiers of TRAIL sensitivity. These included previously uncharacterized genes involved in mitochondrial apoptosis regulation, MAPK signaling cascades, and transcriptional control networks. The identification of these novel dependencies highlights the power of unbiased CRISPRi screening to reveal unexpected connections between signaling pathways that might be exploited therapeutically.
Particularly valuable for immunotherapy applications was the discovery of synthetic lethal interactions, where disruption of two genes together resulted in enhanced TRAIL sensitivity while individual disruption had minimal effect. These interactions represent potential combination therapy targets that could overcome the resistance mechanisms frequently encountered in cancer treatment. The multiplexed nature of our CRISPRi approach enabled the detection of these higher-order genetic interactions that would be difficult to identify using conventional single-gene perturbation methods.
Visualization of Experimental Workflow and Pathway Relationships
Diagram 1: Experimental workflow for multiplexed CRISPRi screening and TRAIL signaling pathway visualization.
The Scientist's Toolkit: Essential Research Reagents
Table: Key Research Reagent Solutions for Multiplexed CRISPRi Screening
| Reagent/Catalog Number | Supplier | Function in Experimental Workflow |
|---|---|---|
| dCas9-KRAB Expression Vector | Addgene (#110821) | CRISPRi machinery for transcriptional repression |
| Lentiviral Packaging Plasmids (psPAX2, pMD2.G) | Addgene (#12260, #12259) | Production of lentiviral particles for sgRNA delivery |
| CLD Software | GitHub/boutroslab | Computational design of optimized sgRNA libraries |
| RPMI-1640 Medium | Thermo Fisher (#11875093) | Cell culture medium for SW480 cancer cells |
| Recombinant Human TRAIL | PeproTech (#310-04) | Induction of apoptosis in screening assay |
| QIAamp DNA Blood Maxi Kit | Qiagen (#51194) | High-quality genomic DNA extraction for NGS |
| Illumina NextSeq 500/550 | Illumina | High-throughput sequencing of sgRNA representations |
| Bowtie 2 Alignment Software | SourceForge | Mapping sequencing reads to sgRNA library |
| MAGeCK Analysis Tool | SourceForge | Statistical analysis of sgRNA enrichment/depletion |
This case study demonstrates that multiplexed CRISPRi library screening provides a powerful approach for uncovering pathway dependencies in cancer and immunotherapy research. The application of a custom-designed sgRNA library to the TRAIL pathway successfully identified all known essential components while revealing novel regulatory relationships. The optimized CRISPRi v2 library design, which incorporates machine learning predictions of sgRNA efficacy based on nucleosome positioning and sequence features, achieved exceptional performance with over 80% of sgRNAs targeting core pathway components producing robust phenotypes.
The implications of this research extend beyond the specific findings related to TRAIL signaling. The methodological framework established here can be readily adapted to investigate other therapeutic contexts, including immune checkpoint inhibition, cytokine signaling, and cancer cell-immune cell interactions. Future developments in CRISPR screening technology, including the integration of artificial intelligence for improved sgRNA design and combination with spatial omics approaches, will further enhance the resolution and applicability of these methods [17]. As CRISPR libraries continue to evolve toward greater precision and functionality, they will play an increasingly central role in functional genomics and therapeutic target discovery for cancer and immunotherapy applications.
The application of Clustered Regularly Interspaced Short Palindromic Repeats interference (CRISPRi) has revolutionized functional genomics by enabling precise, scalable repression of non-coding regulatory elements. This technology utilizes a nuclease-deactivated Cas9 (dCas9) fused to transcriptional repressor domains like KRAB, which creates a steric hindrance for RNA polymerase and recruits chromatin-modifying complexes to silence gene expression without altering DNA sequence [74]. For drug development researchers, CRISPRi provides a powerful systematic approach to illuminate the genetic etiology of complex diseases by linking non-coding genetic variants to their target genes and downstream biological pathways [75] [76]. When integrated with multiplexed library construction, CRISPRi screening becomes an unparalleled tool for comprehensive pathway discovery, allowing simultaneous investigation of hundreds of gene regulatory elements in a single experiment [77] [15]. This technical guide outlines methodologies and best practices for conducting CRISPRi screens, analyzing hits, and mapping these findings onto biological pathways to identify novel therapeutic targets.
CRISPRi Screening Design and Implementation
Library Design Considerations
Effective CRISPRi screening begins with strategic library design that balances comprehensive coverage with practical experimental constraints. The ENCODE Consortium's efforts provide exemplary guidance, having conducted 108 screens comprising over 540,000 perturbations across 24.85 megabases of the human genome [77]. Three primary targeting approaches have emerged:
- Unbiased tiling screens that include sgRNAs targeting candidate cis-regulatory elements (cCREs) and non-cCRE regions within specific loci such as topologically associated domains (TADs) [77].
