CAR-T Cell Engineering: From Foundational Principles to Next-Generation Clinical Protocols
This article provides a comprehensive overview of chimeric antigen receptor (CAR)-T cell engineering protocols, tracing the evolution from foundational concepts to cutting-edge clinical applications.
CAR-T Cell Engineering: From Foundational Principles to Next-Generation Clinical Protocols
Abstract
This article provides a comprehensive overview of chimeric antigen receptor (CAR)-T cell engineering protocols, tracing the evolution from foundational concepts to cutting-edge clinical applications. It details the structural components and modular design of synthetic CAR receptors, established manufacturing workflows for hematologic malignancies, and critical troubleshooting for challenges like T-cell exhaustion and cytokine release syndrome. The content further explores advanced validation techniques and comparative analyses of novel engineering strategies—including logic-gated, armored, and allogeneic CAR-T cells—that are pushing the boundaries in solid tumors. Aimed at researchers, scientists, and drug development professionals, this review synthesizes current literature and clinical trial data to serve as a methodological guide and innovation roadmap for the next generation of cancer immunotherapies.
The Architecture and Evolution of CAR-T Cells: Deconstructing the Engineering Blueprint
Chimeric Antigen Receptors (CARs) are synthetic receptors designed to redirect immune effector cells, primarily T cells, to recognize and eliminate cancer cells with specified surface antigens. The modular architecture of a CAR allows for the customization of its functional properties, making the understanding of its core structural domains fundamental to cancer immunotherapy research [1] [2]. These engineered receptors typically fuse an extracellular antigen-recognition domain to intracellular T-cell signaling modules, enabling Major Histocompatibility Complex (MHC)-independent T-cell activation [3]. This application note details the core structural domains of a CAR, provides protocols for their evaluation, and visualizes the critical relationships and workflows to aid in the rational design of CAR constructs for preclinical research.
Core Structural Domains of a CAR
The functional efficacy of a CAR is dictated by the synergistic operation of its four core domains: the antigen-binding domain, the hinge region, the transmembrane domain, and the intracellular signaling domain [2]. The structure and selection of each component directly influence the stability, signaling potency, and ultimate success of the CAR-T cell product.
Antigen-Binding Domain
The antigen-binding domain, typically a single-chain variable fragment (scFv), confers specificity to the target antigen. Derived from the variable heavy (VH) and variable light (VL) chains of a monoclonal antibody connected by a flexible linker, the scFv enables the CAR to bind directly to cell surface antigens without the need for antigen presentation by MHC molecules [1] [2]. Beyond simple recognition, the affinity of the scFv is a critical parameter. A high affinity must be balanced to ensure effective tumor cell recognition while avoiding activation-induced cell death or excessive on-target, off-tumor toxicities that can occur when the target antigen is expressed on healthy tissues [2]. The epitope location and target antigen density on the tumor cell surface are also key considerations that influence CAR functionality [2].
Hinge/Spacer Region
The hinge or spacer is an extracellular structural region that links the antigen-binding domain to the transmembrane domain. Its primary functions are to provide flexibility, overcome steric hindrance from the cell membrane or complex glycans, and allow sufficient intercellular distance for the formation of a stable immunological synapse [2]. The length and composition of the hinge (commonly derived from CD8α, CD28, or IgG Fc regions) significantly impact CAR expression, signaling strength, and epitope recognition [2]. The optimal spacer length is often determined empirically and is dependent on the position of the target epitope; for instance, membrane-proximal epitopes may require long spacers, while membrane-distal epitopes are more effectively targeted with short hinges [2]. Researchers should note that IgG-derived spacers can interact with Fcγ receptors on innate immune cells, potentially leading to off-target activation and CAR-T cell depletion in vivo; this can be mitigated by selecting alternative spacers or engineering the Fc portion to eliminate binding [2].
Transmembrane Domain
The transmembrane (TM) domain is a hydrophobic alpha helix that anchors the CAR to the T cell membrane. While its primary role is structural, the choice of TM domain can influence CAR expression levels, stability, and function [1] [2]. The TM domain can mediate CAR dimerization and functional interaction with endogenous signaling molecules. For example, a CD3ζ-derived TM domain may facilitate incorporation of the CAR into the native TCR complex, potentially enhancing signaling but possibly at the cost of decreased receptor stability compared to a CD28 TM domain [2]. The TM domain is frequently selected to match the extracellular spacer or intracellular co-stimulatory domains for compatibility, but its impact on overall CAR function warrants careful consideration during the design phase [2].
Intracellular Signaling Domain
The intracellular domain is the functional engine of the CAR, responsible for transmitting activation signals upon antigen binding. Its design has evolved through several generations, outlined in the table below.
Table 1: Evolution of CAR Intracellular Signaling Domains
| Generation | Signaling Components | Key Features & Functional Consequences | Representative Targets in Clinical Use |
|---|---|---|---|
| First | CD3ζ only [1] | Provides Signal 1 (primary T-cell activation); limited persistence & cytokine secretion; requires exogenous IL-2 [1] | CD19 [1] |
| Second | CD3ζ + one co-stimulatory domain (e.g., CD28 or 4-1BB) [1] | Provides both Signal 1 and Signal 2; enhances proliferation, cytokine production, cytotoxicity, and in vivo persistence [1] [2]. CD28ζ favors potent activation; 4-1BBζ favors persistence [1]. | CD19 (Kymriah, Yescarta) [4] |
| Third | CD3ζ + two or more co-stimulatory domains (e.g., CD28+4-1BB or CD28+OX40) [1] | Aims to augment potency with stronger cytokine production and killing ability; clinical outcomes not consistently superior to 2nd gen [1]. | CD20, HER2 [1] |
| Fourth (TRUCK) | Second-gen base + inducible cytokine (e.g., IL-12) [1] | "Armored CARs" modify the tumor microenvironment (TME); recruit/activate innate immune cells; can target antigen-negative cancer cells [1]. | In clinical trials for solid tumors [5] |
Advanced Engineering and Universal CAR Systems
To address challenges like antigen escape, toxicity, and complex manufacturing, advanced universal CAR (UniCAR) systems have been developed. These platforms split the conventional CAR into two separate components: a universal signaling module expressed on the T cell and a soluble switch module that directs specificity [6]. This design allows for control over the timing, intensity, and target of the CAR-T cell response. The switch molecule serves as a bridge between the UniCAR T cell and the tumor cell, and its administration can be titrated or interrupted to fine-tune anti-tumor activity or mitigate side effects [6]. Examples include the SUPRA CAR system, which uses leucine zipper pairing, and the biotin-binding immunoreceptor (BBIR) system, which utilizes high-affinity biotin-avidin interaction [6]. This modular approach enables one batch of UniCAR T cells to be redirected against multiple tumor antigens without genetic re-engineering, offering a versatile and potentially more cost-effective strategy [6].
Experimental Protocols for CAR-T Cell Evaluation
Protocol: In Vitro Cytotoxicity Assay (Standard Chromium-51 Release Assay)
This protocol measures the ability of CAR-T cells to lyse antigen-expressing target cells.
Preparation of Target Cells:
- Harvest and wash antigen-positive and antigen-negative (control) tumor cell lines (e.g., MC38-CEA vs. MC38).
- Resuspend cells at 1x10^6 cells/mL in culture medium and label with 100 µCi of Na₂⁵¹CrO₄ for 1 hour at 37°C with occasional gentle mixing.
- Wash cells three times with PBS to remove unincorporated radioactivity and resuspend in complete RPMI-1640 medium at 1x10^5 cells/mL.
Preparation of Effector CAR-T Cells:
- Thaw or harvest expanded CAR-T cells and control T cells (e.g., non-transduced or mock-transduced).
- Count and serially dilute the cells in complete medium to create effector-to-target (E:T) ratios (e.g., 40:1, 20:1, 10:1, 5:1) in a 96-well U-bottom plate.
Co-culture and Assay Execution:
- Add 100 µL of labeled target cells (1x10^4 cells) to each well containing effector cells.
- Include controls for spontaneous release (target cells + medium only) and maximum release (target cells + 1% Triton X-100).
- Centrifuge the plate briefly and incubate for 4-6 hours at 37°C, 5% CO₂.
- After incubation, centrifuge the plate and transfer 50 µL of supernatant from each well to a LumaPlate.
- Measure radioactivity using a gamma counter.
Data Analysis:
- Calculate the percentage of specific lysis using the formula:
% Specific Lysis = (Experimental Release – Spontaneous Release) / (Maximum Release – Spontaneous Release) x 100. - Plot % specific lysis against E:T ratios for CAR-T and control cells. Effective CAR-T cells should show potent, antigen-specific lysis.
- Calculate the percentage of specific lysis using the formula:
Protocol: Flow Cytometric Analysis of CAR Expression and T-cell Phenotype
This protocol is used to determine transduction efficiency and characterize the resulting CAR-T cell product.
Staining for CAR Expression:
- For CARs with an extracellular tag (e.g., a truncated EGFR), use a biotinylated or fluorophore-conjugated protein (e.g., recombinant EGF or an anti-tag antibody) followed by a secondary staining reagent if needed [3].
- For CARs where the scFv is accessible, use a recombinant target antigen protein fused to a marker like Fc or fluorescent protein.
- Resuspend 0.5-1x10^6 CAR-T cells in FACS buffer (PBS + 2% FBS). Incubate with the detection reagent for 30 minutes on ice in the dark. Wash twice with FACS buffer.
Immunophenotyping of T-cell Subsets:
- Perform surface staining with antibodies against CD3, CD4, CD8, and memory markers (e.g., CD45RO, CD62L, CCR7).
- For intracellular staining of exhaustion markers (e.g., PD-1, TIM-3, LAG-3), use a fixation/permeabilization kit according to the manufacturer's instructions after surface staining.
- Incubate with antibodies for 30 minutes on ice, wash, and resuspend in FACS buffer for acquisition.
Data Acquisition and Analysis:
- Acquire data on a flow cytometer. Use fluorescence-minus-one (FMO) controls to set positive gates accurately.
- Analyze data to determine the percentage of CAR-positive cells and the distribution of T-cell subsets (naive, central memory, effector memory, terminally differentiated) and exhaustion markers within the CAR+ population.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for CAR-T Cell Research
| Research Reagent / Tool | Function in CAR-T Cell Development | Example Applications |
|---|---|---|
| Viral Vectors (Lentivirus/Retrovirus) [1] [3] | Stable genomic integration and long-term CAR expression. | Standard method for generating persistent clinical CAR-T products. |
| mRNA Electroporation [3] | Transient CAR expression; mitigates genotoxicity risk. | Ideal for testing novel scFvs in vitro or for targets where safety concerns are high. |
| Sleeping Beauty Transposon System [3] | Non-viral, cost-effective method for stable gene transfer. | An alternative to viral transduction; requires optimization for high efficiency. |
| Recombinant Antigen Protein | Detection of CAR surface expression via flow cytometry. | Quality control of CAR-T product pre-infusion. |
| Cytokine Detection Assays (ELISA/ELISpot) | Quantification of cytokine secretion (IFN-γ, IL-2) upon antigen stimulation. | Measurement of CAR-T cell functional activation. |
| Automated Manufacturing Platforms (e.g., Cocoon, CliniMACS Prodigy) [7] | Closed, automated system for cell washing, activation, transduction, and expansion. | Standardizing and scaling CAR-T production for clinical applications. |
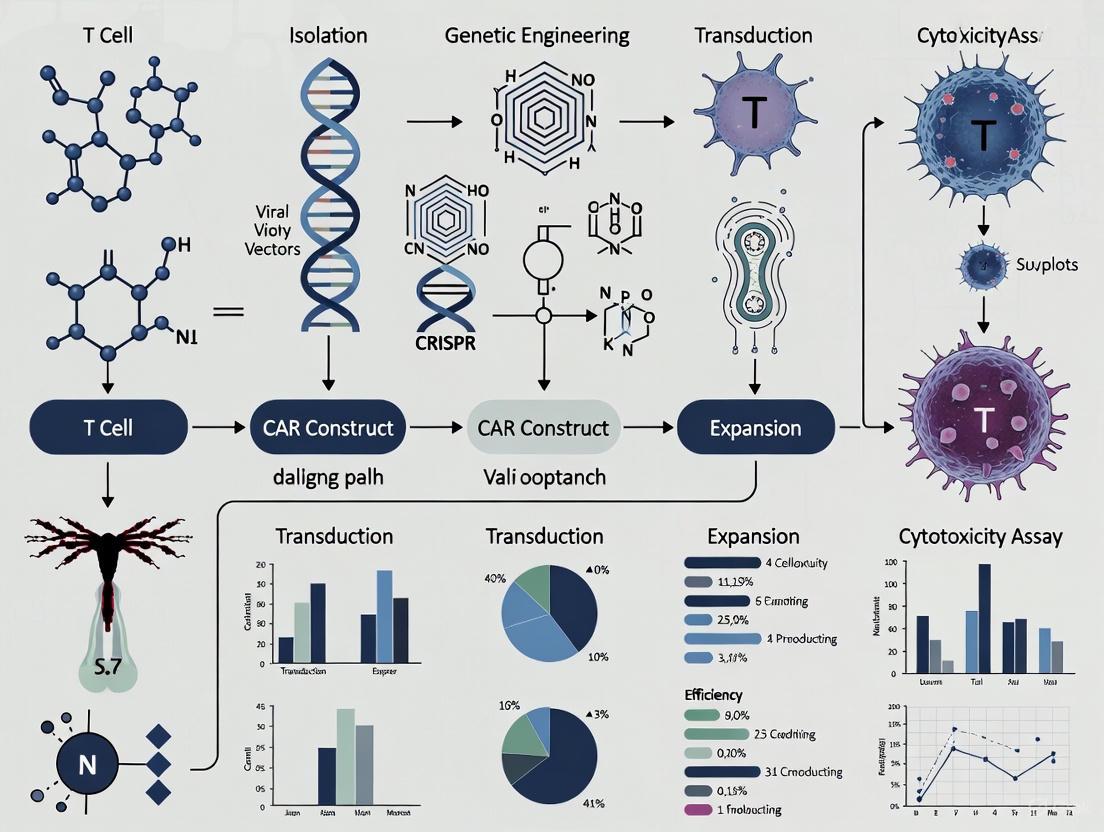
CAR Structure and Engineering Workflow Visualizations
The following diagrams illustrate the core concepts of CAR architecture and the experimental workflow for their evaluation.
CAR Domain Architecture and Signaling
Diagram 1: Core CAR Structure and Signaling Output. This diagram depicts the modular domains of a second-generation CAR. Antigen binding initiates intracellular signaling through primary (CD3ζ) and co-stimulatory (e.g., CD28/4-1BB) domains, leading to T-cell effector functions.
CAR-T Cell Functional Validation Workflow
Diagram 2: CAR-T Cell Engineering and Validation Workflow. This flowchart outlines the key stages in generating and validating CAR-T cells, from construct design to in vitro and in vivo functional assessment.
Chimeric Antigen Receptor (CAR) T-cell therapy represents a paradigm shift in cancer immunotherapy, enabling the direct targeting of tumor antigens through engineered synthetic receptors. Since the concept was first proposed in 1987 and independently illustrated in 1989, CAR technology has undergone remarkable evolution through five distinct generations, each addressing critical limitations in persistence, efficacy, and safety [8] [9]. This evolution has transformed CAR-T cells from research curiosities to powerful clinical tools with proven efficacy against hematological malignancies, culminating in the first FDA approvals in 2017 [8] [10]. The structural refinements across generations have progressively enhanced T-cell activation, persistence, and tumor eradication capabilities while introducing sophisticated control mechanisms. This application note provides a comprehensive technical overview of CAR design generations, detailing their structural characteristics, signaling mechanisms, and experimental protocols to support researchers in developing next-generation CAR-T therapies.
Structural Foundations of CAR Design
The foundational architecture of all CAR constructs consists of three core domains: an extracellular antigen recognition domain, a transmembrane domain, and an intracellular signaling domain [9]. The extracellular domain typically features a single-chain variable fragment (scFv) derived from monoclonal antibodies, though alternative binding scaffolds like nanobodies, DARPins, and natural ligands are increasingly employed [10] [9]. This domain is connected via a hinge region that provides flexibility and access to target epitopes. The transmembrane domain anchors the receptor in the cell membrane and is often derived from CD8α, CD28, or CD4. The intracellular signaling domain initiates T-cell activation and has been systematically enhanced across generations with additional costimulatory domains and signaling modules [8] [9].
Table 1: Core Structural Components of CAR Constructs
| Domain | Key Components | Function | Common Sources |
|---|---|---|---|
| Extracellular | scFv, Nanobodies, Ligands | Antigen recognition | Antibody VH and VL regions [9] |
| Hinge/Spacer | Immunoglobulin domains | Flexibility, epitope access | CD8α, CD28, IgG [9] |
| Transmembrane | Hydrophobic alpha-helices | Membrane anchoring | CD8α, CD28, CD3ζ [9] |
| Intracellular | Signaling domains | T-cell activation, persistence | CD3ζ, CD28, 4-1BB, ICOS [8] |
The Generational Progression of CAR Designs
First-Generation CARs: Proof of Concept
First-generation CARs established the fundamental principle of redirecting T-cell specificity through a simple architecture consisting of an extracellular scFv fused directly to an intracellular CD3ζ signaling domain [8]. These receptors demonstrated that T cells could be engineered to recognize surface antigens independent of MHC restriction, triggering cytotoxicity upon target engagement [8]. However, clinical applications revealed critical limitations including poor persistence, limited expansion, and inadequate antitumor activity in vivo due to the absence of costimulatory signaling [8]. The CD3ζ domain alone provided primary activation signal but failed to sustain long-term T-cell function, leading to activation-induced cell death and limited therapeutic efficacy in early clinical trials [8] [10].
Second-Generation CARs: Clinical Breakthrough
Second-generation CARs addressed the persistence limitation by incorporating a single costimulatory domain (CD28 or 4-1BB) alongside the CD3ζ signaling domain [8] [10]. This architectural innovation provided both Signal 1 (activation) and Signal 2 (costimulation) through a single receptor, dramatically enhancing T-cell expansion, persistence, and antitumor efficacy [8]. The specific costimulatory domain incorporated significantly influenced functional properties: CD28 domains promoted rapid, robust expansion and effector function, while 4-1BB domains enhanced mitochondrial biogenesis, memory formation, and long-term persistence [11] [10]. This generation achieved remarkable clinical success in B-cell malignancies, leading to the first FDA-approved CAR-T therapies (Kymriah, Yescarta) targeting CD19 [8] [10].
Third-Generation CARs: Signal Amplification
Third-generation CARs further amplified T-cell signaling by incorporating two distinct costimulatory domains (typically CD28 plus 4-1BB or OX40) in tandem with CD3ζ [8] [10]. This design aimed to synergize the advantages of different costimulatory pathways to enhance potency, persistence, and cytokine production [10]. Preclinical studies demonstrated that third-generation CARs exhibited superior expansion and prolonged persistence compared to second-generation constructs in some CD19-targeted therapies [10]. However, concerns emerged regarding potential overactivation leading to accelerated T-cell exhaustion and increased risk of severe adverse effects, highlighting the delicate balance between enhanced efficacy and toxicity [10].
Fourth-Generation CARs (TRUCKs): Armored Engineering
Fourth-generation CARs, also termed "TRUCKs" (T cells Redirected for Universal Cytokine Killing) or "armored CARs," represent a paradigm shift from simply enhancing T-cell signaling to modifying the tumor microenvironment [11] [10]. These constructs are based on second-generation CARs but incorporate inducible cytokine expression cassettes (e.g., IL-12, IL-15, IL-18) that activate upon antigen engagement [11] [10]. These engineered cells function as "mini pharmacies" that locally release immunomodulatory cytokines to enhance the antitumor response by recruiting and activating endogenous immune cells, countering immunosuppressive factors in the tumor microenvironment, and promoting CAR-T cell survival [11]. This approach shows particular promise for solid tumors, where the immunosuppressive microenvironment presents a major barrier to CAR-T efficacy [11] [12].
Fifth-Generation CARs: Precision Control
Fifth-generation CARs represent the cutting edge of CAR engineering, incorporating cytokine receptor signaling domains (e.g., IL-2Rβ) that activate the JAK/STAT pathway upon antigen engagement [12] [10]. These constructs create an autocrine loop that enhances cell proliferation and survival while maintaining the costimulatory advantages of earlier generations [12]. Additionally, fifth-generation designs increasingly incorporate logic-gated systems, inducible suicide switches, and synthetic regulatory circuits that enable precise control over T-cell activity, potentially mitigating toxicity risks while enhancing antitumor efficacy [10]. These advanced systems represent the transition from constantly active receptors to smart therapeutics that can integrate multiple environmental signals [10].
Table 2: Comparative Analysis of CAR Generations
| Generation | Intracellular Domains | Key Innovations | Clinical Status | Advantages | Limitations |
|---|---|---|---|---|---|
| First | CD3ζ [8] | MHC-independent targeting | Historical significance | Proof of concept | Poor persistence, limited efficacy [8] |
| Second | CD3ζ + 1 costimulatory domain (CD28 or 4-1BB) [8] [10] | Integrated costimulation | FDA-approved for hematologic malignancies [8] | Enhanced persistence and efficacy | Limited solid tumor activity, toxicity concerns [11] |
| Third | CD3ζ + 2 costimulatory domains (e.g., CD28+4-1BB) [8] [10] | Multiple costimulatory signals | Clinical trials [10] | Potentially enhanced potency | Risk of overactivation and exhaustion [10] |
| Fourth | Second-generation base + inducible cytokines [11] [10] | Local cytokine delivery | Clinical trials for solid tumors [11] | Modifies tumor microenvironment | Complex manufacturing, potential off-tissue effects [10] |
| Fifth | Second-generation base + cytokine receptor domains (e.g., IL-2Rβ) [12] [10] | JAK/STAT activation, logic gates | Preclinical and early clinical development [12] [10] | Enhanced proliferation, precision control | High design complexity, safety validation needed [10] ``` |
CAR Signaling Pathways
The intracellular signaling architecture of CAR constructs has evolved considerably across generations, with each addition enhancing T-cell activation, persistence, and functionality. First-generation CARs relied exclusively on CD3ζ-derived immunoreceptor tyrosine-based activation motifs (ITAMs) that initiate the canonical T-cell receptor signaling cascade [8] [9]. Second-generation constructs incorporated costimulatory domains that activate distinct signaling pathways: CD28 enhances PI3K/AKT and NF-κB signaling for rapid effector function, while 4-1BB promotes TRAF/NF-κB signaling that supports mitochondrial biogenesis and long-term persistence [8] [10]. Third-generation CARs combine multiple costimulatory signals to potentially synergize these advantages. Fourth and fifth-generation designs introduce cytokine signaling and inducible gene expression, creating more complex regulatory networks that integrate environmental cues [11] [12] [10].
Experimental Protocols for CAR-T Cell Generation
T-Cell Isolation and Activation
Purpose: To obtain high-quality T cells from donor blood and activate them for genetic modification. Procedure: (1) Collect peripheral blood mononuclear cells (PBMCs) from patient or donor via leukapheresis; (2) Isolate T cells using negative selection (Pan T Cell Isolation Kit) or positive selection (CD3/CD28 beads); (3) Activate T cells using anti-CD3/CD28 antibodies (1 µg/mL) or Dynabeads (bead-to-cell ratio 3:1) in complete media (RPMI-1640 + 10% FBS + 2 mM L-glutamine + 1% penicillin/streptomycin + 10 mM HEPES) supplemented with IL-2 (100 IU/mL); (4) Culture at 37°C, 5% CO₂ for 24-48 hours before transduction [13] [10].
CAR Gene Delivery Methods
Viral Transduction (Lentiviral/Adenoviral) Purpose: Achieve stable genomic integration and long-term CAR expression. Procedure: (1) Preload RetroNectin-coated plates (10 µg/mL) with lentiviral vectors (MOI 5-20) by centrifugation (2000 × g, 90 minutes, 32°C); (2) Seed activated T cells (1×10⁶ cells/mL) in viral supernatant; (3) Centrifuge (1000 × g, 90 minutes, 32°C); (4) Incubate at 37°C, 5% CO₂ for 24 hours; (5) Replace with fresh complete media with IL-2; (6) Repeat transduction if necessary [13] [10].
Non-Viral Transfection (Electroporation) Purpose: Avoid viral vector limitations using transposon systems or mRNA. Procedure: (1) Use Sleeping Beauty or PiggyBac transposon systems with corresponding transposase; (2) Resuspend activated T cells in electroporation buffer with DNA plasmids (CAR transposon + transposase at 4:1 ratio); (3) Electroporate using optimized program (e.g., 500V, 5ms pulse for Neon system); (4) Immediately transfer to pre-warmed complete media; (5) For mRNA electroporation, use 2-5 µg mRNA with similar parameters for transient expression [13] [10].
CAR-T Cell Expansion and Validation
Purpose: Expand transduced T cells to therapeutic doses and validate CAR expression and function. Expansion Protocol: (1) Culture transduced T cells at 0.5-1×10⁶ cells/mL in complete media with IL-2 (50-100 IU/mL); (2) Monitor cell density and split every 2-3 days to maintain optimal concentration; (3) Expand for 7-14 days to achieve target cell numbers (typically 1-10×10⁸ CAR-T cells for infusion); (4) Perform analytical assessments throughout expansion [13].
Validation Assays: (1) Flow cytometry for CAR expression using protein L or target antigen staining; (2) Coculture assays with target cells to measure cytokine production (ELISA for IFN-γ, IL-2) and cytotoxicity (LDH release, real-time cell analysis); (3) Proliferation assays (CFSE dilution) following antigen stimulation; (4) Memory phenotype characterization (CD45RO, CD62L, CD27) by flow cytometry [13] [10].
Research Reagent Solutions
Table 3: Essential Research Reagents for CAR-T Cell Development
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Gene Delivery Systems | Lentiviral vectors, Gamma-retroviral vectors, Sleeping Beauty transposon, PiggyBac transposon, mRNA [13] [10] | CAR gene transfer | Lentiviral: High efficiency, stable integration [10]; mRNA: Transient, safer profile [13] |
| T-cell Activation | Anti-CD3/CD28 antibodies, IL-2, IL-7, IL-15, Dynabeads [13] | T-cell stimulation and expansion | CD3/CD28 beads provide strong activation signal; Cytokines influence differentiation [13] |
| Culture Media | X-VIVO 15, TexMACS, RPMI-1640 with 10% FBS, Serum-free media [13] | Cell maintenance and expansion | Serum-free media preferred for clinical applications [13] |
| Detection Reagents | Protein L, Recombinant target antigen, Anti-Fab antibodies [13] | CAR expression detection | Protein L detects most scFvs without interfering with antigen binding [13] |
| Functional Assays | LDH release kit, IFN-γ ELISA, CFSE, Real-time cell analyzers (e.g., xCelligence) [13] | Efficacy assessment | Multiple assays recommended for comprehensive functional characterization [13] |
Emerging Applications and Future Directions
The evolution of CAR designs has enabled expansion into novel therapeutic areas beyond hematological malignancies. In solid tumors, targets such as MSLN (mesothelin), GPC3, HER2, and GD2 are being investigated in clinical trials, though challenges remain with the immunosuppressive tumor microenvironment [12]. For autoimmune diseases, CD19-targeted CAR-T cells have demonstrated remarkable efficacy in eliminating pathogenic B cells in systemic lupus erythematosus, with sustained remissions in 80% of treated cases in clinical trials [11]. Similar approaches are being explored for multiple sclerosis, systemic sclerosis, and autoimmune neuropathies [11]. Emerging delivery technologies including in vivo CAR generation using targeted viral vectors or lipid nanoparticles could revolutionize manufacturing by eliminating complex ex vivo processes [13]. Additionally, advanced gene editing tools like CRISPR/Cas9 enable precise integration of CAR constructs into safe harbor loci, potentially enhancing safety and efficacy profiles [8] [10].
