Bacterial vs. Yeast vs. Mammalian Cell Hosts: A Strategic Guide to Heterologous Expression Systems
Selecting the optimal heterologous expression system is a critical decision that impacts the success of recombinant protein production for research, therapeutic, and industrial applications.
Bacterial vs. Yeast vs. Mammalian Cell Hosts: A Strategic Guide to Heterologous Expression Systems
Abstract
Selecting the optimal heterologous expression system is a critical decision that impacts the success of recombinant protein production for research, therapeutic, and industrial applications. This article provides a comprehensive comparison of the three dominant platforms—bacterial, yeast, and mammalian cell hosts—catering to the needs of researchers, scientists, and drug development professionals. It covers foundational principles, practical methodologies, advanced troubleshooting strategies, and a direct comparative analysis of cost, yield, and post-translational modification capabilities. By synthesizing current research and engineering advances, this guide delivers a strategic framework for system selection, optimization, and validation to efficiently produce high-quality functional proteins.
Understanding Heterologous Expression Systems: Core Principles and Host Organism Profiles
Defining Heterologous Expression and Its Role in Modern Biotechnology
Heterologous expression is a fundamental genetic engineering technique that involves the expression of a gene or part of a gene in a host organism that does not naturally possess that gene fragment [1]. This recombinant DNA technology provides scientists with a powerful pathway to efficiently express and experiment with combinations of genes and mutants that do not naturally occur, enabling the study of protein function, the effects of mutations, and differential interactions [1]. In modern biotechnology, this methodology has become indispensable for both basic research and industrial applications, from deciphering fundamental biological mechanisms to producing therapeutic proteins and novel natural products. The strategic selection of an appropriate host system—whether bacterial, yeast, or mammalian—represents a critical decision point that directly influences the success and functionality of the expressed recombinant protein, forming the core thesis of this comparative analysis.
Key Host Systems for Heterologous Expression
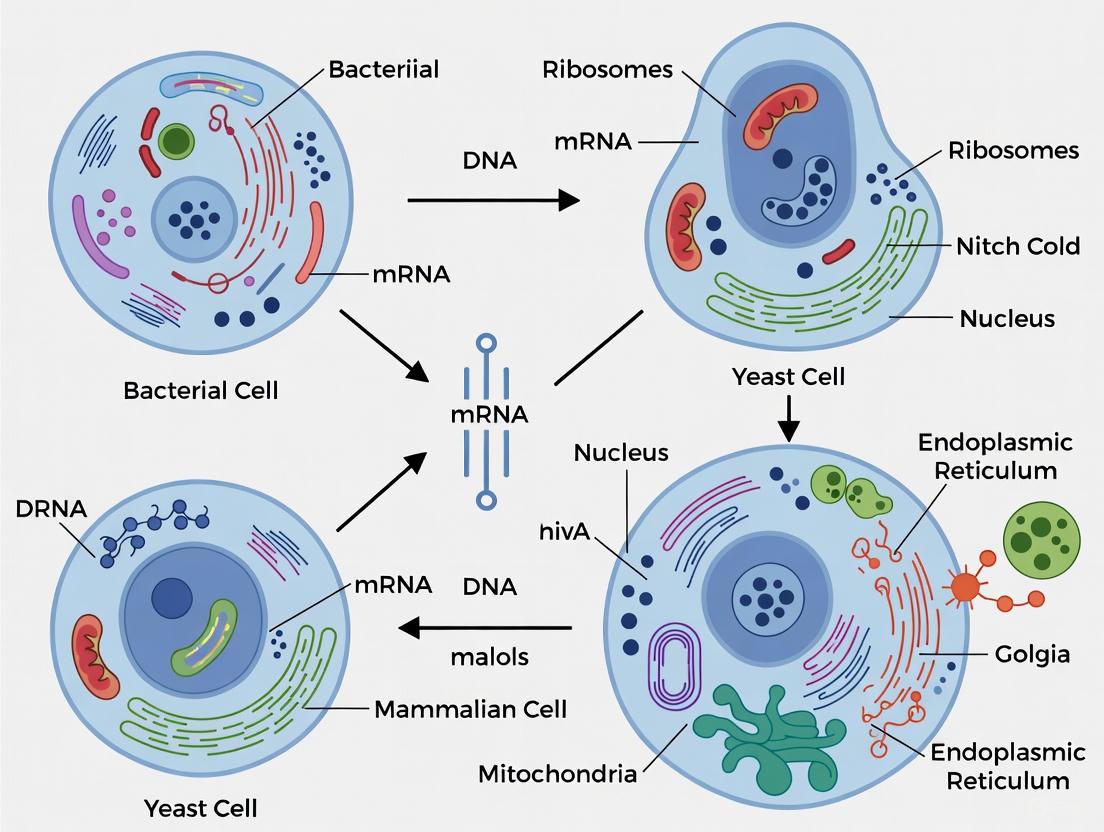
The choice of host organism for heterologous expression creates significant trade-offs between simplicity, cost, yield, and the ability to produce properly modified and folded proteins. The three primary systems—bacterial, yeast, and mammalian cells—each possess distinct advantages and limitations that make them suitable for different applications.
Table 1: Comparison of Major Heterologous Expression Host Systems
| Parameter | Bacterial (E. coli) | Yeast (P. pastoris, S. cerevisiae) | Mammalian (CHO, HEK) |
|---|---|---|---|
| Growth Rate | Very fast (~20-30 min doubling time) [1] | Fast (~90 min doubling time) [1] | Slow (24-48 hr doubling time) |
| Cost | Low [2] | Moderate [1] | High |
| Yield | High [2] | High (up to 30% of total protein) [1] | Low to moderate |
| Post-Translational Modifications | Limited or none [3] | Basic modifications, hypermannosylation issues [1] [4] | Complex, human-like [3] |
| Protein Folding | Often improper, inclusion body formation [3] | Generally correct [2] | Generally correct [3] |
| Secretion Efficiency | Variable | High [2] | Moderate |
| Typical Applications | Non-glycosylated proteins, research enzymes, antibody fragments [2] | Industrial enzymes, biofuels, protein interaction studies [3] | Therapeutic proteins, complex mammalian proteins, antibodies [3] |
Bacterial Expression Systems
Escherichia coli remains the most widely used heterologous expression system due to its rapid growth rate, well-characterized genetics, and low-cost cultivation requirements [1] [2]. The ability to achieve high cell densities with minimal technical requirements makes bacterial systems particularly attractive for high-throughput applications and large-scale production of non-eukaryotic proteins [4]. However, the absence of sophisticated post-translational modification machinery in prokaryotic systems presents a significant limitation for expressing functional eukaryotic proteins [1] [3]. Additionally, proteins expressed in large quantities in E. coli frequently precipitate and form inclusion bodies, necessitating complex denaturation and renaturation procedures to recover functional activity [1]. Beyond E. coli, other bacterial hosts like Bacillus subtilis offer advantages such as direct secretion of proteins into the culture medium and absence of lipopolysaccharides (which can cause inflammatory responses), though they face challenges with extracellular proteases that can degrade heterologous proteins [1].
Yeast Expression Systems
Yeast systems, particularly Saccharomyces cerevisiae and Pichia pastoris, represent an effective compromise between bacterial and mammalian systems, combining the growth advantages of microorganisms with eukaryotic processing capabilities [1] [2]. As eukaryotes, yeast cells provide advanced protein folding pathways and can secrete correctly folded and processed heterologous proteins into the culture media [2]. This makes them particularly valuable for industrial enzyme production and functional studies of eukaryotic proteins [3]. However, yeast systems have limitations in their glycosylation patterns, often resulting in hyper-mannosylation—the addition of excessive mannose residues—that can hinder proper protein folding and function, potentially limiting their suitability for therapeutic applications [1] [4]. Despite this limitation, yeast systems have been successfully employed to produce vaccines for hepatitis B and Hantavirus, demonstrating their pharmaceutical relevance [1].
Mammalian Cell Expression Systems
Mammalian expression systems, such as Chinese Hamster Ovary (CHO) and Human Embryonic Kidney (HEK) cells, represent the gold standard for producing complex human therapeutic proteins due to their ability to perform authentic post-translational modifications [3]. These systems properly and efficiently recognize the signals for synthesis, processing, and secretion of eukaryotic proteins, resulting in products with the most native structure and activity [2] [3]. This capability is particularly crucial for therapeutic proteins like monoclonal antibodies, hormones, and cytokines, where precise glycosylation patterns can directly impact biological activity, stability, and immunogenicity [3]. The main disadvantages of mammalian systems include their demanding culture conditions, slow growth rates, technical complexity, and high cost, making them the least economical option among the three systems [4]. Additionally, subtle differences in glycosylation patterns between species must be considered, as murine cells may add galactose-α(1,3)-galactose epitopes that are recognized by human xenoreactive antibodies, potentially reducing the half-life of therapeutics in humans [2].
Methodologies and Experimental Workflows
Successful heterologous expression requires a systematic approach encompassing gene isolation, vector construction, host transformation, and protein expression. The experimental workflow varies depending on the host system but follows a consistent conceptual framework.
Gene Isolation and Vector Construction
The process begins with isolation of the target gene, which can be accomplished through various methods depending on whether the genomic sequence is known. For known sequences, polymerase chain reaction (PCR) serves as the primary method for gene amplification and isolation [1]. PCR involves sequential phases of denaturation (strand separation at ~95°C), annealing (primer binding to complementary sequences), and extension (DNA polymerase-mediated replication) to specifically amplify the gene of interest [1]. For unknown sequences, restriction enzyme-based approaches or modern metagenomic techniques can be employed to identify and isolate novel genes from environmental samples [1] [5].
Once isolated, the gene is cloned into an expression vector containing essential regulatory elements: a promoter to drive transcription, a ribosomal binding site for translation initiation, selectable markers for host selection, and appropriate termination sequences [4]. Different host systems require specialized vector components, with bacterial systems utilizing promoters like tac or T7, yeast systems employing promoters such as AOX1 in P. pastoris, and mammalian systems often using viral promoters like CMV or SV40 [2] [4].
Host Transformation and Gene Delivery
Introducing foreign DNA into host cells employs distinct methodologies tailored to each host system:
Electroporation utilizes high-voltage electrical pulses to create transient pores in cell membranes, allowing DNA entry into the cell. This technique works with almost any tissue type and demonstrates high gene delivery efficiency with minimal host cell damage when appropriate field strengths are applied [1]. Electroporation is effective for both short-term and long-term transfection across bacterial, yeast, and mammalian systems [1].
Lipofection employs lipid-based vesicles (liposomes) that encapsulate DNA and either directly fuse with the cell membrane or undergo endocytosis, subsequently releasing DNA into the cell. This method works with numerous cell types, offers high reproducibility, and serves as a rapid technique for both stable and transient expression [1].
Viral Transduction uses engineered viral vectors (particularly lentiviruses or adenoviruses) to deliver genetic material into host cells. Lentiviral vectors are particularly valuable because they can transduce non-dividing cells and integrate DNA into the host genome, enabling stable expression across diverse cell types [1].
Gene Gun Delivery (biolistics) represents a physical method that uses helium propulsion to deliver DNA-coated gold particles directly into cells. This technique has been traditionally used for transgenic plant generation but has also proven successful for animal cells at lower helium pressures [1].
Table 2: Common Gene Delivery Methods Across Host Systems
| Method | Mechanism | Host Compatibility | Expression Type |
|---|---|---|---|
| Electroporation | Electrical pulses create membrane pores [1] | Bacterial, yeast, mammalian [1] | Transient and stable [1] |
| Lipofection | Liposome fusion or endocytosis [1] | Mammalian, some yeast [1] | Primarily transient [1] |
| Viral Transduction | Viral vector infection [1] | Mammalian, insect [1] | Stable (lentivirus) or transient (adenovirus) [1] |
| Gene Gun/Biolistics | Helium propulsion of DNA-coated particles [1] | Plant, mammalian [1] | Stable and transient [1] |
Advanced Applications in Biotechnology
Heterologous expression technologies have enabled groundbreaking applications across multiple biotechnology sectors, particularly in natural product discovery and therapeutic protein production.
Natural Product Discovery
The activation of silent biosynthetic gene clusters (BGCs) through heterologous expression has revolutionized natural product discovery, especially for compounds from difficult-to-culture marine microorganisms and environmental samples [6]. Metagenomic approaches that extract community DNA directly from environmental samples and express BGCs in tractable host organisms have provided access to previously inaccessible chemical diversity [5] [6]. This strategy has proven particularly valuable for discovering novel antibiotics at a time when drug resistance poses a serious and growing threat to global health [7]. For example, heterologous expression of BGCs from marine actinomycetes and cyanobacteria in engineered chassis strains has yielded new bioactive compounds with pharmaceutical potential [6]. Similarly, Burkholderia species have emerged as promising heterologous hosts for natural product expression due to their intrinsic biosynthetic capabilities, enabling the production of novel small molecules in titers sufficient for drug development [7].
Therapeutic Protein Production
The production of biopharmaceuticals represents one of the most significant industrial applications of heterologous expression technology. Mammalian cell lines remain the preferred system for producing complex therapeutic proteins like monoclonal antibodies, hormones, and vaccines that require authentic human-like post-translational modifications for optimal efficacy and safety [3]. The global market for biopharmaceutical proteins approaches $400 billion annually, driving continuous optimization of expression platforms [8]. Recent advances in fungal expression systems, particularly engineered Aspergillus niger strains, demonstrate how strategic genetic modifications can create robust platforms for high-yield protein production. One study achieved yields ranging from 110.8 to 416.8 mg/L for diverse proteins including glucose oxidase, pectate lyase, and the immunomodulatory protein LZ-8 by deleting background glucoamylase genes and integrating target genes into native high-expression loci [8].