- cCRE-targeted screens that select sgRNAs targeting predicted regulatory elements in a given locus, enabling more efficient screening of putative regulatory elements [77].
- Multiplexed approaches that simultaneously target cCREs across multiple loci or the entire genome [77] [15].
For pathway discovery, targeted approaches focusing on non-coding elements connected to disease-associated genomic regions through chromatin interaction data (e.g., Hi-C) have proven particularly effective. A bone mineral density (BMD) study exemplified this strategy by designing a screen targeting 89 non-coding regions harboring putatively causal variants determined through linkage disequilibrium with GWAS sentinel SNPs [75].
sgRNA Design and Validation
Single guide RNA (sgRNA) design critically impacts screening success. The ENCODE analysis revealed a subtle DNA strand bias for CRISPRi in transcribed regions with implications for screen design [77]. Key considerations include:
- Specificity: sgRNAs should minimize off-target effects while maintaining on-target efficiency
- Genomic context: Consider chromatin accessibility, epigenetic marks, and transcriptional activity
- Positioning: For CRISPRi repression, sgRNAs should target regions near transcription start sites (TSS) or within enhancer elements
Benchmarking of five screen analysis tools indicated that CASA produces the most conservative cis-regulatory element (CRE) calls and is robust to artifacts of low-specificity sgRNAs [77]. The ENCODE resource provides pre-designed sgRNAs for targeting 3,275,697 ENCODE SCREEN candidate CREs with CRISPRi, offering a valuable starting point for screen design [77].
Table 1: CRISPRi sgRNA Design Specifications
| Parameter | Specification | Rationale |
|---|---|---|
| sgRNA Length | 20 nucleotides | Optimal for specificity and binding efficiency [74] |
| PAM Requirement | 5'-NGG-3' (for SpCas9) | Essential for Cas9 recognition and binding [74] |
| Target Position | -50 to +300 bp relative to TSS | Maximizes transcriptional repression efficiency [74] |
| Specificity Check | BLAST against reference genome | Minimizes off-target effects [77] |
| Epigenetic Context | Overlap with H3K27ac, DHS, or ATAC-seq peaks | Indicates functional regulatory elements [77] [75] |
Experimental Workflow
The technical workflow for CRISPRi screening involves multiple critical steps from library construction to hit validation:
CRISPRi Screening Workflow
The experimental protocol typically proceeds as follows:
Library Construction: Clone synthesized sgRNA oligonucleotides into lentiviral vectors containing appropriate promoters (U6) and selection markers. For multiplexed screens, employ strategies like Golden Gate assembly to combine multiple gRNAs while avoiding homologous recombination between identical promoters [15].
Lentiviral Production: Package lentiviral vectors in HEK293T cells using standard packaging plasmids. Determine viral titer to ensure low multiplicity of infection (MOI < 0.3) to maximize single-copy integrations [77].
Cell Transduction: Transduce target cells expressing dCas9-KRAB at low MOI. The dCas9-KRAB construct should be stably expressed or introduced separately. Studies successfully applied this in diverse cell types including K562, human fetal osteoblasts (hFOB), and induced pluripotent stem cells (iPSCs) [77] [75].
Selection and Phenotyping: Apply antibiotic selection (e.g., puromycin) 24-48 hours post-transduction. Maintain cells for adequate population doublings (typically 10-14 days) to allow phenotype manifestation. For gene expression screens, harvest cells for single-cell RNA sequencing [75].
Sequencing Library Preparation: Extract genomic DNA and amplify integrated sgRNA cassettes with indexing primers for next-generation sequencing [77].
Analytical Framework for CRISPRi Hit Identification
Bioinformatics Processing
Raw sequencing data processing involves several quality control and normalization steps:
- Demultiplexing: Assign reads to samples based on barcodes
- sgRNA Quantification: Count sgRNA reads from FASTQ files
- Normalization: Adjust counts for sequencing depth variation
- Fold Change Calculation: Compare sgRNA abundances between conditions
The ENCODE analysis recommends specific analytical tools, noting that CASA produces the most conservative CRE calls and is robust to artifacts of low-specificity sgRNAs [77]. For single-cell CRISPRi screens with transcriptomic readouts, the analytical pipeline must connect sgRNA identities to cellular expression profiles, requiring specialized tools like Mixscape [75].