The generational evolution of CAR designs represents a remarkable convergence of immunology, synthetic biology, and genetic engineering. From the simple first-generation constructs to the sophisticated fifth-generation systems, each iteration has addressed critical limitations while expanding therapeutic possibilities. The progressive incorporation of costimulatory domains, cytokine signaling modules, and regulatory circuits has transformed CAR-T cells from simple cytotoxic agents to intelligent therapeutic systems capable of complex environmental integration. For researchers developing next-generation CAR therapies, careful consideration of structural components, signaling architecture, and safety controls remains paramount. As the field advances toward more universal, controllable, and persistent CAR therapies, these fundamental principles of CAR design will continue to guide innovation in this transformative cancer immunotherapy modality.
Key Historical Milestones and Paradigm Shifts in CAR-T Therapy
Chimeric antigen receptor (CAR) T-cell therapy represents a transformative approach in cancer immunotherapy, marking a significant paradigm shift from conventional cancer treatments. This groundbreaking modality involves genetically engineering a patient's own T lymphocytes to express synthetic receptors that recognize specific tumor-associated antigens, thereby redirecting the immune system to target and eliminate malignant cells. The development of CAR-T therapy spans several decades of intensive research, culminating in unprecedented clinical success for patients with relapsed or refractory hematologic malignancies. This application note details the key historical milestones, provides detailed experimental protocols central to CAR-T development and manufacturing, and outlines advanced computational approaches that are shaping the next generation of CAR-T therapies. Framed within the broader context of CAR T-cell engineering protocols for cancer immunotherapy research, this resource provides scientists and drug development professionals with both foundational knowledge and cutting-edge methodologies driving the field forward.
Historical Milestones and Paradigm Shifts
The evolution of CAR-T therapy is characterized by several distinct phases of innovation, each representing a fundamental shift in how researchers approach cancer treatment. Table 1 summarizes the key historical milestones that have defined this revolutionary therapeutic field.
Table 1: Key Historical Milestones in CAR-T Therapy Development
| Year | Milestone | Significance | Key Researchers/Entities |
|---|---|---|---|
| 1860s-1890s | Early observations of infection-induced tumor regression | Foundation of cancer immunotherapy; established that immune system can fight cancer | Busch, Fehleisen, Coley [14] |
| 1987 | First concept of chimeric T-cell receptor | Proof-of-concept that antibody-derived variable regions could be fused to TCR constant regions to activate T cells | Kurosawa et al. [14] |
| 1989 | Redirection of T cells to specific antigens | Demonstrated engineered receptors could redirect T-cell specificity | Eshhar et al. [14] |
| 2017 | First FDA approval of CAR-T therapy (tisagenlecleucel) | Landmark regulatory approval for pediatric and young adult r/r B-cell ALL | FDA, Novartis [14] |
| 2023 | Six approved CAR-T therapies | Demonstrated unprecedented efficacy in B-cell malignancies and multiple myeloma | Various pharmaceutical companies [14] |
| 2024-2025 | Expansion into autoimmune diseases (e.g., SLE) | Paradigm shift beyond oncology; early success in refractory autoimmune conditions | Various academic and industry groups [14] [15] |
The conceptual foundation for CAR-T therapy emerged from early observations in the 1860s-1890s by German physicians Wilhelm Busch and Friedrich Fehleisen, and later by Dr. William B. Coley at New York Hospital, who noted tumor regression in patients with concurrent bacterial infections [14]. This established the fundamental principle that the immune system could be harnessed to fight cancer, laying the groundwork for modern cancer immunotherapy.
The modern era of CAR-T therapy began with groundbreaking work in the late 1980s. In 1987, Japanese immunologist Dr. Yoshikazu Kurosawa and his team reported the first chimeric T-cell receptor, combining antibody-derived variable regions (VH/VL) with T-cell receptor constant regions [14]. This landmark study demonstrated that modified receptors could activate T cells in response to specific antigens. Two years later, Dr. Zelig Eshhar and colleagues at the Weizmann Institute of Science described a similar approach to redirect T cells to recognize predefined antigens, establishing the basic architecture of modern CAR constructs [14].
The most significant translational milestone occurred in 2017 when the FDA approved tisagenlecleucel for pediatric and young adult patients with relapsed or refractory acute lymphoblastic leukemia [14]. This landmark approval validated decades of research and established CAR-T therapy as a viable treatment modality. By 2023, six CAR-T therapies had received regulatory approval, demonstrating remarkable efficacy against various B-cell malignancies and multiple myeloma [14].
A recent paradigm shift has been the application of CAR-T therapy beyond oncology. Early studies have demonstrated promising results in autoimmune diseases including systemic lupus erythematosus (SLE) and antisynthetase syndrome [14]. As of 2025, dual-target CAR-T products such as CTA313 (targeting both CD19 and BCMA) have shown encouraging early results in patients with refractory active SLE, potentially offering a more comprehensive immune reset and drug-free remission for some patients [15].
Experimental Protocols in CAR-T Therapy
CAR-T Cell Manufacturing Workflow
The manufacturing of CAR-T cells is a multi-step process that requires strict adherence to protocol to ensure product quality, efficacy, and safety. Figure 1 illustrates the complete workflow, from leukapheresis to final infusion.
Figure 1: CAR-T Cell Manufacturing Workflow. This diagram outlines the key steps in producing clinical-grade CAR-T cells, from initial T-cell collection through activation, genetic modification, expansion, and final product formulation.
Detailed Manufacturing Protocol
Step 1: Leukapheresis and T-cell Collection
- Procedure: Collect patient's peripheral blood mononuclear cells (PBMCs) via leukapheresis. Process within 24 hours of collection.
- Critical Parameters: Target yield of 1-10 × 10^9 PBMCs; maintain sterility throughout processing.
- Quality Check: Cell viability >90% by trypan blue exclusion; enumerate CD3+ T-cell percentage by flow cytometry (target >40% of PBMCs).
Step 2: T-cell Activation
- Procedure: Resuspend cells at 1-2 × 10^6 cells/mL in appropriate media (X-VIVO 15 or TexMACS). Add T-cell activator (e.g., CD3/CD28 antibodies conjugated to magnetic beads) at 1:1 bead-to-cell ratio.
- Culture Conditions: Incubate at 37°C, 5% CO2 for 24-48 hours.
- Media Composition: Supplement with 5-10% human AB serum or serum-free replacements; add IL-2 (100-300 IU/mL) to promote T-cell survival and proliferation [16] [17].
Step 3: Genetic Modification
- Procedure: Transduce activated T cells with lentiviral or retroviral vectors encoding CAR construct at multiplicity of infection (MOI) of 5-20. Use centrifugation (2000 × g, 90-120 minutes at 32°C) or static transduction with retronectin to enhance transduction efficiency.
- Critical Parameters: Determine transduction efficiency 48-72 hours post-transduction by flow cytometry for CAR expression or surface markers (e.g., LNGFR for truncated selection marker).
- Alternative Approaches: For non-viral methods, use transposon/transposase systems (e.g., Sleeping Beauty) or CRISPR-based gene editing for targeted integration [16].
Step 4: Ex Vivo Expansion
- Procedure: Culture transduced cells in gas-permeable culture bags or G-Rex bioreactors at 0.5-1 × 10^6 cells/mL. Maintain culture for 7-14 days with regular feeding (50-70% media exchange every 2-3 days).
- Cytokine Support: Include IL-2 (50-100 IU/mL) or cytokine combinations (IL-7/IL-15 at 10-20 ng/mL each) to promote central memory phenotype and enhance persistence [16] [17].
- Monitoring: Monitor cell density, viability, and CAR expression daily. Target expansion fold of 20-100× from initial T-cell count.
Step 5: Formulation and Cryopreservation
- Procedure: Harvest cells when viability >80% and target cell number achieved. Wash cells and resuspend in cryopreservation medium (e.g., 5% DMSO, 40% human AB serum, 55% saline).
- Final Product: Fill in infusion bags; cryopreserve using controlled-rate freezer. Store in vapor phase liquid nitrogen until infusion.
- Release Testing: Perform sterility (bacterial/fungal culture), mycoplasma, endotoxin, and identity/potency assays before release [16].
In Vitro Functional Assessment Protocols
Cytotoxicity Assay (xCELLigence System)
Purpose: To quantitatively measure CAR-T cell-mediated killing of tumor cells in real-time.
Materials:
- xCELLigence RTCA instrument (ACEA Biosciences)
- E-Plate 16 or 96
- Target tumor cells expressing cognate antigen
- CAR-T cells and untransduced T-cell controls
- Appropriate cell culture medium
Procedure:
- Background Measurement: Add 50 μL of culture medium to each well of the E-Plate and measure background impedance.
- Seed Target Cells: Add 50,000 tumor cells in 100 μL medium per well (for 96-well format). Allow cells to adhere and proliferate for 24 hours.
- Establish Baseline: Monitor Cell Index (CI) every 15 minutes until plateau phase is reached (typically 18-24 hours).
- Add Effector Cells: Add CAR-T cells at various effector-to-target (E:T) ratios (e.g., 1:1, 3:1, 10:1) in 50 μL medium. Include controls (target cells alone; untransduced T cells).
- Continuous Monitoring: Monitor CI every 15 minutes for 3-7 days. Normalize CI to time of effector cell addition.
- Data Analysis: Calculate specific lysis using formula: % Specific Lysis = [1 - (CI{sample}/CI{target alone})] × 100 [18].
Troubleshooting:
- Low CI values may indicate poor cell adhesion; optimize seeding density.
- Edge effect in plates can cause well-to-well variation; use interior wells for critical comparisons.
- For non-adherent target cells, use alternate systems (e.g., flow cytometry-based killing assays).
Cytokine Release Assay
Purpose: To quantify T-cell activation by measuring cytokine secretion following antigen engagement.
Materials:
- CAR-T cells and target cells
- Culture plates (96-well U-bottom recommended)
- Cytokine detection kit (Luminex or ELISA for IFN-γ, IL-2, TNF-α)
- Plate reader or Luminex instrument
Procedure:
- Co-culture Setup: Seed 100,000 CAR-T cells with 100,000 target cells in 200 μL complete medium per well. Include controls (CAR-T cells alone; target cells alone; untransduced T cells with targets).
- Incubation: Incubate for 18-24 hours at 37°C, 5% CO2.
- Supernatant Collection: Centrifuge plates at 300 × g for 5 minutes; carefully transfer 150 μL supernatant to new plate.
- Cytokine Quantification: Analyze supernatants using multiplex bead array (Luminex) or ELISA according to manufacturer's protocols.
- Data Analysis: Calculate antigen-specific cytokine production by subtracting background from control wells. Report as pg/mL/10^6 cells [16].
Table 2: Research Reagent Solutions for CAR-T Development
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Cell Culture Media | X-VIVO 15, TexMACS, RPMI-1640 with supplements | Supports T-cell growth and maintains optimal phenotype during expansion | Different media yield varied T-cell phenotypes and functions [16] [17] |
| Activation Reagents | CD3/CD28 antibodies conjugated to magnetic beads, soluble agonists | Activates T-cell signaling pathways prior to genetic modification | Bead-to-cell ratio and activation duration impact differentiation state [16] |
| Gene Delivery Vectors | Lentiviral vectors, retroviral vectors, transposon systems | Mediates stable integration of CAR transgene into T-cell genome | Vector choice affects transduction efficiency, insertion site profile, and safety [16] |
| Cytokine Supplements | IL-2, IL-7, IL-15, IL-21 | Promotes T-cell survival, expansion, and modulates differentiation | IL-7/IL-15 combination favors less differentiated memory phenotypes [16] [17] |
| Characterization Antibodies | Anti-CAR detection reagents, CD3, CD4, CD8, CD45RA, CCR7 | Assesses transduction efficiency, phenotype, and differentiation status | Central memory (CD45RA-CCR7+) and naive (CD45RA+CCR7+) subsets correlate with persistence [16] |
Computational and Modeling Approaches
The complexity of CAR-T cell behavior in vivo has prompted the development of sophisticated computational models to understand and predict therapy performance. These approaches are particularly valuable for addressing challenges in solid tumors and optimizing dosing strategies.
Quantitative Systems Pharmacology (QSP) Modeling
Background: QSP modeling integrates multiscale data to characterize CAR-T cell fate and antitumor cytotoxicity, from cellular interactions to clinical-level patient responses [19].
Application Protocol:
- Model Framework Establishment:
- Define key model components: CAR-T cell trafficking, tumor cell dynamics, antigen expression heterogeneity, and tumor microenvironment factors.
- Incorporate known biological parameters: CAR-T proliferation rates, tumor doubling times, antigen density, and immunosuppressive factors.
Data Integration:
- Input preclinical data: in vitro cytotoxicity, CAR-T proliferation kinetics, cytokine secretion profiles.
- Incorporate patient-specific parameters: tumor burden, antigen expression patterns, immune cell infiltration.
Virtual Patient Population Generation:
- Create in silico cohorts reflecting physiological and pathophysiological variability.
- Define parameter distributions based on clinical data from relevant patient populations.
Simulation and Prediction:
- Simulate clinical responses to different CAR-T dosing regimens (e.g., fractionated dosing vs. flat dosing).
- Identify critical factors driving response variability and potential resistance mechanisms [19].
Case Example: A recent multiscale QSP model for CLDN18.2-targeted CAR-T therapy in gastric cancer integrated in vitro and in vivo data to project clinical efficacy and optimize dosing strategies. The model successfully characterized the paired cellular kinetics-cytotoxicity response and informed clinical trial design [19].
SINDy Algorithm for Model Discovery
Background: Sparse Identification of Nonlinear Dynamics (SINDy) is a data-driven approach that discovers governing equations from time-series data without pre-specified model structures [18].
Application Protocol:
- Data Preparation:
- Collect high-temporal resolution data on cancer cell and CAR-T cell populations from in vitro killing assays.
- Preprocess data: smooth noise, interpolate missing time points if necessary.
Library Construction:
- Build a comprehensive library of candidate mathematical terms that could describe the interaction dynamics (e.g., linear, quadratic, cubic terms; predator-prey interaction terms).
Sparse Regression:
- Apply sequential thresholded least-squares algorithm to identify the minimal set of terms that accurately describe the dynamics.
- Validate discovered models against held-out data.
Biological Interpretation:
- Relate identified mathematical terms to biological mechanisms (e.g., single vs. double CAR-T-cancer cell binding; functional responses; density-dependent growth) [18].
Implementation Example: In a recent application to CAR T-cell killing of glioblastoma cells, SINDy revealed interaction dynamics that included terms suggestive of single CAR T-cell binding and logistic growth constraints, providing unique insight into the mechanisms governing CAR T-cell efficacy [18].
Figure 2 illustrates the integration of computational and experimental approaches in CAR-T therapy development.
Figure 2: Computational and Experimental Workflow Integration. This diagram shows how experimental data feeds into both mechanistic (QSP) and data-driven (SINDy) modeling approaches to generate predictions that inform therapy optimization.
CAR-T therapy has undergone remarkable evolution from conceptual beginnings to established therapeutic modality, with ongoing paradigm shifts expanding its application into autoimmune diseases. The historical milestones outlined in this application note demonstrate how fundamental scientific discoveries have been translated into clinically impactful treatments. The detailed experimental protocols provide researchers with standardized methodologies for CAR-T cell manufacturing and functional assessment, while the computational approaches offer powerful tools to overcome persistent challenges, particularly in solid tumors. As the field continues to advance, the integration of sophisticated engineering strategies with multiscale computational modeling will be essential for developing next-generation CAR-T therapies with enhanced efficacy, improved safety profiles, and broader applicability across diverse disease indications.
The integration of costimulatory domains is a critical advancement in chimeric antigen receptor (CAR) T cell engineering, moving beyond first-generation designs that relied solely on the CD3ζ activation signal [20] [21]. Second-generation CARs incorporate a single costimulatory domain, with CD28 and 4-1BB emerging as the most prevalent and clinically successful choices [20] [21]. All currently marketed CAR-T cell products are of this second generation, and the choice between these two costimulatory domains profoundly influences the clinical behavior of the resulting cellular therapy, impacting everything from initial cytotoxic potency to long-term persistence and safety profile [22] [23] [21]. This application note provides a structured, comparative analysis of CD28 and 4-1BB to inform research and development protocols in cancer immunotherapy.
Functional and Clinical Comparison
The selection of a costimulatory domain dictates the functional phenotype, metabolic programming, and clinical performance of CAR-T cells. The table below summarizes the core characteristics and comparative outcomes associated with CD28 and 4-1BB.
Table 1: Functional and Clinical Characteristics of CD28 vs. 4-1BB Costimulatory Domains
| Characteristic | CD28 | 4-1BB |
|---|---|---|
| Signaling Kinetics | Rapid, high-amplitude signaling [24] | Slower, lower-amplitude signaling [24] |
| Metabolic Profile | Glycolysis-biased metabolism; supports an effector phenotype [23] | Enhanced mitochondrial fitness and oxidative metabolism; supports memory phenotype [23] |
| In Vivo Persistence | Shorter persistence; prone to exhaustion [22] [25] [24] | Superior long-term persistence [26] [27] [21] |
| T cell Differentiation | Promotes effector/effector memory differentiation [23] [25] | Favors central memory differentiation [26] [23] |
| Clinical Safety Profile | Associated with more severe CRS and ICANS in a B-NHL study [22] | Better tolerated; lower incidence of severe CRS and ICANS in a B-NHL study [22] |
| Representative Products | Axicabtagene ciloleucel (Yescarta), Brexucabtagene autoleucel (Tecartus) [21] | Tisagenlecleucel (Kymriah), Lisocabtagene maraleucel (Breyanzi) [21] |
Signaling Mechanisms and Downstream Pathways
The distinct clinical profiles of CD28 and 4-1BB are rooted in their differential engagement of downstream signaling cascades.
CD28 Signaling Motifs and Pathways
The cytoplasmic tail of CD28 contains key signaling motifs—YMNM, PRRP, and PYAP—that activate downstream pathways by recruiting specific kinases and adaptor proteins [25]. Upon engagement, these motifs:
- YMNM: Recruits the p85 subunit of PI3K, activating the PI3K-AKT pathway and promoting glucose uptake and glycolytic metabolism [25].
- PYAP/PRRP: Binds LCK and GRB2-GADS, activating the Ras-MAPK pathway and regulating actin polymerization and immune synapse formation [25]. This robust signaling drives potent effector functions but can also lead to terminal differentiation and exhaustion [25] [24].
4-1BB Signaling Pathway
The 4-1BB costimulatory domain signals primarily through TNF receptor-associated factors (TRAFs), leading to the activation of the NF-κB pathway [26] [27]. This signaling axis enhances cell survival, promotes the development of long-lived memory T cells, and sustains mitochondrial biogenesis and fitness, which underpin the superior persistence of 4-1BB-co-stimulated CAR-T cells [26] [23].
Comparative Signaling Pathway Diagram
The following diagram illustrates the key signaling pathways and transcriptional outcomes initiated by the CD28 and 4-1BB costimulatory domains, integrating with the core CD3ζ activation signal.
Experimental Protocols for In Vitro Evaluation
A standardized in vitro evaluation is essential for directly comparing the function of CAR-T cells incorporating different costimulatory domains.
CAR-T Cell Manufacturing Protocol
Objective: To generate human CAR-T cells expressing either CD28 or 4-1BB costimulatory domains. Key Reagents:
- Source of T cells: Human peripheral blood mononuclear cells (PBMCs) from healthy donors or patients [22] [27].
- T cell Activation: Stimulate purified T cells with plate-bound anti-CD3 (e.g., 0.25 μg/ml) and anti-CD28 (e.g., 1 μg/ml) antibodies for 72 hours [27].
- Genetic Transduction: Transduce activated T cells with lentiviral vectors encoding the CAR constructs at a predetermined multiplicity of infection (MOI; e.g., MOI 10) [27].
- Cell Culture: Culture transduced T cells in RPMI-1640 medium supplemented with IL-2 (e.g., 200 IU/ml) [27]. For persistent stimulation, cells can be re-stimulated weekly with irradiated tumor cells [27].
Cytotoxic Killing Assay
Objective: To quantify the specific cytotoxic potency of CAR-T cells against target tumor cells. Procedure:
- Coculture Setup: Seed target tumor cells (e.g., Raji or NALM-6 for CD19+ targets) in a 96-well plate. Add CAR-T cells at various effector-to-target (E:T) ratios (e.g., 1:0.5, 1:1, 1:2) [27].
- Incubation: Coculture cells for 12-24 hours at 37°C.
- Analysis: Harvest cells and analyze tumor cell lysis by flow cytometry. Use antibodies against markers like CD3 (for T cells) and CD19 (for target B cells) to distinguish populations and quantify specific killing [27].
Cytokine Release Assay
Objective: To measure T cell activation strength by quantifying secreted cytokines. Procedure:
- Stimulation: Coculture CAR-T cells with target tumor cells at specified E:T ratios for 12-24 hours.
- Supernatant Collection: Centrifuge the coculture plates and collect the cell-free supernatant.
- Quantification: Analyze supernatant concentrations of key cytokines like IFN-γ, TNF-α, and IL-2 using a Cytometric Bead Array (CBA) or ELISA, following manufacturer protocols [27]. CD28-CAR T cells typically exhibit more rapid and robust cytokine secretion [22] [24].
Experimental Workflow Diagram
The standard workflow for generating and functionally testing CAR-T cells with different costimulatory domains is outlined below.
The Scientist's Toolkit: Key Research Reagents
Successful evaluation of costimulatory domains requires a suite of reliable reagents and tools. The following table lists essential materials for the protocols described in this note.
Table 2: Essential Research Reagents for Costimulatory Domain Evaluation
| Reagent / Material | Function / Application | Examples / Specifications |
|---|---|---|
| Lentiviral Vectors | Delivery of CAR constructs into T cells. | Constructs with identical scFv, hinge/TM, and CD3ζ, differing only in CD28 vs. 4-1BB costimulatory domain [22] [21]. |
| Anti-CD3/Anti-CD28 Antibodies | Polyclonal T cell activation during manufacturing. | Plate-bound or bead-bound antibodies for initial T cell stimulation [27]. |
| Recombinant Human IL-2 | Supports T cell survival and expansion in culture. | Used at 200 IU/ml during ex vivo expansion [27]. |
| Flow Cytometry Antibodies | Cell phenotyping and cytotoxicity analysis. | Anti-CD3, anti-CD19 (for target cells), anti-CD45RO/RA, anti-CD62L (memory phenotyping), viability dyes [27]. |
| Cytometric Bead Array (CBA) | Multiplexed quantification of cytokine secretion. | Commercial kits for human IFN-γ, TNF-α, IL-2, etc. [27]. |
| Target Cell Lines | Antigen-positive cells for functional assays. | CD19+ lines: Raji, Daudi, NALM-6 [27]. Ensure consistent antigen density. |
| NSG Mice | In vivo model for evaluating persistence and anti-tumor efficacy. | NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice for xenograft studies [27]. |
Emerging Strategies and Combinatorial Approaches
Given the complementary strengths of CD28 and 4-1BB, advanced engineering strategies are being explored to harness the benefits of both.
- Third-Generation CARs: These constructs incorporate both CD28 and 4-1BB costimulatory domains in tandem within the same CAR molecule. Research indicates that the specific order and structure of these combined domains are critical for optimal function, influencing LCK recruitment and enhancing expansion and persistence [26].
- Optimized CD28 Mutants: Rather than using the full native domain, researchers are engineering mutated versions of CD28 (e.g., mut06 with a modified PYAP motif) to reduce exhaustion and improve signaling quality. Combining this optimized CD28 with 4-1BB in a third-generation design has shown enhanced anti-tumor activity in preclinical models [26].
- Constitutive 4-1BB Co-expression: An alternative to domain stacking is to express a canonical CD28-based CAR alongside a separate, constitutively expressed full-length 4-1BB receptor. This strategy has been shown to improve proliferation, reduce apoptosis, and enhance in vivo tumor control compared to the CD28-CAR alone [27].
The choice between CD28 and 4-1BB is not a matter of identifying a universally superior domain, but rather of selecting the right tool for the specific therapeutic context. CD28 drives powerful, rapid effector responses suitable for aggressive malignancies but carries a higher risk of toxicity and limited durability. 4-1BB promotes long-term persistence and a favorable safety profile, which may be critical for controlling relapsing disease. The future of costimulatory engineering lies in sophisticated combination strategies—such as third-generation CARs and optimized domain mutants—that aim to synthetically engineer T cells with the rapid strike capability of CD28 and the enduring persistence of 4-1BB, thereby overcoming the limitations of each individual domain.
From Bench to Bedside: CAR-T Cell Manufacturing and Clinical Translation Protocols
Chimeric antigen receptor (CAR) T-cell therapy represents a groundbreaking advancement in cancer immunotherapy, demonstrating remarkable efficacy in treating hematological malignancies. The manufacturing of CAR-T cells is a multi-step process that begins with the collection of a patient's immune cells and culminates with the infusion of engineered T cells capable of selectively targeting and destroying tumor cells. This application note delineates a standardized, robust workflow for the production of research-grade CAR-T cells, encompassing the critical stages of leukapresis processing, T-cell activation, viral transduction, and in vitro expansion. The protocols and data presented herein are framed within the context of advancing CAR T-cell engineering protocols for cancer immunotherapy research, providing researchers and drug development professionals with detailed methodologies to enhance reproducibility and efficacy in pre-clinical studies.
The journey from patient leukapheresis to CAR-T cell infusion product is a meticulously orchestrated sequence. Figure 1 below provides a holistic overview of this complex process, highlighting the key stages and their temporal relationships.
A critical initial decision in the workflow is the selection and processing of the starting material. The choice between fresh and cryopreserved leukapheresis, or the use of PBMCs, can significantly impact initial cell quality and downstream manufacturing success. Table 1 summarizes key quantitative benchmarks for different starting materials, providing researchers with critical quality attributes to target.