Enzyme Engineering and Industrial Biotechnology
Heterologous expression enables the production of industrial enzymes for applications in biofuel production, bioremediation, food processing, and textile manufacturing [9] [8]. The cellulase enzyme system for lignocellulosic biomass degradation provides a compelling example of how heterologous expression can optimize enzyme cocktails by balancing the activities of multiple enzyme components [9]. For instance, expressing β-glucosidase genes from Penicillium decumbens or Periconia sp. in Trichoderma reesei strains significantly enhanced cellulose degradation efficiency by addressing the native strain's limited β-glucosidase activity [9]. Consolidated bioprocessing (CBP), which combines cellulose hydrolysis and fermentation in a single step without externally supplied enzymes, represents an emerging application that relies on heterologous expression of complete cellulase systems in non-cellulolytic organisms [9].
The Scientist's Toolkit: Essential Research Reagents
Successful heterologous expression experiments require carefully selected reagents and genetic tools tailored to each host system.
Table 3: Essential Research Reagents for Heterologous Expression
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Expression Vectors | pET series (bacterial), pPICZ (yeast), pcDNA3.1 (mammalian) | Carry gene of interest with host-specific regulatory elements [2] [4] |
| Enzymes for Cloning | Restriction enzymes, DNA ligase, polymerases | Gene fragment isolation and vector construction [1] |
| Transfection Reagents | Lipofectamine, polyethyleneimine (PEI) | Facilitate DNA entry into host cells [1] |
| Selection Antibiotics | Ampicillin, kanamycin (bacterial), zeocin, geneticin (eukaryotic) | Select for successfully transformed hosts [2] |
| Induction Compounds | IPTG (bacterial), methanol (P. pastoris), tetracycline (mammalian) | Regulate expression of target gene [2] |
| Protease Inhibitors | PMSF, complete protease inhibitor cocktails | Prevent degradation of expressed proteins [1] |
| Chromatography Resins | Ni-NTA, glutathione sepharose, protein A/G | Purify tagged recombinant proteins |
Heterologous expression stands as a cornerstone technology in modern biotechnology, enabling the functional characterization of genes and the production of valuable proteins across research, industrial, and therapeutic domains. The strategic selection of an appropriate host system—balancing the simplicity and yield of bacterial systems, the eukaryotic processing capability of yeast, and the authentic post-translational modification capacity of mammalian cells—remains a critical determinant of experimental and commercial success. As synthetic biology and genetic engineering technologies continue to advance, emerging hosts like engineered Aspergillus niger and Burkholderia species are expanding the toolbox available to scientists, offering new pathways to access novel natural products and optimize recombinant protein yields. These developments promise to further accelerate drug discovery and industrial biotechnology applications, reinforcing the central role of heterologous expression in addressing some of the most pressing challenges in human health and sustainable technology.
Within the field of heterologous protein production, the selection of an appropriate expression host is a critical determinant of success for research, therapeutic, and industrial applications. The primary systems—bacterial, yeast, and mammalian cells—each present a unique profile of capabilities and constraints [10]. Escherichia coli, a gram-negative prokaryote, stands as one of the most established and widely utilized hosts in this landscape [11]. This guide provides an objective comparison of the E. coli expression system against yeast and mammalian alternatives, framing its performance within the broader context of available hosts. We summarize supporting experimental data and delineate the specific scenarios for which E. coli is the most suitable platform, providing researchers with a clear framework for host selection.
Performance Comparison of Expression Systems
The choice of an expression system often involves balancing cost, speed, and the ability to produce a complex, functional protein. The table below provides a comparative overview of the three major host systems based on key performance metrics.
Table 1: Comparative analysis of heterologous protein expression systems.
| Feature | E. coli | Yeast | Mammalian Cells |
|---|---|---|---|
| Speed & Cost | Very fast growth (hours), low cost [10] [3] | Fast growth, low cost [12] [3] | Slow growth (days), very high cost [11] [10] |
| Post-Translational Modifications | Limited; lacks glycosylation machinery and other complex PTMs [10] [13] | Capable of N- and O-glycosylation (high-mannose type) [12] [10] | Complex, human-like glycosylation; extensive PTM capability [10] |
| Typical Yield | High yields for soluble, non-complex proteins [11] [3] | High secretion titers, suitable for scale-up [12] | Variable yields; lower than microbial systems for non-complex proteins [11] |
| Handling & Scale-Up | Simple genetic manipulation and fermentation [13] [3] | Simple fermentation, easy scale-up [12] [3] | Complex culture requirements, difficult scale-up [10] [3] |
| Ideal Protein Type | Prokaryotic proteins, simple eukaryotic proteins (<30-100 kDa), non-glycosylated proteins [11] [13] | Secreted eukaryotic proteins, proteins requiring simple glycosylation [12] [10] | Complex proteins requiring authentic human PTMs (e.g., antibodies, growth factors) [10] [3] |
| Key Limitations | Formation of inclusion bodies, metabolic burden, endotoxin contamination [11] [13] [14] | Hyper-mannosylation can be immunogenic [12] [10] | Risk of viral contamination, high cost, technical complexity [13] |
Advantages of the E. coli Expression System
Well-Characterized Genetics and Speed
E. coli remains the most well-understood expression system, with a fully sequenced and annotated genome for common lab strains [13]. This extensive genetic knowledge base, combined with the availability of a vast collection of expression vectors and engineered host strains, allows for straightforward genetic manipulation [11] [13]. Furthermore, its rapid cellular proliferation (doubling in as little as 20 minutes) enables the production of recombinant protein in a matter of hours, significantly accelerating research and development timelines compared to eukaryotic systems [10] [3].
Cost-Effectiveness and High Yield
The cultivation of E. coli is remarkably cost-effective. It requires inexpensive growth media and uncomplicated fermentation procedures, leading to high cell densities and, consequently, high yields of the target recombinant protein [13]. This cost structure is far more economical than the complex and expensive media required for mammalian cell culture [11]. For proteins that it can express well, E. coli often delivers the highest yield per unit of cost, making it the system of choice for industrial-scale production of non-complex proteins like hormones and cytokines [11] [13].
Absence of Human Viral Contaminants
A significant advantage for therapeutic protein production is the absence of a risk from human-pathogenic viral contaminants. Unlike mammalian cell lines, which can harbor endogenous retroviruses or require extensive viral clearance validation, E. coli presents no such safety concerns, simplifying the downstream regulatory pathway for biologic drugs [13].
Inherent Limitations of E. coli
Post-Translational Modifications and Protein Folding
A principal limitation of E. coli is its inability to perform complex post-translational modifications, most notably human-like glycosylation [10] [13]. This restricts its use for producing many therapeutic proteins, such as monoclonal antibodies, where specific glycan structures are critical for stability, half-life, and biological activity [13]. Additionally, the reducing environment of the E. coli cytoplasm often prevents the correct formation of disulfide bonds, which are essential for the proper folding and function of many eukaryotic proteins [15]. This can lead to misfolded, inactive products.
Inclusion Body Formation and Solubility
The overexpression of recombinant proteins, particularly those from eukaryotic sources, frequently results in the formation of insoluble aggregates known as inclusion bodies (IBs) [11] [13]. While IBs can contain high concentrations of the protein, recovering active, soluble protein requires tedious and often inefficient processes of solubilization with denaturants and subsequent refolding [13] [16]. This adds significant complexity and cost to the production process.
Metabolic Burden and Cellular Stress
High-level expression of heterologous genes places a substantial metabolic burden on the host cell [14]. This burden arises from the competition for the cell's resources, such as energy, precursors, and translational machinery, between the recombinant process and native cellular functions [16]. The consequences include reduced growth rates, downregulation of essential metabolic pathways, and activation of stress responses, which can ultimately lead to reduced protein yields and genetic instability [14]. The plasmid copy number and promoter strength are key factors influencing this burden [11] [14].
Endotoxin Contamination
As a gram-negative bacterium, E. coli produces endotoxins (lipopolysaccharides, LPS) in its outer membrane [13]. These pyrogenic molecules can cause severe immune reactions in humans and must be completely removed from any therapeutic protein destined for in vivo use. The purification process to remove endotoxins adds an additional, often challenging, validation step for pharmaceuticals produced in E. coli [13].
Experimental Data and Workflows
A High-Throughput Screening Pipeline
Modern structural genomics programs rely on high-throughput (HTP) pipelines to rapidly screen numerous protein targets. One such protocol for E. coli involves a 96-well plate format that can test up to 96 proteins in parallel within one week [17]. The workflow begins with commercially synthesized, codon-optimized genes cloned into an expression vector (e.g., pMCSG53 with a cleavable hexa-histidine tag). Following transformation, expression is tested under various conditions (e.g., media, temperature). Solubility is then assessed via high-throughput methods. Targets that show promising expression and solubility can be advanced to large-scale purification [17]. This approach allows for efficient optimization and is highly scalable for functional genomics.
Diagram 1: HTP protein expression screening pipeline.
Quantitative Impact of Expression Systems
A study investigating the production of a Kringle yellow fluorescent protein (KrYFP) in E. coli BL21(DE3) quantified the impact of promoter strength and plasmid copy number on protein yield and cell growth—a direct measure of metabolic burden [14]. Researchers compared four promoters of different strengths (PT7lac, Ptrc, Ptac, PBAD) and two replication origins (high-copy pMB1' and low-copy p15A) in both wild-type and engineered E. coli strains.
The results demonstrated that the very strong PT7lac promoter, combined with a high-copy origin, generated the highest transcriptional load. This did not always correlate with the highest soluble protein yield, as the associated metabolic burden could overwhelm the host cell, diverting resources from growth and proper protein folding [14]. A balance between plasmid copy number and promoter strength was found to be essential for maximizing the yield of soluble, functional recombinant protein while minimizing detrimental cellular effects [14].
Table 2: Key reagents for recombinant protein expression in E. coli.
| Reagent / Tool | Function / Explanation |
|---|---|
| Expression Vectors (e.g., pET, pBAD) | Plasmids containing origin of replication, promoter, MCS, and selectable marker [11] [10]. |
| E. coli Strains (e.g., BL21(DE3), Origami, SHuffle) | Specialized hosts for T7 polymerase expression, disulfide bond formation, or toxic protein production [15] [13]. |
| Fusion Tags (e.g., His-tag, MBP, SUMO) | Affinity tags for purification; solubility enhancers to prevent aggregation [11] [13] [18]. |
| Chaperone Plasmids | Co-expression vectors for proteins like GroEL/GroES that assist in proper folding [11] [13]. |
| Inducers (e.g., IPTG, L-Arabinose) | Chemicals used to trigger the transcription of the target gene from inducible promoters [14]. |
Escherichia coli rightfully maintains its status as a prokaryotic workhorse for heterologous protein expression, offering unparalleled speed, cost-effectiveness, and yield for a wide range of protein targets. Its well-characterized genetics and simplicity of use make it the ideal first choice for many laboratories. However, its inherent limitations in performing complex post-translational modifications and a tendency to produce insoluble aggregates or induce metabolic burden are significant constraints. The decision to use E. coli must therefore be guided by the nature of the target protein and the requirements of the downstream application. For simple, non-glycosylated prokaryotic or eukaryotic proteins, E. coli is often unmatched. For complex proteins requiring authentic eukaryotic folding and PTMs, yeast or mammalian systems, despite their higher cost and complexity, become the necessary choice. A comprehensive understanding of this performance landscape allows researchers to strategically select the most appropriate host, ensuring successful and efficient recombinant protein production.
The selection of an appropriate host system is a critical first step in heterologous protein production, framing a fundamental trade-off between simplicity and processing capability. Bacterial systems such as E. coli offer rapid growth and simplicity but lack the cellular machinery for complex post-translational modifications essential for many eukaryotic proteins [19] [4]. Mammalian cells provide these advanced modifications but come with high costs, complex nutritional requirements, and viral contamination risks [19] [10]. Yeast systems, particularly Saccharomyces cerevisiae and Pichia pastoris, strategically occupy the middle ground, offering the eukaryotic processing capabilities that bacteria lack, while maintaining the simplicity and cost-effectiveness that mammalian cells lack [19] [20] [4]. This review provides a comprehensive comparative analysis of these two yeast workhorses, examining their distinct advantages, limitations, and optimal applications within the broader context of expression host selection.
Saccharomyces cerevisiae: The Model Eukaryote
S. cerevisiae, a genetically well-characterized and Generally Recognized As Safe (GRAS) organism, has served as a foundational tool in biotechnology for decades [21]. Its key advantages include exceptionally clear genetics, extensive availability of molecular biology tools, and a long history of use in pharmaceutical production, including for hepatitis B and human papillomavirus vaccines [19] [21]. As a eukaryotic host, it performs essential post-translational modifications such as glycosylation, disulfide bond formation, and protein secretion, though its N-glycosylation pattern is of the high-mannose type, which can be immunogenic in therapeutic applications [19] [10]. It can achieve high cell densities and expresses recombinant proteins at up to 49.3% (w/w) of its own cellular protein content [21].
Pichia pastoris: The Methylotrophic Workhorse
P. pastoris (syn. Komagataella phaffii), another GRAS organism, has gained prominence as a powerful platform for recombinant protein production [19] [22]. This methylotrophic yeast can utilize methanol as its sole carbon source, employing the strong, tightly regulated alcohol oxidase 1 (AOX1) promoter to drive high-level protein expression [23] [20]. Its significant advantages include an exceptional capacity for high-cell-density fermentation (>150 g dry cell weight/liter), very high protein titers (exceeding 10 g/L for some proteins), and efficient secretion of recombinant proteins into the culture medium with limited endogenous secretory proteins, greatly simplifying downstream purification [19] [22]. While it also performs glycosylation, its N-linked glycans are shorter and more similar to mammalian patterns than those of S. cerevisiae [19] [20].