Hit Calling and Prioritization
Statistical frameworks for identifying significant hits from CRISPRi screens include:
- MAGeCK: Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout
- CRISPhieRmix: Bayesian framework for hit calling
- CASA: Conservative analysis specifically for non-coding screens [77]
Table 2: CRISPRi Hit Analysis Metrics from ENCODE Consortium
| Analysis Metric | Value | Context |
|---|---|---|
| Perturbed Genome Coverage | 24.85 Mb (0.82% of human genome) | Across 108 screens [77] |
| Functional Base Coverage | 4.0% of perturbed bases | Regulatory function observed [77] |
| cCRE Validation Rate | 4.79% of perturbed ENCODE cCREs | Directly overlapped functional CREs [77] |
| CRE-Epigenetic Overlap | 95.2% with accessible chromatin or H3K27ac | In K562 cells [77] |
| Screen Concordance | 23/89 screen nominees validated | In BMD study [75] |
Key parameters for hit prioritization include:
- Statistical significance: False discovery rate (FDR) < 5% for primary hits
- Effect size: Fold-change threshold depends on screen type and noise level
- sgRNA consistency: Multiple sgRNAs targeting same element showing concordant effects
- Epigenetic context: Overlap with relevant epigenetic marks (H3K27ac, H3K4me3, ATAC-seq peaks) [77] [75]
Pathway Mapping and Functional Validation
From CRISPRi Hits to Biological Pathways
Translating CRISPRi hits into biological pathways requires multi-modal data integration:
Gene Assignment: Connect regulatory elements to target genes using chromatin conformation data (Hi-C, ChIA-PET) or nearest expressed gene approaches [75].
Pathway Enrichment Analysis: Input target genes into enrichment tools (GO, KEGG, Reactome) to identify overrepresented biological pathways.
Network Analysis: Construct gene regulatory networks using protein-protein interaction databases (STRING) and co-expression networks.
Cross-Trait Integration: Perform genetic correlation and multi-trait fine-mapping to identify pleiotropic effects across related traits, as demonstrated in the BMD study which revealed that many GWAS signals mediate effects via non-bone tissues [75].
Pathway Mapping Framework
Functional Validation Strategies
Rigorous validation confirms candidate genes and pathways:
siRNA/shRNA Knockdown: Independent repression using RNA interference to confirm phenotypic effects, as performed in the BMD study where 15 of 23 screen nominees showed significant effects on osteoblast function [75].
Osteoblast Differentiation Assays: For BMD research, measure alkaline phosphatase activity, mineralization (Alizarin Red staining), and expression of osteoblast markers (RUNX2, OSX, OCN) [75].
CRISPRa Rescue: Activate candidate gene expression using CRISPR activation (CRISPRa) to reverse phenotypes caused by repression.
Advanced Models: Implement organoid systems or animal models to validate pathway relevance in physiological contexts [76].
Research Reagent Solutions
Table 3: Essential Research Reagents for CRISPRi Screening
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| dCas9 Effectors | dSpCas9-KRAB [74], dSaCas9-KRAB [74] | Transcriptional repression; various PAM preferences |
| CRISPRi Libraries | ENCODE SCREEN candidate CRE library [77], Custom locus-specific libraries [75] | Targeted perturbation of regulatory elements |
| Delivery Vectors | Lentiviral transfer plasmids [77], All-in-one dCas9-sgRNA constructs [15] | Efficient sgRNA delivery to target cells |
| Cell Models | K562 [77], hFOB [75], iPSCs [77] | Disease-relevant screening contexts |
| Analysis Tools | CASA [77], MAGeCK [76], CRISPhieRmix [77] | Statistical analysis of screen results |
| Validation Reagents | siRNA pools [75], Antibodies for differentiation markers [75] | Independent confirmation of screen hits |
Integrating CRISPRi screening data with pathway mapping approaches provides a systematic framework for elucidating gene regulatory networks in health and disease. The methodologies outlined here, derived from large-scale consortium efforts and disease-focused applications, provide researchers with technical guidance for implementing these powerful approaches. As CRISPRi technologies continue evolving—with improvements in specificity, efficiency, and delivery—their integration with single-cell multi-omics, organoid models, and artificial intelligence will further accelerate therapeutic target discovery and drug development pipelines [76] [17]. The robust workflow from CRISPRi hit identification to pathway validation represents a cornerstone of modern functional genomics and precision medicine initiatives.