Table 1: Comparative Analysis of CAR-T Starting Materials [28] [29]
| Parameter | Fresh Leukapheresis | Cryopreserved Leukapheresis | PBMCs |
|---|---|---|---|
| Initial Viability | >95% | 90-97% | >90% |
| Post-Thaw Viability | N/A | ≥90% (Target) | Variable |
| Lymphocyte Proportion | ~68.7% | ~66.6% | ~52.2% |
| T Cell (CD3+) Proportion | High | 42-51% | Variable |
| Key Advantage | Maximum initial viability | Supply chain flexibility, scheduling freedom | Established protocol |
| Primary Challenge | Strict 24-72hr processing window [29] | Optimized freeze/thaw protocol required | Potential cell loss during isolation |
Detailed Experimental Protocols
Leukapheresis Processing and PBMC Isolation
The initial step in CAR-T manufacturing involves obtaining a sufficient number of peripheral blood mononuclear cells (PBMCs), which include T lymphocytes, from a leukapheresis product [28].
Principle: Density gradient centrifugation separates PBMCs from other blood components based on density differences [30].
Materials:
- Leukapheresis product (fresh or cryopreserved)
- Phosphate Buffered Saline (PBS)
- Density Gradient Medium (e.g., Ficoll-Paque or Lymphoprep)
- Centrifuge
- SepMate tubes (optional, for process acceleration)
Procedure:
- Dilution: Dilute the leukapheresis product 1:1 to 1:2 with PBS [28].
- Layering: Carefully layer the diluted blood over the density gradient medium in a centrifuge tube. Maintain a sharp interface [30] [28].
- Centrifugation: Centrifuge at 400 × g for 30 minutes at room temperature with the brake disengaged [28].
- Collection: After centrifugation, carefully aspirate the plasma and PBMC layers. The PBMCs form a distinct white ring at the plasma/gradient medium interface. Collect this layer into a new tube [28].
- Washing: Wash the collected PBMCs with PBS or a balanced salt solution and centrifuge to pellet the cells. Perform a cell count and viability assessment.
T Cell Activation
Isolated T cells must be activated to enter the cell cycle and become receptive to genetic modification via transduction.
Principle: T-cell receptor (TCR) complex engagement with co-stimulatory signals initiates T-cell proliferation and cytokine production.
Materials:
- Isolated PBMCs
- Complete Medium (e.g., AIM-V medium supplemented with 2-10% human AB serum [28])
- T-Cell Activator (e.g., anti-CD3/CD28 antibodies)
- Recombinant Human IL-2
Procedure:
- Resuspension: Resuspend PBMCs in complete medium at a concentration of 1-2 × 10^6 cells/mL [28].
- Stimulation: Add T-cell activation reagents. A common method is using a combination of 50 ng/mL anti-CD3 monoclonal antibody (e.g., OKT-3) and 300 IU/mL recombinant human IL-2 [28]. Alternatives include coated magnetic beads or soluble antibodies.
- Incubation: Culture cells in a humidified incubator at 37°C and 5% CO2 for 24-48 hours prior to transduction.
CAR Transduction
This critical step introduces the CAR gene into activated T cells, enabling them to express the chimeric antigen receptor.
Principle: Viral vectors, such as gamma-retroviruses or lentiviruses, are engineered to carry the CAR transgene and facilitate its integration into the host T-cell genome, leading to stable CAR expression [31] [28].
Materials:
- Activated T cells
- CAR Viral Vector Supernatant (Lentiviral or Retroviral)
- Retronectin or other transduction enhancers
- Polybrene (for lentiviral transduction)
- Centrifuge
Procedure (Retronectin-assisted Retroviral Transduction):
- Coating: Coat non-tissue culture treated plates with Retronectin (10 µg/mL in PBS) for 2 hours at room temperature or overnight at 4°C [28].
- Blocking: Block the plates with 2.5% human albumin in PBS for 30 minutes, then wash [28].
- Vector Loading: Load the diluted retroviral supernatant onto the coated plates. Centrifuge at 2000 × g for 0.5-2 hours at 32°C, then aspirate the supernatant [28].
- Transduction: Seed the activated T cells onto the vector-coated plates. Centrifuge the plates at 1000 × g for 15 minutes (spinoculation) and incubate at 37°C overnight [28].
- Harvest: Approximately 24 hours post-transduction, harvest the cells and transfer them to expansion vessels [28].
Table 2 compares the primary viral vector systems used in CAR-T research, highlighting their distinct characteristics to guide experimental design.
Table 2: Comparison of CAR Transduction Methods [31]
| Vector System | Mechanism | Advantages | Disadvantages |
|---|---|---|---|
| Lentiviral Vector | Integrates into host genome, including non-dividing cells. | High transduction efficiency; broad tropism. | Complex production; safety concerns regarding insertional mutagenesis. |
| Retroviral Vector | Integrates into host genome (requires cell division). | Robust, well-established methodology. | Requires cell division; safety concerns regarding insertional mutagenesis. |
| Non-Viral Methods (e.g., Transposon Systems, mRNA Electroporation) | Genome integration via transposase (Sleeping Beauty) or transient cytoplasmic expression (mRNA). | Reduced risk of insertional mutagenesis; often more cost-effective. | Historically lower efficiency than viral methods (transposons); transient expression (mRNA). |
In Vitro Expansion
Following transduction, CAR-T cells are expanded ex vivo to achieve the target cell dose required for therapeutic efficacy.
Principle: Transduced T cells are cultured in cytokine-supplemented media to promote proliferation and achieve the desired cell numbers while maintaining a less differentiated, more therapeutically potent cell phenotype.
Materials:
- Transduced T cells
- Complete Medium
- Recombinant Human IL-2
- Culture Flasks or Bioreactors (e.g., G-Rex flasks)
Procedure:
- Initiation of Culture: Resuspend the harvested, transduced cells in complete medium supplemented with IL-2 (e.g., 300 IU/mL) at a density of 0.5 × 10^6 cells/mL [28].
- Maintenance: Culture the cells in a humidified incubator at 37°C and 5% CO2 for 7-14 days. Maintain cell concentration between 0.5-2.0 × 10^6 cells/mL by diluting with fresh medium + IL-2 as needed [28].
- Monitoring: Monitor cell density, viability, and CAR expression regularly via flow cytometry.
- Harvest: Once the target cell number is achieved and viability is confirmed to be >90%, harvest the cells for downstream assays, cryopreservation, or infusion.
The Scientist's Toolkit: Research Reagent Solutions
Successful execution of the CAR-T workflow relies on a suite of specialized reagents and tools. The table below details essential components and their functions.
Research Reagent Solutions
| Item | Function | Example Products / Components |
|---|---|---|
| Cell Separation Kits | Isolation of specific cell subsets (e.g., T cells) from complex starting materials like leukopaks with high purity and viability. | EasySep [30], Akadeum Microbubble Kits [31] [32] |
| T Cell Activators | Provides signal 1 (TCR engagement via anti-CD3) and signal 2 (co-stimulation via anti-CD28) for robust T cell activation and proliferation. | Anti-CD3/CD28 antibodies, TransAct |
| CAR Viral Vectors | Delivery vehicle for the stable integration of the CAR transgene into the host T cell genome. | Lentiviral/CD19 CAR retroviral supernatant [28] |
| Transduction Enhancers | Increases transduction efficiency by co-localizing viral particles and target cells. | Retronectin [28], Polybrene |
| Cell Culture Media | Provides nutrients and essential components for T cell growth, expansion, and maintenance of function. | AIM-V medium [28], X-VIVO, TexMACS |
| Cytokines | Supports T cell survival, proliferation, and can influence differentiation state (e.g., memory vs. effector phenotypes). | Recombinant Human IL-2 [28], IL-7, IL-15 |
| Cryopreservation Media | Protects cells from damage during freezing and long-term storage, preserving viability and function. | CS10 (10% DMSO) [29] |
Signaling Pathways in CAR-T Cell Activation
Understanding the intracellular signaling that governs CAR-T cell function is essential for optimizing their therapeutic potential. The core signaling architecture of a second-generation CAR, the most clinically advanced design, integrates signals from both the TCR complex and co-stimulatory receptors. Figure 2 illustrates this integrated signaling pathway, which is triggered upon CAR engagement with its target antigen.
This application note provides a detailed, standardized framework for the key manufacturing stages of research-grade CAR-T cells: leukapheresis processing, T-cell activation, transduction, and expansion. By adhering to the protocols, quantitative benchmarks, and reagent specifications outlined herein, researchers can enhance the consistency, efficacy, and safety profiling of their CAR-T cell products. The continued refinement of these workflows, informed by a deep understanding of CAR signaling and cell biology, is paramount for advancing the next generation of cancer immunotherapies and translating pre-clinical success into broader clinical application.
The engineering of chimeric antigen receptor (CAR) T cells represents a paradigm shift in cancer immunotherapy, showing remarkable efficacy against hematological malignancies. The success of this cellular therapy is fundamentally dependent on the methods used for gene delivery, which can be broadly categorized into viral and non-viral approaches [33]. Viral vectors, particularly lentiviruses and gammaretroviruses, have been the historical workhorses for CAR gene delivery, offering high transduction efficiency and stable long-term transgene expression. However, concerns regarding immunogenicity, insertional mutagenesis, complex manufacturing, and high costs have prompted the development of non-viral alternatives [34]. Non-viral methods, including electroporation of mRNA or DNA and nanoparticle-based delivery, are emerging as promising strategies that offer enhanced safety profiles, greater payload capacity, reduced manufacturing timelines, and lower costs [35] [36]. This application note provides a detailed comparison of these platforms and outlines optimized protocols for their implementation in CAR T cell research, framed within the context of a broader thesis on CAR T cell engineering protocols.
Vector Platform Comparison
The choice between viral and non-viral vector systems involves critical trade-offs between transduction efficiency, safety, manufacturability, and clinical performance. Table 1 summarizes the key characteristics of the predominant vector platforms used in CAR T cell engineering.
Table 1: Quantitative Comparison of Viral and Non-Viral Vector Platforms for CAR T Cell Engineering
| Feature | Lentiviral (LV) Vectors | Adenoviral (Ad) Vectors | Electroporation (mRNA) | Electroporation (DNA/CRISPR) |
|---|---|---|---|---|
| Transduction/Transfection Efficiency | High (>70%) [33] | High [37] | High (CAR+ levels ~70%) [38] | High (Knockin efficiency 35-89%) [36] |
| Integration Profile | Semi-random integration [34] | Non-integrating [37] | Non-integrating (transient) [38] | Targeted integration (with CRISPR) [36] |
| Cargo Capacity | ~8 kb [34] | Large capacity [37] [34] | Limited by mRNA size [38] | Limited by DNA plasmid size [36] |
| Stability of Expression | Stable, long-term [33] | Transient [37] | Transient (2-3 days) [38] | Stable (with targeted knockin) [36] |
| Key Advantages | Stable expression, well-characterized [33] [34] | High transgene expression, large cargo capacity [37] [34] | Rapid production, high safety, no genomic integration [38] [33] | Precise genome editing, "off-the-shelf" potential [33] [36] |
| Key Limitations | Risk of insertional mutagenesis, high cost, complex GMP manufacturing [34] | High immunogenicity, transient expression [34] | Transient CAR expression, potential for repeated dosing [38] | Technical complexity, requires optimization of HDR [36] |
Detailed Experimental Protocols
Protocol 1: Non-Viral CRISPR-Mediated CAR Knockin via Electroporation
This protocol, adapted from a presentation by MaxCyte, details the optimized steps for achieving precise, high-efficiency knockin of a CAR construct into the TRAC locus of primary human T cells using CRISPR/Cas9 and electroporation [36].
Key Reagents and Materials
- Primary Human T Cells: Isolated from leukapheresis product or PBMCs of healthy donors.
- T Cell Activation Reagent: Anti-CD3/anti-CD28 antibodies immobilized on a polymeric nanomatrix (e.g., for protocol B from [38]).
- Culture Medium: TheraPEAK T-VIVO medium supplemented with IL-2 [38].
- CRISPR Components:
- Cas9 Nuclease: High-quality, research-grade.
- sgRNA: Targeting the TRAC locus.
- HDR Template: A Nanoplasmid DNA vector containing the CAR expression cassette flanked by homology arms to the TRAC locus. The inclusion of a Cas9-targeting sequence (CTS) within the template is recommended to enhance knockin efficiency [36].
- Electroporation System: MaxCyte ExPERT GTx instrument with corresponding electroporation buffers [36].
- HDR Enhancer: Small molecule M3814 (2 µM final concentration), added post-electroporation [36].
Step-by-Step Workflow
- T Cell Isolation and Activation: Isolate T cells from PBMCs using a standard density gradient centrifugation or magnetic bead-based negative selection kit. Activate the isolated T cells (1-2 x 10^6 cells/mL) using the immobilized anti-CD3/anti-CD28 nanomatrix in TheraPEAK T-VIVO medium supplemented with IL-2 (e.g., 100 IU/mL) for 48 hours at 37°C and 5% CO2 [38] [36].
- Pre-Electroporation Preparation: Harvest the activated T cells and wash them with PBS. Resuspend the cell pellet in MaxCyte electroporation buffer at a concentration of 1-2 x 10^8 cells/mL [36].
- Complex Formation and Electroporation:
- Pre-complex the TRAC-targeting RNP by incubating 0.5 µM Cas9 protein with a 2x molar ratio (1.0 µM) of sgRNA for 10-15 minutes at room temperature.
- Mix the cell suspension with the pre-complexed RNP and 200 µg/mL of the Nanoplasmid HDR template.
- Immediately transfer the mixture to an electroporation cuvette and electroporate using the Expanded T Cell Four (ETC4) protocol on the MaxCyte ExPERT GTx platform [36].
- Post-Electroporation Recovery and Expansion:
- Transfer the electroporated cells to pre-warmed culture medium.
- Add the HDR enhancer M3814 to a final concentration of 2 µM.
- Culture the cells at 37°C and 5% CO2, expanding them for 14 days with periodic medium changes and IL-2 supplementation. Monitor cell density, viability, and CAR expression regularly [36].
Expected Outcomes and Quality Control
- Cell Viability: A significant drop in viability is expected immediately post-electroporation, but cells should recover to >80% within 48 hours [36].
- Knockin Efficiency: Using this optimized workflow, CAR knockin efficiencies of 67% to 89% can be achieved by day 7-14 post-electroporation, as measured by flow cytometry for the CAR protein [36].
- Functional Assessment: On day 14, co-culture engineered CAR-T cells with CD19-expressing target cells (e.g., Ramos cells) at various Effector:Target (E:T) ratios. Assess specific target cell killing and the production of effector molecules (Granzyme B, IFN-γ) via cytotoxicity assays and ELISA, respectively [36].
Protocol 2: mRNA-Based Transient CAR T Cell Generation
This protocol is ideal for applications requiring rapid testing of CAR designs or for generating transient CAR T cells for research purposes, avoiding genomic integration [38].
Key Reagents and Materials
- Culture Medium: TheraPEAK T-VIVO medium (Protocol B), which supports higher expansion rates and viability compared to some alternatives [38].
- T Cell Activator: Immobilized anti-CD3/anti-CD28 antibodies (for Protocol B) [38].
- CAR-mRNA: In vitro transcribed (IVT) mRNA encoding the full CAR construct, capped and polyadenylated.
- Electroporation System: A research-grade electroporator with optimized settings for primary T cells (e.g., Pulse Code "CM-138" as identified in [38]).
Step-by-Step Workflow
- T Cell Expansion: Isolate and activate T cells as described in Protocol 1, step 1. Expand the activated T cells for 6-10 days. Days 6-10 post-activation have been identified as the most suitable window for mRNA transfection, balancing cell number, viability, and activation status [38].
- Electroporation:
- Harvest expanded T cells and wash with PBS.
- Resuspend cells in appropriate electroporation buffer.
- Mix cells with CAR-mRNA (typically 5-10 µg per 1x10^6 cells) and electroporate using the pre-optimized pulse code.
- Recovery and Analysis:
- Immediately transfer cells to culture medium.
- Analyze CAR expression and viability 18-24 hours post-electroporation. CAR expression is typically transient, peaking at 24 hours and declining significantly by 48-72 hours [38].
Expected Outcomes and Quality Control
- CAR Expression: >70% CAR-positive T cells at 24 hours post-transfection, dropping by at least 50% at 48 hours [38].
- Cell Viability: >80% viability at 24 hours post-transfection [38].
- Phenotype: Protocol B tends to promote a higher fraction of CD8+ cytotoxic T cells, which may be beneficial for antitumor cytotoxicity [38].
The Scientist's Toolkit: Essential Research Reagents
Successful CAR T cell engineering relies on a suite of critical reagents. Table 2 lists key materials and their functions based on the cited protocols.
Table 2: Essential Research Reagents for CAR T Cell Engineering
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| T Cell Medium | Supports ex vivo T cell viability, activation, and expansion. | TheraPEAK T-VIVO; ImmunoCult-XF T Cell Expansion Medium [38] |
| T Cell Activator | Provides signal 1 (CD3) and signal 2 (CD28) for T cell activation. | Immobilized anti-CD3/anti-CD28 on nanomatrix; soluble antibody mixtures [38] |
| CRISPR-Cas9 System | Enables precise genomic editing for targeted CAR integration. | Cas9 protein, TRAC-targeting sgRNA, HDR template plasmid/Nanoplasmid [36] |
| HDR Enhancer | Small molecule inhibitor that improves Homology-Directed Repair efficiency. | M3814; used post-electroporation to boost knockin rates [36] |
| Electroporation System | Enables efficient delivery of nucleic acids (mRNA, DNA, RNP) into T cells. | MaxCyte ExPERT GTx; optimized protocols (e.g., ETC4) are critical for high viability and efficiency [36] |
| mRNA Construct | Template for transient CAR expression without genomic integration. | In vitro transcribed (IVT) CAR-mRNA; used for rapid testing/transient expression [38] |
Workflow Visualization
The following diagrams illustrate the key procedural and mechanistic workflows described in the protocols.
Non-Viral CAR-T Engineering Workflow
CAR Signaling & Engineering Evolution
Clinical Application and Efficacy in Hematologic Malignancies (ALL, NHL, MM)
Chimeric Antigen Receptor T-cell (CAR-T) therapy has revolutionized the treatment of relapsed and refractory hematologic malignancies by engineering a patient's own T-cells to recognize and eliminate cancer cells. This adoptive cell transfer approach has demonstrated remarkable success across various blood cancers, particularly B-cell acute lymphoblastic leukemia (B-ALL), non-Hodgkin lymphoma (NHL), and multiple myeloma (MM). [39] [40]
The therapy involves extracting T-cells from a patient, genetically modifying them to express chimeric antigen receptors specific to tumor antigens, expanding the population ex vivo, and reinfusing them back into the patient. These engineered cells then recognize and destroy malignant cells expressing the target antigen, producing durable responses in many patients who have exhausted conventional treatment options. [40] [41]
Table 1: Clinical Efficacy of CAR-T Therapy Across Hematologic Malignancies
| Malignancy | Molecular Target | Representative Products | Efficacy Metrics | Patient Population |
|---|---|---|---|---|
| B-ALL | CD19 | Kymriah (tisagenlecleucel) | High remission rates (60-80%) in refractory patients [40] | Relapsed/refractory B-ALL |
| NHL | CD19 | Yescarta (axicabtagene ciloleucel) | 91.7% ORR, 75% CR in novel AT101 trial [42] | Relapsed/refractory DLBCL, FL, MCL |
| Multiple Myeloma | BCMA | Abecma (ide-cel), Carvykti (cilta-cel) | High rates of MRD negativity; improved PFS/OS [43] | ≥1 prior line of therapy (updated 2024) |
The field continues to evolve with improvements in CAR design, expansion techniques, and combination approaches. Recent advances include targeting novel antigens, optimizing costimulatory domains, and developing strategies to enhance CAR-T cell persistence and functionality within the immunosuppressive tumor microenvironment. [44] [45]
Detailed Application Protocols by Malignancy
B-Cell Acute Lymphoblastic Leukemia (B-ALL)
CD19-directed CAR-T therapy has demonstrated exceptional efficacy in B-ALL, with remission rates exceeding 60-80% in some patient populations who had limited treatment options remaining. [40] The critical success factor is targeting the CD19 antigen ubiquitously expressed on B-lineage cells.
Key Protocol Parameters:
- Target Antigen: CD19
- CAR Construct: Anti-CD19 scFv (typically FMC63-derived) coupled with CD3ζ signaling domain and costimulatory domains (CD28 or 4-1BB)
- Manufacturing Time: 2-3 weeks
- Lymphodepletion: Fludarabine/cyclophosphamide regimen 3-5 days pre-infusion
- Cell Dose: 1-5×10⁶ CAR-T cells/kg (for children) or 0.5-2.5×10⁸ CAR-T cells (fixed dose for adults)
Clinical Management Considerations: Patients require intensive monitoring for cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). Tocilizumab (IL-6 receptor antagonist) is standard first-line intervention for severe CRS. The therapy induces profound B-cell aplasia, requiring immunoglobulin replacement when necessary. [39] [40]
Non-Hodgkin Lymphoma (NHL)
CAR-T therapy has shown significant efficacy against various NHL subtypes, including diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), and mantle cell lymphoma (MCL). Recent innovations include novel CD19-targeting approaches to overcome resistance mechanisms.
Breakthrough Approach: AT101 Novel CD19 Targeting A first-in-human phase I trial investigated AT101, a novel CAR-T product utilizing a humanized chicken antibody (h1218) that targets a membrane-proximal epitope of CD19 (amino acids 51-63 in exon 2) distinct from the FMC63 epitope used in all commercial CD19 CAR-T products. [42]
Table 2: AT101 Clinical Trial Results in Relapsed/Refractory NHL
| Parameter | Dose Level 1 | Dose Level 2 & 3 | Overall Cohort |
|---|---|---|---|
| Patient Number | 3 | 9 | 12 |
| NHL Subtypes | DLBCL, FL, MCL, MZL | DLBCL, FL, MCL, MZL | 7 DLBCL, 3 FL, 1 MCL, 1 MZL |
| Overall Response Rate | 66.7% | 100% | 91.7% |
| Complete Response Rate | 33.3% | 100% | 75.0% |
| Grade ≥3 CRS | 0% | 11.1% | 8.3% |
| Grade ≥3 ICANS | 0% | 11.1% | 8.3% |
Mechanistic Advantages of AT101:
- Overcomes FMC63-epitope loss mutations (L174V, R163L)
- Avoids epitope masking from previous FMC63-CAR19 treatment
- Faster on/off rates reduce activation-induced cell death (AICD)
- Enhanced expansion and persistence compared to FMC63-CAR19
- Effective against FMC63-CAR19-resistant lymphomas [42]
Technical Protocol:
- Lymphodepletion: Fludarabine (25 mg/m²/day × 3 days) + cyclophosphamide (250 mg/m²/day × 3 days)
- Dosing: Three dose levels evaluated (0.5-2.5×10⁶ CAR-T cells/kg)
- Response Assessment: PET-CT at day 28, month 3, and every 3 months thereafter
- Monitoring: CAR-T expansion by qPCR, B-cell aplasia as pharmacodynamic marker
Multiple Myeloma
BCMA (B-cell maturation antigen)-targeted CAR-T therapies have transformed multiple myeloma treatment, with two commercially available products showing significant efficacy in heavily pretreated patients.
Approved Products and Indications:
- Idecabtagene vicleucel (ide-cel, Abecma): Approved for patients after ≥2 prior lines of therapy
- Ciltacabtagene autoleucel (cilta-cel, Carvykti): Approved for patients after ≥1 prior line of therapy [43]
Current Clinical Trial Landscape:
- CAR-PRISM Trial (NCT05767359): Investigating cilta-cel in high-risk smoldering myeloma at two dose levels to determine safety and efficacy in this precursor population
- CARTITUDE-6 Trial (NCT05257083): Comparing head-to-head cilta-cel versus autologous stem cell transplantation (ASCT) after induction therapy with daratumumab, lenalidomide, bortezomib, and dexamethasone in newly diagnosed multiple myeloma [43]
Novel Target Development: GPRC5D
- GPRC5D is a novel target highly expressed on plasma cells discovered by Dana-Farber researchers
- Early-phase trials with GPRC5D-targeted CAR-T (arlocabtagene autoleucel) show high response rates in heavily pre-treated patients, including those with prior BCMA-directed therapies
- Favorable toxicity profile with lower rates of on-target, off-tumor toxicities compared to talquetamab (GPRC5D-targeted bispecific T-cell engager) [43]
CAR-T Cell Engineering and Signaling Pathways
The fundamental CAR construct consists of an extracellular antigen-recognition domain (typically scFv derived from monoclonal antibodies), a hinge region, transmembrane domain, and intracellular signaling domains comprising costimulatory molecules and CD3ζ activation domain.
CAR Signaling Pathway and T-cell Activation
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents for CAR-T Development
| Reagent Category | Specific Examples | Research Function | Application Notes |
|---|---|---|---|
| Antigen-Targeting Domains | FMC63 scFv (CD19), h1218 scFv (CD19), BCMA scFv | Target recognition | h1218 binds membrane-proximal CD19 epitope with faster on/off rates [42] |
| Signaling Domains | CD3ζ, 4-1BB (CD137), CD28 | T-cell activation and costimulation | 4-1BB enhances persistence; CD28 boosts initial expansion [44] |
| Gene Delivery Systems | Lentiviral vectors, Retroviral vectors, mRNA electroporation | CAR gene transfer | mRNA enables transient expression; viral vectors provide genomic integration [46] |
| Cell Culture Media | IL-2, IL-7, IL-15 | T-cell expansion and maintenance | Cytokine combination affects final T-cell differentiation state [39] |
| Quality Control Assays | Flow cytometry, qPCR, Cytotoxicity assays | Product characterization | Critical for assessing identity, purity, potency, and safety [47] |
Experimental Protocols
mRNA CAR-T Cell Production Protocol
The mRNA electroporation approach enables transient CAR expression, which may be advantageous for managing toxicity profiles, particularly in early-phase clinical trials or for targets with on-tumor, off-cancer toxicity concerns. [46]
Step-by-Step Protocol:
T-cell Isolation and Activation:
- Isolate PBMCs from leukapheresis product via density gradient centrifugation
- Isolate T-cells using negative selection (Pan T Cell Isolation Kit)
- Activate T-cells with CD3/CD28 beads (25 µL beads per 1×10⁶ cells) in complete media (RPMI-1640 + 10% FBS + 2 mM L-glutamine)
- Add IL-2 (100 IU/mL) to culture media
mRNA Production:
- Clone CAR sequence into mRNA expression vector containing 5' and 3' UTRs for enhanced stability
- Perform in vitro transcription using T7 RNA polymerase, including 5' cap analog and base-modified nucleotides
- Purify mRNA using cellulose-based methods
- Quality control: assess integrity (Agilent Bioanalyzer), concentration (spectrophotometry), and sterility
mRNA Electroporation:
- Harvest activated T-cells 48-72 hours post-activation
- Wash and resuspend in electroporation buffer at 50-100×10⁶ cells/mL
- Mix cells with mRNA (2-5 µg per 1×10⁶ cells)
- Electroporate using square-wave protocol (500 V, 5 ms pulse)
- Immediately transfer to pre-warmed complete media with IL-2 (100 IU/mL)
Expansion and Formulation:
- Culture electroporated cells at 1×10⁶ cells/mL
- Maintain for 7-14 days, monitoring cell density and viability
- Harvest when viability >80% and expansion reaches target dose
- Formulate in infusion solution (often containing human serum albumin and DMSO) for cryopreservation or immediate infusion [46]
In Vitro Potency Assessment Protocol
Comprehensive potency assays are critical for evaluating CAR-T product functionality and predicting clinical performance.