Table 1: Fundamental Characteristics of S. cerevisiae and P. pastoris
| Characteristic | S. cerevisiae | P. pastoris |
|---|---|---|
| Classification | Crabtree-Positive Yeast | Methylotrophic Yeast |
| GRAS Status | Yes [21] | Yes [23] |
| Doubling Time | ~90 minutes [21] | 60-120 minutes [19] |
| Common Promoters | Constitutive (e.g., PGAP, PTEF1) [21] | Inducible (e.g., PAOX1, PGAP) [23] [22] |
| Glycosylation Type | High-mannose (Hypermannosylation) [19] [10] | High-mannose, but shorter chains [19] [20] |
| Secretion Efficiency | High [21] | Very High [19] |
| Therapeutic Proteins | Hepatitis B vaccine, HPV vaccine [19] | Human insulin, interferon [20] |
Critical Comparative Analysis: Performance and Experimental Data
Quantitative Performance Metrics
Direct comparison of protein production data highlights the distinct performance profiles of each system. P. pastoris is renowned for achieving extremely high protein titers, in some cases exceeding 10 g/L, which can represent up to 30% of total cellular protein [22]. A recent biotechnological application demonstrated the production of 5.79 g/L of a steroid drug intermediate using an engineered P. pastoris strain in a fed-batch bioreactor [23]. While S. cerevisiae also achieves high expression levels, its yields for industrial enzymes and therapeutic proteins are generally lower on a volumetric basis, though it can still generate recombinant proteins at nearly half of its own cellular protein mass [21]. Furthermore, P. pastoris can typically reach higher cell densities in bioreactors compared to S. cerevisiae, a key factor for industrial-scale production [20].
Protein Processing and Glycosylation
A critical differentiator between these yeast systems and their suitability for human therapeutics lies in their glycosylation patterns. Both yeasts perform N- and O-linked glycosylation, but the structures differ. S. cerevisiae tends to produce hypermannosylated N-glycans, which can increase immunogenicity in humans and reduce the efficacy of therapeutic proteins [19] [10]. P. pastoris also produces high-mannose glycans, but the chains are typically shorter and more akin to the core oligosaccharides found in mammals, making them less immunogenic [19] [20]. This key difference has driven extensive engineering efforts in both hosts, particularly in S. cerevisiae, to humanize their glycosylation pathways for producing biologics like antibodies [21].
Table 2: Direct Comparison of S. cerevisiae and P. pastoris for Recombinant Protein Production
| Parameter | S. cerevisiae | P. pastoris |
|---|---|---|
| Typical Protein Titer | High (up to 49.3% of cellular protein) [21] | Very High (can exceed >10 g/L) [22] |
| Inducible Expression System | Available (e.g., GAL1 promoter) | Strong, methanol-inducible AOX1 system [23] |
| Secretion Background | Moderate | Low, simplifying purification [19] |
| Glycosylation Similarity to Humans | Lower (Hypermannosylation) [10] | Higher (Shorter Mannose Chains) [19] [20] |
| Genetic Tool Availability | Extensive and mature [21] | Growing rapidly (e.g., CRISPR/Cas9) [22] |
| Metabolic Engineering | Highly advanced, genome-scale models [21] | Developing, but robust tools available [20] [22] |
| Typical Carbon Sources | Glucose, Glycerol, Galactose [21] | Glucose, Glycerol, Methanol [20] |
Experimental Design and Methodologies
Standard Protein Production Workflow
A generalized experimental workflow for producing recombinant proteins in either yeast system involves common stages from gene design to protein purification. The process begins with codon optimization of the target gene to match the host's bias, followed by cloning into an appropriate expression vector [21]. The constructed vector is then integrated into the yeast genome or maintained episomally. Cultivation typically progresses from small-scale shake flasks to controlled bioreactors for high-cell-density fermentation [24]. For P. pastoris, induction is typically achieved by adding methanol to shift the culture from a growth phase to a production phase [23]. Finally, the protein is harvested from the supernatant (if secreted) or from cell lysates (if intracellular) and purified.
A Representative Protocol: Production in P. pastoris
The following detailed protocol, adapted from a recent study producing a steroid intermediate, exemplifies a high-efficiency process in P. pastoris [23].
Objective: To produce 15α-hydroxy-D-ethylgonendione (15α-OH-DE) using an engineered P. pastoris strain co-expressing a steroid 15α-hydroxylase (PRH) and a glucose-6-phosphate dehydrogenase (ZWF1) gene.
Strains and Vectors:
- Host Strain: P. pastoris GS115 (his4, aox1::ARG4, arg4) [23].
- Expression Vectors: pPIC3.5K (for intracellular PRH expression) and pPICZαA (modified for intracellular ZWF1 expression), providing G418 and Zeocin resistance, respectively [23].
Methodology:
- Strain Engineering: The PRH gene from Penicillium raistrickii was cloned into pPIC3.5K, and the ZWF1 gene from S. cerevisiae was cloned into the modified pPICZαA vector. Plasmids were linearized and sequentially integrated into the P. pastoris GS115 genome by electroporation. Positive clones were selected on MD and YPD plates with appropriate antibiotics [23].
- Shake-Flask Cultivation: A single colony was used to inoculate BMGY medium (10 g/L glycerol) and cultured at 30°C until the OD600 reached ~10. Cells were harvested and resuspended in BMMY medium (10 g/L methanol) to induce expression. The culture was continued for 120 h with 10 g/L methanol added every 24 h to maintain induction [23].
- Bioreactor Fermentation: The process was scaled up in a 5-L stirred-tank bioreactor. The fermentation lasted 196 h total, with a glycerol batch phase, a glycerol-fed-batch phase for cell growth, and a 170 h methanol-fed-batch phase for induction and biotransformation. The substrate (DE) was fed at 10 g/L [23].
- Analysis and Validation: Product (15α-OH-DE) titer was quantified to reach 5.79 g/L, the highest reported titer for this compound, demonstrating the success of the engineered strain and optimized process [23].
Pathway and Cellular Machinery
The efficiency of yeast as cell factories hinges on their internal cellular machinery. The diagram below illustrates the key pathways involved in protein expression, folding, and secretion, which are common to both S. cerevisiae and P. pastoris, though with noted differences in efficiency and glycosylation details.
The Scientist's Toolkit: Essential Research Reagents
Successful recombinant protein production in yeast relies on a suite of specialized reagents and genetic tools. The following table details key components for working with S. cerevisiae and P. pastoris.
Table 3: Essential Research Reagents for Yeast-Based Protein Expression
| Reagent / Tool | Function | Example Host/Application |
|---|---|---|
| pPIC3.5K / pPICZαA | Expression vectors for chromosomal integration in P. pastoris; offer G418 and Zeocin resistance, respectively [23]. | P. pastoris |
| AOX1 Promoter (PAOX1) | Strong, methanol-inducible promoter for high-level protein expression in P. pastoris [23] [22]. | P. pastoris |
| GAP Promoter (PGAP) | Strong, constitutive promoter used in both S. cerevisiae and P. pastoris [21] [22]. | Both |
| CRISPR/Cas9 System | Genome editing tool for precise gene knock-outs, knock-ins, and other genetic modifications [21] [22]. | Both |
| BMGY / BMMY Media | Complex media for growth (BMGY) and methanol-induced expression (BMMY) in P. pastoris [23]. | P. pastoris |
| YPD / SC Media | Standard complex (YPD) and defined minimal (SC) media for S. cerevisiae cultivation [21]. | S. cerevisiae |
| HIS4 / ARG4 Selectable Markers | Auxotrophic markers for selection of transformed cells without antibiotics [23] [21]. | Both |
S. cerevisiae and P. pastoris both provide an effective eukaryotic compromise for recombinant protein production, yet they serve distinct optimal applications. S. cerevisiae is ideal for research requiring a vast, well-established genetic toolbox, for targets where its hypermannosylation is not prohibitive, and for production processes that benefit from its long history of industrial use and GRAS status [19] [21]. P. pastoris is often superior when the primary objectives are maximizing protein titer, achieving high cell densities in a bioreactor, or secreting proteins into a clean background for easier purification [19] [22]. Its shorter glycosylation chains also make it preferable for many therapeutic proteins, though both systems may require glyco-engineering for fully humanized glycosylation.
The choice between these two powerful yeast systems ultimately depends on the specific protein of interest, the required yield and quality, the available fermentation infrastructure, and the intended final application of the recombinant product.
The selection of an appropriate host system is a foundational decision in biopharmaceutical development, influencing the structural fidelity, biological activity, and ultimately, the efficacy and safety of a therapeutic protein. While bacterial and yeast systems offer advantages for simpler proteins, mammalian cell systems have emerged as the indispensable platform for producing complex human therapeutics, particularly monoclonal antibodies and other proteins requiring sophisticated post-translational modifications. This guide provides an objective comparison of host systems and details the experimental methodologies that establish mammalian cells as the gold standard.
Host System Comparison: A Multi-Parameter Analysis
The choice of an expression system involves balancing yield, cost, scalability,, and most critically, the ability to produce a biologically functional product. The table below provides a structured comparison of the four primary host systems used in heterologous protein expression.
Table 1: Comprehensive Comparison of Protein Expression Systems
| Parameter | Bacterial (E. coli) | Yeast (P. pastoris, S. cerevisiae) | Insect (Baculovirus/Sf9) | Mammalian (CHO, HEK293) |
|---|---|---|---|---|
| Growth Speed & Cost | Very fast (doubling time ~20 min), inexpensive [2] [25] [4] | Fast, inexpensive [2] [3] | Moderate speed, moderately expensive [2] | Slow, highest cost [2] [26] |
| Typical Yield | High for simple proteins [3] | Up to several mg/L [4] | 10-100 mg/L, up to 1 g/L reported [4] | >1-3 g/L for transient; >3 g/L for stable systems [27] [28] |
| Post-Translational Modifications (PTMs) | Limited; lacks eukaryotic glycosylation, disulfide bond formation can be inefficient [3] [4] | Hypermannosylation (high mannose); non-human pattern [2] [4] | Simple glycosylation (paucimannose); lacks complex human patterns [2] [4] | Complex, human-like glycosylation (e.g., incorporation of galactose, sialic acid) [26] [3] [4] |
| Protein Folding & Complexity | Prone to insoluble inclusion bodies; unsuitable for multi-domain eukaryotic proteins [3] [4] | Capable of disulfide bond formation and secretion of folded proteins [2] [3] | Proper folding and assembly for many complex proteins [2] | Superior folding, assembly of complex multi-subunit proteins (e.g., full-length antibodies) [3] [27] |
| Key Advantages | Easy genetic manipulation, high yield for simple proteins, low cost [2] [25] | Eukaryotic protein folding and secretion, rapid growth, scalable [2] [3] | High yields of complex, functional eukaryotic proteins [4] | Most physiologically relevant PTMs, highest product quality for human therapeutics [26] [3] [27] |
| Primary Limitations | Lack of complex PTMs, frequent formation of inclusion bodies [3] [4] | Non-human, immunogenic glycosylation patterns [4] | Non-human glycosylation; baculovirus production is time-consuming [2] | Technically demanding, expensive, slow growth, risk of viral contamination [26] |
Experimental Validation: Protocols and Data
The superiority of mammalian systems is demonstrated through direct comparative experiments, particularly when analyzing glycosylation and functionality of therapeutics like monoclonal antibodies (mAbs).
Experimental Protocol: Transient Transfection in Mammalian Cells
The following detailed protocol is standard for rapid protein production in Human Embryonic Kidney (HEK293) or Chinese Hamster Ovary (CHO) cells [27].
Day 1: Cell Seeding
- Culture HEK293 or CHO cells in appropriate serum-free medium (e.g., FreeStyle 293 or ExpiCHO Expression Medium) to maintain logarithmic growth.
- One day before transfection, seed cells at a density of 0.5 - 1.0 x 10^6 viable cells/mL in a vented shaker flask. The culture volume should not exceed 50% of the flask's total capacity to ensure proper aeration.
Day 2: Transfection Complex Formation
- Step A (DNA Dilution): Dilute the plasmid DNA encoding the gene of interest (e.g., an antibody heavy and light chain) in a pre-determined volume of Opti-MEM or similar reduced-serum medium.
- Step B (Transfection Reagent Dilution): In a separate tube, dilute the cationic lipid-based transfection reagent (e.g., ExpiFectamine 293) in the same volume of Opti-MEM.
- Step C (Complexation): Combine the diluted DNA with the diluted transfection reagent. Mix gently and incubate for 10-20 minutes at room temperature to allow for DNA-lipid complex formation.
Day 2: Transfection and Enhancement
- Add the DNA-lipid complexes dropwise to the cell culture while gently swirling the flask.
- 18-22 hours post-transfection, add transfection enhancers (e.g., ExpiFectamine 293 Transfection Enhancer). These solutions contain components like sugars and lipids that improve plasmid delivery and cell health, boosting protein yield.
Day 4-7: Harvest
- Monitor cell viability and productivity. Harvest the culture supernatant 3-5 days post-transfection by centrifugation at 4,000 x g for 30 minutes to remove cells and debris.
- The supernatant containing the secreted recombinant protein is now ready for purification.
This workflow is summarized in the following diagram:
Key Experimental Data: Glycosylation and Yield
Quantitative data from optimized systems highlights the performance of mammalian cells. For instance, the ExpiCHO Expression System can achieve titers of up to 3 g/L for human IgG proteins, significantly outperforming other systems in both yield and quality [27]. A critical comparative experiment involves analyzing the glycosylation profile of an antibody produced in different hosts.
Table 2: Glycosylation Profile Comparison of a Recombinant IgG [27]
| Expression System | Glycosylation Pattern | Therapeutic Relevance |
|---|---|---|
| Stable CHO (Reference) | Complex, human-like glycoforms with low mannose | Establishes the benchmark for product quality. |
| ExpiCHO Transient | Glycosylation profile highly similar to stable CHO | Provides high correlation between early-stage and production-scale material. |
| Expi293 Transient | Altered glycosylation profile compared to stable CHO | May require further engineering for optimal glycan patterns. |
| Yeast | High-mannose, non-human pattern [4] | Can be immunogenic in humans; unsuitable for most therapeutics without extensive engineering. |
| Insect | Paucimannose (simple) structures; lacks sialic acid [4] | Non-human pattern can affect serum half-life and bioactivity. |
This data demonstrates that mammalian cells, particularly CHO-based systems, are uniquely capable of reproducing the complex glycosylation critical for the stability, bioactivity, and pharmacokinetics of therapeutic proteins [26] [4]. Non-human glycosylation patterns can lead to rapid clearance from the bloodstream or unwanted immune responses [2].
The Scientist's Toolkit: Essential Reagents for Mammalian Expression
Successful recombinant protein production in mammalian cells relies on a suite of specialized reagents and tools.