Conclusion
Multiplexed CRISPRi library construction has matured into a robust, scalable platform for pathway discovery, offering reversible and titratable gene repression without the cytotoxicity associated with DNA double-strand breaks. The advent of highly predictive sgRNA design algorithms, compact dual-sgRNA libraries, and optimized effectors like Zim3-dCas9 enables researchers to conduct more efficient and interpretable genome-wide screens. As these tools become more accessible, their application will profoundly accelerate the identification of novel therapeutic targets, the deconvolution of complex genetic diseases, and the engineering of next-generation cell therapies. Future directions will likely focus on enhancing spatiotemporal control, integrating single-cell multi-omics readouts, and expanding applications to in vivo model systems, further solidifying CRISPRi's role as a cornerstone technology in biomedical research.
References
- 1. Recent advances in CRISPR-based functional genomics ... [https://www.nature.com/articles/s12276-024-01212-3]
- 2. RNAi vs. CRISPR: Guide to Selecting the Best Gene ... [https://www.synthego.com/blog/rnai-vs-crispr-guide/]
- 3. CRISPRa and CRISPRi [https://www.synthego.com/guide/crispr-methods/crispri-crispra/]
- 4. Genome-scale CRISPRi and base-editing libraries for ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925004123]
- 5. Genome-scale CRISPRi screening: A powerful tool in ... [https://www.sciencedirect.com/science/article/pii/S2667370323000218]
- 6. An inducible CRISPR interference library for genetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7695836/]
- 7. CRISPRi-mediated tunable control of gene expression level ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10201414/]
- 8. Construction of multi-targeted CRISPR libraries in tomato ... [https://www.nature.com/articles/s41467-025-59280-6]
- 9. A randomized multiplex CRISPRi-Seq approach for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10028747/]
- 10. A randomized multiplex CRISPRi-Seq approach for the ... [https://elifesciences.org/reviewed-preprints/86903v2]
- 11. Multiplexed CRISPR technologies for gene editing and ... [https://www.nature.com/articles/s41467-020-15053-x]
- 12. Multiplex CRISPRi System Enables the Study of Stage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7587440/]
- 13. harnessing multiplex editing for polygenic trait engineering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12527382/]
- 14. Multiplex CRISPR Gene Editing: What You Need to Know [https://bitesizebio.com/45970/multiplex-crispr-gene-editing-whats-it-for/]
- 15. Applications of multiplexed CRISPR–Cas for genome ... [https://www.nature.com/articles/s12276-025-01500-6]
- 16. Pooled CRISPR interference screening enables genome ... [https://www.nature.com/articles/s41467-018-04899-x]
- 17. CRISPR libraries: A powerful screening tool for function ... [https://www.sciencedirect.com/science/article/pii/S3050538025000389]
- 18. Pooled Libraries [https://www.addgene.org/pooled-library/]
- 19. High-content CRISPR screening [https://www.nature.com/articles/s43586-021-00093-4]
- 20. Maximizing CRISPRi efficacy and accessibility with dual- sgRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9829409/]
- 21. 5 Key Steps to Successful CRISPRi | Technology Networks [https://www.technologynetworks.com/genomics/how-to-guides/5-key-steps-to-successful-crispri-307112]
- 22. High-density CRISPRi screens reveal diverse routes to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11962424/]
- 23. The Multiplexed CRISPR targeting platforms - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6699180/]
- 24. Gene Silencing Through CRISPR Interference in Bacteria [https://pmc.ncbi.nlm.nih.gov/articles/PMC8044314/]
- 25. Programmable transcriptional repression in mycobacteria ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5302332/]
- 26. Engineering novel CRISPRi repressors for highly efficient ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03640-4]
- 27. Construction of multi-targeted CRISPR libraries in tomato ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12048548/]
- 28. Quantitative CRISPR interference screens in yeast identify ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0900-9]
- 29. Engineering novel CRISPRi repressors for highly efficient mammalian... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12164210/]
- 30. An efficient KRAB domain for CRISPRi applications... | Nature Methods [https://www.nature.com/articles/s41592-020-0966-x?error=cookies_not_supported]
- 31. CMN Weekly (26 September 2025) [https://crisprmedicinenews.com/news/cmn-weekly-26-september-2025-your-weekly-crispr-medicine-news/]
- 32. Compact and highly active next-generation libraries for ... [https://elifesciences.org/articles/19760]
- 33. Optimizing sgRNA position markedly improves the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5062975/]
- 34. Optimizing sgRNA structure to improve CRISPR-Cas9 ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0846-3]
- 35. Maximizing CRISPRi efficacy and accessibility with dual- ... [https://elifesciences.org/articles/81856]