Cytotoxic Activity Assessment:
- Target Cell Preparation:
- Label target cells (CD19+ for B-ALL/NHL, BCMA+ for myeloma) with cell tracker dye (e.g., CFSE)
- Alternatively, use engineered target cells expressing luciferase or GFP for automated readouts
Co-culture Establishment:
- Seed target cells in 96-well plates at 5×10⁴ cells/well
- Add CAR-T cells at effector:target ratios (10:1, 5:1, 1:1, 0.5:1)
- Include controls: target cells alone, untransduced T-cells
- Incubate for 24-48 hours at 37°C, 5% CO₂
Viability Measurement:
- Flow Cytometry: Add counting beads and viability dye to quantify specific lysis
- Impedance-based: Use xCELLigence system for real-time killing kinetics
- Luciferase-based: Measure luminescence after adding substrate
- Chromium-51 Release: Traditional gold standard, but declining use due to radioactivity concerns [47]
Cytokine Release Profiling:
- Collect supernatant from co-culture at 24 hours
- Quantify IFN-γ, IL-2, TNF-α, IL-6 using multiplex ELISA or Luminex technology
- Compare cytokine profile to reference standards [47]
CAR-T cell therapy has fundamentally transformed the treatment landscape for hematologic malignancies, providing life-saving options for patients with otherwise limited alternatives. The continued optimization of CAR constructs, including novel targeting approaches like the h1218 scFv for CD19, combined with advances in manufacturing and patient management, promise to enhance efficacy while reducing toxicities. [42]
The field is rapidly evolving toward earlier lines of therapy, as evidenced by the recent approval of BCMA-targeted CAR-T products for earlier-stage multiple myeloma. [43] Ongoing clinical trials investigating CAR-T therapy in high-risk smoldering myeloma and as potential alternatives to stem cell transplantation represent the next frontier in maximizing patient benefit from this revolutionary immunotherapeutic approach.
Emerging Clinical Targets and Trials for Solid Tumors
Chimeric antigen receptor (CAR)-T cell therapy represents a revolutionary modality in cancer immunotherapy, demonstrating remarkable success in treating hematologic malignancies. Complete remission rates range from 60% to 90% for relapsed/refractory B-cell leukemia, lymphoma, and multiple myeloma [48]. However, solid tumors constitute approximately 90% of clinically diagnosed cancers, creating a significant unmet clinical need [49] [48]. The translation of CAR-T therapy to solid tumors has faced substantial biological challenges, including physical barriers that limit T-cell infiltration, a highly immunosuppressive tumor microenvironment (TME), and tumor antigen heterogeneity that promotes antigen escape [48]. Recent advances in target selection, CAR design, and delivery strategies are now yielding promising clinical results, heralding a new era for CAR-T therapy in solid oncology.
This application note provides researchers and drug development professionals with a comprehensive overview of the current landscape, focusing on emergent targets, clinical trial data, and detailed experimental methodologies. We synthesize findings from recent key studies presented at major oncology conferences in 2025, including the American Society of Clinical Oncology (ASCO) Annual Meeting and the European Society for Medical Oncology (ESMO) Congress, to deliver actionable insights for advancing CAR-T research and development for solid tumors.
Current Clinical Landscape of Solid Tumor Targets
The target landscape for CAR-T therapy in solid tumors has diversified significantly, with candidates advancing across multiple cancer types. Successful target selection must account for tumor specificity, expression homogeneity, and safety profile to minimize on-target, off-tumor toxicity. The following table summarizes key emerging targets with recent clinical validation.
Table 1: Emerging Clinical Targets in CAR-T Therapy for Solid Tumors
| Target Antigen | Key Solid Tumor Indications | Clinical Trial Stage | Notable Findings |
|---|---|---|---|
| Claudin 18.2 | Gastric, Gastroesophageal Junction (GEJ) | Early Clinical Trials | LB1908 demonstrated lesion shrinkage in 83% of patients; manageable safety profile [5]. |
| B7-H3 | Glioblastoma (GBM) | Phase I | B7H3-CAR-T showed median OS of 14.6 months; delivered via intratumoral/ intraventricular route [5]. |
| IL13Rα2 | Glioblastoma, Melanoma | Phase I | Tumor shrinkage in 85% of evaluable GBM patients; Phase I trial ongoing for melanoma (NCT04119024) [5] [50]. |
| MSLN (Mesothelin) | Malignant Pleural Mesothelioma, NSCLC, Ovarian, Pancreatic | Phase I | JL-Lightning CAR-T (aPD1-MSLN) showed 100% ORR in mesothelioma at Dose Level 2, including a complete response [5]. |
| DLL3 | Small Cell Lung Cancer (SCLC) | Phase I | LB2102 (with dnTGFβRII) demonstrated dose-dependent activity; benchmarks against FDA-approved tarlatamab [5]. |
| GPC3 | Hepatocellular Carcinoma (HCC) | Phase I | Ori-C101 (armored CAR-T) yielded objective responses and an ongoing complete response at 9 months in one patient [5]. |
| CEA (Carcinoembryonic Antigen) | Colorectal Cancer (with liver mets) | Phase I | Prolonged relapse-free survival post-resection; 57% recurrence-free at high dose [5]. |
| PRAME | Uveal Melanoma, other solid tumors | Phase Ib/II | TCR T-cell therapy (anzutresgene autoleucel) showed 67% confirmed ORR in metastatic uveal melanoma [50]. |
| EGFR/IL13Rα2 (Bivalent) | Glioblastoma (GBM) | Phase I | CART-EGFR-IL13Rα2 induced tumor shrinkage (1-62%, median 35%) in 85% of evaluable patients [5]. |
Recent breakthroughs are particularly notable in gastrointestinal cancers and glioblastoma. The world's first randomized controlled trial of Car T-cell therapy in solid tumors focused on advanced gastric or gastro-oesophageal junction (GEJ) cancer, demonstrating that patients receiving CAR-T therapy lived an average of 7.9 months compared to 5.5 months with standard care—a statistically significant 40% improvement in overall survival [49]. For glioblastoma, which has a historically dismal median overall survival of 6-9 months upon recurrence, multiple targets including B7-H3, IL13Rα2, and EGFR are showing promising signals of efficacy, particularly with localized delivery strategies [5].
Detailed Target Biology and Signaling Pathways
Understanding the biological function of emerging targets and their role in oncogenic signaling is crucial for rational CAR design and predicting mechanisms of response and resistance.
Claudin 18.2
Claudin 18.2 is a tight junction protein that becomes abnormally exposed on the surface of gastric, pancreatic, and other epithelial cancer cells, making it an ideal target. Its normal expression is restricted to gastric mucosa, limiting off-tumor toxicity. CAR-T cells targeting Claudin 18.2 recognize the extracellular domain of the protein, initiating a cytotoxic response independent of its native biological function [5].
Diagram 1: Claudin 18.2 CAR-T Cell Activation Pathway. This diagram illustrates the molecular mechanism by which Claudin 18.2-specific CAR-T cells recognize and initiate the destruction of target tumor cells. The binding of the single-chain variable fragment (scFv) to the Claudin 18.2 antigen triggers intracellular signaling through the CD3ζ and costimulatory domains, leading to T-cell activation and tumor cell lysis.
B7-H3 (CD276)
B7-H3 is an immune checkpoint molecule belonging to the B7 superfamily that is overexpressed in various solid tumors, including glioblastoma, neuroblastoma, and prostate cancer. While its exact physiological ligand remains unclear, B7-H3 is known to modulate T-cell function, often contributing to an immunosuppressive tumor microenvironment. Targeting B7-H3 allows CAR-T cells to simultaneously directly kill tumor cells and potentially counteract local immune suppression [5] [48].
Logic-Gated Targeting Strategies
To enhance tumor specificity and reduce on-target, off-tumor toxicity, advanced logic-gated CAR systems are in development. For instance, the A2B694 CAR-T product is designed to attack tumor cells expressing mesothelin (MSLN) but lacking human leukocyte antigen A02 (HLA-A02). This "AND-NOT" logic gate prevents toxicity in normal cells that express HLA-A*02, even if they have low levels of MSLN, providing a critical safety mechanism for targets with heterogeneous normal tissue expression [5].
Quantitative Clinical Trial Data Analysis
The efficacy of CAR-T therapy in solid tumors is demonstrated through standardized oncology endpoints. The following table synthesizes key efficacy data from recent clinical trials presented at the 2025 ASCO Annual Meeting.
Table 2: Efficacy Outcomes from Recent CAR-T Clinical Trials in Solid Tumors
| Trial / Target | Cancer Type | Overall Survival (OS) | Progression-Free Survival (PFS) | Overall Response Rate (ORR) | Disease Control Rate (DCR) |
|---|---|---|---|---|---|
| Satri-cel (CLDN18.2) | Advanced Gastric/GEJ | 7.9 months (vs 5.5 mo control) [49] | 3.3 months (vs 1.8 mo control) [49] | Not specified | Not specified |
| B7-H3 CAR-T | Recurrent GBM | Median OS: 14.6 months (95% CI: 2.3-26.8) [5] | Not specified | Not specified | Not specified |
| CART-EGFR-IL13Rα2 | Recurrent GBM | Not specified | Not specified | Not specified | 85% with tumor shrinkage [5] |
| JL-Lightning (aPD1-MSLN) | Malignant Pleural Mesothelioma (DL2) | Not specified | Not specified | 100% (3/3 patients; 1 CR) [5] | 100% [5] |
| PRAME TCR-T (anzu-cel) | Metastatic Uveal Melanoma | Not specified | Median PFS: 8.5 months [50] | 67% (confirmed) [50] | Not specified |
| GCC19CART (DL2) | Metastatic Colorectal Cancer | Not specified | Not specified | 80% [5] | Not specified |
Quantitative systems pharmacology (QSP) modeling is emerging as a powerful tool to facilitate clinical translation. These mechanistic models integrate multiscale data—from CAR-antigen interaction kinetics to in vivo CAR-T biodistribution and patient tumor heterogeneity—to simulate clinical outcomes and optimize dosing strategies. For example, QSP models have been used to prospectively simulate response to Claudin 18.2-targeted CAR-T therapies under different dosing regimens, including step-fractionated dosing [51].
Detailed Experimental Protocols
Protocol: Manufacturing mRNA CAR-T Cells via Electroporation
This protocol outlines the medium-scale production of messenger RNA (mRNA) CAR-T cells using electroporation, suitable for both hematological and solid tumor targets [46].
Key Materials and Reagents
- Source T-cells: Isolated from patient peripheral blood mononuclear cells (PBMCs) via leukapheresis.
- mRNA Construct: In vitro-transcribed mRNA encoding the CAR construct (e.g., anti-CD19 or solid tumor target).
- Electroporation System: Such as Gene Pulser Xcell (Bio-Rad) or Neon (Thermo Fisher).
- Cell Culture Media: X-VIVO 15 or TexMACS medium supplemented with IL-2 (100-300 IU/mL).
- Activation Beads: Anti-CD3/CD28 magnetic beads.
- Quality Control Assays: Flow cytometry for CAR expression, cytotoxicity assays.
Step-by-Step Procedure
- T-cell Isolation and Activation: Isolate PBMCs from leukapheresis product using Ficoll density gradient centrifugation. Activate T-cells using anti-CD3/CD28 beads at a 3:1 bead-to-cell ratio in complete media supplemented with IL-2 (100 IU/mL). Culture for 24-48 hours at 37°C, 5% CO₂.
- mRNA Electroporation: Harvest activated T-cells and resuspend in electroporation buffer at a concentration of 1-2 × 10⁷ cells/mL. Mix cells with 5-10 µg of CAR-encoding mRNA per 10⁶ cells. Transfer to an electroporation cuvette. Perform electroporation using optimized parameters (e.g., 500 V for 5 ms for a single pulse).
- Post-Transfection Recovery: Immediately transfer electroporated cells to pre-warmed complete media. Incubate at 37°C for 15-30 minutes before diluting further. This recovery step is critical for cell viability.
- Large-Scale Expansion: Culture the transfected T-cells in complete media with IL-2 (100-300 IU/mL). Maintain cell density between 0.5-2 × 10⁶ cells/mL, performing media exchanges or splitting as needed. Expand cells for 7-14 days to achieve the required therapeutic dose.
- Quality Assessment and Harvest: On day 7-10, determine CAR expression percentage via flow cytometry using a target antigen-specific protein or detection tag. Assess cell viability by trypan blue exclusion. Functionally validate cells via cytokine release or cytotoxicity assays against antigen-positive target cells. Harvest cells, wash, and formulate in infusion buffer for cryopreservation or immediate administration.
Protocol: Intracranial Administration of CAR-T Cells for Glioblastoma
Local delivery is critical for overcoming the blood-brain barrier in CNS malignancies. This protocol details the intracerebroventricular administration of CAR-T cells via an Ommaya reservoir [5].
Key Materials and Reagents
- CAR-T Cell Product: Formulated in appropriate infusion buffer at prescribed concentration.
- Ommaya Reservoir: Pre-implanted subcutaneous reservoir and catheter system.
- Lymphodepleting Chemotherapy: Fludarabine (30 mg/m²/day × 3 days) and cyclophosphamide (500 mg/m²/day × 2 days) may be administered prior to infusion, depending on the trial protocol [5].
- Administration Supplies: 25-gauge butterfly needle, sterile drapes, antiseptic solution.
- Toxicity Management Agents: Anakinra and dexamethasone on hand for management of therapy-related neurotoxicity [5].
Step-by-Step Procedure
- Pre-Infusion Preparation: Confirm patient eligibility and absence of intracranial hypertension or active infection. Verify Ommaya reservoir patency. Pre-medicate the patient per institutional and trial protocol (e.g., with acetaminophen and diphenhydramine).
- CAR-T Cell Preparation: Thaw the cryopreserved CAR-T cell product (if applicable) and bring to room temperature. Gently mix the product to ensure homogeneity. Confirm cell count and viability.
- Aseptic Preparation: Position the patient supine with head turned appropriately. Prepare the scalp over the Ommaya reservoir using aseptic technique with chlorhexidine or iodine solution. Apply sterile drapes to create a sterile field.
- Reservoir Access: Palpate the reservoir dome. Stabilize it with non-dominant hand. Using a 25-gauge butterfly needle, aseptically puncture the center of the reservoir dome. Aspirate a small volume of cerebrospinal fluid (CSF) to confirm correct needle placement.
- CAR-T Cell Infusion: Slowly instill the prepared CAR-T cell suspension into the reservoir. The total volume is typically 5-10 mL, infused slowly over 5-10 minutes. Follow the infusion with a flush of preservative-free normal saline to clear the catheter dead space.
- Post-Infusion Management: Withdraw the needle and apply pressure if needed. Monitor the patient closely for immediate adverse events, particularly changes in neurological status. Schedule subsequent doses or assessments per the clinical trial protocol, which may involve repeated administrations.
Workflow for CAR-T Therapy Development and Administration
The end-to-end process, from cell collection to patient monitoring, involves multiple critical stages as visualized below.
Diagram 2: CAR-T Therapy Clinical Workflow. This diagram outlines the key stages in the clinical application of CAR-T therapy, from initial cell collection from the patient through manufacturing, quality control, infusion, and long-term patient follow-up.
The Scientist's Toolkit: Essential Research Reagents
Successful development and implementation of CAR-T therapies for solid tumors requires a comprehensive suite of specialized research reagents and tools.
Table 3: Essential Research Reagents for Solid Tumor CAR-T Development
| Reagent / Tool Category | Specific Examples | Primary Function in R&D |
|---|---|---|
| CAR Construct Components | scFv domains, CD3ζ signaling domain, 4-1BB/CD28 costimulatory domains | Forms the core CAR architecture for T-cell activation upon antigen binding [46]. |
| Gene Delivery Vectors | Lentiviral vectors, Retroviral vectors, mRNA for electroporation | Mediates stable or transient genetic modification of patient T-cells to express the CAR [46]. |
| T-cell Culture Supplements | Recombinant IL-2, IL-7, IL-15, Anti-CD3/CD28 activation beads | Supports T-cell activation, ex vivo expansion, and promotes persistence phenotypes [46]. |
| Target Antigen Tools | Recombinant proteins (e.g., Claudin 18.2, B7-H3), Antigen-positive cell lines | Validates CAR binding specificity and function in pre-clinical assays. |
| Tumor Microenvironment Modulators | TGF-β inhibitors, PD-1/PD-L1 blocking antibodies, IL-7/CCL19 cytokines | Counteracts immunosuppression; armored CAR-T cells may be engineered to secrete these [48]. |
| In Vivo Model Systems | Immunodeficient mice (NSG), Patient-derived xenografts (PDX) | Provides physiologically relevant systems for evaluating CAR-T efficacy and safety [51]. |
| Toxicity Management Agents | Anakinra (IL-1R antagonist), Corticosteroids (e.g., Dexamethasone) | Mitigates cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) [5]. |
The field of CAR-T therapy for solid tumors is rapidly evolving beyond its hematologic origins, driven by innovative targets like Claudin 18.2, B7-H3, and logic-gated approaches. Recent clinical data, particularly from 2025 conference presentations, provide compelling evidence that these therapies can deliver meaningful survival benefits for patients with advanced gastric cancer, glioblastoma, and other challenging solid malignancies.
Future progress hinges on overcoming the remaining hurdles of the immunosuppressive tumor microenvironment, antigen heterogeneity, and treatment-related toxicities. Key strategic directions include the development of armored CARs that secrete immunomodulatory cytokines, rational combination therapies with immune checkpoint inhibitors, and advanced delivery techniques such as localized administration. Furthermore, the integration of quantitative systems pharmacology models and the exploration of "off-the-shelf" allogeneic products will be instrumental in optimizing dosing and improving accessibility [48] [51]. As these sophisticated engineering and clinical strategies converge, CAR-T therapy is poised to redefine the treatment paradigm for a broad spectrum of solid tumors, ultimately offering new hope for patients with limited therapeutic options.
Overcoming Clinical Hurdles: Optimization Strategies for Efficacy and Safety
Addressing Tumor Microenvironment Suppression and Poor Trafficking
Chimeric Antigen Receptor (CAR) T-cell therapy has revolutionized the treatment of hematological malignancies, with multiple FDA-approved products achieving remarkable response rates in patients with B-cell lymphomas, leukemias, and multiple myeloma [52] [53]. However, the translational success of this innovative immunotherapy in solid tumors remains limited, primarily due to two interconnected biological barriers: the immunosuppressive tumor microenvironment (TME) and inefficient cellular trafficking to tumor sites [54] [12]. The TME in solid tumors establishes a formidable fortress through multiple mechanisms, including physical barriers that impede T-cell infiltration, soluble immunosuppressive factors that inhibit T-cell function, and suppressive immune populations that actively disable cytotoxic activity [53] [55]. Simultaneously, inefficient trafficking resulting from aberrant tumor vasculature and chemokine mismatch prevents sufficient numbers of CAR-T cells from reaching their targets [54]. This application note details the latest protocols and strategic approaches to overcome these challenges, providing researchers with methodologies to enhance CAR-T cell efficacy against solid tumors.
Key Challenges and Quantitative Evidence
Clinical Response Rates Across Malignancies
Table 1: Clinical Response Rates of CAR-T Cell Therapy Across Cancer Types
| Cancer Type | Target Antigen | Clinical Response Rate | Reference |
|---|---|---|---|
| B-cell Acute Lymphoblastic Leukemia | CD19 | 81% Complete Remission | [53] |
| Diffuse Large B-cell Lymphoma | CD19 | 51% Complete Response | [53] |
| Multiple Myeloma | BCMA | 73-97% Overall Response Rate | [53] |
| Solid Tumors (Various) | Multiple Targets | Limited Success; Mostly Preclinical/Phase I | [12] |
Resistance Mechanisms in Solid Tumors
Table 2: Identified Resistance Mechanisms to CAR-T Cell Therapy in Solid Tumors
| Resistance Mechanism | Key Findings | Experimental Evidence |
|---|---|---|
| Tumor-Associated Macrophages (TAMs) | CSF1R+/TREM2+ macrophages associated with CAR-T dysfunction and relapse in DLBCL | Analysis of 80 tumor samples from 55 DLBCL patients [55] |
| Antigen Escape | Loss of target antigen expression in 15% of relapsed lymphoma cases | Clinical biopsy analysis post-CAR-T treatment [55] |
| Physical Barriers | Dense extracellular matrix (ECM) and abnormal vasculature impede infiltration | Preclinical models of prostate, pancreatic, and brain tumors [54] [53] |
| Soluble Immunosuppressive Factors | TGF-β, IL-10, and other cytokines inhibit CAR-T cell function | In vitro T-cell suppression assays [56] |
Experimental Protocols for TME Modification and Enhanced Trafficking
Protocol 1: Chemotherapy-Mediated TME Priming for CAR-T Cell Enhancement
Principle: Low-dose chemotherapy can remodel the TME to augment CAR-T cell infiltration and efficacy by altering cancer-associated fibroblast phenotype, enhancing extracellular matrix degradation, and promoting pro-inflammatory macrophage differentiation [57].
Materials:
- Patient-derived xenograft (PDX) models of human cancer
- Lewis Y (LeY)-specific CAR-T cells or other antigen-specific CAR-T cells
- Carboplatin (preferred) or other platinum-based chemotherapeutic
- Flow cytometry antibodies for immune profiling (CD45, CD3, CD8, CD4, CD11b, F4/80, CD206, CSF1R, TREM2)
- Histochemistry reagents for extracellular matrix analysis (Masson's Trichrome, collagen hybridizing peptides)
Methodology:
- Tumor Establishment: Implant PDX tumors subcutaneously in immunodeficient mice and allow to reach approximately 100-150 mm³.
- Chemotherapy Pre-treatment: Administer a single dose of carboplatin (50 mg/kg, intraperitoneal) one week prior to CAR-T cell infusion [57].
- CAR-T Cell Administration: Infuse 5-10 × 10⁶ CAR-T cells intravenously 7 days post-chemotherapy.
- Tumor Monitoring: Measure tumor dimensions 2-3 times weekly using calipers. Calculate volume using the formula: V = (length × width²)/2.
- Endpoint Analysis: At designated endpoints (typically 4-6 weeks post-treatment):
a. Immune Profiling: Harvest tumors, process to single-cell suspensions, and perform flow cytometry to quantify CAR-T cell infiltration and characterize macrophage polarization (M1 vs M2).
b. Histopathological Analysis: Fix tumor tissue in 10% neutral buffered formalin, embed in paraffin, section, and stain for:
- ECM components (Masson's Trichrome for collagen)
- Proliferation markers (phospho-histone H3)
- Target antigen expression (LeY or other relevant antigen)
- T-cell markers (CD3) to visualize infiltration patterns
- Functional Assessment: Isolate tumor-infiltrating lymphocytes and perform ex vivo cytotoxicity assays or cytokine production measurements to assess CAR-T cell functional status.
Expected Results: The combination of carboplatin pre-treatment with CAR-T cell therapy should significantly enhance tumor regression compared to either treatment alone, with corresponding increases in CAR-T cell infiltration, shifts in macrophage polarization toward M1 phenotype, and reduced ECM density [57].
Protocol 2: Oncolytic Virus-Mediated TME Remodeling
Principle: Oncolytic Newcastle Disease Virus (NDV) mediates direct tumor cell lysis and immunogenic cell death, while simultaneously reprogramming the immunosuppressive TME through dendritic cell activation, macrophage repolarization, and enhanced immune cell recruitment [56].
Materials:
- Engineered oncolytic NDV (preferably armed with immunostimulatory transgenes)
- Solid tumor models (syngeneic or human xenograft in humanized mice)
- CAR-T cells targeting appropriate tumor antigen
- Viral titer quantification kit (plaque assay or TCID50)
- Cytokine/chemokine detection multiplex assays
- Immunogenic cell death markers: calreticulin, ATP, HMGB1
Methodology:
- Virus Preparation and Titration: Propagate NDV in specific pathogen-free chicken embryos or Vero cells. Purify and determine viral titer using plaque assay.
- In Vivo Treatment Schedule: a. Viral Administration: Inject NDV intratumorally (1×10⁸ PFU in 50 μL PBS) when tumors reach ~100 mm³. b. CAR-T Cell Therapy: Administer CAR-T cells (5-10×10⁶ cells) intravenously 3-5 days post-viral injection to coincide with peak TME remodeling.
- TME Analysis: a. Immunogenic Cell Death Assessment: Harvest tumors 24-48 hours post-NDV treatment and analyze for surface calreticulin exposure by flow cytometry, and extracellular ATP/HMGB1 release by ELISA. b. Chemokine Profile: Use multiplex ELISA to measure CXCL9, CXCL10, CCL5, and other T-cell attracting chemokines in tumor homogenates. c. Immune Cell Recruitment: Analyze tumor digests by flow cytometry for dendritic cell maturation (CD80/CD86), macrophage polarization, and endogenous T-cell activation.
- CAR-T Cell Functional Assessment: Isolate tumor-infiltrating CAR-T cells and evaluate:
- Phenotype: Expression of exhaustion markers (PD-1, TIM-3, LAG-3)
- Function: Cytokine production (IFN-γ, TNF-α) upon restimulation
- Proliferation: CFSE dilution or Ki67 expression
Expected Results: NDV pre-treatment should create a more permissive TME characterized by increased T-cell chemokine production, reduced immunosuppressive cell populations, enhanced CAR-T cell infiltration and persistence, and decreased exhaustion markers [56].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Studying TME Suppression and Trafficking
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| TME-Modifying Agents | Low-dose carboplatin [57], Oncolytic NDV [56] | Pre-conditioning tumor microenvironment to enhance CAR-T cell infiltration and function |
| Engineered CAR-T Cells | "Armored" CARs with cytokine secretion (IL-12) [53], TRUCK cells (4th gen) [52] | Equip CAR-T cells to resist suppression and modify their own microenvironment |
| Myeloid-Targeting Agents | CSF1R inhibitors, TREM2 blockers [55] | Counteract tumor-associated macrophage-mediated suppression |
| ECM-Targeting Reagents | Collagenase, hyaluronidase, TGF-β inhibitors [53] | Disrupt physical barriers to improve CAR-T cell tumor penetration |
| Analytical Tools | Spatial transcriptomics, single-cell RNA sequencing [55] | Comprehensive analysis of TME composition and cell-cell interactions |
| Chemokine Receptors | Engineered CXCR2, CCR4 [54] | Enhance homing of CAR-T cells to tumor sites |
Strategic Framework for Combination Therapies
The following diagram illustrates the strategic approach to combining TME-modifying agents with CAR-T cell therapy, highlighting the key mechanisms and temporal sequence for optimal efficacy:
Overcoming the dual challenges of TME suppression and poor trafficking represents the next frontier in expanding CAR-T cell therapy to solid tumors. The protocols and strategic frameworks presented herein provide researchers with validated methodologies to dismantle the tumor's defensive fortifications and enhance CAR-T cell recruitment and function. The combination approaches detailed—particularly chemotherapy pre-conditioning and oncolytic virotherapy—demonstrate that temporal sequencing of TME-modifying agents with CAR-T cell administration is critical for achieving synergistic efficacy. As the field advances, the integration of these strategies with next-generation "armored" CAR constructs capable of resisting suppression will likely be essential for achieving the durable responses in solid tumors that have thus far remained elusive. Future research directions should focus on identifying predictive biomarkers for specific TME subtypes and developing personalized combination regimens matched to individual tumor microenvironmental profiles.