Table 3: Key Research Reagent Solutions for Mammalian Cell Expression
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Expression Vectors (e.g., pcDNA) | Plasmid DNA containing the gene of interest, promoter (e.g., CMV), and selectable marker. | Delivering the genetic blueprint for the recombinant protein to the host cell [27]. |
| Specialized Media (e.g., Expi293, ExpiCHO) | Chemically defined, serum-free media optimized for high-density culture and transfection. | Supporting robust cell growth and high-level protein production in suspension cultures [27]. |
| Transfection Reagents (e.g., Lipids, Polymers) | Cationic lipids or polymers that complex with DNA to facilitate its entry into cells. | Enabling high-efficiency delivery of plasmid DNA into mammalian cells in suspension [27]. |
| Selection Antibiotics (e.g., Geneticin/G418, Puromycin) | Toxic compounds that eliminate untransfected cells, allowing for the selection of stable cell lines. | Selecting and maintaining pools of cells that have stably integrated the expression construct into their genome [27]. |
| Transfection Enhancers | Supplements that improve transfection efficiency and/or boost recombinant protein secretion. | Increasing volumetric yield in transient transfection experiments by improving cell health and productivity [27]. |
While bacterial and yeast systems remain excellent choices for producing a wide range of enzymes and non-glycosylated proteins, the data from glycosylation analysis and productivity benchmarks firmly establish mammalian cell systems as the gold standard for complex human therapeutics. Their unparalleled ability to perform human-like post-translational modifications and correctly fold intricate proteins ensures that biopharmaceuticals, especially monoclonal antibodies, exhibit the necessary safety, efficacy, and stability for clinical use. As engineering advances continue to push yields higher and reduce production costs, the central role of mammalian systems in biopharmaceutical manufacturing is set to strengthen further.
The selection of an optimal heterologous expression host is a critical first step in the successful production of recombinant proteins, a process fundamental to modern biologics research and drug development. This choice is governed by a balance of four key criteria: yield, cost, scalability, and the capacity for essential post-translational modifications (PTMs). The most commonly employed host systems—bacterial (e.g., E. coli), yeast (e.g., P. pastoris), and mammalian cells (e.g., CHO, HEK293)—each present a distinct profile of advantages and limitations against these benchmarks [10] [29]. Bacterial systems are prized for their simplicity and low cost but often fail to produce functional complex eukaryotic proteins. Mammalian cells support the most complex PTMs but incur higher costs and longer timelines. Yeast systems offer a middle ground, providing eukaryotic folding and secretion pathways with prokaryotic-like scalability [30]. This guide provides a structured comparison of these systems, equipping researchers with the data necessary to align their project goals with the most suitable expression platform.
Comparative Analysis of Major Expression Systems
The table below summarizes the core characteristics of the three primary heterologous expression hosts, providing a direct comparison based on the key selection criteria.
Table 1: Key Characteristics of Major Heterologous Protein Expression Systems
| Criterion | E. coli (Bacterial) | Yeast (e.g., P. pastoris) | Mammalian Cells (e.g., CHO, HEK293) |
|---|---|---|---|
| Typical Yield | High for simple, soluble proteins [29] | High cell densities; high yields for secreted proteins [29] | Lower volumetric yield than microbial systems [29] |
| Cost & Speed | Low cost; rapid growth (2-3 weeks) [30] | Cost-effective; faster than mammalian cells [29] | High cost; longer timelines (4-6 weeks) [30] |
| Scalability | Excellent, straightforward scale-up [30] | High, cost-effective fermentation [29] | Moderate, complex and expensive scale-up [29] [30] |
| PTM Capability | Limited; no glycosylation, simple disulfide bonds possible [10] | Hyper-mannose glycosylation; disulfide bonds [10] [29] | Complex, human-like PTMs including sialylation [10] [31] |
| Ideal Protein Types | Non-glycosylated proteins, single domains, proteins for structural biology [10] [29] | Secreted proteins, enzymes with simple glycosylation needs [29] | Complex proteins, antibodies, targets requiring human-like glycosylation [10] [31] |
| Key Limitations | Formation of inclusion bodies, no native glycosylation [10] [29] | Non-human, immunogenic glycosylation patterns [10] | High cost, technical complexity, longer development times [31] [29] |
The Critical Role of Post-Translational Modifications (PTMs)
Post-translational modifications are covalent processing events that dramatically expand the functional diversity of the proteome, influencing almost all aspects of normal cell biology and pathogenesis [32] [33]. Over 650 types of PTMs have been described, including phosphorylation, glycosylation, ubiquitination, and acetylation [33]. These modifications are essential for proper protein folding, conformation, stability, and biological activity [32]. The capacity of an expression system to perform the necessary PTMs is often the deciding factor for producing a biologically active recombinant protein.
Glycosylation: A Decisive Factor
Among PTMs, glycosylation is one of the most critical for therapeutic proteins due to its profound effects on pharmacokinetics, stability, and immunogenicity [32] [33]. The type of glycosylation varies significantly between expression hosts:
- Mammalian Cells (e.g., CHO, HEK293): Produce complex, terminally sialylated N-glycans that are most similar to human glycoproteins, making them the preferred choice for therapeutics [10] [31].
- Insect Cells: Typically produce paucimannose or oligomannose N-glycans, which lack sialic acid and may contain immunogenic core α(1,3)-fucose modifications [10].
- Yeast: Perform N- and O-linked glycosylation, but the patterns are quite different from mammals. Yeast N-glycosylation is of the high-mannose type, which can be immunogenic in humans [10] [29].
- E. coli: Lacks the cellular machinery for eukaryotic glycosylation, making it unsuitable for producing glycoproteins [10].
The following diagram illustrates the decision-making workflow for selecting an expression system based on protein characteristics and PTM requirements.
Experimental Evidence: The Impact of PTMs on Expression Success
The critical influence of PTMs on heterologous protein production has been demonstrated through systematic studies. One comprehensive analysis expressed 1,488 human proteins in a bacterial cell-free system (E. coli S30 extracts) that has a limited capacity for eukaryotic PTMs [34]. The study revealed statistically significant correlations between the predicted presence of certain PTM sites and the success of soluble protein expression.
Table 2: Correlation Between Predicted PTMs and Soluble Expression in a Bacterial System
| Post-Translational Modification | Correlation with Soluble Expression | Potential Rationale |
|---|---|---|
| Myristoylation | Negative [34] | Incorrect membrane targeting in a prokaryotic environment. |
| Glycosylation (N-linked) | Negative [34] | Lack of glycosylation machinery leads to improper folding and aggregation. |
| Disulfide Bond Formation | Negative [34] | The reducing cytoplasm of E. coli hinders correct bond formation. |
| Palmitoylation | Negative [34] | Disruption of membrane association and protein function. |
| Phosphorylation | Positive [34] | Phosphorylation sites may correlate with structural disorder or regulatory regions that are more soluble. |
| Ubiquitination | Positive [34] | Sites may be surface-exposed and located in unstructured regions. |
These findings underscore that the inability of a host system to support required PTMs is a major cause of low yield, poor solubility, and loss of biological activity in recombinant proteins [34]. The experimental protocol for such studies typically involves:
- Cell-Free Expression: Using E. coli S30 extracts for coupled transcription/translation from linear DNA templates under uniform conditions [34].
- Fractionation: Separating soluble and insoluble reaction products by centrifugation at 10,000 × g.
- Analysis: Evaluating yields via SDS-PAGE and protein staining, categorizing proteins as soluble (A), insoluble (C), or not expressed (N).
- Bioinformatic Prediction: Using tools like PROSITE, CSS-Palm, and UbPred to predict PTM sites in the expressed sequences [34].
- Statistical Analysis: Applying categorical data analysis (e.g., Fisher's exact test) to determine significant correlations between PTM prediction and expression success [34].
The Scientist's Toolkit: Essential Reagents for Heterologous Expression
Successful recombinant protein production relies on a suite of specialized reagents and genetic tools. The following table details key solutions for constructing and optimizing expression in different hosts.
Table 3: Key Research Reagent Solutions for Heterologous Expression
| Reagent / Tool | Function | Application Notes |
|---|---|---|
| Expression Vectors | Plasmids carrying regulatory elements (promoter, origin, tag) to control target gene expression [10] [11]. | Choice of promoter (e.g., T7, AOX1, CMV) is host-specific and critical for yield and regulation [11] [29]. |
| Specialized Host Strains | Engineered cells optimized for specific challenges like codon usage, disulfide bond formation, or toxic protein expression [29]. | E. coli BL21(DE3) derivatives (e.g., Rosetta for rare codons, Origami for disulfide bonds) are widely used [29]. |
| Affinity Tags | Short peptide sequences (e.g., His-tag, GST-tag) fused to the target protein to facilitate purification [11]. | Can influence protein solubility and yield. Removal may require a subsequent cleavage step [11]. |
| Culture Media | Optimized formulations providing nutrients, buffers, and inducers for cell growth and protein production. | Critical for achieving high cell density and yield; cost varies significantly between systems (low for bacteria, high for mammalian) [30]. |
| Transfection Reagents | Chemical or polymer-based agents to introduce DNA into mammalian or insect cells. | Essential for transient expression in mammalian cells (e.g., HEK293); efficiency is key for high yield [29]. |
The selection of a heterologous expression host is a strategic decision that balances practical constraints against biological requirements. E. coli remains the system of choice for high-yield, low-cost production of proteins that are small, soluble, and do not require eukaryotic PTMs. Mammalian cells are indispensable for producing the most complex therapeutic proteins, such as monoclonal antibodies, where authentic glycosylation is a prerequisite for biological activity and regulatory approval. Yeast systems effectively bridge the gap, offering a robust and scalable platform for proteins that benefit from eukaryotic secretion and folding mechanisms but are tolerant of non-human glycosylation.
There is no single "best" system; the optimal choice is entirely dependent on the characteristics of the target protein and the ultimate application of the final product. By applying the key criteria of yield, cost, scalability, and PTMs, researchers can make an informed selection that maximizes the likelihood of successful recombinant protein production.
Implementation in Practice: Expression Vectors, Cultivation, and Host-Specific Workflows
The selection of an appropriate host organism—bacterial, yeast, or mammalian cells—is a foundational decision in heterologous protein expression research. This choice directly dictates the design of the expression vector, a critical tool for delivering and maintaining the gene of interest within the host. The performance of a vector is governed by its key components: the promoter to drive transcription, selectable markers to maintain plasmid pressure, and signal peptides to direct protein localization. This guide provides a objective comparison of these essential elements across the three primary host systems, equipping researchers and drug development professionals with the data needed to optimize their experimental outcomes.
Core Components of Expression Vectors: A Comparative Analysis
The table below summarizes the characteristics of essential vector components across different host systems.
Table 1: Comparison of Core Vector Components Across Host Systems
| Vector Component | Bacterial Systems (E. coli) | Yeast Systems (e.g., S. cerevisiae, P. pastoris) | Mammalian Systems (e.g., HEK293, CHO) |
|---|---|---|---|
| Common Promoters | T7, lac, trp, tac [10] | GAL1, AOX1 (P. pastoris), GAP [35] | CMV, EF-1α, SV40 [35] |
| Induction Method | IPTG (for T7/lac), Temperature | Galactose (for GAL1), Methanol (for AOX1) | No induction required for constitutive promoters; Tetracycline for Tet-On/Off systems |
| Common Selectable Markers | Antibiotic resistance (Ampicillin, Kanamycin) [10] | Amino acid prototrophy (URA3, LEU2), Antibiotic resistance (G418, Zeocin) [35] | Antibiotic resistance (Puromycin, G418/Geneticin), Metabolic (DHFR, GS) [35] |
| Common Signal Peptides | PelB, OmpA, DsbA (for periplasmic secretion) [10] | α-factor (S. cerevisiae), PHO1 (P. pastoris) | Native leader sequences (e.g., for Antibodies) |
| Typical Secretion Pathway | Sec (post-translational) or SRP (co-translational) to periplasm [10] | ER → Golgi → Extracellular medium | ER → Golgi → Extracellular medium |
Experimental Performance Data and Protocols
The selection of a host system and vector design has a direct and measurable impact on protein yield and quality. The following section presents experimental data and detailed methodologies for key studies.
Quantitative Yield Comparison Across Host Systems
Table 2: Representative Protein Yields from Different Host Systems
| Host System | Example Protein | Yield | Experimental Notes | Source |
|---|---|---|---|---|
| Bacterial (E. coli) | Not Specified | Varies widely | Well-suited for prokaryotic proteins and simple eukaryotic proteins without complex PTMs; can form insoluble aggregates. [10] | [10] |
| Yeast (P. pastoris) | Not Specified | High (multi-gram/L scale) | Scalable with simple growth media; suitable for large-scale production. [35] | [35] |
| Insect Cells (Baculovirus) | Recombinant Proteins | Up to 500 mg/L | Robust system for complex proteins and virus-like particles (VLPs). [35] | [35] |
| Mammalian (CHO/HEK293) | Complex Biologics (e.g., mAbs) | Good, can be optimized | Essential for proteins requiring human-like PTMs; yield can be improved via vector and cell line engineering. [35] | [35] |
| Plant (N. benthamiana) | GFP (via optimized PVX vector) | 0.50 mg/g Fresh Weight | Achieved with a viral vector engineered to co-express a silencing suppressor; represents a 3-4 fold increase over the base system. [36] | [36] |
Detailed Experimental Protocol: Enhancing Plant-Based Expression with Viral Vectors
A 2025 study provides a clear example of how vector engineering can dramatically enhance protein yield by addressing a key host defense mechanism. The following workflow and protocol detail this approach [36].
Title: Workflow for Engineering Enhanced PVX Expression Vectors
Key Reagents and Materials:
- Plant Material: Nicotiana benthamiana plants, 4-5 weeks old.
- Vector Backbone: Deconstructed Potato Virus X (PVX) vectors (e.g., pP1, pP2, pP3).
- Viral Suppressors of RNA Silencing (VSRs): Genes for P19 (Tomato bushy stunt virus), P38 (Turnip crinkle virus), or NSs (Tomato zonate spot virus).