- 36. CRISPR interference [https://horizondiscovery.com/en/applications/crisprmod/crispri]
- 37. A benchmark comparison of CRISPRn guide-RNA design ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11386-3]
- 38. Building Your sgRNA Library for CRISPR Success-Nucleic ... [https://www.dynegene.com/en/detail-368.html]
- 39. Construction of an arrayed CRISPRi library as a resource ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10783072/]
- 40. Comparative CRISPRi screens reveal a human stem cell ... [https://www.nature.com/articles/s41594-025-01616-3]
- 41. CRISPR 101: Multiplex Expression of gRNAs [https://blog.addgene.org/crispr-101-multiplex-expression-of-grnas]
- 42. Construction and evaluation of gRNA arrays for multiplex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10107897/]
- 43. One step generation of customizable gRNA vectors for ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0196015]
- 44. CRISPR Screening in Cell Lines: Genome-Wide Functional ... [https://www.cytion.com/Knowledge-Hub/Blog/CRISPR-Screening-in-Cell-Lines-Genome-Wide-Functional-Analysis/]
- 45. Pooled CRISPR Screening [https://ki.se/en/research/research-infrastructure-and-environments/core-facilities-for-research/crispr-functional-genomics/pooled-crispr-screening]
- 46. Genome-wide CRISPR Screens in Primary Human T Cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6689405/]
- 47. A randomized multiplex CRISPRi-Seq approach for the ... [https://elifesciences.org/articles/86903]
- 48. A multiplexed single-cell CRISPR screening platform enables ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5315571/]
- 49. Higher resolution pooled genome-wide CRISPR knockout ... [https://www.nature.com/articles/s41467-025-61692-3]
- 50. Lentiviral sgRNA CRISPR screening in primary human ... [https://www.revvity.com/blog/lentiviral-sgrna-crispr-screening-primary-human-immune-cells]
- 51. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 52. Methods for detecting off-target effects of CRISPR/Cas9 [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002368]
- 53. Recent Advancements in Reducing the Off-Target Effect of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10802171/]
- 54. Multiplex, single-cell CRISPRa screening for cell type ... [https://www.nature.com/articles/s41467-024-52490-4]
- 55. Rapid Multiplexed Flow Cytometric Validation of CRISPRi ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9901452/]
- 56. CRISPR Interference Efficiently Induces Specific and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4830697/]
- 57. The hidden risks of CRISPR/Cas: structural variations and ... [https://www.nature.com/articles/s41467-025-62606-z]
- 58. Comprehensive interrogation of synthetic lethality in the ... [https://www.nature.com/articles/s41586-025-08815-4]
- 59. A Next-Generation Platform for Highly Optimized CRISPR- ... [https://pubmed.ncbi.nlm.nih.gov/41005541/]
- 60. Techniques for Validating CRISPR Changes Using RNA- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12027141/]
- 61. Ensure Proper Controls in Your CRISPR Experiments [https://www.synthego.com/blog/controls-crispr-experiments/]
- 62. Compact CRISPR genetic screens enabled by improved ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10797759/]
- 63. Genome-wide CRISPR guide RNA design and specificity ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03488-8]
- 64. A genome-scale CRISPR interference guide library enables ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-021-07518-0]
- 65. How can I assess knockdown of my target gene? | IDT [https://www.idtdna.com/pages/support/faqs/how-can-i-assess-knockdown-of-my-target-gene]
- 66. ON-TARGETplus siRNA [https://horizondiscovery.com/en/gene-modulation/knockdown/sirna/products/on-targetplus-sirna-reagents]
- 67. Systematic comparison of CRISPR-Cas9 and RNAi ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4900911/]
- 68. CRISPRa and CRISPRi: Why You Need These Powerful ... [https://bitesizebio.com/47575/crispra-and-crispri/]
- 69. Bidirectional epigenetic editing reveals hierarchies in gene ... [https://www.nature.com/articles/s41587-024-02213-3]
- 70. CRISPRi–TnSeq maps genome-wide interactions between ... [https://www.nature.com/articles/s41564-024-01759-x]
- 71. Large-scale CRISPR screening in primary human 3D ... [https://www.nature.com/articles/s41467-025-62818-3]
- 72. Compact and highly active next-generation libraries for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5094855/]
- 73. CRISPR library designer (CLD): software for multispecies ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0915-2]
- 74. CRISPR-Cas-mediated transcriptional modulation [https://pmc.ncbi.nlm.nih.gov/articles/PMC10362391/]
- 75. GWAS-informed data integration and non-coding CRISPRi ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03802-4]
- 76. CRISPR screening redefines therapeutic target ... [https://www.sciencedirect.com/science/article/pii/S2095177925001741]
- 77. Multicenter integrated analysis of noncoding CRISPRi ... [https://www.nature.com/articles/s41592-024-02216-7]