Managing Antigen Escape and Tumor Heterogeneity with Dual-Targeting CARs
Antigen escape and tumor heterogeneity represent two of the most significant barriers to durable responses following chimeric antigen receptor (CAR) T-cell therapy. Whereas single-targeted CAR-T cells can achieve remarkable initial response rates in hematological malignancies, tumor cells frequently evade immune surveillance through antigen loss, downregulation, or modulation [58] [59]. Dual-targeting CAR strategies have emerged as a promising approach to overcome these limitations by enabling simultaneous recognition of multiple tumor-associated antigens (TAAs), thereby reducing the probability of relapse due to antigen escape [60] [58]. This Application Note provides a structured framework for the design, evaluation, and implementation of dual-targeted CAR therapies, featuring standardized protocols for in vitro assessment and quantitative modeling of CAR-T cell pharmacology.
Dual-Targeting CAR Strategies: Design and Implementation
The selection of an appropriate dual-targeting strategy is fundamental to project success. The primary architectural configurations each present distinct advantages and manufacturing considerations, which must be aligned with the intended clinical application [58].
Table 1: Comparison of Dual-Targeting CAR Strategies
| CAR Strategy | Key Feature | Mechanism of Action | Advantages | Disadvantages/Limitations |
|---|---|---|---|---|
| Cocktail/Sequential Infusion | Two separate single-CAR T-cell products | Co-administration or sequential infusion of two distinct products [58] | Precisely defined dose for each product; uses established single CAR constructs [58] | High manufacturing cost; potential for uneven in vivo expansion [58] |
| Co-transduction | Heterogeneous product from two vectors | T cells transduced with two separate vectors encoding different CARs [58] | Utilizes existing, optimized CAR vectors [58] | Mixed final product (single- and dual-CAR T cells); complex manufacturing [58] |
| Bicistronic CAR | Single vector, two distinct CARs | One vector encodes two separate CAR structures via a bicistronic design [60] [58] | Homogeneous product; potential for dual co-stimulation [58] | Large vector size can challenge packaging; may have lower transduction efficiency [58] |
| Bivalent Tandem CAR | Single CAR with two scFvs | One CAR protein with two antigen-binding domains in tandem [58] | Homogeneous product; reduced manufacturing cost [58] | Requires complex protein engineering to optimize binding [58] |
| Bivalent Loop CAR | Single CAR with looped scFvs | One CAR protein with two antigen-binding domains in a loop configuration [58] | Homogeneous product; potentially higher potency than tandem design [58] | Most complex protein design; requires extensive optimization [58] |
The following diagram illustrates the structural and functional relationships between the primary dual-targeting CAR strategies.
Quantitative Pharmacology of Dual-Targeted CAR-T Cells
Understanding the dose-exposure-response relationship is critical for optimizing dual-targeted CAR-T cell therapies. A recently developed multiscale cellular kinetic-pharmacodynamic (CK-PD) model, validated with data from CD19/CD22 and GPRC5D/BCMA bicistronic CAR-Ts, provides a quantitative framework for predicting therapy outcomes [60].
Table 2: Key Parameters from a Multiscale CK-PD Model of Dual-Targeted CAR-T Cells
| Parameter Category | Specific Parameter | Impact on Therapy Exposure/Outcome | Experimental Notes |
|---|---|---|---|
| Drug/Product Factors | CAR binder affinity | Determines overall potency and specificity [60] | Characterized via in vitro killing assays across E:T ratios [60] |
| CAR-T expansion rate constant | Major determinant of in vivo CAR-T cell exposure [60] | Derived from clinical qPCR/FCM data; influenced by product phenotype [60] | |
| System/Tumor Factors | Relative target antigen expression | Governs tumor recognition and dictates CAR-T cell activation [60] | Measured on patient tumor cells pre-lymphodepletion [60] |
| Maximum tumor killing rate (kmax) | Directly impacts rate of tumor cell clearance [60] | Estimated from in vitro cytotoxicity time-course data [60] | |
| Dosing & Composition | Initial phenotypic composition (Memory/Effector) | Affects persistence and expansion potential [60] | Input as % memory (Naïve+CM+EM) and % effector (TEMRA) cells in product [60] |
| Tumor burden | Influences the level and duration of CAR-T cell expansion [60] | Model input based on baseline disease measurements [60] |
Global sensitivity analysis from this model identified relative antigen expression, the maximum killing rate constant, and the CAR-T expansion rate constant as the most prominent determinants of therapy exposure, highlighting key parameters for optimization [60].
Experimental Protocols
Protocol: In Vitro Cytotoxicity Assay for Dual-Targeted CAR-T Cell Potency
This protocol details the procedure for assessing the cytotoxic activity of dual-targeted CAR-T cells against tumor cell lines with varying antigen expression profiles, a critical step in evaluating product functionality and the risk of antigen escape [60].
1. Key Research Reagent Solutions
Table 3: Essential Reagents for In Vitro Cytotoxicity Assessment
| Reagent/Cell Type | Specifications/Function | Example & Notes |
|---|---|---|
| Effector Cells | Dual-targeted CAR-T cells | e.g., Bicistronic CD19/CD22 CAR-T (AUTO3) or GPRC5D/BCMA CAR-T [60]. Include untransduced (NT) T cells as a negative control. |
| Target Cells | Tumor cell lines with defined antigen profiles | Utilize a panel: e.g., SupT1WT, CD19+ SupT1, CD22+ SupT1, RajiWT, RajiCD19KO (to model antigen escape) [60]. |
| Culture Medium | Supports co-culture of effector and target cells | Use appropriate base medium (e.g., RPMI-1640) supplemented with serum/cytokines as required. |
| Viability Assay | Quantifies live/dead target cells | e.g., Flow cytometry-based cytotoxicity assay (Annexin V/PI), or real-time cell analysis (RTCA) systems. |
| Cytokine Detection | Measures T-cell activation | ELISA or multiplex bead-based assays for IFN-γ, Granzyme B, IL-2, etc. |
2. Procedure
- Preparation: Thaw and rest CAR-T cells according to established protocols. Maintain tumor cell lines in logarithmic growth phase. For the assay, consider using a panel of target cells, including wild-type lines and genetically engineered variants with single or double antigen knockouts to rigorously test the dual-targeting capability [60].
- Effector:Target (E:T) Co-culture: Seed target cells in a 96-well U-bottom plate. Add CAR-T cells at specified E:T ratios (e.g., 0:1, 0.3:1, 1:1, 3:1). Include wells with target cells alone (spontaneous death) and effector cells alone as controls. Perform technical replicates for statistical rigor.
- Incubation: Co-culture cells for a predetermined duration (e.g., 24 or 72 hours) in a humidified incubator at 37°C and 5% CO₂ [60].
- Endpoint Measurement:
- Viability Assessment: Harvest co-culture cells and stain with viability dye (e.g., propidium iodide) and a cell counting standard (e.g., counting beads). Analyze by flow cytometry. Calculate specific lysis using the formula:
% Specific Lysis = [1 - (Count of viable target cells in test well / Count of viable target cells in control well)] × 100. - Functional Assessment: Collect supernatant from centrifuged co-culture plates. Quantify secreted cytokines (e.g., IFN-γ, Granzyme B) using ELISA or a multiplex immunoassay.
- Viability Assessment: Harvest co-culture cells and stain with viability dye (e.g., propidium iodide) and a cell counting standard (e.g., counting beads). Analyze by flow cytometry. Calculate specific lysis using the formula:
- Data Analysis: Plot dose-response curves (specific lysis vs. E:T ratio) and compare potency across different target cell lines. Superior killing of single-antigen expressing lines by dual-CARs versus single-CARs demonstrates functionality and confirms the ability to mitigate antigen escape [60].
Protocol: Constructing a Cellular Kinetic-Pharmacodynamic (CK-PD) Model
This protocol outlines the development of a mechanistic mathematical model to integrate preclinical and clinical data, enabling the prediction of dual-targeted CAR-T cell expansion, persistence, and anti-tumor activity in patients [60].
1. Key Research Reagent Solutions
- Clinical PK/PD Data: Longitudinal measurements of CAR-T cell concentrations in peripheral blood (via qPCR and flow cytometry) and tumor burden [60].
- Product Characterization Data: Phenotypic composition (CD4:CD8 ratio, memory/effector subsets) of the infused CAR-T product [60].
- Patient Baseline Data: Tumor-associated antigen (TAA) density on patient tumor cells and total tumor burden before lymphodepletion [60].
- Software: Modeling and simulation software (e.g., R, NONMEM, MATLAB).
2. Procedure
- Model Structure Definition: Develop a system of ordinary differential equations (ODEs) capturing key biological processes. A foundational three-compartment model can describe interactions between tumor cells (T), effector CAR-T cells (CT), and memory CAR-T cells (CM) [61] [62].
- Tumor Dynamics:
dT/dt = f(T) - γ·T·C_Twheref(T)is tumor growth (e.g., logistic) andγis the killing rate by effectors [61]. - Effector CAR-T Dynamics:
dC_T/dt = φ·C_T - ρ·C_T + θ·T·C_M - α·T·C_Taccounting for proliferation (φ), decay/differentiation (ρ), memory-to-effector conversion (θ), and tumor-mediated inhibition (α) [61]. - Memory CAR-T Dynamics:
dC_M/dt = ε·ρ·C_T - μ·C_Mrepresenting formation from effectors (ε) and natural decay (μ) [61].
- Tumor Dynamics:
- Data Integration & Parameter Estimation:
- In Vitro-In Vivo Extrapolation: Use parameters derived from in vitro cytotoxicity assays (e.g., maximum killing rate
γ) as initial estimates for the in vivo model [60]. - Clinical Calibration: Calibrate the model using individual-patient clinical CK data. Incorporate patient-specific inputs, such as the initial memory/effector composition of the product and baseline TAA levels, to account for population heterogeneity [60].
- In Vitro-In Vivo Extrapolation: Use parameters derived from in vitro cytotoxicity assays (e.g., maximum killing rate
- Model Validation & Application:
- Sensitivity Analysis: Perform a Global Sensitivity Analysis (GSA) to identify the model parameters (e.g., CAR-T expansion rate, antigen expression, killing rate) to which the outcome (e.g., CAR-T exposure) is most sensitive [60].
- Simulation: Use the validated model to perform in silico trials for dose optimization, predict responses in new patient populations, or guide reverse translation efforts to refine CAR binder affinities [60].
The following diagram visualizes the structure and key interactions within a basic CK-PD model for CAR-T cell therapy.
Dual-targeting CAR-T cell therapies represent a formidable strategy to counter tumor antigen escape and heterogeneity. The successful implementation of these advanced therapies relies on the rational selection of a CAR architecture, rigorous in vitro functional validation using standardized potency assays, and the application of quantitative pharmacological models to decipher complex dose-exposure-response relationships. The experimental frameworks and protocols detailed herein provide a foundational roadmap for researchers to systematically develop, characterize, and optimize next-generation dual-targeted CAR-T cell products, thereby accelerating their translation into clinically effective treatments for refractory cancers.
Cytokine Release Syndrome (CRS) and Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS) are the most common and potentially severe toxicities associated with T-cell engaging immunotherapies, including Chimeric Antigen Receptor (CAR) T-cell therapy and bispecific antibodies [63]. These adverse events result from the potent immune activation central to the therapeutic mechanism of these treatments. In CAR T-cell therapy, a patient's T-cells are genetically engineered to express synthetic receptors that target specific tumor antigens, creating a "living drug" that can persist in the body and provide long-term anti-cancer activity [4]. However, this robust immune activation triggers a cascade of inflammatory mediators that can lead to systemic toxicities requiring precise management protocols.
The development of CAR T-cells has evolved through multiple generations, with current approved therapies utilizing second-generation constructs containing either CD28 or 4-1BB (CD137) co-stimulatory domains [52]. These costimulatory signals enhance T-cell proliferation, cytotoxicity, and persistence, but also contribute to the intensity of immune activation and associated toxicities. The management of CRS and ICANS has therefore become an integral component of treatment protocols for these innovative cancer immunotherapies, requiring standardized grading systems and evidence-based intervention strategies to ensure patient safety while preserving therapeutic efficacy.
Pathophysiology and Clinical Presentation
Cytokine Release Syndrome (CRS)
CRS is a systemic inflammatory response characterized by excessive immune activation and elevated circulating cytokine levels. The syndrome typically occurs within days to weeks following CAR T-cell infusion or bispecific antibody administration [63]. The pathophysiology involves widespread T-cell activation leading to a massive release of proinflammatory cytokines including interleukin-6 (IL-6), interferon-gamma (IFN-γ), granulocyte-macrophage colony-stimulating factor (GM-CSF), and others [63] [4]. This cytokine storm can affect multiple organ systems and range in severity from mild flu-like symptoms to life-threatening circulatory collapse and multi-organ failure.
The clinical presentation of CRS includes fever (often the first sign), hypotension, hypoxia, and potential end-organ dysfunction. The American Society for Transplantation and Cellular Therapy (ASTCT) has established a consensus grading system that classifies CRS severity based on three clinical parameters: fever, hypotension, and hypoxia [63]. This standardized grading system enables consistent toxicity assessment across different institutions and clinical trials, facilitating appropriate management interventions.
Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS)
ICANS represents a distinct toxicity syndrome characterized by neurological symptoms that typically follow or coincide with CRS. The pathophysiology of ICANS is multifactorial, involving endothelial activation, blood-brain barrier disruption, and cerebral edema, though the exact mechanisms remain under investigation [63] [4]. Notably, elevated levels of inflammatory cytokines, particularly IL-6, are observed in cerebrospinal fluid during ICANS episodes, suggesting central nervous system involvement in the systemic inflammatory response.
Clinical manifestations of ICANS encompass a spectrum of neurological symptoms, including encephalopathy (ranging from mild confusion to coma), expressive aphasia, impaired motor skills, headache, seizures, and cerebral edema [63]. The ASTCT grading system for ICANS incorporates multiple assessment domains, including the Immune Effector Cell-Associated Encephalopathy (ICE) score, level of consciousness, seizure activity, motor findings, and evidence of elevated intracranial pressure [63]. This comprehensive assessment approach ensures accurate grading and appropriate management of neurotoxicity.
Standardized Toxicity Grading Systems
Consistent and accurate grading of CRS and ICANS is fundamental to appropriate management. The ASTCT consensus grading systems provide standardized criteria that guide therapeutic decisions and escalation of care.
Table 1: ASTCT Consensus Grading for Cytokine Release Syndrome (CRS)
| Grade | Fever | Hypotension | Hypoxia |
|---|---|---|---|
| Grade 1 | Temperature ≥38°C | None | None |
| Grade 2 | Temperature ≥38°C | Not requiring vasopressors | Requiring low-flow nasal cannula or blow-by |
| Grade 3 | Temperature ≥38°C | Requiring a vasopressor (with or without vasopressin) | Requiring high-flow oxygen, non-rebreather mask, or Venturi mask |
| Grade 4 | Temperature ≥38°C | Requiring multiple vasopressors (excluding vasopressin) | Requiring positive pressure (CPAP, BiPAP, intubation, mechanical ventilation) |
Table 2: ASTCT Consensus Grading for Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS)
| Grade | ICE Score | Level of Consciousness | Seizures | Motor Findings | Elevated ICP/Cerebral Edema |
|---|---|---|---|---|---|
| Grade 1 | 7-9 | Awakens spontaneously | None | None | None |
| Grade 2 | 3-6 | Awakens to voice | None | None | None |
| Grade 3 | 0-2 | Awakens only to tactile stimulus | Any clinical seizure that resolves rapidly; or non-convulsive seizures on EEG that resolve with intervention | None | Focal/local edema on neuroimaging |
| Grade 4 | 0 (unarousable) | Stupor or coma | Life-threatening prolonged seizure (>5 minutes); or repetitive clinical or electrical seizures without return to baseline | Deep focal motor weakness | Diffuse cerebral edema; decerebrate or decorticate posturing; cranial nerve VI palsy; papilledema; Cushing's triad |
The following diagram illustrates the clinical decision pathway for managing CRS and ICANS based on standardized grading:
Comprehensive Management Protocols
CRS Management Protocol
The management of CRS follows a graded approach based on severity, with prompt recognition and early intervention being critical to preventing progression to severe stages.
Table 3: Evidence-Based CRS Management Protocol
| CRS Grade | First-Line Interventions | Second-Line Interventions | Supportive Care Measures | Therapy Administration |
|---|---|---|---|---|
| Grade 1 | Supportive care only | Not applicable | Antipyretics for fever, IV fluids for dehydration | Continue CAR T-cell therapy or bispecific antibodies |
| Grade 2 | Tocilizumab 8 mg/kg (max 800 mg) IV; may repeat every 8 hours if no clinical improvement | Consider corticosteroids if no improvement after 2-3 doses of tocilizumab | IV fluids, consider low-flow oxygen, cardiac monitoring | Withhold therapy until CRS resolves to Grade 1; consider hospitalization for next dose [64] |
| Grade 3 | Tocilizumab + corticosteroids (methylprednisolone 1-2 mg/kg/day or dexamethasone 10-20 mg every 6-24 hours) | Consider alternative anticytokine therapy (anakinra, siltuximab) for refractory cases | High-flow oxygen, ICU level care, vasopressor support | Withhold therapy until CRS resolves to Grade 1; hospitalize for next dose [64] |
| Grade 4 | Aggressive tocilizumab + high-dose corticosteroids (methylprednisolone 1000 mg/day) | Anakinra, siltuximab, or other immunomodulators | ICU transfer, mechanical ventilation, multiple vasopressors | Permanently discontinue therapy for recurrent Grade 3 or any Grade 4 CRS [64] |
Detailed Management Considerations:
Tocilizumab Administration: The IL-6 receptor antagonist tocilizumab is FDA-approved for severe CRS and is recommended for Grade 2 or higher CRS. The standard dose is 8 mg/kg (maximum 800 mg) intravenously over one hour, which may be repeated every 8 hours for persistent or worsening symptoms [63]. Early administration of tocilizumab has been associated with reduced progression to severe CRS without negatively impacting treatment efficacy [63].
Corticosteroid Therapy: For severe or life-threatening CRS (Grade ≥3), corticosteroids should be initiated concurrently with tocilizumab. Methylprednisolone (1-2 mg/kg/day) or dexamethasone (10-20 mg every 6-24 hours) are commonly used. Corticosteroids should be tapered gradually once CRS resolves to Grade 1, typically over several days to prevent rebound inflammation [63].
Refractory CRS Management: For cases refractory to tocilizumab and corticosteroids, alternative agents include the IL-1 receptor antagonist anakinra (100 mg subcutaneous or IV every 6-12 hours) or the anti-IL-6 monoclonal antibody siltuximab [63]. The optimal timing and sequencing of these agents continues to be investigated in clinical trials.
ICANS Management Protocol
Management of ICANS requires a distinct approach, with corticosteroids serving as the primary therapeutic intervention, particularly in the absence of concurrent CRS.
Table 4: Evidence-Based ICANS Management Protocol
| ICANS Grade | Neurological Assessment | Pharmacological Management | Supportive Care & Monitoring | Therapy Administration |
|---|---|---|---|---|
| Grade 1 | ICE score every 8-12 hours, frequent neurological checks | Supportive care only; no pharmacological intervention required | Seizure precautions, fall precautions, neurologist consultation | Withhold therapy until ICANS resolves [64] |
| Grade 2 | ICE score every 4-8 hours, frequent neurological checks | Dexamethasone 10 mg IV every 6 hours; continue until improvement to Grade 1, then taper | Consider non-sedating anti-seizure prophylaxis, neurological consultation | Withhold therapy until ICANS resolves [64] |
| Grade 3 | Continuous neurological monitoring, ICE score every 4 hours | Dexamethasone 10 mg IV every 6 hours; if no improvement, consider methylprednisolone 1000 mg/day | ICU level care, EEG monitoring, seizure management, neuroimaging | Withhold therapy until ICANS resolves; permanent discontinuation for recurrent Grade 3 [64] |
| Grade 4 | Continuous neurological monitoring in ICU | High-dose methylprednisolone 1000 mg/day for 2-5 days; consider intrathecal chemotherapy for severe cases | Aggressive management of cerebral edema, mechanical ventilation for compromised airway | Permanently discontinue therapy [64] |
Detailed Management Considerations:
Corticosteroid Administration: Dexamethasone is preferred for ICANS management due to its excellent CNS penetration. For Grade 2 ICANS, dexamethasone 10 mg IV every 6 hours should be initiated and continued until improvement to Grade 1, followed by a rapid taper over several days [64] [63]. For severe or life-threatening ICANS (Grade ≥3), high-dose methylprednisolone (1000 mg/day) is recommended.
Tocilizumab Caution in ICANS: Current guidelines recommend against using tocilizumab for isolated ICANS without concurrent CRS, as it may potentially exacerbate neurotoxicity by increasing circulating IL-6 levels that cannot cross the blood-brain barrier, thereby creating a concentration gradient that favors IL-6 entry into the CNS [63]. For patients with concurrent CRS and ICANS, tocilizumab should be administered according to CRS management guidelines.
Supportive Care Measures: Seizure prophylaxis with non-sedating antiepileptics (e.g., levetiracetam) should be considered for Grade 2 or higher ICANS. Maintenance of adequate cerebral perfusion pressure, management of increased intracranial pressure, and prevention of complications related to immobilization are essential components of care.
Experimental Models and Research Methodologies
In Vitro Models for CRS/ICANS Investigation
Research to elucidate the mechanisms of CRS and ICANS relies on sophisticated experimental models that replicate critical aspects of the human immune response:
Primary Human T-cell Coculture Systems: Isolated CD3+ T-cells from healthy donors are transduced with CAR constructs using lentiviral or retroviral vectors [52] [65]. These engineered T-cells are then cocultured with target antigen-expressing tumor cells at various effector-to-target ratios (typically 1:1 to 10:1) to simulate therapeutic activation. Supernatants are collected at multiple timepoints (6, 24, 48, 72 hours) for cytokine profiling via Luminex or ELISA to quantify key inflammatory mediators including IL-6, IFN-γ, IL-2, TNF-α, GM-CSF, and IL-10.
Endothelial Transwell Models: To investigate ICANS pathophysiology, human cerebral microvascular endothelial cells are cultured on transwell inserts to establish blood-brain barrier (BBB) models. Following stimulation with cytokines or patient serum, measurements of transendothelial electrical resistance (TEER) and permeability to fluorescent dextran assess BBB integrity. T-cell migration across the endothelial barrier is quantified to evaluate neurotropism.
Macrophage and Microglia Coculture Systems: Monocyte-derived macrophages or induced microglia-like cells are incorporated into these models to investigate the role of innate immune cells in CRS/ICANS pathogenesis. These systems enable assessment of cell-cell contact dependencies and soluble mediator effects through conditioned media transfer experiments.
In Vivo Models for Toxicity Assessment
Animal models remain essential for evaluating CRS and ICANS pathophysiology and testing novel management strategies:
Immunodeficient Mouse Models with Human Tumor Xenografts: NSG (NOD-scid-IL2Rγnull) mice engrafted with human tumor cell lines (e.g., Nalm6 for B-ALL) subsequently receive human CAR T-cells via intravenous injection. Comprehensive toxicity assessment includes daily weights, clinical scoring, serum cytokine measurements, and histological analysis of major organs. This model effectively recapitulates CRS manifestations and enables evaluation of therapeutic interventions.
Humanized Mouse Models: NSG mice engrafted with human CD34+ hematopoietic stem cells develop a functional human immune system, enabling study of CRS/ICANS in the context of a more complete human immune microenvironment. These models permit investigation of donor-specific factors and human-specific immune pathways.
CRS/ICANS Biomarker Monitoring: Serial blood collection enables quantification of human cytokine levels (IL-6, IFN-γ, GM-CSF, IL-10) and CAR T-cell expansion kinetics via flow cytometry. For neurotoxicity assessment, behavioral tests (rotarod, open field), brain immunohistochemistry for microglial activation, and measurement of cerebrovascular permeability are performed.
The following diagram illustrates the key signaling pathways involved in CRS and ICANS pathogenesis and their pharmacological targeting:
The Scientist's Toolkit: Research Reagent Solutions
Table 5: Essential Research Reagents for CRS/ICANS Investigation
| Reagent Category | Specific Examples | Research Application | Commercial Sources |
|---|---|---|---|
| Human Immune Cells | Primary human T-cells, CD34+ hematopoietic stem cells, peripheral blood mononuclear cells (PBMCs) | In vitro and in vivo modeling of human immune responses | StemCell Technologies, Lonza, AllCells |
| CAR Constructs | Second-generation CARs (CD28 or 4-1BB costimulatory domains), viral vectors (lentivirus, retrovirus) | Engineering of T-cells for functional studies | Addgene, academic collaborations, custom synthesis |
| Cytokine Detection | Luminex multiplex panels, ELISA kits for IL-6, IFN-γ, GM-CSF, IL-10 | Quantification of inflammatory mediators in supernatants and serum | R&D Systems, BioLegend, Thermo Fisher |
| Cell Culture Reagents | T-cell activation reagents (anti-CD3/CD28 beads), serum-free media, cytokine supplements (IL-2, IL-7, IL-15) | Maintenance and expansion of primary T-cells | Gibco, Miltenyi Biotec, STEMCELL Technologies |
| Immunoassay Kits | Phospho-flow cytometry antibodies, apoptosis detection kits, cytotoxicity assays | Mechanism of action studies and toxicity assessment | BD Biosciences, BioLegend, Abcam |
| Animal Models | NSG mice, human cytokine knock-in models, tumor xenograft models | In vivo toxicity and efficacy testing | Jackson Laboratory, Charles River |
Emerging Research and Future Directions
The field of CRS and ICANS management continues to evolve with several promising research directions:
Novel Immunomodulatory Approaches: Next-generation CAR designs incorporating "safety switches" (e.g., inducible caspase 9) or co-expression of cytokine-neutralizing constructs (e.g., membrane-bound IL-6 scavengers) aim to mitigate toxicity while preserving antitumor efficacy [66]. These engineered cells may provide greater control over the immune response and enable rapid shutdown in cases of severe toxicity.
Biomarker Discovery and Risk Stratification: Research efforts are focusing on identifying predictive biomarkers for severe toxicity, including genetic polymorphisms in cytokine genes, pre-existing inflammatory states, and tumor burden metrics [63]. Such biomarkers could enable preemptive intervention and personalized management strategies.