- Agrobacterium tumefaciens: Strain GV3101.
- Culture Media: LB broth with appropriate antibiotics (e.g., Kanamycin, Rifampicin).
- Infiltration Buffer: 10 mM MES, 10 mM MgCl₂, 150 µM Acetosyringone, pH 5.6.
- Analysis Reagents: SDS-PAGE gels, primary and secondary antibodies for Western blot, GFP-specific ELISA kits.
Detailed Methodology [36]:
- Vector Construction: The heterologous VSR gene (P19, P38, or NSs) is cloned into a deconstructed PVX vector under the control of the CaMV 35S promoter. The nopaline synthase (NOS) terminator is used. Critically, the VSR cassette is placed in a reverse orientation relative to the target gene to minimize transcriptional interference.
- Agrobacterium Preparation: The constructed plasmid is transformed into Agrobacterium tumefaciens. A single colony is used to inoculate a starter culture, which is grown overnight. The cells are then pelleted and resuspended in infiltration buffer to an optical density (OD₆₀₀) of 0.5-1.0. The culture is incubated for 2-4 hours at room temperature.
- Plant Infiltration: The Agrobacterium suspension is infiltrated into the abaxial side of N. benthamiana leaves using a needleless syringe.
- Incubation and Harvest: Infiltrated plants are maintained in growth chambers for 3-5 days. The infiltrated leaf tissue is harvested, flash-frozen in liquid nitrogen, and stored at -80°C until analysis.
- Protein Analysis: Total soluble protein is extracted from the ground leaf tissue. The concentration of the target recombinant protein (e.g., GFP, VP1, S2) is quantified using Western blot analysis and/or antigen-specific ELISA.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Heterologous Expression Research
| Item | Function in Research | Example Applications |
|---|---|---|
| VSRs (Viral Suppressors of RNAi) | Enhance recombinant protein yield by inhibiting the host's RNA silencing machinery. | Boosting antigen expression in plant systems (e.g., using P19 or NSs) [36]. |
| PEI (Polyethylenimine) | A chemical transfection reagent for delivering DNA into mammalian cells. | Transient gene expression in HEK293 cells for rapid protein production [35]. |
| Acetosyringone | A phenolic compound that induces the Vir genes in Agrobacterium tumefaciens. | Essential for efficient T-DNA transfer during agro-infiltration of plants [36]. |
| IPTG (Isopropyl β-D-1-thiogalactopyranoside) | A molecular mimic of allolactose that induces the lac and T7 lac promoters. | Triggering protein expression in E. coli expression systems [10]. |
| Geneticin (G418) | An aminoglycoside antibiotic that inhibits protein synthesis in eukaryotic cells. | Selection of stable mammalian and yeast cell lines expressing the neomycin resistance gene [35]. |
The choice between bacterial, yeast, and mammalian hosts for heterologous expression is not a one-size-fits-all decision but a strategic trade-off. Bacterial systems offer unmatched speed and cost-effectiveness for simple proteins. Yeast systems strike a balance, providing eukaryotic processing capabilities at a prokaryotic scale. Mammalian cells remain the gold standard for producing the most complex therapeutic proteins requiring authentic human post-translational modifications. As demonstrated by advanced plant expression systems, yield limitations in any host can be overcome through sophisticated vector engineering, such as the incorporation of VSRs. The most successful expression strategy is therefore one that aligns the target protein's biochemical requirements with the host's inherent strengths, guided by the rational design of its expression vector.
The selection of an appropriate gene delivery method is a critical step in heterologous protein expression, directly influencing the success and efficiency of downstream research and therapeutic development. These techniques form the essential bridge between genetic engineering and functional protein production, enabling scientists to introduce foreign DNA into host organisms ranging from simple bacteria to complex mammalian cells. The choice of method is intrinsically linked to the selected host system—bacterial, yeast, or mammalian—each presenting unique cellular barriers and requirements. This guide provides a comparative analysis of foundational and advanced gene delivery technologies, offering objective performance data and detailed protocols to inform researchers' experimental design. By examining techniques from classical heat shock to sophisticated viral transduction, we aim to equip scientists with the knowledge to select the optimal strategy for their specific expression host and research goals.
Core Techniques and Their Mechanisms
Bacterial Transformation: Heat Shock and Electroporation
In bacterial systems, such as E. coli, transformation introduces plasmid DNA into cells. Heat shock remains a cornerstone technique, utilizing a brief 42°C thermal pulse to create a temperature gradient that induces membrane fluidity and DNA uptake [12]. The process relies on chemically competent cells treated with calcium chloride to neutralize DNA charge and facilitate binding. Alternatively, electroporation uses a high-voltage electrical pulse to create transient pores in the cell membrane, allowing DNA entry. This method is highly efficient for large DNA constructs and requires cells to be prepared in a low-conductivity buffer to prevent arcing [12].
Yeast Transformation: Lithium Acetate and Electroporation
Yeast transformation techniques must overcome the robust cell wall. The lithium acetate (LiAc) method involves incubating cells with LiAc, which alters membrane structure, followed by a heat shock in the presence of single-stranded carrier DNA that competes with genomic DNA for non-specific binding sites [37]. This is effective for both replicating plasmids and genomic integration. Electroporation is also highly effective in yeast, often yielding high transformation efficiencies, particularly for laborious library constructions [37]. For specialized applications, PEG-mediated spheroplast fusion is used, where the cell wall is enzymatically removed with Zymolyase, and the resulting spheroplasts are fused with other cells or organelles using polyethylene glycol (PEG) to deliver entire chromosomes or large DNA cargoes [38].
Mammalian Cell Transfection: Chemical and Physical Methods
Mammalian cell transfection is more complex due to the absence of a cell wall and the presence of a nucleus. Lipofection uses cationic lipids that encapsulate nucleic acids to form liposomes, which fuse with the plasma membrane and release their cargo into the cytoplasm [35] [27]. Calcium phosphate co-precipitation involves mixing DNA with calcium chloride and adding it to a phosphate-buffered solution, forming a fine precipitate that settles onto cells and is internalized by endocytosis [39]. Polyethyleneimine (PEI) is a synthetic polymer that condenses DNA into positively charged nanoparticles, which adhere to the cell surface and enter via endocytosis [39]. Electroporation is also widely used for mammalian cells, especially those difficult to transfect with chemical methods, by applying a controlled electrical field to create nanopores [27].
Viral Transduction in Mammalian Systems
Viral transduction uses engineered viruses to achieve high-efficiency gene delivery, even in non-dividing cells. Key viral vectors include:
- Lentiviruses (LVs): RNA viruses that provide stable genomic integration in both dividing and non-dividing cells, enabling long-term transgene expression. Modern self-inactivating (SIN) designs have improved safety [40].
- Adenoviruses (AVs): DNA viruses that remain episomal, resulting in high-level but transient transgene expression. Their pronounced immunogenicity and limited payload capacity (~8 kb) can be constraints [40].
- Adeno-Associated Viruses (AAVs): Small, non-integrating viruses with a favorable safety profile, suitable for transducing delicate immune cells, though they have a small payload capacity (~4.7 kb) [40].
- BacMam System: A hybrid system utilizing modified baculoviruses, which are engineered with mammalian promoters to deliver genes to mammalian cells. This safe system is unable to replicate in human cells and is used for both transient and stable expression [35].
Table 1: Summary of Core Gene Delivery Techniques by Host System
| Host System | Technique | Mechanism of Action | Primary Use Case |
|---|---|---|---|
| Bacterial | Heat Shock | Calcium chloride pre-treatment creates membrane competence; heat pulse drives DNA uptake [12]. | Routine plasmid propagation in E. coli. |
| Electroporation | Electrical pulse creates transient pores in cell membrane [12]. | Large plasmids or library construction. | |
| Yeast | Lithium Acetate (LiAc) | Alkali cation alters cell wall & membrane; heat shock drives DNA uptake [37]. | Standard plasmid introduction and genomic integration. |
| Electroporation | Electrical pulse creates transient pores in cell wall and membrane [37]. | High-efficiency transformation, especially for libraries. | |
| PEG-mediated Spheroplast Fusion | Cell wall is enzymatically removed; PEG fuses spheroplasts to deliver cargo [38]. | Delivery of very large DNA constructs (e.g., entire chromosomes). | |
| Mammalian | Lipofection | Cationic lipids form liposomes that fuse with plasma membrane [35] [27]. | Broadly applicable transient or stable transfection. |
| Calcium Phosphate | DNA-calcium phosphate precipitate is internalized by endocytosis [39]. | Cost-effective transient transfection, particularly of HEK293 cells. | |
| Polyethyleneimine (PEI) | Cationic polymer condenses DNA into nanoparticles for endocytosis [39]. | Large-scale transient transfection (e.g., in bioreactors). | |
| Electroporation | Electrical pulse creates transient pores in plasma membrane [27]. | Hard-to-transfect cells (e.g., primary cells, immune cells). | |
| Viral Transduction (LV, AV, AAV) | Engineered virus particles bind cell surface receptors and deliver genetic material via viral entry pathways [40]. | High-efficiency gene delivery, stable cell line generation, and hard-to-transfect cells. |
Comparative Performance Data
The efficiency of a gene delivery method is a key determinant for experimental success, but it must be balanced against practical considerations like cost, scalability, and technical accessibility. Performance is highly dependent on the host cell system.
In bacterial and yeast systems, transformation efficiencies are typically quantified as colony-forming units (CFUs) per microgram of DNA. Electroporation generally surpasses chemical methods, often yielding efficiencies exceeding 10⁸ CFU/µg in optimized E. coli strains and 10⁵ to 10⁶ transformants/µg in yeast [12] [37]. These microbial systems offer rapid turnaround, with transformed colonies often obtained within 24 hours.
For mammalian cells, performance metrics are more varied. Standard chemical transfections (e.g., lipofection, PEI) in HEK293 cells can achieve high efficiency, with 50-80% of cells expressing a transgene like GFP [39]. However, viral transduction consistently delivers superior efficiency, particularly for challenging primary cells. In clinical CAR-T cell manufacturing, lentiviral transduction efficiencies typically range from 30% to 70% [40]. Advanced methods like virus-free PASSIGE (prime-editing-assisted site-specific integrase gene editing) with evolved recombinases have reported targeted integration efficiencies of up to 60% in human cell lines and over 30% in primary human fibroblasts [41].
Table 2: Experimental Performance and Practical Considerations
| Technique | Typical Efficiency | Timeline | Cost & Scalability | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Heat Shock | ~10⁷ - 10⁸ CFU/µg (Bacteria) [12] | 1-2 days | Low cost; highly scalable. | Simplicity, reliability, low cost. | Lower efficiency for large plasmids. |
| LiAc Yeast | ~10⁴ - 10⁶ transformants/µg [37] | 2-3 days | Low cost; scalable. | Amenable to genomic integration. | Requires optimized protocol. |
| Lipofection | 50-80% (e.g., HEK293) [39] | 1-3 days (transient) | Moderate cost; scalable with optimized reagents. | Broad cell type applicability. | Cytotoxicity at high doses; cost for large scale. |
| PEI Transfection | High in suspension HEK293 | 1-3 days (transient) | Low cost; excellent for large-scale transient transfection [39]. | Cost-effective for liter-scale production. | Can be cytotoxic; requires optimization. |
| Electroporation (Mammalian) | Varies by cell type | 1-3 days | High equipment cost; scalable with specialized devices. | Works on hard-to-transfect cells. | High cell death if not optimized; specialized equipment. |
| Lentiviral Transduction | 30-70% (e.g., T cells) [40] | Weeks (incl. virus production) | High cost; scalable production possible but complex. | Stable integration in dividing & non-dividing cells. | Biosafety level 2+; insertional mutagenesis risk (low with SIN designs). |
| BacMam System | High in many mammalian lines [35] | 1-2 weeks (incl. virus production) | Moderate cost; scalable. | Safe (non-replicating in mammals); high protein yields reported. | Transient expression; requires baculovirus production. |
Detailed Experimental Protocols
Protocol 1: Lithium Acetate Transformation ofS. cerevisiae
This is a standard chemical method for introducing DNA into yeast cells [37].
- Growth of Competent Cells: Inoculate a fresh colony of S. cerevisiae into 5 mL of YPD or selective medium. Grow overnight at 30°C with shaking until the OD600 reaches 0.5-1.0.
- Cell Harvesting and Washing: Pellet cells by centrifugation at 3000 × g for 5 minutes. Wash the pellet first with 5 mL of sterile water, then with 5 mL of 100 mM lithium acetate (LiAc), and resuspend the final pellet in 500 µL of 100 mM LiAc.
- Preparation of DNA Mix: For a single transformation, mix 100-500 ng of plasmid DNA and 100 µg of denatured, single-stranded carrier DNA (e.g., salmon sperm DNA) in a sterile microcentrifuge tube.
- Incubation with Cells: Add 50 µL of the competent cell suspension to the DNA mix. Vortex to mix.
- PEG Treatment: Add 300 µL of 40% polyethylene glycol (PEG) 3350 in 100 mM LiAc. Mix thoroughly by vortexing and incubate at 30°C for 30 minutes.
- Heat Shock: Transfer the tube to a 42°C water bath for 15-25 minutes.
- Plating and Selection: Pellet the cells briefly, remove the supernatant, and resuspend in 100-200 µL of sterile water. Plate the entire suspension onto the appropriate selective agar medium and incubate at 30°C for 2-4 days until colonies appear.
Protocol 2: Viral Transduction of Human T Cells for Cell Therapy
This protocol outlines the key steps for genetically modifying immune cells, such as T cells, using lentiviral vectors [40].
- Cell Activation: Isolate primary T cells from donor blood or leukapheresis product. Activate the T cells by culturing with anti-CD3/CD28 antibodies for 24-48 hours. This upregulates receptor expression and increases susceptibility to transduction.
- Optimization of Parameters: Prior to the main experiment, titrate the Multiplicity of Infection (MOI), which is the ratio of infectious viral particles to target cells. An optimal MOI (often between 1 and 10) balances high transduction efficiency with low cytotoxicity and ensures a safe Vector Copy Number (VCN) below 5 copies per cell [40].
- Transduction Process: Seed the activated T cells in retronectin-coated plates or with a transduction enhancer like Polybrene. Add the calculated volume of lentiviral vector supernatant to the cells. To enhance cell-virus contact, perform spinoculation by centrifuging the plate at 800-1200 × g for 30-90 minutes at 32°C.
- Post-Transduction Culture: After incubation (typically 6-24 hours), remove the virus-containing medium and replace it with fresh culture medium supplemented with cytokines (e.g., IL-2, IL-7, IL-15) to support cell survival, expansion, and function.
- Analysis: 48-96 hours post-transduction, analyze transduction efficiency by flow cytometry for surface marker expression or GFP expression. Quantify VCN by droplet digital PCR (ddPCR) to ensure safety specifications are met [40].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Kits for Gene Delivery
| Reagent/Kits | Function | Example Applications |
|---|---|---|
| Zymolyase | An enzyme complex (β-1,3-glucanase) that digests the yeast cell wall to generate spheroplasts for fusion-based delivery [38]. | PEG-mediated spheroplast fusion for delivering large DNA cargo. |
| Polyethyleneimine (PEI) | A cationic polymer that condenses DNA into nanoparticles, facilitating cellular uptake via endocytosis. A cost-effective transfection reagent [39]. | Large-scale transient protein production in HEK293 or CHO suspension cells. |
| Lentiviral Vectors (VSV-G pseudotyped) | Engineered lentiviruses with a broad tropism envelope protein (VSV-G) that enables efficient gene delivery to a wide range of mammalian cell types, including non-dividing cells [40]. | Creating stable cell lines, gene delivery to primary cells (e.g., T cells, NK cells), and gene function studies. |
| BacMam Technology | A baculovirus-based vector system engineered to carry a gene of interest under a mammalian promoter for efficient transduction of mammalian cells [35]. | Safe and high-yield protein production in a variety of mammalian cell lines without viral replication. |
| ExpiFectamine 293 Transfection Kit | A proprietary, cationic lipid-based transfection reagent system optimized for high-density suspension cultures of HEK293 cells [27]. | High-yield transient protein expression for research and pre-clinical biologics production. |
| Jump-In T-REx System | A suite of technologies for creating mammalian cell lines with targeted, single-copy integration of a gene of interest, coupled with inducible expression [27]. | Production of toxic proteins or tightly regulated, consistent expression for functional studies. |
Decision Workflow for Technique Selection
The following diagram illustrates a logical workflow for selecting the most appropriate gene delivery technique based on key experimental parameters.
Technique Selection Workflow
The landscape of transformation and transfection techniques offers a diverse toolkit for heterologous expression across bacterial, yeast, and mammalian hosts. The optimal choice is not a one-size-fits-all solution but a strategic decision based on the host system, the nature of the genetic cargo, the requirement for transient or stable expression, and the desired throughput and efficiency. While microbial systems provide speed and simplicity, mammalian systems, empowered by advanced chemical and viral methods, are indispensable for producing complex, therapeutically relevant proteins with proper post-translational modifications. As the field progresses, emerging technologies like PASSIGE with evolved recombinases are pushing the boundaries of efficiency and precision for large DNA integration [41]. By understanding the principles, performance data, and protocols outlined in this guide, researchers can rationally select and optimize the most effective gene delivery method to advance their scientific and therapeutic objectives.
Transitioning from small-scale shake flasks to controlled bioreactors represents a critical juncture in bioprocess development, particularly within the context of selecting appropriate hosts for heterologous protein expression. This scale-up is essential for translating laboratory research into commercially viable processes in the biopharmaceutical, biofuel, and industrial enzyme sectors. The selection of an expression host—bacterial, yeast, or mammalian cells—profoundly influences the strategy and success of this scale-up, as each system presents unique metabolic, physiological, and biosynthetic challenges. While shake flasks are indispensable for initial screening and media optimization, they lack the controlled environment necessary to predict performance in large-scale production bioreactors accurately. Understanding the technical distinctions between these cultivation systems enables scientists and drug development professionals to design more efficient and predictive scale-up workflows, ultimately accelerating the development timeline for new biologics and recombinant products.
Shake Flasks vs. Bioreactors: A Systematic Comparison
The fundamental differences between shake flasks and bioreactors extend beyond simple volume increase. They represent a shift from a largely uncontrolled environment to a highly monitored and regulated one, directly impacting cell physiology and product yield.
Table 1: Key Parameter Comparison Between Shake Flasks and Bioreactors
| Parameter | Shake Flask | Bioreactor |
|---|---|---|
| Temperature Control | ✓ (Incubator-level, all flasks) | ✓ (Individual vessel) |
| Agitation | ✓ (Orbital shaking) | ✓ (Impeller stirring) |
| pH Control | (✓) (Requires additional equipment) | ✓ (Direct, automated) |
| Dissolved Oxygen (pO₂) | (✓) (Limited, surface aeration) | ✓ (Direct, via sparging & agitation) |
| Gas Flow Control | (✓) (Limited) | ✓ (Precise O₂, N₂, CO₂, air blending) |
| Feed Strategies | (✓) (Manual, batch) | ✓ (Automated fed-batch, perfusion) |
| Exhaust Gas Analysis | (✓) (Rare) | ✓ (For metabolic monitoring) |
| Working Volume | Typically < 1 L | Millilitres to thousands of litres |
| Scale-Up Relevance | Low (Different mixing/O₂ principles) | High (Mimics production-scale STRs) |
Table 2: Comparative Performance Metrics for Different Host Cells
| Host System / Condition | Maximum Cell Density (Cells/mL) or OD | Key Scale-Up Finding | Source |
|---|---|---|---|
| E. coli (Shake Flask) | OD₆₀₀ ~ 4-6 | Baseline for high-growth prokaryotes. | [42] |
| E. coli (Bioreactor, Batch) | OD₆₀₀ ~ 14-20 | Superior mixing and aeration in a bioreactor. | [42] |
| CHO Cells (Shake Flask) | ~0.94 x 10⁷ cells/mL | Lower maximum density vs. bioreactors. | [43] |
| CHO Cells (Bioreactor) | ~1.5 x 10⁷ cells/mL | 60% higher max cell density achieved. | [43] |
| DuckCelt-T17 (Avian, Fed-Batch) | Significant improvement | Fed-batch strategy improved growth & viability. | [44] |
| Pichia pastoris (Bioreactor, High Aeration) | OD >20 | High O₂ transfer enables very high densities. | [45] |
The data reveals a consistent trend across diverse host systems: bioreactors facilitate significantly higher cell densities. This is primarily due to superior oxygen mass transfer (kLa) and advanced process control. For instance, Chinese Hamster Ovary (CHO) cells, a cornerstone for therapeutic protein production, achieved a 60% higher maximum cell density in bioreactors compared to shake flasks [43]. Similarly, E. coli cultures can reach optical densities (OD₆₀₀) several times greater in a controlled bioreactor environment than in shake flasks [42].
Beyond quantitative yield, the culture environment fundamentally alters cell physiology. A proteomic study demonstrated that CHO cells in shake flasks and bioreactors present different host cell protein (HCP) profiles in the supernatant, a critical consideration for downstream purification in drug manufacturing [43]. This implies that data from flask cultures, while valuable for early development, may not fully predict the impurity profile at commercial scale.
Expression Hosts and Their Scale-Up Characteristics
The choice of host organism—bacteria, yeast, or mammalian cells—dictates the complexity of the scale-up process, driven by differences in cellular structure, metabolic pathways, and product requirements.
Table 3: Heterologous Expression Hosts: Advantages and Scale-Up Challenges
| Host System | Key Advantages | Primary Scale-Up Challenges | Example Product |
|---|---|---|---|
| Bacterial (e.g., E. coli) | Rapid growth, high yields, simple media, extensive genetic tools. [4] | Inclusion body formation, endotoxin removal, lack of complex PTMs. [4] | Human insulin [12] |
| Yeast (e.g., S. cerevisiae, K. phaffii) | Eukaryotic PTMs (glycosylation), high-density growth, Crabtree-negative species allow efficient respiration. [12] | Hypermannosylation (non-human glycosylation), protease activity, oxygen demand at high cell density. [45] [12] | Hepatitis B vaccine, Human serum albumin [12] |
| Mammalian (e.g., CHO, HEK293) | Most complex & human-like PTMs, correct folding for complex biologics. [4] | Low volumetric yield, expensive media, shear sensitivity, viral contamination risk. [43] | Monoclonal antibodies [43] |
- Bacterial Systems: E. coli remains a workhorse due to its simplicity and productivity. Scale-up in bioreactors focuses on achieving very high cell densities through tight control of nutrient feeding and oxygen supply to prevent acetate formation [42].
- Yeast Systems: Crabtree-negative yeasts like Komagataella phaffii (Pichia pastoris) are particularly suited for bioreactor cultivation because they do not ferment glucose to ethanol under oxygen-limited conditions, enabling efficient respiratory metabolism and very high cell densities [12]. This makes them excellent hosts for recombinant protein production, with processes requiring high oxygen transfer rates, as demonstrated with P. pastoris, where increasing the aeration rate directly boosted final culture density [45].
- Mammalian Systems: Scale-up is driven by the need for complex post-translational modifications. The move to bioreactors is crucial not only for increasing yield but also for ensuring consistent product quality. Bioreactors allow for control over parameters like dissolved CO₂ and pH, which can significantly impact glycosylation patterns and the HCP profile, critical quality attributes for therapeutics [46] [43].
Methodologies: From Flask to Bioreactor Experiments
A successful scale-up requires a methodical and data-driven approach. The following workflow and experimental strategies are commonly employed.
Experimental Workflow for Process Development
Key Experimental Protocols
1. Shake Flask Supplementation Studies: As performed for the DuckCelt-T17 avian cell line, this involves culturing cells in shake flasks with various nutrient supplements. For example, L-glutamine can be compared to more stable alternatives like GlutaMAX, or fed-batch strategies can be mimicked by bolus feeding on days 3 and 6. Cultures are monitored daily for growth, viability, and metabolite consumption (glucose, glutamine) and production (lactate, ammonium) to identify optimal feeding strategies before moving to a bioreactor [44].
2. Bioreactor Scale-Up with Parameter Control: A typical lab-scale bioreactor experiment (e.g., in a 3L vessel) involves inoculating the optimized culture from shake flasks. Key parameters like temperature, pH, and dissolved oxygen (dO₂) are tightly controlled. The dO₂ is often maintained via cascades that adjust agitation, gas flow, and oxygen blending. The impact of aeration strategy is critical; for example, reducing the initial sparge rate in a 3L bioreactor was shown to better mimic large-scale conditions by avoiding excessively low pCO₂ levels [46].
3. Perfusion Feasibility Testing: At the lab-scale, a perfusion test can be conducted where fresh media is continuously added, and spent media is harvested while cells are retained. This strategy, which achieved ~3 times the maximum viable cell count of batch cultures in one study, is investigated for its potential to enable continuous virus harvesting or to maintain high cell densities for continuous production [44].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Reagents and Materials for Cell Culture Scale-Up
| Reagent/Material | Function in Scale-Up | Example |
|---|---|---|
| Serum-Free Medium | Defined, animal-origin-free base medium supporting growth and production; essential for therapeutic protein consistency. | OptiPRO SFM [44] |
| Stable Glutamine Source | Provides a essential amino acid for energy and biosynthesis; more stable alternatives prevent ammonia buildup. | GlutaMAX [44] |
| Antifoam Agent | Suppresses foam formation caused by sparging and agitation in bioreactors, preventing overflow and contamination. | Pluronic F-68 [44] |
| pH Control Solutions | Acids and bases for automated, two-sided pH control to maintain optimal physiological range for the host. | Sodium carbonate, NaHCO₃ [42] |
| Supplemental Nutrients | Concentrated feeds for fed-batch processes to extend culture duration and increase cell density and productivity. | Glucose solutions, Yeast Extract [44] [45] |
| Single-Use Bioreactor Vessel | Pre-sterilized, disposable bag for a single batch; eliminates cleaning validation and cross-contamination risk. | CellexusBag [45] |
The journey from shake flasks to industrial bioreactors is a cornerstone of modern bioprocess development. This transition is not merely an increase in volume but a fundamental shift towards a controlled, monitored, and automated environment that unlocks the full potential of bacterial, yeast, and mammalian host systems. While shake flasks remain invaluable for initial strain screening and basic optimization, bioreactors are indispensable for achieving the high cell densities and, more importantly, the consistent product quality required for commercial-scale manufacturing. The increasing adoption of single-use systems and advanced feeding strategies like perfusion further enhances the efficiency and flexibility of scaled-up processes. For researchers and drug developers, a deep understanding of the principles governing this scale-up is essential for successfully translating promising laboratory discoveries into life-saving and market-ready biotechnological products.
Heterologous expression serves as a fundamental technology platform across biotechnology, enabling the production of complex biological products by engineering host organisms to express genes from foreign sources. The selection of an appropriate expression host—whether bacterial, yeast, or mammalian cell systems—represents a critical decision point that profoundly influences the yield, functionality, and scalability of the resulting product. Each host system offers distinct advantages and limitations based on its cellular machinery, post-translational modification capabilities, and scalability. This comparison guide objectively evaluates the performance of these heterologous expression platforms through three key application areas: industrial enzymes, subunit vaccines, and monoclonal antibodies. By examining successful case studies and supporting experimental data, we provide researchers, scientists, and drug development professionals with a practical framework for selecting expression systems based on empirical evidence rather than theoretical considerations alone.
Bacterial Expression Systems
Bacterial systems, particularly Escherichia coli and various Burkholderia species, represent the most established and widely utilized platforms for heterologous protein production due to their rapid growth, well-characterized genetics, and cost-effective cultivation. The simplicity of bacterial systems makes them ideal for producing a wide range of industrial enzymes and simple protein therapeutics that do not require complex eukaryotic post-translational modifications. Recent advances in synthetic biology and metabolic engineering have further expanded their capabilities, enabling the production of more complex natural products and biomolecules through sophisticated engineering approaches [47] [48].