In Vivo CAR T-cell Engineering: Emerging platforms that generate CAR T-cells directly within the patient's body using viral vectors or nanoparticle delivery systems may alter the toxicity profile of these therapies [65]. Early preclinical data suggest that in vivo generated CAR T-cells might exhibit reduced incidence and severity of CRS and ICANS, potentially due to more controlled expansion kinetics.
Advanced Neurotoxicity Management: Investigation of targeted therapies for ICANS includes agents that specifically protect endothelial integrity, modulate neuroinflammation, or prevent seizure activity without compromising antitumor immunity. The exploration of intrathecal drug delivery for severe neurotoxicity represents another active area of clinical investigation.
As the field of cellular immunotherapy expands to include solid tumors and non-oncological indications, the development of refined toxicity management protocols will remain essential for maximizing the therapeutic potential of these powerful treatments while ensuring patient safety.
Combating T-cell Exhaustion through Epigenetic and Cytokine Engineering
T-cell exhaustion represents a fundamental barrier to durable responses in cancer immunotherapy, particularly for chimeric antigen receptor (CAR)-T cell therapies. This dysfunctional state, driven by chronic antigen exposure within the immunosuppressive tumor microenvironment (TME), is characterized by progressive loss of effector functions, sustained expression of inhibitory receptors, and impaired tumor-killing capacity [67] [68]. This Application Note details two synergistic engineering strategies—epigenetic reprogramming and cytokine engineering—to prevent or reverse T-cell exhaustion, thereby enhancing the persistence and efficacy of adoptive cell therapies. These protocols are designed for researchers and drug development professionals working to overcome the limitations of current CAR-T cell platforms in both hematological malignancies and solid tumors.
Key Engineering Strategies and Quantitative Evidence
Epigenetic Reprogramming to Reverse Exhaustion
The exhausted T-cell state is established and maintained through stable epigenetic modifications that create a barrier to reinvigoration by conventional approaches alone [67]. Targeting these epigenetic regulators enables rewriting of the transcriptional program to preserve T-cell function.
Table 1: Epigenetic Modifiers for Combating T-Cell Exhaustion
| Epigenetic Target | Compound Class | Mechanism of Action | Effect on T-cell Function |
|---|---|---|---|
| DNMT | Azacitidine (AZA), Decitabine (DAC) [67] | DNA methyltransferase inhibitor; promotes DNA hypomethylation | Upregulates TAAs and co-stimulatory molecules on tumor cells [69] |
| EZH2 | EZH2 Inhibitors (e.g., GSK126, Tazemetostat) [69] | Inhibits H3K27 trimethylation; reduces repressive chromatin marks | Induces surface expression of sparse antigens like GD2 in Ewing sarcoma [69] |
| BET Proteins | JQ1 [69] | Bromodomain inhibitor; disrupts reading of acetylated histones | Reduces PD-L1 expression on tumor and immune cells in the TME [69] |
| HDAC | HDAC Inhibitors (e.g., Vorinostat) [70] [67] | Histone deacetylase inhibitor; promotes open chromatin state | Reverses suppressive macrophage activity and enhances T-cell anti-tumor activity [69] |
Cytokine Engineering to Reinforce T-cell Fitness
Cytokine engineering provides a cell-intrinsic method to shield CAR-T cells from the immunosuppressive TME. A prominent example involves the constitutive or inducible expression of pro-inflammatory cytokines like Interleukin-18 (IL-18).
Table 2: Engineered Cytokine Strategies to Counter Exhaustion
| Strategy | Engineering Approach | Key Outcomes | Clinical/Preclinical Evidence |
|---|---|---|---|
| IL-18 "Armored" CAR-T | CAR-T cells engineered to secrete IL-18 (e.g., huCART19-IL18) [71] | Recruits additional immune cells; supports CAR-T persistence and function [71] | 52% complete remission rate in R/R lymphoma patients; durable remissions >2 years [71] |
| IL-7/IL-15 Co-expression | GD2 CAR-T cells modified to express IL-7 and IL-15 [69] | Enhances T-cell survival and memory formation; reduces PD-L1 levels on cancer cells [69] | Preclinical models show reduced exhaustion and improved efficacy [69] |
| JAK/STAT Pathway Engagement | 5th-gen CARs incorporating IL-2R fragment [52] [69] | Enables antigen-dependent JAK/STAT activation for enhanced persistence | Promotes memory formation and sustains CAR-T activity [52] |
Diagram 1: Synergistic pathways of epigenetic and cytokine engineering in combating T-cell exhaustion. These independent interventions converge to enhance T-cell fitness and promote durable tumor control.
Detailed Experimental Protocols
Protocol: Combinatorial Testing of Epigenetic Modifiers with CAR-T Cells
Objective: To evaluate the ability of epigenetic compounds to enhance CAR-T cell function by upregulating target antigens on tumor cells and reducing T-cell exhaustion.
Materials:
- Tumor Cell Lines: Applicable to the CAR target (e.g., AML cell lines for AML-CAR-T) [69].
- CAR-T Cells: Second-generation CAR-T cells targeting the antigen of interest.
- Epigenetic Compounds: DNMTi (Azacitidine, 1µM), EZH2i (GSK126, 500nM), HDACi (Vorinostat, 250nM) [67] [69].
- Culture Media: RPMI-1640 or DMEM supplemented with 10% FBS.
- Flow Cytometry Antibodies: Antibodies against the target antigen (e.g., anti-GD2 for Ewing sarcoma), exhaustion markers (anti-PD-1, anti-TIM-3), and activation markers (anti-CD69) [69].
Methodology:
- Tumor Cell Pre-treatment:
- Culture tumor cells at 60-70% confluence.
- Treat cells with respective epigenetic compounds or vehicle control (DMSO) for 72 hours [69].
- Harvest cells and confirm viability >90% via trypan blue exclusion.
Co-culture Assay:
- Seed pre-treated tumor cells in 96-well U-bottom plates at 1x10^4 cells/well.
- Add CAR-T cells at an Effector:Target (E:T) ratio of 1:1. Include controls for tumor cells alone and CAR-T cells alone.
- Centrifuge plates at 300 x g for 3 minutes to initiate cell contact.
- Incubate at 37°C, 5% CO2 for 24-48 hours.
Functional and Phenotypic Analysis:
- Cytotoxicity: After 24 hours, measure specific lysis using a real-time cell analyzer (e.g., xCelligence) or lactate dehydrogenase (LDH) release assay.
- Cytokine Production: After 24 hours, collect supernatant and quantify IFN-γ and IL-2 levels by ELISA.
- Exhaustion Marker Expression: After 48 hours, harvest CAR-T cells and stain for surface (e.g., PD-1, TIM-3) and intracellular exhaustion markers for flow cytometry analysis.
- Tumor Antigen Modulation: Analyze target antigen density on pre-treated tumor cells via flow cytometry.
Protocol: Evaluating IL-18 Armored CAR-T Cell Efficacy
Objective: To assess the anti-tumor function and persistence of CAR-T cells engineered to constitutively secrete IL-18.
Materials:
- IL-18 Armored CAR-T Cells: huCART19-IL18 (CD19-targeting CAR with IL-18 transgene) [71].
- Control CAR-T Cells: Standard second-generation CD19 CAR-T cells.
- Target Cells: CD19+ lymphoma cell line (e.g., SEM) [72].
- Mouse Model: NSG mice for in vivo studies.
- ELISA Kits: Mouse and human IL-18, IFN-γ.
Methodology:
- In Vitro Cytotoxicity and Cytokine Profiling:
- Co-culture CAR-T cells with target cells at various E:T ratios (e.g., 1:1 to 1:10) for 24 hours.
- Measure cytotoxicity via Annexin V/Propidium Iodide staining and flow cytometry [72].
- Analyze supernatant for IL-18, IFN-γ, and Granzyme B levels by multiplex ELISA.
- In Vivo Persistence and Anti-Tumor Efficacy:
- Tumor Engraftment: Inject 5x10^5 CD19+ tumor cells subcutaneously into NSG mice. Allow tumors to establish (~100 mm³).
- Treatment: Randomize mice into groups (n=5/group) and administer a single intravenous dose of 5x10^6 IL-18 armored CAR-T cells or control CAR-T cells.
- Tumor Monitoring: Measure tumor volume by caliper 2-3 times weekly.
- Persistence Analysis: Collect peripheral blood weekly. Isolate PBMCs and quantify human T-cell persistence via flow cytometry using anti-human CD3/CD8 antibodies.
- Tumor Microenvironment Interrogation: At endpoint, harvest tumors, process into single-cell suspensions, and analyze immune cell infiltration (e.g., endogenous NK cells, macrophages) and exhaustion markers on recovered CAR-T cells.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Epigenetic and Cytokine Engineering Research
| Research Reagent / Tool | Function/Application | Example Use-Case |
|---|---|---|
| DNMT Inhibitors (Azacitidine) | Induces DNA hypomethylation, upregulating tumor antigen expression [67] [69] | Pre-treatment of AML cells to enhance CAR-T targeting [69] |
| EZH2 Inhibitors (GSK126) | Reduces H3K27me3 repressive mark to promote antigen expression [69] | Increases GD2 antigen density on Ewing sarcoma cells for GD2-CAR T therapy [69] |
| IL-18 Armored CAR Construct | Enables constitutive IL-18 secretion to modulate the TME and enhance CAR-T fitness [71] | huCART19-IL18 for treating relapsed/refractory lymphoma [71] |
| Anti-PD-1 / Anti-TIM-3 Antibodies | Immune checkpoint blockade to reverse T-cell exhaustion [67] [68] | Used in combination with CAR-T therapy to improve long-term remission [69] |
| CRISPR-Cas9 System | Gene editing to knockout exhaustion-associated genes (e.g., PD-1) in CAR-T cells [69] | Generation of PD-1 deficient CAR-T cells to resist TME-mediated exhaustion [69] |
| Flow Cytometry Panels (Exhaustion) | Multiparametric phenotyping of T-cell dysfunction [72] | Quantifying PD-1, TIM-3, LAG-3 expression on CAR-T cells post-activation |
Diagram 2: Logical workflow from identifying T-cell exhaustion to achieving a functional cure via targeted engineering strategies and reagent solutions.
Concluding Remarks
The convergence of epigenetic reprogramming and cytokine engineering represents a transformative approach to overcoming T-cell exhaustion in adoptive cell therapy. The protocols outlined herein provide a validated roadmap for researchers to systematically enhance CAR-T cell function and persistence. As the field advances, the combination of these strategies with other modalities, such as metabolic engineering (e.g., GLUT1 overexpression to enhance nutrient competition [73]) and optimized CAR designs (e.g., transmembrane domain engineering to reduce exhaustion [72]), is poised to unlock the full potential of CAR-T cell therapy for a broader range of cancers, particularly solid tumors where the immunosuppressive TME remains a formidable challenge.
Benchmarking Next-Generation Constructs: Validation and Comparative Efficacy
In Vitro and In Vivo Preclinical Models for CAR-T Cell Validation
Chimeric Antigen Receptor T-cell therapy represents a breakthrough in cancer immunotherapy, demonstrating remarkable efficacy against hematological malignancies. The preclinical evaluation of CAR-T cells is a critical step in clinical translation, requiring comprehensive assessment of antitumor potency, persistence, and safety profiles. This document provides detailed application notes and experimental protocols for the standardized preclinical validation of CAR-T cell products, framed within the broader context of CAR-T cell engineering protocols for cancer immunotherapy research. We summarize established methodologies and emerging technologies that enable researchers to generate reliable, clinically predictive data on CAR-T cell function, from in vitro cytotoxicity assays to complex in vivo models that recapitulate human tumor microenvironment interactions [74].
Critical Parameters for CAR-T Cell Evaluation
Before selecting specific models, researchers must define the key functional parameters requiring assessment. Comprehensive preclinical evaluation should investigate multiple dimensions of CAR-T cell biology and therapeutic potential [74].
Table 1: Essential Parameters for Preclinical CAR-T Cell Evaluation
| Evaluation Category | Specific Parameters | Significance |
|---|---|---|
| Cytotoxic Activity | Target cell killing (specific lysis), degranulation (CD107a expression), cytotoxic molecule production (granzyme B, perforin) | Primary effector function; direct antitumor potency |
| Cellular Expansion & Persistence | Proliferation kinetics, peak expansion, long-term persistence | Correlates with clinical response durability; indicates T cell fitness |
| Cytokine Profile | Effector cytokines (IFN-γ, TNF-α, IL-2), inflammatory cytokines (IL-6, IL-10), chemokines | Indicates activation potency; predicts efficacy and toxicity risk (e.g., CRS) |
| Cell Phenotype & Differentiation | Memory markers (CD62L, CCR7, CD45RA/RO), exhaustion markers (PD-1, TIM-3, LAG-3), activation markers (CD69) | Predicts persistence capacity and functional durability; identifies dysfunctional states |
| Tumor Microenvironment Interaction | Immunosuppressive factor resistance, chemokine receptor-mediated trafficking, stromal component engagement | Critical for solid tumor applications; indicates tissue penetrance capability |
| Safety & Specificity | On-target/off-tumor toxicity, cytokine release syndrome potential, neurotoxicity indicators | Essential for clinical risk assessment; identifies therapeutic window |
The activation profile and functional status of CAR-T cells can be monitored through surface markers that dynamically change during differentiation, activation, and memory formation. CAR-T cells with a memory-like phenotype (CD62L+, CCR7+) typically mediate superior clinical efficacy, while an exhaustion-like phenotype (PD-1+, TIM-3+, LAG-3+) limits long-term potency [74]. Polyfunctional CAR-T cells capable of simultaneously secreting multiple immune-stimulatory cytokines and cytotoxic molecules at the single-cell level represent a particularly promising biomarker for clinical outcomes [74].
In Vitro Models for CAR-T Cell Functional Evaluation
In vitro models provide controlled, reductionist systems for initial CAR-T cell functional assessment. These platforms are particularly valuable for high-throughput screening of CAR constructs, mechanism-of-action studies, and combination therapy testing.
TAA-Dependent Cytotoxicity Assessment
Protocol: Co-culture Cytotoxicity Assay Using TAA-Expressing Cells
Principle: Measure CAR-T cell-mediated killing of antigen-expressing target cells over time, quantifying specific lysis and activation parameters.
Materials:
- CAR-T cells (effector cells)
- Target cells (TAA-expressing tumor cells)
- Control cells (antigen-negative or irrelevant antigen)
- Culture plates (96-well U-bottom recommended)
- Flow cytometer with appropriate antibodies
- Cytokine analysis platform (ELISA, Luminex, or MSD)
Procedure:
- Effector and Target Cell Preparation:
- Harvest and count CAR-T cells and target cells. Ensure >90% viability for both populations.
- Label target cells with fluorescent membrane dyes (e.g., CFSE, CellTrace Violet) according to manufacturer protocols at 1-5 μM for 10-20 minutes at 37°C.
- Include antigen-negative control cells to assess antigen-specificity.
Co-culture Setup:
- Plate target cells at 5,000-50,000 cells/well in complete medium.
- Add CAR-T cells at effector-to-target (E:T) ratios ranging from 1:1 to 25:1.
- Include target cells alone (spontaneous death control) and target cells with detergent (maximum death control).
- Centrifuge plates briefly (500 × g, 1 minute) to initiate cell contact.
- Incubate at 37°C, 5% CO₂ for 6-48 hours depending on assay objectives.
Viability Assessment:
- Add viability dye (e.g., 7-AAD, propidium iodide, or Annexin V) according to manufacturer instructions.
- Analyze by flow cytometry within 1 hour of staining.
- Calculate specific lysis: % Specific Lysis = [(% Death in experimental - % Spontaneous death) / (100 - % Spontaneous death)] × 100
Concurrent Readouts:
- CD107a Degranulation: Add anti-CD107a antibody during co-culture with protein transport inhibitor.
- Cytokine Production: Collect supernatant for multiplex cytokine analysis at 12-24 hours.
- CAR-T Cell Phenotype: Stain for activation (CD69), memory, and exhaustion markers after co-culture.
Technical Notes:
- For comparing CAR constructs targeting different TAAs, engineer a consistent parental cell line (e.g., Nalm-6 leukemia cells) to express each TAA at similar densities to minimize confounding variables [74].
- Include blockade controls with excess soluble antigen or anti-target antibodies to confirm specificity.
- For kinetic real-time killing assessment, consider impedance-based systems (e.g., xCelligence) or continuous viability indicator systems.
Antigen-Specific Activation Assays
Protocol: Plate-Bound or Bead-Bound Antigen Stimulation
Principle: Isolate CAR signaling through controlled presentation of recombinant antigen without confounding cellular interactions.
Materials:
- Recombinant TAA protein or anti-idiotype antibody
- Anti-Fc capture antibody (for plate-bound)
- Streptavidin-coated nanobeads (for bead-bound)
- Protein L (alternative for antibody-based CAR activation)
Procedure:
- Antigen Immobilization:
- Plate-bound: Coat plates with anti-Fc antibody (1-10 μg/mL), then add recombinant TAA-Fc fusion protein (1-5 μg/mL).
- Bead-bound: Incubate streptavidin beads with biotinylated TAA according to manufacturer recommendations.
- Wash to remove unbound protein.
CAR-T Cell Stimulation:
- Add CAR-T cells to antigen-coated surfaces or beads at 1:1 to 1:5 cell:bead ratio.
- Incubate for 6-24 hours depending on readouts.
Analysis:
- Assess early activation markers (CD69) at 6-12 hours.
- Measure cytokine production in supernatant at 12-24 hours.
- Evaluate proliferation by dye dilution over 3-5 days.
Technical Notes:
- This system provides reduced complexity for studying CAR signaling pathways without interference from co-stimulatory or inhibitory ligands on target cells.
- Particularly useful for biochemical assays requiring large cell numbers, such as phosphorylation analysis or RNA sequencing [74].
In Vivo Models for CAR-T Cell Validation
In vivo models provide indispensable assessment of CAR-T cell function within physiological systems, evaluating trafficking, expansion, persistence, and toxicity in biologically relevant contexts.
Table 2: In Vivo Models for CAR-T Cell Preclinical Evaluation
| Model Type | Key Features | Applications | Limitations |
|---|---|---|---|
| Immunodeficient Mouse Models (e.g., NSG, NOG) | Human tumor xenografts + human CAR-T cells; no murine immune system | Basic antitumor efficacy, trafficking, expansion kinetics, persistence | Lack functional immune context; cannot assess CRS or endogenous immunity |
| Immunocompetent Mouse Models (Syngeneic) | Murine CAR-T cells + murine tumors; intact immune system | Assessment of endogenous immune modulation, CRS modeling, TME interactions | Species-specific antigen differences; murine vs. human T cell biology disparities |
| Humanized Mouse Models (e.g., NSG with human immune system) | Human tumor + human CAR-T cells + human immune components | CRS/ICANS toxicity assessment, antigen spreading, tumor-immune ecosystem | Technical complexity, cost, variability in human immune reconstitution |
| Non-Human Primate (NHP) Models | Phylogenetically close to humans; similar immune biology | Safety/toxicology studies, pharmacokinetics, biodistribution | Extreme cost, ethical considerations, limited availability |
Standard Xenograft Model Protocol
Protocol: Antitumor Efficacy Assessment in Immunodeficient Mice
Principle: Establish human tumors in immunodeficient mice, administer CAR-T cells, and monitor tumor growth and animal survival to evaluate therapeutic potential.
Materials:
- Immunodeficient mice (NSG, NOG, or similar, 6-12 weeks old)
- Human tumor cell line (validated for TAA expression)
- CAR-T cells and appropriate control T cells
- Matrigel (for solid tumor models)
- IVIS imaging system (if using luciferase-expressing cells)
- Flow cytometer with human-specific antibodies
Procedure:
- Tumor Engraftment:
- Hematological Malignancies: Inject 0.5-5×10⁶ tumor cells intravenously via tail vein.
- Solid Tumors: Inject 1-5×10⁶ tumor cells subcutaneously in 50-100 μL PBS/Matrigel (1:1).
- Allow 7-14 days for tumor establishment (verify by bioluminescence, caliper measurement, or clinical signs).
Lymphodepletion (Optional):
- Administer cyclophosphamide (100-200 mg/kg) or irradiation (1-2 Gy) 1-2 days before CAR-T cell injection to enhance engraftment.
CAR-T Cell Administration:
- Inject 2-10×10⁶ CAR-T cells intravenously via tail vein in 100-200 μL PBS.
- Include control groups (untreated, mock T cells, non-transduced T cells).
Monitoring:
- Tumor Growth: Measure solid tumors 2-3 times weekly by caliper; monitor disseminated disease by bioluminescence weekly.
- CAR-T Cell Kinetics: Collect peripheral blood periodically (5-50 μL) for flow cytometry (human CD3+CAR+) or qPCR/ddPCR for CAR transgene.
- Clinical Observations: Monitor weight, activity, and signs of toxicity (e.g., hunched posture, piloerection) daily.
- Endpoint Analysis: Harvest tumors, organs for immunohistochemistry, and blood for complete analysis at study endpoint.
Technical Notes:
- For solid tumors, consider orthotopic implantation when possible for more relevant microenvironment.
- Include antigen-negative tumor control groups to assess on-target/off-tumor toxicity.
- For serial monitoring of CAR-T expansion, implement tail vein blood collection methods (10-50 μL weekly) with minimal animal distress.
Advanced In Vivo Modeling Considerations
Humanized Mouse Models for Toxicity Assessment: To evaluate cytokine release syndrome (CRS) and neurotoxicity (ICANS), humanized models incorporating human immune components provide valuable preclinical data:
- Human Hematopoietic Stem Cell Engraftment: Inject CD34+ human cord blood cells into conditioned neonatal or adult NSG mice.
- Tumor and CAR-T Cell Administration: After 12-16 weeks of immune reconstitution, establish tumors followed by CAR-T cells.
- Toxicity Monitoring: Measure human cytokines (IL-6, IFN-γ, IL-10) in serum, monitor body temperature, activity, and neurological symptoms.
Dual-Flank Tumor Models: To assess antigen escape and heterogeneous targeting:
- Implant two tumor types: One expressing primary target antigen, another expressing different or no antigen.
- Administer CAR-T cells and monitor differential tumor responses.
- Evaluate antigen loss in resistant tumors by flow cytometry or immunohistochemistry.
Analytical Methods for CAR-T Cell Monitoring
Accurate quantification of CAR-T cell expansion and persistence is critical for correlating cellular kinetics with efficacy and toxicity outcomes.
Flow Cytometry Detection
Protocol: Surface CAR Detection by Flow Cytometry
Principle: Use CAR-specific reagents (recombinant antigen, anti-idiotype antibodies, or Protein L) to detect CAR expression on transduced T cells.
Materials:
- Biotinylated target antigen or anti-CAR antibody
- Fluorescent streptavidin conjugates
- Anti-human CD3, CD4, CD8 antibodies
- Viability dye
- Flow cytometry staining buffer (PBS + 2% FBS)
Procedure:
- Cell Preparation: Wash cells twice with flow cytometry buffer.
- Surface Staining:
- Incubate cells with biotinylated antigen (1-5 μg/mL) or anti-CAR antibody for 30 minutes on ice.
- Wash twice.
- Incubate with fluorescent streptavidin and surface antibody cocktail for 20 minutes on ice.
- Wash twice and resuspend in buffer with viability dye.
- Acquisition and Analysis: Collect data on flow cytometer; analyze using appropriate gating strategy (live/singlets/CD3+/CAR+).
Technical Notes:
- Include untransduced T cells as negative control for gating.
- For intracellular cytokine staining, add protein transport inhibitor during stimulation, then perform fixation/permeabilization before cytokine antibody staining.
Molecular Monitoring Methods
Table 3: Comparison of CAR-T Cell Quantification Methods
| Method | Principle | Sensitivity | Key Applications | Advantages | Limitations |
|---|---|---|---|---|---|
| Flow Cytometry | Surface CAR detection with labeled antigens/antibodies | Moderate (0.1-1%) | Phenotype analysis, subpopulation characterization, protein expression | Multi-parameter, functional capacity, viable cells | Requires specific detection reagent, affected by CAR density |
| qPCR | CAR transgene amplification with reference gene normalization | High (0.01-0.1%) | Expansion kinetics, biodistribution, persistence | Sensitive, established protocols, quantitative | Affected by gDNA variability, relative quantification only |
| Digital Droplet PCR (ddPCR) | Partitioned PCR for absolute target quantification without standard curve | Very high (0.001-0.01%) | Precise kinetics, minimal residual disease detection, low-level persistence | Absolute quantification, high precision, resistant to PCR inhibitors | Specialized equipment, higher cost per sample |
| Volume-Based qPCR | Spike-in calibration with external control for blood volume normalization | High (0.01-0.1%) | Accurate pharmacokinetics in lymphodepleted hosts | Eliminates gDNA variability effects, true concentration in blood | More complex sample processing, validation required |
Protocol: Digital Droplet PCR for CAR Transgene Quantification
Principle: Partition PCR reaction into thousands of nanodroplets for absolute quantification of CAR transgene copies without standard curves.
Materials:
- ddPCR system (Bio-Rad QX200 or equivalent)
- CAR-specific primers and probe
- Genomic DNA extraction kit
- Restriction enzyme (optional, for complex gDNA)
- Droplet generation oil and DG8 cartridges
Procedure:
- DNA Extraction:
- Extract gDNA from blood or tissue samples using silica column method.
- Quantify DNA concentration and quality (A260/A280 ratio 1.8-2.0).
- Adjust to working concentration (10-100 ng/μL).
Droplet Generation:
- Prepare 20 μL reaction mix: 10 μL ddPCR Supermix, 1 μL each primer (900 nM final), 0.5 μL probe (250 nM final), 50-100 ng gDNA, nuclease-free water to volume.
- Load sample and oil into droplet generator cartridge.
- Process according to manufacturer protocol to generate ~20,000 droplets.
PCR Amplification:
- Transfer droplets to 96-well PCR plate.
- Seal plate and run thermal cycling: 95°C for 10 minutes; 40 cycles of 94°C for 30 seconds and 60°C for 60 seconds; 98°C for 10 minutes; 4°C hold.
- Use ramp rate of 2°C/second.
Droplet Reading and Analysis:
- Place plate in droplet reader.
- Analyze using manufacturer's software with amplitude threshold set based on negative controls.
- Calculate copies/μg gDNA: (Positive droplets × Dilution factor) / (DNA amount in μg × 0.00085*)
- *0.00085 represents the portion of the reaction volume (20 μL) occupied by 1 μg DNA
Technical Notes:
- For clinical correlation, studies show that patients with peak CAR transcript levels >5000 copies/μg gDNA ("good expanders") are more likely to achieve favorable responses and experience severe toxicity, supporting ddPCR's predictive value [75].
- Include no-template controls and positive controls (CAR plasmid) in each run.
- For longitudinal studies, use consistent DNA extraction methods and batch samples when possible.
Engineering Next-Generation CAR-T Cells with Enhanced Specificity
Beyond conventional CAR designs, emerging engineering strategies incorporate sophisticated regulatory mechanisms to enhance safety and efficacy, particularly for solid tumor applications.