Case Study: Natural Product Production in EngineeredBurkholderiaSpecies
Burkholderia bacteria have emerged as particularly promising hosts for expressing complex natural products due to their intrinsic biosynthetic capabilities and metabolic versatility. These organisms naturally produce a diverse array of bioactive compounds and can be engineered to express biosynthetic gene clusters (BGCs) from related species.
Experimental Protocol:
- Strain Engineering: The chassis strain Burkholderia thailandensis E264 was generated through targeted deletions of endogenous biosynthetic gene clusters (Δtdp::attB) and efflux systems (ΔBAC::attB and ΔoprC::attB) to minimize background interference and enhance product accumulation [47].
- Vector Design: ϕC31 integrative vectors containing constitutive promoters (Pgenta) and native E264 promoters were constructed to drive expression of heterologous biosynthetic pathways [47].
- Culture Conditions: Fermentation was conducted in optimized media with continuous monitoring of metabolic precursors to maximize titers.
- Product Analysis: Natural product isolation and quantification were performed using HPLC-MS/MS with comparison to authentic standards.
Performance Data: The platform achieved remarkable production levels, including 985 mg/L of FK228 (romidepsin), a histone deacetylase inhibitor used in T-cell lymphoma treatment [47]. This represents one of the highest reported titers for this complex natural product in any heterologous system.
Advantages and Limitations:
- Advantages: High precursor availability for natural product biosynthesis; ability to harbor and express large gene clusters; established synthetic biology tools [47].
- Limitations: Potential pathogenicity concerns with some species; less developed for eukaryotic protein production; limited glycosylation capabilities [47].
Case Study: Streptomyces Platform for Natural Product Discovery
The Micro-HEP platform utilizes engineered Streptomyces coelicolor A3(2)-2023 as a chassis for expressing cryptic biosynthetic gene clusters discovered through genome mining.
Experimental Protocol:
- Host Engineering: Four endogenous BGCs were deleted from S. coelicolor to reduce metabolic competition, and multiple recombinase-mediated cassette exchange sites were introduced to facilitate stable integration of heterologous gene clusters [48].
- DNA Assembly: BGCs were cloned and modified in specialized E. coli strains containing a rhamnose-inducible Redαβγ recombination system before transfer to Streptomyces via conjugation [48].
- Expression Optimization: Copy number optimization was achieved through recombinase-mediated cassette exchange, with 2-4 copies of the xiamenmycin BGC integrated, demonstrating a direct correlation between copy number and product yield [48].
Performance Data: The platform successfully produced xiamenmycin (anti-fibrotic compound) and identified the new natural product griseorhodin H, demonstrating its utility in natural product discovery [48].
Figure 1: Bacterial Natural Product Expression Workflow. This diagram illustrates the multi-stage process for heterologous expression of natural products in engineered bacterial hosts, from biosynthetic gene cluster identification to optimized production.
Yeast Expression Systems
Yeast expression systems, particularly Saccharomyces cerevisiae, occupy a unique niche between prokaryotic simplicity and eukaryotic complexity. As generally recognized as safe (GRAS) organisms, yeast platforms combine the advantages of rapid growth and easy scale-up with the ability to perform many eukaryotic post-translational modifications. This makes them particularly valuable for producing proteins that require proper folding, disulfide bond formation, or basic glycosylation but do not demand the complex human-like glycosylation patterns necessary for certain therapeutic proteins [49].
Case Study: Industrial Enzyme Production inS. cerevisiae
S. cerevisiae has been extensively engineered for high-level production of industrial enzymes, leveraging its strong secretion capacity and well-developed genetic tools.
Experimental Protocol:
- Hyperexpression System Design: Strong constitutive or inducible promoters (e.g., PGK1, GPD) were employed to drive high-level transcription of heterologous genes [49].
- Codon Optimization: Gene sequences were optimized to match the preferred codon usage of S. cerevisiae to enhance translational efficiency [49].
- Secretion Engineering: Native signal peptides (e.g., α-factor mating pheromone) were fused to target proteins to facilitate extracellular secretion, simplifying downstream purification [49].
- Glycosylation Pathway Engineering: Humanization of glycosylation pathways was achieved through knockout of endogenous glycosyltransferases (e.g., och1Δ) and introduction of human glycosylation enzymes [49].
Performance Data: Table 1: Representative Heterologous Protein Production in S. cerevisiae
| Protein Type | Specific Product | Titer/Activity | Production Scale | Reference |
|---|---|---|---|---|
| Medicinal Protein | Transferrin | 2.33 g/L | Fed-batch, 10L bioreactor | [49] |
| Food Protein | Brazzein | 9 mg/L | Shake flask | [49] |
| Industrial Enzyme | Lipase | 11,000 U/L | Fed-batch, 5L bioreactor | [49] |
| Industrial Enzyme | Laccase3 | 1176.04 U/L | Shake flask | [49] |
Advantages and Limitations:
- Advantages: GRAS status; strong secretion capacity; well-developed genetic tools; eukaryotic protein processing capabilities [49].
- Limitations: Tendency for hypermannosylation; lower yields for some complex proteins compared to specialized systems; limited capacity for certain human-like post-translational modifications [49].
Case Study: Protein Subunit Vaccine Production
Protein subunit vaccines represent a rapidly advancing application of yeast expression systems, particularly for viral antigens like the SARS-CoV-2 spike protein.
Experimental Protocol:
- Antigen Design: DNA sequences encoding the SARS-CoV-2 spike protein or its receptor-binding domain were codon-optimized for yeast expression [50].
- Strain Engineering: Engineered S. cerevisiae strains with enhanced protein folding capacity and reduced protease activity were employed as hosts [50].
- Fermentation Process: High-cell density fed-batch fermentation was conducted under controlled conditions to maximize antigen yield [50].
- Purification and Formulation: Recombinant antigens were purified using chromatographic methods and combined with appropriate adjuvants (e.g., AS03, CpG/Alum) [50].
Performance Data: The SCB-2019 vaccine developed by Clover Biopharmaceuticals utilizes a trimeric SARS-CoV-2 spike protein (S-Trimer) produced in Chinese hamster ovary (CHO) cells (not yeast, despite initial consideration of yeast platforms), demonstrating the flexibility of eukaryotic systems for complex antigen production [50]. When adjuvanted with either AS03 or CpG/Alum, the vaccine candidate induced potent humoral and cellular immune responses with high virus-neutralizing activity in preclinical models [50].
Mammalian Cell Expression Systems
Mammalian cell systems, primarily Chinese Hamster Ovary (CHO) cells, represent the gold standard for producing complex therapeutic proteins that require authentic human-like post-translational modifications, particularly sophisticated glycosylation patterns. While historically used for monoclonal antibody production, these platforms have expanded to include other complex biologics such as bispecific antibodies, antibody-drug conjugates, and viral antigens for subunit vaccines [50] [51].
Case Study: Monoclonal Antibody Production for Biosimilars
The production of biosimilar monoclonal antibodies requires precise replication of the innovator product's higher-order structure (HOS) to ensure comparable efficacy and safety profiles.
Experimental Protocol:
- Cell Line Development: CHO cells were engineered to express the heavy and light chains of the target monoclonal antibody using strong viral promoters [51].
- Process Optimization: Fed-batch bioreactor processes were optimized for temperature, pH, dissolved oxygen, and nutrient feeding strategies to maximize titer and quality [51].
- HOS Characterization: Antibody array ELISA technology utilizing >30 polyclonal antibodies covering different regions of the mAb molecule was employed to systematically measure surface-epitope distribution and detect conformational differences as small as 0.1% [51].
- Analytical Comparability: Comprehensive analysis included glycosylation profiling, bioassays, and stability studies to demonstrate biosimilarity to the reference product [51].
Performance Data: Table 2: Biosimilar Monoclonal Antibody Higher-Order Structure Comparability
| Case Study | Reference Product | Biosimilar Conformational Similarity | Key Findings | Reference |
|---|---|---|---|---|
| 1 | Trastuzumab | High similarity | No differences >15% RSD across 34 antibody coverage areas; ≤0.1% conformational impurity | [51] |
| 2 | Bevacizumab | Good similarity with minor differences | 0.1-0.2% new epitope exposure; no efficacy difference in bioassays | [51] |
| 3 | Adalimumab | Batch-dependent variation | One batch matched reference; two batches showed 0.1-0.2% unfolding | [51] |
Advantages and Limitations:
- Advantages: Authentic human-like post-translational modifications; proper complex protein folding; established regulatory track record [51].
- Limitations: Higher production costs; longer development timelines; technical complexity of cell culture processes; potential viral contamination risks [51].
Case Study: SARS-CoV-2 Subunit Vaccine Production
Protein subunit vaccines against SARS-CoV-2 represent a significant success story for mammalian expression systems, particularly in responding rapidly to the global pandemic.
Experimental Protocol:
- Antigen Design: DNA sequences encoding the SARS-CoV-2 spike protein or receptor-binding domain were cloned into expression vectors optimized for mammalian cells [50].
- Cell Culture Production: Recombinant proteins were expressed in mammalian cell systems (e.g., CHO cells) using controlled bioreactor processes [50].
- Purification: Target antigens were purified using affinity and chromatographic methods to achieve high purity levels [50].
- Formulation: Purified antigens were combined with appropriate adjuvants (e.g., AS03 for the Sanofi-GSK vaccine) to enhance immunogenicity [50].
Performance Data: The Sanofi-GSK VAT00002 vaccine candidate, containing a recombinant SARS-CoV-2 spike protein produced in insect cells (baculovirus system), demonstrated 95-100% seroconversion rates across all adult age categories in Phase 2 trials, with high neutralizing antibody levels after a single injection in previously infected individuals [50].
Figure 2: Heterologous Expression Host Selection Algorithm. This decision tree guides researchers in selecting appropriate expression systems based on protein requirements, production scale, and post-translational modification needs.
Comparative Performance Analysis
Cross-Platform Performance Metrics
Direct comparison of different expression systems reveals distinctive performance patterns across key metrics including yield, production timeline, cost structure, and product authenticity.
Table 3: Expression System Performance Comparison
| Performance Metric | Bacterial Systems | Yeast Systems | Mammalian Systems |
|---|---|---|---|
| Typical Yield | High (g/L range for many proteins) | Moderate to High (mg to g/L) | Moderate (mg to g/L for complex proteins) |
| Development Timeline | Shortest (weeks to months) | Short (months) | Longest (6-18 months) |
| Production Cost | Lowest | Low to Moderate | Highest |
| Glycosylation Capability | None | High-mannose type | Complex human-like |
| Scale-up Feasibility | Excellent | Excellent | Good to Excellent |
| Regulatory Acceptance | Established | Well-established | Gold standard for therapeutics |
Technical Considerations for Platform Selection
Product Complexity should guide initial platform selection. Bacterial systems excel with simple proteins lacking post-translational modifications, such as many industrial enzymes and non-glycosylated therapeutic proteins [47] [49]. Yeast systems provide a balanced solution for proteins requiring eukaryotic folding and secretion but tolerant of non-human glycosylation [49]. Mammalian systems remain essential for complex glycosylated proteins like monoclonal antibodies and certain viral antigens [50] [51].
Timeline and Resource Constraints significantly influence platform choice. Bacterial and yeast systems offer rapid development cycles and lower capital investment, making them ideal for research phase production and products with thin profit margins [47] [49]. Mammalian systems require substantial upfront investment and longer development timelines but deliver the authentic post-translational modifications necessary for many therapeutic applications [51].
Scalability and Production Costs vary substantially across platforms. Microbial systems generally offer more straightforward scale-up and lower production costs, while mammalian cell culture involves complex media requirements and sophisticated bioreactor systems [49] [51]. However, continuing advances in mammalian cell culture technology have dramatically increased titers, partially offsetting the cost differential for high-value therapeutics.
The Scientist's Toolkit
Essential Research Reagent Solutions
Table 4: Key Reagents for Heterologous Expression Research
| Reagent/Category | Function/Purpose | Example Applications |
|---|---|---|
| ϕC31 Integrative Vectors | Site-specific chromosomal integration | Stable expression in Burkholderia and Streptomyces systems [47] |
| CRISPR/Cas9 Systems | Precise genome editing | Gene knockouts, promoter replacements, pathway engineering [8] [48] |
| Redαβγ Recombination System | Homologous recombination in E. coli | BAC modification, pathway engineering [48] |
| RMCE Cassettes (Cre-lox, Vika-vox) | Recombinase-mediated cassette exchange | Marker-free genomic integration, multi-copy expression [48] |
| Antibody Array ELISA | Higher-order structure analysis | Biosimilar comparability assessment [51] |
| Specialized Promoters | Transcriptional control | Constitutive (Pgenta) and inducible (araC/PBAD) expression [47] |
| Optimized Signal Peptides | Protein secretion enhancement | Extracellular production simplification [49] |
The selection of an appropriate heterologous expression system represents a critical strategic decision that balances multiple factors including product complexity, required yield, timeline constraints, and available resources. Bacterial systems offer compelling advantages for simple proteins and natural products where rapid, cost-effective production is paramount. Yeast platforms provide an optimal balance between eukaryotic functionality and microbial practicality for many industrial enzymes and simpler biologics. Mammalian cell systems remain indispensable for complex therapeutic proteins requiring authentic human-like post-translational modifications. The continuing advancement of genetic engineering tools and bioprocess optimization across all platforms promises to further blur the traditional boundaries between these systems, enabling researchers to select or even combine platforms based on precise product requirements rather than historical precedent. As the case studies presented demonstrate, empirical performance data rather than theoretical considerations should guide platform selection for both research and commercial applications.
The selection of an optimal heterologous expression host is a critical decision in biopharmaceutical and industrial enzyme production. While traditional systems like E. coli, yeast, and mammalian cells each occupy important niches, emerging microbial platforms offer compelling advantages for specific applications. Aspergillus niger, a filamentous fungus, demonstrates exceptional protein secretion capacity, while Brevibacillus species, Gram-positive bacteria, provide a simplified yet efficient platform for prokaryotic expression. This guide provides an objective comparison of these two emerging systems, contextualized within the broader landscape of heterologous expression technologies, to support researchers in selecting the optimal platform for their specific protein production needs.