Tumor Microenvironment-Gated Inducible CAR-T Cells
Protocol: Design of TME-Gated CAR-T Cells with Combinatorial Control
Principle: Engineer CAR-T cells requiring multiple tumor-specific signals for full activation, minimizing on-target/off-tumor toxicity while maintaining antitumor efficacy.
Design Strategy:
- Split CAR System:
- Separate CAR into two inactive subunits: extracellular antigen-binding domain (p1) and intracellular signaling domain (p2).
- Fuse each subunit to heterodimerizing plant-derived proteins (ABI and PYL).
TME-Activated Inducer:
- Design small molecule inducer (abscisic acid, ABA) conjugated to TME-sensitive moieties (e.g., nitroreductase-sensitive linkers).
- In hypoxic TME, nitroreductases cleave linker, releasing active ABA.
Combinatorial Activation:
- CAR activation requires: (1) Tumor antigen binding, (2) Free ABA from TME cleavage, (3) ABA-induced dimerization of split CAR subunits.
- Normal tissues lacking appropriate TME signals cannot activate CAR-T cells.
Experimental Validation:
- In Vitro Specificity Testing:
- Co-culture TME-iCAR-T cells with target cells under normoxic vs. hypoxic conditions.
- Measure cytokine production and cytotoxicity only in the presence of both antigen and hypoxia.
- Confirm lack of activation against antigen-positive normal cells under normoxia.
- In Vivo Efficacy and Safety:
- Establish dual-flank tumors with antigen-positive cells.
- Administer TME-iCAR-T cells and monitor tumor-specific control.
- Assess off-tumor toxicity in normal antigen-expressing tissues.
Technical Notes:
- This approach demonstrates how synthetic biology principles can be applied to enhance the therapeutic window of CAR-T cells for solid tumors [76].
- Alternative TME signals include overexpressed proteases (MMP-cleavable linkers) or abnormal metabolites.
Research Reagent Solutions
Table 4: Essential Research Reagents for CAR-T Cell Preclinical Evaluation
| Reagent Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| CAR Detection Reagents | Biotinylated recombinant antigens, anti-idiotype antibodies, Protein L | Flow cytometry monitoring, CAR expression validation | Specificity validation required; consider affinity and staining conditions |
| TAA-Expressing Cell Lines | Nalm-6 (leukemia), K562 (CML), Raji (lymphoma), solid tumor lines engineered with TAAs | Standardized cytotoxicity assays, antigen-specific activation | Engineered lines should be validated for consistent, stable antigen expression |
| Cytokine Analysis Panels | Multiplex arrays for IFN-γ, IL-2, IL-6, TNF-α, IL-10, granzyme B | Functional potency assessment, toxicity prediction | Match sensitivity to expected ranges; include both stimulatory and inflammatory cytokines |
| T Cell Phenotype Antibodies | CD62L, CCR7, CD45RA/RO (memory), PD-1, TIM-3, LAG-3 (exhaustion), CD69 (activation) | Differentiation status, functional capacity, persistence potential | Multi-panel design requires compensation controls; validate clone specificity |
| Molecular Quantification Reagents | CAR-specific primers/probes, gDNA extraction kits, reference gene assays | Cellular kinetics, biodistribution, persistence monitoring | Validate primer specificity; optimize extraction efficiency; select stable reference genes |
| In Vivo Model Components | Immunodeficient mice (NSG, NOG), luciferase-expressing tumor cells, Matrigel | Preclinical efficacy, toxicity, biodistribution studies | Follow ethical guidelines; monitor engraftment; consider orthotopic models for relevance |
Visualization of CAR-T Cell Signaling and Experimental Workflows
CAR-T Cell Activation Signaling Pathway: This diagram illustrates the sequential signaling events following CAR engagement with tumor-associated antigen (TAA), leading to diverse functional outcomes including both therapeutic effector responses and potential exhaustion pathways.
Preclinical Validation Workflow: Comprehensive pathway for CAR-T cell validation integrating in vitro functional screens with in vivo efficacy and safety assessments, supported by robust analytical methodologies throughout the development process.
Chimeric Antigen Receptor (CAR) T-cell therapy has revolutionized cancer treatment, particularly for hematologic malignancies. While second-generation CARs form the backbone of currently approved therapies, challenges such as on-target/off-tumor toxicity, immunosuppressive tumor microenvironments (TME), and antigen heterogeneity have limited their efficacy, especially in solid tumors [77] [76] [52]. This has driven the development of advanced CAR platforms with enhanced specificity and functionality. This application note provides a comparative analysis and detailed protocols for three innovative platforms: armored CARs, logic-gated CARs, and TME-gated CARs, providing researchers with practical methodologies for their implementation in cancer immunotherapy research.
Technology Classifications and Definitions
Armored CARs: Second-generation CARs engineered to secrete cytokines or express additional stimulatory ligands to enhance persistence and counteract immunosuppressive TMEs [77] [78]. Also classified as fourth-generation CARs or TRUCKs (T cells Redirected for Universal Cytokine-mediated Killing) [79] [52].
Logic-Gated CARs: CAR systems incorporating Boolean logic operations (AND, OR, NOT) requiring multiple antigen recognition events for T-cell activation, thereby improving tumor specificity [77] [80].
TME-Gated CARs: Inducible CAR systems activated by tumor microenvironment-specific signals (e.g., hypoxia, enzymes) combined with tumor antigens, providing spatial control of T-cell activity [76].
Quantitative Comparison of Platform Characteristics
Table 1: Comparative Analysis of Advanced CAR-T Platforms
| Platform Feature | Armored CARs | Logic-Gated CARs | TME-Gated CARs |
|---|---|---|---|
| Primary Mechanism | Cytokine secretion (e.g., IL-18) or co-stimulatory ligand expression [78] | Boolean antigen recognition via split signaling or inhibitory receptors [77] [80] | TME signal (e.g., hypoxia) + antigen via small molecule inducers [76] |
| Key Advantage | Enhanced persistence & tumor control [78] | Improved specificity; reduced on-target/off-tumor toxicity [77] | Spatial restriction to tumor sites [76] |
| Clinical Status | Phase 1 trials (e.g., huCART19-IL18) [78] | Phase 1/2 trials (e.g., A2B530, IMPT-314) [77] | Preclinical development [76] |
| Manufacturing Complexity | Moderate (single CAR construct) [78] | High (multiple receptors requiring stoichiometric expression) [77] | High (inducible system + prodrug administration) [76] |
| Therapeutic Index | Moderate improvement | High improvement | Potentially high improvement |
| Best Application | Hostile TME; post-CAR-T failure [78] | Tumors with heterogeneous antigen expression [77] | Solid tumors with distinct TME signatures [76] |
Signaling Pathways and Engineering Approaches
Armored CAR Signaling and Enhancement Mechanisms
Diagram 1: Armored CAR (huCART19-IL18) enhanced activation mechanism.
Logic-Gated CAR Circuit Designs
Diagram 2: Logic-gated CAR Boolean recognition circuits.
TME-Gated Inducible Activation System
Diagram 3: TME-gated inducible CAR activation mechanism.
Experimental Protocols
Protocol 1: Armored CAR (huCART19-IL18) Functional Validation
Objective: Evaluate enhanced anti-tumor efficacy and cytokine-mediated bystander activation of IL-18-secreting armored CAR-T cells.
Materials:
- huCART19-IL18 construct (CD19-scFv-4-1BB-CD3ζ with IL-18 expression cassette)
- Primary human T-cells from healthy donors
- CD19+ tumor cell lines (e.g., Nalm6, Raji)
- Lymphodepletion chemotherapy agents (fludarabine, cyclophosphamide)
- IL-18 ELISA kit
- Flow cytometry antibodies (CD3, CD4, CD8, CD19, CD107a, IFN-γ, granzyme B)
Methodology:
- CAR T-cell Manufacturing:
- Isolate PBMCs from healthy donor blood using Ficoll density gradient centrifugation
- Activate T-cells with anti-CD3/CD28 beads for 24 hours
- Transduce with lentiviral vector encoding huCART19-IL18 at MOI 10-20
- Expand cells in complete media with IL-2 (100 IU/mL) for 10-14 days
- Validate CAR expression by flow cytometry and IL-18 secretion by ELISA
In Vitro Cytotoxicity Assay:
- Co-culture CAR-T cells with CD19+ tumor cells at E:T ratios (1:1 to 20:1) for 24-48 hours
- Measure specific lysis using flow cytometry-based killing assay:
- Label target cells with CellTrace Violet
- Add 7-AAD after co-culture to distinguish live/dead cells
- Analyze by flow cytometry; calculate % specific lysis = (% dead target - % spontaneous death) / (100% - % spontaneous death) × 100
- Assess activation markers (CD107a) and cytokine production (IFN-γ) by intracellular staining
In Vivo Efficacy Study:
- Utilize NSG mice engrafted with CD19+ human tumor cells (1×10^6 cells, IV)
- After 7 days, lymphodeplete with fludarabine (25 mg/kg) and cyclophosphamide (25 mg/kg) for 2 days
- Administer CAR-T cells (1-5×10^6 cells/mouse, IV)
- Monitor tumor burden weekly by bioluminescence imaging
- Assess CAR-T persistence in blood and tissues by flow cytometry
- Evaluate toxicity via body weight tracking and clinical scoring [78]
Protocol 2: AND-Gated LINK CAR Functional Characterization
Objective: Validate Boolean logic AND-gated tumor recognition using the LINK CAR system to prevent on-target/off-tumor toxicity.
Materials:
- LINK CAR constructs (LAT-based CAR-A, SLP-76-based CAR-B)
- Tumor cell lines with defined antigen expression profiles
- Primary human T-cells from healthy donors
- CRISPR-Cas9 system for endogenous TCR knockout
- Phospho-flow antibodies (pZAP-70, pERK, pS6)
Methodology:
- LINK CAR Engineering:
- Design CAR-A: scFv-A-CD8TM-LATICD
- Design CAR-B: scFv-B-CD8TM-SLP76ICD
- Clone into separate lentiviral vectors with different selection markers
- Transduce primary T-cells sequentially or simultaneously
- Sort double-positive population by FACS
Selective Activation Profiling:
- Co-culture LINK CAR-T cells with target cells expressing:
- Antigen A only
- Antigen B only
- Both antigens A and B
- Neither antigen
- Measure early activation (CD69, CD25) at 6 hours by flow cytometry
- Assess cytokine production (IL-2, IFN-γ, TNF-α) after 24 hours by multiplex ELISA
- Evaluate phosphorylation of signaling molecules (ZAP-70, ERK) by phospho-flow cytometry
- Co-culture LINK CAR-T cells with target cells expressing:
Precision Cytotoxicity Assessment:
- Set up 3D co-culture with antigen-heterogeneous tumor spheroids
- Monitor real-time killing using impedance-based systems (e.g., xCelligence)
- Compare killing efficiency against mixed populations of target and non-target cells
- Validate specificity in humanized mouse models with mixed tumor implants [80]
Protocol 3: TME-Gated Inducible CAR (TME-iCAR) Activation Profiling
Objective: Characterize hypoxia-dependent CAR activation using engineered ABA-inducible system.
Materials:
- Split CAR constructs (p1: ABI-fused, p2: PYL/PYR-fused)
- Hypoxia-activated ABA prodrugs (nitroimidazole or nitrobenzyl conjugates)
- Hypoxia chamber (1% O₂)
- Primary human T-cells
- HPLC system for prodrug activation analysis
Methodology:
- Split CAR Assembly Validation:
- Transduce T-cells with both split CAR components
- Verify surface expression of both parts by flow cytometry
- Test CAR assembly with active ABA (positive control)
Hypoxia-Dependent Activation:
- Culture TME-iCAR T-cells with target cells under:
- Normoxia (21% O₂) + ABA prodrug
- Hypoxia (1% O₂) + ABA prodrug
- Hypoxia without prodrug
- Normoxia with active ABA (control)
- Incubate for 24-48 hours
- Measure T-cell activation (CD69, CD25) by flow cytometry
- Quantify cytokine production (IL-2, IFN-γ) by ELISA
- Culture TME-iCAR T-cells with target cells under:
Prodrug Activation Kinetics:
- Analyze ABA release from prodrugs using HPLC:
- Incubate prodrugs in PBS under normoxia or hypoxia
- Collect samples at 0, 2, 4, 8, 12, 24, 48 hours
- Separate using reverse-phase C18 column
- Quantify free ABA by comparison to standard curve
- Correlate ABA concentration with CAR-T activation threshold [76]
- Analyze ABA release from prodrugs using HPLC:
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for Advanced CAR-T Development
| Reagent/Category | Specific Examples | Research Function | Considerations |
|---|---|---|---|
| Signaling Domain Variants | ZAP-70, PLCγ1 CAR endodomains [80] | Replace CD3ζ to reduce tonic signaling & enable logic gating | Alters activation threshold; requires validation |
| Inducible Dimerization Systems | ABA-CIP system [76] | Controlled CAR assembly via small molecule inducers | Prodrug design critical for TME specificity |
| Cytokine Payloads | IL-18, IL-12 [77] [78] | Armor CARs to enhance persistence & remodel TME | Monitor for potential systemic toxicity |
| Inhibitory CAR (iCAR) Domains | PD-1, CTLA-4 intracellular domains [77] | Implement NOT gate logic for safety | Balance inhibition strength to maintain efficacy |
| Hypoxia-Activated Prodrugs | Nitroimidazole-ABA, Nitrobenzyl-ABA [76] | Activate CARs specifically in hypoxic TME | Optimize reduction potential for tumor specificity |
| Advanced Cytotoxicity Assays | Impedance-based, Flow cytometry killing assays [81] | Measure real-time & antigen-specific killing | Choose based on throughput and resolution needs |
The advanced CAR platforms detailed herein represent the forefront of T-cell engineering, each addressing distinct challenges in cancer immunotherapy. Armored CARs enhance persistence and overcome immunosuppressive environments, showing particular promise in patients with prior CAR-T treatment failure [78]. Logic-gated CARs employ Boolean recognition circuits to dramatically improve tumor specificity, with several candidates now in clinical trials [77] [80]. TME-gated CARs represent an emerging approach that restricts activation to tumor sites using microenvironmental cues, offering potential for solid tumor applications [76].
The experimental protocols provided enable researchers to functionally validate these sophisticated systems, with particular attention to precision activation controls and comprehensive safety profiling. As these platforms mature, their integration with emerging technologies like in vivo CAR delivery and computational modeling of CAR-T dynamics will further accelerate the development of safer, more effective cellular therapies for cancer [79] [82].
Evaluating Allogeneic 'Off-the-Shelf' CAR-T Cells vs. Autologous Products
Chimeric Antigen Receptor T-cell (CAR-T) therapy represents a groundbreaking advancement in cancer immunotherapy, leveraging genetically engineered T-cells to target and eliminate malignant cells [52]. This field is primarily dominated by two distinct approaches: autologous therapy, which utilizes the patient's own T-cells, and allogeneic therapy, which employs T-cells from healthy donors to create "off-the-shelf" products [83] [84]. Autologous CAR-T products have demonstrated remarkable success in treating hematologic malignancies, with six FDA-approved products currently available [85] [52]. These therapies have achieved response rates of 35-40% in patients with relapsed or refractory disease, establishing a new standard of care for conditions such as B-cell acute lymphoblastic leukemia and large B-cell lymphoma [85] [86].
The fundamental distinction between these modalities lies in their cell sourcing and manufacturing paradigms. Autologous CAR-T therapy involves a personalized manufacturing process where T-cells are collected from the patient via leukapheresis, genetically modified to express CARs targeting tumor-associated antigens (such as CD19 or BCMA), expanded ex vivo, and then reinfused into the same patient [83] [85]. This personalized approach minimizes immunologic incompatibility but faces challenges related to manufacturing complexity and time constraints. In contrast, allogeneic CAR-T therapy sources T-cells from healthy donors, enabling large-scale production of cryopreserved, readily available doses that can treat multiple patients [87] [85]. This "off-the-shelf" approach requires sophisticated genetic engineering to mitigate immune-mediated rejection and graft-versus-host disease (GVHD), typically through gene editing technologies that disrupt T-cell receptor (TCR) signaling and human leukocyte antigen (HLA) expression [85] [86].
Table 1: Core Characteristics of Autologous vs. Allogeneic CAR-T Therapies
| Parameter | Autologous CAR-T | Allogeneic CAR-T |
|---|---|---|
| Cell Source | Patient's own T-cells | Healthy donor T-cells |
| Manufacturing Timeline | 3-4 weeks [85] | 4 days from enrollment to therapy [88] |
| Key Advantages | Minimal risk of immunologic rejection and GVHD [85] | Immediate availability, standardized product quality, scalable production [87] [85] |
| Major Challenges | Manufacturing delays, variable T-cell quality, high cost [85] [84] | Risk of GVHD and host-versus-graft rejection, potential need for repeat dosing [89] [85] |
| Primary Genetic Modifications | CAR gene insertion via viral vectors [52] | CAR insertion plus TCR ablation and often HLA modification [85] [86] |
| Clinical Status | Multiple FDA-approved products [52] | Investigational (Phase 1/2 trials) [88] |
Comparative Performance and Clinical Metrics
When evaluating autologous versus allogeneic CAR-T products, critical performance metrics include efficacy, durability, safety profiles, and manufacturing considerations. Autologous CAR-T therapies have established impressive clinical benchmarks, with products like axicabtagene ciloleucel (Yescarta) and brexucabtagene autoleucel (Tecartus) demonstrating robust response rates in B-cell malignancies [52]. These therapies benefit from using patient-derived T-cells that naturally persist in the recipient's immune system, potentially providing long-term surveillance against disease recurrence [89]. However, their personalized nature introduces significant variability in product quality and potency, particularly because cancer patients often have T-cells compromised by prior therapies [85].
Allogeneic CAR-T products aim to overcome these limitations by utilizing healthy donor cells that typically exhibit superior expansion capacity and functionality. Recent clinical data from investigational allogeneic products show promising efficacy, with the anti-CD70 allogeneic CAR-T candidate ALLO-316 demonstrating a 31% confirmed response rate in patients with advanced renal cell carcinoma who had exhausted standard treatment options [88]. Notably, four of five confirmed responders maintained ongoing responses, with one patient in remission for over 12 months, suggesting the potential for durable activity despite concerns about allogeneic cell persistence [88]. The median duration of response has not yet been reached in this trial, indicating sustained disease control in responding patients [88].
Table 2: Clinical Performance and Manufacturing Metrics
| Metric | Autologous CAR-T | Allogeneic CAR-T |
|---|---|---|
| Manufacturing Failure Rate | 2-10% [85] | Not fully characterized |
| Durability Concerns | T-cell exhaustion from chronic antigen exposure [85] | Host-versus-graft rejection potentially limiting persistence [89] [85] |
| Response Rates in Hematologic Malignancies | Established efficacy with multiple approved products [52] | Emerging data showing promise in early-phase trials [83] [88] |
| Solid Tumor Applications | Limited success to date [52] | Emerging activity (e.g., 31% ORR in RCC with CD70 targeting) [88] |
| Characteristic Safety Profile | CRS, ICANS, cytopenias [85] [52] | CRS, ICANS, plus risk of GVHD and unique toxicities like IEC-HS [88] |
| Production Scalability | Limited by patient-specific manufacturing [84] | High potential for scale using master cell banks [87] [85] |
From a safety perspective, both approaches share class-effect toxicities including cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) [85] [52]. However, allogeneic CAR-T products introduce additional risks, particularly graft-versus-host disease (GVHD), where donor T-cells attack host tissues [85]. Recent clinical data with ALLO-316 reported no GVHD cases, suggesting that modern gene-editing approaches can effectively mitigate this risk [88]. Allogeneic products may also trigger host-versus-graft responses, where the recipient's immune system recognizes and eliminates the donor-derived cells, potentially limiting their persistence and efficacy [89] [85]. Additionally, allogeneic CAR-T therapies have been associated with emerging toxicities such as immune effector cell-associated hemophagocytic lymphohistiocytosis-like syndrome (IEC-HS), which occurred in 36% of patients receiving ALLO-316, with 9% experiencing grade 3-4 events [88].
Engineering and Manufacturing Protocols
Autologous CAR-T Manufacturing Workflow
The production of autologous CAR-T cells follows a standardized multi-step process that begins with leukapheresis to collect peripheral blood mononuclear cells (PBMCs) from the patient [85]. This initial step is particularly challenging for heavily pretreated patients who may have lymphopenia or functionally impaired T-cells due to prior therapies. Following collection, T-cells are isolated and activated using methods such as anti-CD3/CD28 magnetic beads or recombinant cytokines. The critical genetic modification step involves transducing activated T-cells with viral vectors (typically lentiviral or gamma-retroviral) encoding the CAR construct [85] [52]. Most FDA-approved autologous products utilize second-generation CAR designs incorporating either CD28 or 4-1BB costimulatory domains alongside the CD3ζ activation domain [52].
Following transduction, CAR-T cells undergo ex vivo expansion in bioreactor systems, typically over 7-10 days, to achieve therapeutically relevant cell doses (ranging from 10^8 to 10^9 cells) [85]. Throughout this process, quality control testing monitors vector copy number, CAR expression, sterility, and potency. The final formulated product is cryopreserved and shipped back to the treatment center, where it is thawed and infused into the patient after lymphodepleting chemotherapy [85]. The entire manufacturing timeline typically spans 3-4 weeks, during which patients with aggressive malignancies may require bridging therapy to control disease progression [85]. Recent innovations have aimed to accelerate this process, with some platforms achieving manufacturing turnaround in 7 days or less while maintaining product quality and efficacy [90].
Allogeneic CAR-T Manufacturing and Engineering Protocols
Allogeneic CAR-T manufacturing shares similarities with autologous approaches but incorporates additional genetic modifications to enable universal application. The process begins with PBMC collection from carefully screened healthy donors, providing a starting population of robust, therapy-naïve T-cells [85]. To mitigate the risk of GVHD, the T-cell receptor alpha constant (TRAC) locus is disrupted using gene-editing technologies such as CRISPR/Cas9, TALENs, or zinc finger nucleases [85] [86]. This critical step eliminates surface expression of the endogenous TCR, preventing alloreactive responses against host tissues. Additional edits may include knockout of HLA class I molecules to evade host CD8+ T-cell recognition and/or incorporation of "stealth" modifications such as overexpression of HLA-G or PD-L1 to enhance persistence [85].
Following gene editing, the manufacturing process parallels autologous approaches with CAR transduction, expansion, and formulation. However, allogeneic platforms enable large-scale production runs yielding hundreds to thousands of doses from a single donor collection [87] [85]. This batch processing facilitates rigorous quality control and product characterization, including confirmation of editing efficiency, CAR expression, and functional potency. The resulting products are cryopreserved as off-the-shelf inventories, available for immediate use when a compatible patient is identified [85]. Alternative cell sources for allogeneic platforms include umbilical cord blood (UCB) and induced pluripotent stem cells (iPSCs) [85] [86]. UCB-derived T-cells offer inherent advantages including antigen-naïvety, reduced alloreactivity, and lower expression of exhaustion markers, while iPSC platforms enable unlimited expansion potential and precise genetic engineering [85].
Protocol for Assessing Allogeneic CAR-T Cell Function and Persistence
Robust characterization of allogeneic CAR-T products requires specialized assays to confirm editing efficiency, function, and potential for alloreactivity. The following protocol outlines key experiments for evaluating investigational allogeneic CAR-T products:
TCR Disruption Confirmation: Flow cytometric analysis using anti-TCRαβ antibodies should demonstrate >95% reduction in surface TCR expression compared to unedited controls [85]. Additional genomic confirmation via next-generation sequencing of the TRAC locus validates editing efficiency and identifies potential chromosomal abnormalities [89].
GVHD Assessment In Vitro: Co-culture allogeneic CAR-T cells with HLA-mismatched PBMCs or primary hematopoietic cells for 72-96 hours. Measure T-cell activation (CD69, CD25), cytokine production (IFN-γ, IL-2), and target cell killing via flow cytometry and luminescence-based cytotoxicity assays [85]. Effective allogeneic CAR-T products should demonstrate significantly reduced alloreactivity compared to unedited controls.
Host-Versus-Graft Response Modeling: Inject allogeneic CAR-T cells into immunocompetent mouse models with partial HLA matching. Monitor CAR-T cell persistence weekly via bioluminescence imaging or flow cytometry of peripheral blood [85]. Compare persistence against autologous CAR-T controls to quantify immune-mediated rejection.
Tumor Killing Efficacy: Evaluate antitumor potency in established xenograft models of hematologic malignancies or solid tumors. Administer allogeneic CAR-T cells following lymphodepletion and monitor tumor burden via bioluminescence or caliper measurements [88]. Include comparator arms with autologous CAR-T products to benchmark efficacy.
Exhaustion Marker Profiling: After repeated antigen stimulation, assess expression of inhibitory receptors (PD-1, TIM-3, LAG-3) and functional capacity via cytokine production and killing assays [89] [85]. Allogeneic products from healthy donors typically exhibit lower exhaustion profiles than autologous products from patients.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Allogeneic CAR-T Development
| Reagent/Category | Function | Examples/Applications |
|---|---|---|
| Gene Editing Systems | TCR disruption and HLA modification | CRISPR/Cas9, TALENs, ZFNs for TRAC locus editing [85] [86] |
| Viral Vectors | CAR gene delivery | Lentiviral, retroviral vectors for stable CAR integration [85] [52] |
| Non-Viral Delivery Systems | CAR gene insertion | Transposon systems (Sleeping Beauty, PiggyBac), mRNA electroporation [85] |
| T-cell Activation Reagents | Ex vivo T-cell stimulation | Anti-CD3/CD28 antibodies, cytokine cocktails (IL-2, IL-7, IL-15) [85] |
| Cell Culture Media | T-cell expansion and maintenance | Serum-free media formulations with optimized nutrients and cytokines [85] |
| Flow Cytometry Antibodies | Phenotypic characterization | Anti-CAR detection reagents, TCRαβ, exhaustion markers (PD-1, LAG-3, TIM-3) [85] |
| Cytokine Detection Assays | Functional assessment | Multiplex immunoassays for IFN-γ, IL-2, IL-6 to quantify activation and CRS potential [85] [52] |
Critical Challenges and Engineering Solutions
Addressing Allogeneic CAR-T Limitations
The development of effective allogeneic CAR-T therapies faces three primary biological challenges: graft-versus-host disease (GVHD), host-versus-graft (HvG) rejection, and limited persistence [85]. GVHD occurs when donor T-cells recognize host tissues as foreign through their endogenous TCRs. The predominant solution involves complete ablation of TCR expression through gene editing of the TRAC locus [85] [86]. Modern approaches using CRISPR/Cas9 or TALENs achieve >95% TCR knockout efficiency, effectively preventing GVHD in preclinical models and early clinical trials [85] [88].
HvG rejection remains a more complex challenge, as recipient immune cells can recognize allogeneic CAR-T cells through multiple pathways, including HLA mismatches [85]. Strategies to circumvent rejection include: (1) Knockout of β-2-microglobulin to eliminate surface HLA class I expression, preventing CD8+ T-cell recognition; (2) Overexpression of non-classical HLA molecules (e.g., HLA-E, HLA-G) that inhibit NK cell and T-cell responses; (3) Incorporation of "self" markers such as CD47 to evade phagocytic clearance [85]. Additionally, selecting HLA-matched donors from pre-established cell banks can minimize immunogenic disparities [85].
Limited persistence of allogeneic CAR-T cells compared to autologous products represents another significant hurdle [89]. This reduced durability may necessitate repeat dosing, potentially increasing treatment costs and complexity. Engineering approaches to enhance persistence include: (1) Knock-in of cytokines (IL-7, IL-15) or cytokine receptors; (2) Dominant-negative TGF-β receptors to overcome immunosuppressive environments; (3) "Safety switches" (e.g., caspase-9-based) to address toxicity concerns that might otherwise limit dose escalation [85].
Emerging Platforms and Future Directions
The allogeneic CAR-T landscape is rapidly evolving with several innovative platforms showing promise. Induced pluripotent stem cell (iPSC)-derived CAR-T products offer potentially unlimited expansion capacity and precise genetic engineering [85] [86]. These platforms enable the generation of master cell lines with multiple engineered features, including enhanced trafficking, resistance to exhaustion, and tissue-specific targeting capabilities. Similarly, cord blood-derived CAR-NK cells provide an alternative allogeneic approach with favorable safety profiles and inherent anti-tumor activity [87] [91].
The future of allogeneic CAR-T therapy will likely involve more sophisticated multi-gene engineering approaches to create increasingly sophisticated products. Key directions include: (1) Logic-gated CAR systems that require multiple antigens for activation, improving tumor specificity; (2) Synthetic cytokine receptors that convert immunosuppressive signals into activating ones; (3) Precision gene editing to knock CARs into endogenous loci (e.g., TRAC, PDCD1) for more physiological expression [52]. Additionally, the application of allogeneic platforms to autoimmune diseases represents an emerging frontier, with early clinical trials exploring CAR-Tregs and CAR-T cells targeting B-cell maturation antigen (BCMA) in conditions like lupus and myasthenia gravis [91].
As these technologies mature, the field is likely to witness a convergence of autologous and allogeneic approaches, with each modality finding its optimal therapeutic niche based on disease indication, patient population, and healthcare setting [84].
Chimeric Antigen Receptor (CAR) T-cell therapy has revolutionized the treatment of hematological malignancies, yet its application, particularly in solid tumors, faces persistent challenges. These limitations include poor intra-tumoral infiltration, an immunosuppressive tumor microenvironment (TME), treatment-related toxicities such as cytokine release syndrome (CRS), and complex, costly manufacturing processes [92]. Consequently, the field is rapidly expanding beyond T-cells to harness the unique biological functions of other immune cell populations. CAR-Natural Killer (CAR-NK), CAR-Macrophage (CAR-M), and CAR-Dendritic Cell (CAR-DC) therapies represent the vanguard of this expansion [93]. These emerging modalities leverage innate immune mechanisms—such as MHC-independent cytotoxicity, phagocytic clearance, and professional antigen presentation—to overcome the barriers that have hindered CAR-T cells. This application note provides a detailed comparison of these platforms, summarizes key quantitative data, and outlines foundational experimental protocols to support research and development in next-generation CAR cell therapies.
Table 1: Comparative Overview of Emerging CAR Cell Therapies
| Feature | CAR-NK | CAR-M | CAR-DC |
|---|---|---|---|
| Core Mechanistic Advantage | Innate, MHC-independent cytotoxicity; multiple activating receptors [92] | Phagocytosis; antigen presentation; TME remodeling [92] | Priming of naive T-cells; orchestration of adaptive immunity [93] |
| Key Target Antigens (Examples) | HER2, MUC1, CD19 [92] | Ongoing research for solid tumor targets [94] | Ongoing research |
| "Off-the-Shelf" Potential | High (Allogeneic use feasible due to low risk of GVHD) [92] | Under investigation [92] | Under investigation |
| Major Safety Advantage | Lower incidence of severe CRS and ICANS [93] | Lower incidence of severe CRS [92] | Data currently limited |
| Persistence | Can be limited (may require engineering) [92] | Can be limited (may require engineering) [94] | Data currently limited |
| Key Challenges | Limited persistence; host immune rejection; antigen heterogeneity [92] [93] | Macrophage plasticity (M1/M2 polarization); short persistence; genetic modification difficulties [92] [94] | Insufficient tissue infiltration; low transduction efficiency [93] |
CAR-NK Cell Therapy
Mechanism and Current Status
CAR-NK cell therapy involves genetically modifying natural killer cells to express synthetic receptors for tumor-specific antigens. A key advantage is their ability to mediate cytotoxicity through MHC-independent mechanisms, allowing them to target malignant cells without prior antigen sensitization [92]. Furthermore, CAR-NK cells can eliminate target cells through native activating receptors and antibody-dependent cellular cytotoxicity (ADCC) via CD16, providing complementary tumor-killing pathways even if antigen escape occurs [95]. Their inherent biology confers a more favorable safety profile, with a reduced risk of severe CRS and neurotoxicity compared to CAR-T cells, and enables the development of "off-the-shelf" allogeneic products from sources like peripheral blood, umbilical cord blood, and stem cells [92] [93].
Detailed Experimental Protocol: CAR-NK Cell Generation
Objective: To generate functional human CAR-NK cells from primary peripheral blood mononuclear cells (PBMCs) using viral transduction. Key Reagent Solutions:
- NK Cell Media: ImmunoCult-XF T Cell Expansion Medium, supplemented with IL-2 (100 IU/mL), IL-15 (10 ng/mL), and IL-21 (10 ng/mL) [95].
- Activation Reagent: Soluble or immobilized antibodies for activation (e.g., anti-CD2, anti-CD3, anti-CD16) [95].
- Genetic Engineering Vector: Lentiviral vector containing the CAR construct under the control of an NK-specific promoter (e.g., EF1α or an endogenous promoter like NKp46 for enhanced specificity) [96].
- Cytotoxicity Assay Reagents: Calcein-AM or LDH-based kits for measuring target cell lysis.
Procedure:
- NK Cell Isolation: Isolate PBMCs from a leukapheresis product or buffy coat using Ficoll density gradient centrifugation. Isolate NK cells by negative selection using a clinical-grade NK cell isolation kit. Assess cell purity via flow cytometry (CD56+/CD3-).
- Activation and Expansion: Resuspend NK cells in pre-warmed NK Cell Media at 1-2x10^6 cells/mL. Add the chosen activation reagent. Culture cells in a humidified incubator at 37°C and 5% CO2 for 48-72 hours prior to transduction.
- Viral Transduction: On day 2-3 of culture, transduce activated NK cells with lentiviral vectors at a pre-optimized Multiplicity of Infection (MOI). Centrifuge plates (e.g., 2000 x g for 90 minutes at 32°C) to enhance transduction efficiency. Return cells to the incubator.
- Expansion and Culture: Replace media every 2-3 days, maintaining cell density between 1-2x10^6 cells/mL. Continue culture for 10-14 days, monitoring cell count and viability.
- Functional Validation:
- CAR Expression: Confirm CAR surface expression by flow cytometry 5-7 days post-transduction using a F(ab')2 anti-mouse IgG antibody or a target antigen-Fc fusion protein.
- Cytotoxicity Assay: Co-culture CAR-NK cells with radiolabeled or Calcein-AM-stained target cells (positive and negative for the target antigen) at various Effector:Target (E:T) ratios for 4-6 hours. Measure specific lysis. A well-designed experiment should show specific lysis of antigen-positive cells only.
- Cytokine Release: Measure IFN-γ and GM-CSF in the co-culture supernatant using ELISA to assess functional activation.
CAR-M Cell Therapy
Mechanism and Current Status
CAR-M therapy leverages the innate phagocytic capacity and immunomodulatory functions of macrophages to address solid tumors [92]. Engineered CAR-Ms directly engulf and clear malignant cells and play a critical role in remodeling the immunosuppressive TME. They secrete pro-inflammatory cytokines (e.g., IL-12, TNF-α) and degrade stromal barriers like the fibrotic extracellular matrix, thereby enhancing the infiltration of other immune cells [92] [94]. Furthermore, CAR-Ms function as potent antigen-presenting cells, phagocytosing tumor material and cross-presenting antigens to T cells to initiate and sustain adaptive antitumor immunity [92]. The primary challenges include the plasticity of macrophages, which can adopt a pro-tumor (M2) phenotype in the TME, and their typically short persistence in vivo [94].
Detailed Experimental Protocol: CAR-M Cell Generation
Objective: To generate human CAR-M cells from primary monocyte-derived macrophages using lentiviral transduction. Key Reagent Solutions:
- Macrophage Differentiation Media: RPMI-1640 supplemented with 10% heat-inactivated human AB serum, 1% GlutaMAX, 1% HEPES, and 50 ng/mL recombinant Human M-CSF. (Use of human serum instead of FBS is recommended for clinical translation).
- Genetic Engineering Vector: Lentiviral vector with a CAR construct driven by a constitutive promoter (e.g., MND or PGK).
- Polarization Cytokines: IFN-γ (20 ng/mL) and LPS (100 ng/mL) for M1 polarization; IL-4 (20 ng/mL) and IL-13 (20 ng/mL) for M2 polarization.
- Phagocytosis Assay Reagents: pHrodo Red-labeled target cells or fluorescent beads.
Procedure:
- Monocyte Isolation: Isulate PBMCs and enrich CD14+ monocytes using positive selection with clinical-grade CD14 microbeads.
- Macrophage Differentiation: Culture isolated monocytes in Macrophage Differentiation Media for 5-7 days to allow differentiation into macrophages (M0). Refresh media with fresh M-CSF every 2-3 days.
- Viral Transduction: On day 3-5 of differentiation, transduce macrophages with lentiviral vectors. A spinoculation protocol is recommended. Polybrene (4-8 µg/mL) can be added to enhance transduction.
- Phenotype and Functional Validation:
- CAR Expression: Confirm CAR expression by flow cytometry or immunofluorescence 5-7 days post-transduction.
- Phagocytosis Assay: Co-culture CAR-M cells with pHrodo Red-labeled target cells (antigen-positive vs. antigen-negative) for 2-4 hours. pHrodo fluorescence increases dramatically in acidic phagolysosomes. Quantify phagocytosis by flow cytometry or fluorescence microscopy.
- Phenotyping: Characterize macrophage polarization by measuring surface markers (e.g., CD80, CD86 for M1; CD206, CD163 for M2) via flow cytometry and by analyzing cytokine secretion profiles (e.g., IL-12, IL-10) using multiplex ELISA.
- In Vivo Assessment: Evaluate tumor infiltration and TME remodeling in immunodeficient mouse models of solid tumors. Analyze tumor sections for human macrophage markers and metrics of immune cell infiltration.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for CAR-NK and CAR-M Research
| Reagent Category | Specific Examples | Function & Importance | Considerations for Protocol Development |
|---|---|---|---|
| Culture Media | ImmunoCult-XF, TheraPEAK T-VIVO, RPMI-1640 + Human AB Serum [38] | Supports viability, expansion, and maintains functional phenotype during ex vivo culture. | Media choice significantly impacts expansion rates and final cell viability. X-VIVO and StemSpan are also common, serum-free options [38]. |
| Cell Activation | Immobilized anti-CD3/anti-CD28, soluble anti-CD2/anti-CD3/anti-CD28, M-CSF [38] [94] | Primes cells for genetic modification and induces proliferation. | Activation method affects surface marker internalization (e.g., CD3) and T cell subset distribution [38]. |
| Genetic Delivery | Lentiviral vectors, mRNA electroporation [38] [96] | Introduces the CAR gene into the host cell genome for stable or transient expression. | Promoter choice (e.g., EF1α, CMV, NK-specific) is critical for controlling CAR expression levels, persistence, and safety profile [96]. |
| Cytokines | IL-2, IL-15, IL-12, IL-18, IL-21, M-CSF, IFN-γ [95] | Enhances expansion, persistence, cytotoxicity, and guides cell differentiation/polarization. | Cytokine cocktails can be used to create "memory-like" NK cells or polarize macrophages to an M1 phenotype [95]. |
| Functional Assays | Flow cytometry kits, pHrodo bioparticles, calcein-AM, LDH kits, multiplex cytokine arrays [95] [94] | Measures CAR expression, phagocytosis, cytotoxicity, and immune activation. | Using antigen-positive and antigen-negative target cells in parallel is essential for demonstrating CAR-specific activity. |
CAR-NK, CAR-M, and CAR-DC therapies represent a logical and promising progression in the field of cellular immunotherapy, each offering unique mechanistic advantages to overcome the limitations of CAR-T cells, especially in solid tumors. CAR-NK cells bring innate cytotoxic versatility and a favorable safety profile, CAR-Ms excel in phagocytosis and TME remodeling, and CAR-DCs hold potential for orchestrating potent adaptive immune responses. The future of these therapies lies in continued optimization of CAR designs, manufacturing processes, and combination treatment strategies. The protocols and data summarized herein provide a foundational toolkit for researchers and drug development professionals to advance these next-generation platforms from promising concepts to transformative clinical realities.
Conclusion
CAR-T cell engineering has fundamentally transformed the therapeutic landscape for hematologic malignancies, establishing a powerful new pillar in cancer immunotherapy. The synthesis of foundational knowledge, refined manufacturing protocols, and innovative troubleshooting strategies has paved the way for clinical success. Future progress hinges on the systematic application of next-generation technologies—including synthetic biology, gene editing, and advanced targeting logic—to overcome the formidable barriers in solid tumors. The convergence of multi-antigen targeting, TME reprogramming, enhanced safety switches, and the development of allogeneic platforms promises to enhance efficacy, improve accessibility, and expand the reach of this living therapy. As the field evolves, the continued collaboration between basic science, translational research, and clinical medicine will be paramount in unlocking the full potential of engineered cell therapies for a broader spectrum of cancers.
References
- 1. - Engineering - PMC CAR T cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC5482931/]
- 2. - CAR cell therapy: current limitations and potential strategies - PMC T [https://pmc.ncbi.nlm.nih.gov/articles/PMC8024391/]
- 3. Cancer immunotherapy using CAR-T cells: from the research bench to the assembly line - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5966018/]
- 4. CAR T Cells: Engineering Immune Cells to Treat Cancer - NCI [https://www.cancer.gov/about-cancer/treatment/research/car-t-cells]
- 5. Emerging advances in CAR - T therapy for solid tumors: latest clinical... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-025-01760-9]
- 6. Engineering switchable and programmable universal CARs for CAR ... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-019-0763-0]
- 7. Surveying local CAR T-cell manufacturing processes to facilitate standardization and expand accessibility - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12057077/]
- 8. Tracing the development of CAR-T cell design [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1615212/full]
- 9. Current updates on generations, approvals, and clinical trials ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9746433/]
- 10. Emerging CAR immunotherapies: broadening therapeutic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12321946/]
- 11. Next-Gen CAR - T Therapy: Expanding Beyond Blood Cancers Cell [https://www.medscape.com/viewarticle/next-gen-car-t-cell-therapy-expanding-beyond-blood-cancers-2025a100077o]
- 12. Advancements and challenges in CAR-T cell therapy for solid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12374352/]
- 13. Strategies for Altering Delivery Technologies to Optimize ... CAR [https://pmc.ncbi.nlm.nih.gov/articles/PMC11989270/]
- 14. From bench to bedside: the history and progress of CAR cell... T [https://pmc.ncbi.nlm.nih.gov/articles/PMC10225594/]
- 15. ACR 2025 : Dual-Target CAR - T for Lupus Shows Early... Therapy [https://www.emjreviews.com/rheumatology/news/acr-2025-car-t-therapies-show-promise-in-autoimmune-disease/]
- 16. Current advances in experimental and computational approaches to... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11285593/]
- 17. Frontiers | Current advances in experimental and computational approaches to enhance CAR T cell manufacturing protocols and improve clinical efficacy [https://www.frontiersin.org/journals/molecular-medicine/articles/10.3389/fmmed.2024.1310002/full]
- 18. Frontiers | Data driven model discovery and interpretation for CAR T-cell killing using sparse identification and latent variables [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1115536/full]
- 19. Mechanistic data-informed multiscale quantitative systems pharmacology modeling framework enables the clinical translation and efficacy assessment of CAR-T therapy in solid tumors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12516987/]
- 20. Advances in Adoptive Cell Therapies in Cancer [https://www.mdpi.com/2076-3271/13/3/190]
- 21. Complexities in comparing the impact of costimulatory domains on... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-023-04372-4]
- 22. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6804784/]
- 23. CAR-T cells containing CD28 versus 4-1BB co-stimulatory ... [https://www.sciencedirect.com/science/article/pii/S2211124725007442]
- 24. Stronger and faster is not always better | Fred Hutchinson Cancer Center [https://www.fredhutch.org/en/news/spotlight/2018/10/vidd_riddell_scisig.html]
- 25. Advances in CAR optimization strategies based on CD28 [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1548772/full]
- 26. - 4 and optimized 1 co-stimulation enhances function of human... BB CD 28 [https://pubmed.ncbi.nlm.nih.gov/34706886/]
- 27. Frontiers | 4 - 1 Boosts the Anti-Tumor Activity of... BB Signaling [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2020.539654/full]
- 28. Impact of cryopreservation on CAR T production and clinical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9582437/]
- 29. Cryopreserved leukapheresis enables scalable and ... [https://www.nature.com/articles/s41598-025-14865-5]
- 30. Isolate Cells from Blood | Cell Separation Techniques [https://www.stemcell.com/cell-separation/isolate-cells-from-blood]
- 31. CAR T Cell Therapy: CAR Construct Transduction Methods [https://www.akadeum.com/blog/car-construct-transduction/?srsltid=AfmBOorl4b4uKgraDboi59whCi8xWBnD6nCmn0-g-05hI7kmKKJuEZwB]
- 32. Procedure and Removing White Blood Cells From... Leukapheresis [https://www.akadeum.com/types-of-cell-separation/leukaphresis/]
- 33. Insights into next-generation immunotherapy designs and tools [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-025-01701-6]
- 34. The State and Future of Viral, Nonviral Vectors for Gene ... [https://www.ajmc.com/view/the-state-and-future-of-viral-non-viral-vectors-for-gene-therapy]
- 35. Viral and non-viral vectors in gene therapy: current state ... [https://www.sciencedirect.com/science/article/pii/S2352396425002786]
- 36. Protocols for Optimized CAR Knockin in Primary Human T ... [https://maxcyte.com/webinar/electroporation-enables-crispr-cas-mediated-car-knockin-in-primary-human-t-cells/]
- 37. Editorial: Vector-based gene delivery in cancer ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1587359/full]
- 38. Comparison of two lab-scale protocols for enhanced ... [https://www.nature.com/articles/s41598-023-45197-x]
- 39. Review: Current clinical of applications ... chimeric antigen receptor [https://pubmed.ncbi.nlm.nih.gov/27592405/]
- 40. - CAR For T in the Real World: 5 Uses... Cell Immunotherapies Cancer [https://www.linkedin.com/pulse/car-t-cell-immunotherapies-cancer-real-pzh2f]
- 41. How CAR - T Therapy Works — In One Simple Flow ( Cell ) 2025 [https://www.linkedin.com/pulse/how-car-t-cell-therapy-works-one-simple-flow-2025-3sjqe]
- 42. Safety and efficacy of a novel anti-CD19 chimeric ... antigen receptor [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-023-01886-9]
- 43. An Update on CAR T-Cell Therapy in Multiple Myeloma | Dana-Farber Cancer Institute [https://www.dana-farber.org/for-physicians/clinical-resources/hematologic-malignancies/advances-newsletter/2025-issue-20/multiple-myeloma-cart-t-cell-therapy-update]
- 44. Engineering CAR T cells for enhanced efficacy and safety - PubMed [https://pubmed.ncbi.nlm.nih.gov/35071966/]
- 45. Advances in CAR-T cell therapy for hematologic and solid malignancies: latest updates from 2024 ESMO Congress - PubMed [https://pubmed.ncbi.nlm.nih.gov/39639359/]
- 46. mRNA Engineering - CAR for T Cells Cancer Immunotherapy [https://pubmed.ncbi.nlm.nih.gov/40877511/]
- 47. Frontiers | The potential of microfluidic cell analysis in CAR ... T cell [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1613836/full]
- 48. Frontiers | Challenges and breakthroughs: current landscape and future... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1652329/full]
- 49. trial helps Immunotherapy patients with... | The Guardian cancer [https://www.theguardian.com/science/2025/may/31/immunotherapy-trial-helps-cancer-patients-with-tumours-live-40-longer]
- 50. Melanoma Clinical Trials to Watch: October 2025 [https://www.curemelanoma.org/blog/melanoma-clinical-trials-to-watch-fall-2025]
- 51. Mechanistic data-informed multiscale quantitative systems ... [https://pubmed.ncbi.nlm.nih.gov/41067881/]
- 52. CAR-T cell therapy for cancer: current challenges and future directions | Signal Transduction and Targeted Therapy [https://www.nature.com/articles/s41392-025-02269-w]
- 53. Frontiers | Breaking barriers: enhancing CAR-armored T cell therapy for solid tumors through microenvironment remodeling [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1638186/full]
- 54. Frontiers | CAR-T cell therapy in brain malignancies: obstacles in the face of cellular trafficking and persistence [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1596499/full]
- 55. ESMO 2025 - When the tumour ... | Gustave Roussy microenvironment [https://www.gustaveroussy.fr/en/esmo-2025-when-tumour-microenvironment-hinders-effectiveness-car-t-cell-therapies]
- 56. the Engineering : oncolytic NDV to facilitate... tumor microenvironment [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-07342-0]
- 57. Low-dose carboplatin modifies the tumor to... microenvironment [https://www.nature.com/articles/s41467-023-40852-3?error=cookies_not_supported&code=dd0848c5-9c32-494d-a645-262041f081bc]
- 58. Current Status and Perspectives of Dual - Targeting Chimeric Antigen... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9265066/]
- 59. Innovative gene engineering strategies to address tumor ... antigen [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-07259-8]
- 60. pharmacology of Quantitative ‐ dual bicistronic targeted ‐T‐cell... CAR [https://pmc.ncbi.nlm.nih.gov/articles/PMC11812944/]
- 61. CARTmath—A Mathematical Model of CAR-T ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8231202/]
- 62. CAR-T cell Goes on a Mathematical Model. [https://www.scientificarchives.com/article/cart-cell-goes-on-a-mathematical-model]
- 63. Anticytokine Therapy and Corticosteroids for Cytokine ... - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK604827/]
- 64. CRS & ICANS Management [https://www.epkinlyhcp.com/crs-icans-management]
- 65. From ex vivo to in vivo chimeric antigen T manufacturing: new... cells [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-06052-3]
- 66. Constructing the cure: engineering the next wave of antibody ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12382549/]
- 67. Targeting the epigenome to reinvigorate T for cells ... cancer [https://mmrjournal.biomedcentral.com/articles/10.1186/s40779-023-00496-2]
- 68. Next-generation T cell immunotherapy [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1662145/full]
- 69. Frontiers | Novel insights into cancer stem cells targeting... [https://www.frontiersin.org/journals/molecular-medicine/articles/10.3389/fmmed.2023.1120090/full]
- 70. Empowering the Potential of CAR - T by... Cell Immunotherapies [https://pmc.ncbi.nlm.nih.gov/articles/PMC10093039/]
- 71. Enhanced CAR therapy offers new strategy for... | ScienceDaily T cell [https://www.sciencedaily.com/releases/2025/05/250507200803.htm]
- 72. Frontiers | Improved CAR internalization and recycling through... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1531344/full]
- 73. Nutrient-gene therapy as a strategy to enhance CAR T cell ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06606-z]
- 74. Preclinical Evaluation of CAR T Cell Function: In Vitro and In Vivo Models - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8955360/]
- 75. Quantitative Assessment of CAR-T Expansion with Digital ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10932155/]
- 76. Engineering TME-gated inducible CAR-T cell therapy for ... [https://www.sciencedirect.com/science/article/pii/S1525001625003168]
- 77. Advancing CAR T Cell Therapy with Logic Gate Engineering [https://www.cellandgene.com/doc/advancing-car-cell-therapy-logic-gate-engineering-0001]
- 78. 'Armored' CAR T-Cell Therapy Shows Promise After Prior ... [https://www.aabb.org/news-resources/news/article/2025/05/14/armored--car-t-cell-therapy-shows-promise-after-prior-car-t-cell-therapy-failure]
- 79. Mathematical models and computational approaches in ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1581210/full]
- 80. Co-opting signalling molecules enables logic-gated control of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10564584/]
- 81. Comparative analysis of assays to measure CAR T cell– ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8064272/]
- 82. Quantitative Insights into CAR-T Cell Therapy [https://www.medrxiv.org/content/10.1101/2025.09.02.25334722v1]
- 83. -Cell Therapy Explained | AJMC Allogeneic vs Autologous CAR T [https://www.ajmc.com/view/allogeneic-vs-autologous-car-t-cell-therapy-explained]
- 84. . Allogeneic - vs : Scalability & Efficacy Autologous CAR T [https://www.grgonline.com/post/allogeneic-vs-autologous-car-t-cell-therapy-overcoming-challenges-in-scalability-and-efficacy]
- 85. Allogeneic CART progress: platforms, current progress and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12198129/]
- 86. Frontiers | Allogeneic CART progress: platforms, current progress and... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1557157/full]
- 87. Emerging trends in clinical allogeneic CAR cell therapy [https://www.sciencedirect.com/science/article/pii/S2666634025001047]
- 88. Press Releases [https://ir.allogene.com/news-releases/news-release-details/allogene-therapeutics-provides-updated-phase-1-data-highlighting]
- 89. between Difference - Autologous Cell Therapy Vs Allogeneic CAR T [https://www.cellandgene.com/doc/autologous-vs-allogeneic-car-t-therapies-time-for-a-second-look-0001]
- 90. The 2025 - CAR Report: Science, Strategy, and the Next Era of T ... Cell [https://www.linkedin.com/pulse/2025-car-t-report-science-strategy-next-era-cell-therapy-bill-gadless-vbvxe]
- 91. Emerging trends in clinical allogeneic CAR cell therapy [https://pubmed.ncbi.nlm.nih.gov/40367950/]
- 92. Frontiers | Beyond CAR-T Cells : exploring CAR-NK, CAR - M , and... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1675807/full]
- 93. Expanding the frontier of CAR : comparative insights into... therapy [https://link.springer.com/article/10.1007/s00277-025-06538-0]
- 94. Unleashing the power of CAR - M in solid tumors... therapy [https://pubmed.ncbi.nlm.nih.gov/40574837/]
- 95. The development and application of chimeric antigen receptor natural killer (CAR-NK) cells for cancer therapy: current state, challenges and emerging therapeutic advances | Experimental Hematology & Oncology | Full Text [https://ehoonline.biomedcentral.com/articles/10.1186/s40164-024-00583-7]
- 96. and prospects of genetic engineering in Application - CAR ... NK cell [https://pmc.ncbi.nlm.nih.gov/articles/PMC12141315/]