Heterologous Expression Systems at a Glance
The table below summarizes the key characteristics of major heterologous expression systems, highlighting how A. niger and Brevibacillus compare to established platforms [52] [53].
| Host System | Optimal Applications | Key Advantages | Major Limitations | Typical Protein Yields |
|---|---|---|---|---|
| Mammalian (e.g., HEK293, CHO) | Complex therapeutic proteins requiring authentic PTMs | Authentic human-like PTMs, proper protein folding | High cost, slow growth, technical complexity | Variable; generally lower than microbial systems |
| Yeast (e.g., S. cerevisiae) | Eukaryotic proteins needing simple glycosylation | Eukaryotic secretion pathway, cost-effective cultivation | Hyperglycosylation, limited PTM complexity | ~g/L scale for many proteins [53] |
| E. coli | Non-glycosylated proteins, industrial enzymes | Rapid growth, high yields, easy genetic manipulation | Formation of inclusion bodies, no native eukaryotic PTMs | Up to 20 g/L for some proteins (e.g., Interferon-α) [52] |
| Aspergillus niger (Emerging) | High-level secretion of industrial enzymes, fungal proteins | Exceptional secretion capacity, GRAS status, strong promoters | Complex genetics, potential for hyperglycosylation | 110-416 mg/L for diverse proteins in R&D [8] |
| Brevibacillus (Emerging) | Secreted bacterial enzymes, non-glycosylated proteins | Minimal extracellular proteases, efficient secretion, simple handling | Limited glycosylation capability, fewer genetic tools | 0.8 g/L for recombinant Riboflavin-binding Protein [52] |
In-Depth Platform Comparison:A. nigervs.Brevibacillus
Aspergillus niger as a High-Secretion Fungal Host
Aspergillus niger is a well-established industrial workhorse for enzyme production, with recent engineering efforts significantly enhancing its capabilities for heterologous protein expression [8] [54].
Key Technological Features:
- Secretory Capacity: Native protein secretion can reach remarkable levels, with industrial glucoamylase production reported up to 30 g/L [8] [54].
- Genetic Tools: Advanced CRISPR/Cas9 systems enable precise genome editing, including targeted integration and multi-gene manipulations [8].
- Engineering Strategies: Successful strain improvement involves deleting background endogenous proteins (e.g., proteases like PepA), overexpressing vesicle trafficking components (e.g., COPI component Cvc2), and exploiting strong native promoters and high-transcription loci [8].
Performance Data: Recent research demonstrates the platform's versatility with the following expression levels for diverse proteins in engineered A. niger chassis strains [8]:
- Glucose oxidase (AnGoxM): ~1276-1328 U/mL
- Thermostable pectate lyase (MtPlyA): ~1627-2106 U/mL
- Bacterial triose phosphate isomerase (TPI): ~1751-1906 U/mg
- Medicinal protein Lingzhi-8 (LZ8): Successfully secreted
Brevibacillus as a Efficient Bacterial Host
Brevibacillus species have emerged as attractive alternatives to E. coli and Bacillus subtilis for producing recombinant proteins, particularly those of bacterial origin [52] [55].
Key Technological Features:
- Secretory Efficiency: Naturally possesses robust Sec secretion pathways for efficient extracellular protein production [52].
- Low Protease Activity: Minimal extracellular protease production reduces recombinant protein degradation, simplifying downstream processing [52].
- Genetic Manipulation: Supports efficient transformation via electroporation and utilizes specially designed E. coli-Brevibacillus shuttle vectors for gene expression [52].
Performance Data: The platform has demonstrated success with various proteins, including [52] [55]:
- Riboflavin-binding protein (rRBP): 0.8 g/L in culture supernatant
- Chitinase (Chi72A): Successful high-activity expression with optimal activity at 60°C
- Antifungal polypeptides: Effective inhibition of ochratoxigenic fungi
Experimental Protocols for Platform Evaluation
Protocol 1: CRISPR/Cas9-Mediated Strain Engineering inA. niger
This protocol outlines the creation of a high-yielding A. niger chassis strain, as described in recent literature [8].
Methodology:
- Parental Strain Selection: Begin with an industrial glucoamylase-producing strain (e.g., AnN1) with robust native secretion machinery.
- Gene Deletion: Use CRISPR/Cas9-assisted marker recycling to:
- Delete multiple copies of highly expressed endogenous genes (e.g., 13 out of 20 copies of the TeGlaA glucoamylase gene) to reduce background protein secretion.
- Disrupt major extracellular protease genes (e.g., PepA) to minimize recombinant protein degradation.
- Target Gene Integration: Integrate heterologous genes into the newly vacated high-expression loci using modular donor plasmids containing strong native promoters (e.g., AAmy promoter) and terminators.
- Secretory Pathway Engineering: Further enhance yield by overexpressing components of the vesicular trafficking system (e.g., COPI component Cvc2), which has been shown to improve production of certain proteins by 18% [8].
Protocol 2: Heterologous Expression inBrevibacillusSystems
This protocol summarizes the standard methodology for expressing recombinant proteins in Brevibacillus [52] [55].
Methodology:
- Vector Construction: Clone the gene of interest into an E. coli-Brevibacillus shuttle vector, ensuring the inclusion of an appropriate signal peptide sequence (e.g., from native Brevibacillus proteins) for efficient secretion.
- Transformation: Introduce the recombinant vector into Brevibacillus host cells (e.g., B. choshinensis) via electroporation.
- Cultivation: Grow transformed cells in suitable media (e.g., modified Luria Bertani). For secretion analysis, culture in minimal salt media to simplify detection of the recombinant protein in the supernatant.
- Protein Analysis: Assess expression levels and functionality through:
- SDS-PAGE and Western blotting of culture supernatants and cell lysates.
- Enzyme activity assays specific to the recombinant protein.
- For antifungal proteins (e.g., chitinases), conduct plate antagonism assays against target phytopathogens [55].
Cellular Machinery and Secretion Pathways
The efficiency of heterologous protein production is largely determined by the cellular machinery and secretion pathways of the host organism. The diagrams below illustrate the key components of this machinery in A. niger and Brevibacillus.
Figure 1: The Eukaryotic Secretion Pathway in A. niger. This complex pathway involves multiple organelles and vesicular transport steps, enabling sophisticated protein processing but also creating potential bottlenecks [8].
Figure 2: The Bacterial Sec Secretion Pathway in Brevibacillus. This simplified, direct pathway facilitates efficient export of proteins across the single cell membrane, minimizing intermediate steps and potential bottlenecks [52].
The Scientist's Toolkit: Essential Research Reagents
The table below catalogues key reagents and materials required for working with A. niger and Brevibacillus expression systems.
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| CRISPR/Cas9 System | Targeted genome editing in A. niger | Cas9 nuclease, gRNA expression cassettes, donor DNA templates [8] |
| Modular Donor Plasmids | Target gene integration in fungi | Vectors with strong promoters (e.g., AAmy, glaA) and terminators [8] |
| E. coli-Brevibacillus Shuttle Vectors | Cloning and expression in Brevibacillus | Plasmids with origins of replication for both hosts [52] [55] |
| Signal Peptides | Directing protein secretion | Native A. niger GlaA signal or Brevibacillus signal sequences [8] [52] |
| Selection Antibiotics | Selective pressure for transformants | Hygromycin B, phleomycin for fungi; kanamycin for bacteria [8] [56] |
| Specialized Growth Media | Optimized culture conditions | Potato dextrose broth for fungi; M9 minimal salts for bacterial expression [8] [57] |
The choice between Aspergillus niger and Brevibacillus expression systems is not a matter of superiority but rather of strategic alignment with project goals.
Select Aspergillus niger when your priority is high-yield secretion of complex eukaryotic proteins, especially industrial enzymes or therapeutic proteins requiring fungal-type post-translational modifications. This system is particularly advantageous when project resources allow for sophisticated strain engineering to maximize protein production [8] [54].
Choose Brevibacillus when working with prokaryotic proteins or enzymes that do not require glycosylation, particularly when seeking a clean supernatant with minimal protease contamination. This platform offers a compelling balance of efficiency and simplicity for appropriate targets [52] [55].
Both platforms demonstrate how understanding and engineering microbial physiology and secretion machinery can create powerful solutions for the expanding needs of recombinant protein production, offering viable alternatives to traditional expression systems in their respective domains of application.
Solving Common Challenges: Strategies for Enhancing Yield, Solubility, and Fidelity
The production of recombinant proteins is a cornerstone of modern biopharmaceuticals, with Escherichia coli remaining one of the most widely used hosts due to its cost-effectiveness, rapid growth, and well-characterized genetics. However, a significant challenge persists: the tendency of overexpressed heterologous proteins to form insoluble aggregates known as inclusion bodies (IBs). These aggregates represent misfolded or partially folded proteins that have lost their biological activity, posing a major hurdle in the production pipeline. Within the broader context of expression host selection—which ranges from bacterial systems to yeast and mammalian cells—each platform presents distinct advantages and limitations. While bacterial systems like E. coli offer high productivity, they often lack the sophisticated folding machinery and post-translational modification capabilities of eukaryotic hosts, making IB formation a particularly prevalent issue. This guide objectively compares two primary strategies—chaperone co-expression and refolding protocols—for recovering functional proteins from IBs, providing supporting experimental data and methodologies to inform decision-making for researchers and drug development professionals.
Understanding Inclusion Bodies: Structure and Formation
Inclusion bodies are dense, refractile particles typically ranging from 0.2 to 1.5 μm in size, often localized at the poles of bacterial cells [58]. Classically considered amorphous aggregates, recent evidence reveals that IBs can contain proteins with native-like secondary structures and even significant biological activity, categorized as "non-classical" inclusion bodies [58] [59]. The formation of IBs is primarily driven by an imbalance between protein synthesis and the host cell's folding capacity, often exacerbated by high expression rates, strong promoters, and the reducing environment of the bacterial cytoplasm which impedes disulfide bond formation [60] [58]. The aggregation process is highly specific, with molecules of the same protein preferentially co-aggregating, and is influenced by factors such as protein hydrophobicity, molecular weight, and the presence of low-complexity regions [60].
Strategy Comparison: Chaperone Co-expression vs. Refolding Protocols
The following table summarizes the core characteristics, applications, and performance data of the two primary strategies for combating inclusion bodies.
Table 1: Comparative Analysis of Strategies for Combating Inclusion Bodies
| Feature | Chaperone Co-expression | Refolding Protocols |
|---|---|---|
| Core Principle | Enhance in vivo folding capacity during protein synthesis [61]. | Solubilize IBs and guide protein renaturation in vitro [58] [62]. |
| Typical Workflow | Co-transform with chaperone plasmid; induce chaperone expression before target protein induction [61]. | Isolate IBs; solubilize with denaturants/detergents; refold via dilution, dialysis, or chromatography [62]. |
| Key Reagents/ Tools | Chaperone plasmids (e.g., pKJE7 for DnaK/DnaJ/GrpE); chemical inducers (L-arabinose) [61]. | Denaturants (Urea, GdnHCl); detergents (N-Lauroylsarcosine); redox agents (GSH/GSSG); arginine [58] [63] [62]. |
| Optimal Use Cases | Proteins prone to misfolding during synthesis; complex multi-domain proteins; high-throughput soluble expression screening. | Proteins that aggregate despite optimization; proteins with complex disulfide bonding patterns. |
| Reported Solubility/Yield Improvement | ~4-fold increase in final yield of soluble anti-HER2 scFv [61]. Up to 100-fold enhancement for some scFvs [61]. | Highly variable (5-80%); depends on protein and protocol. Mild solubilization can yield high activity recovery [59] [58]. |
| Impact on Bioactivity | Generally high, as folding occurs in a cellular environment. Correctly folded protein is often the outcome [61]. | Can be impaired by residual detergents or incorrect refolding [64]. Requires careful optimization to retain activity [59]. |
| Throughput & Scalability | High throughput for expression screening; easily scalable in fermentation [61]. | Can be low-throughput due to empirical optimization; scaling up dilution/dialysis can be challenging [62]. |
| Major Advantages | Preemptive strategy; reduces downstream processing; leverages cellular machinery. | Potentially higher initial protein yield from IBs; necessary when in vivo methods fail. |
| Major Limitations | Metabolic burden on host; does not guarantee solubility for all proteins. | Often empirical and protein-specific; low refolding yields due to aggregation are common [62]. |
Experimental Data and Performance Metrics
- Chaperone Co-expression Efficacy: In a study on anti-HER2 single-chain variable fragment (scFv) production, co-expression with the DnaK/DnaJ/GrpE chaperone system under optimal conditions (0.5 mM IPTG, 30°C) resulted in an approximately four-fold increase in the final yield of purified soluble protein compared to expression without chaperones [61]. SDS-PAGE analysis confirmed the successful co-expression and enhanced solubility of the target protein.
- Refolding Protocol Efficacy: The specific activity of recovered proteins is highly dependent on the solubilization method. Research on antimicrobial proteins showed that while the detergent N-lauroylsarcosine (NLS) solubilized IBs efficiently, it impaired protein activity. In contrast, a spontaneous, detergent-free solubilization strategy in an appropriate buffer (e.g., phosphate buffer or acetic acid) at 37°C for 16-48 hours resulted in the recovery of fully active proteins, as confirmed by antimicrobial and fluorescent activity assays [59] [64].
Detailed Experimental Protocols
Protocol 1: Chaperone Co-expression for Soluble Protein Production
This protocol is adapted from a study demonstrating the enhanced soluble production of anti-HER2 scFv in E. coli [61].
Research Reagent Solutions:
- pKJE7 Chaperone Plasmid: Encodes the DnaK, DnaJ, and GrpE chaperone proteins under an arabinose-inducible promoter [61].
- L-Arabinose: Inducer for the chaperone plasmid expression.
- Isopropyl β-D-1-thiogalactopyranoside (IPTG): Inducer for the target recombinant protein.
- Ni-NTA Resin: For affinity purification of His-tagged recombinant proteins.
Methodology:
- Co-transformation: Co-transform competent E. coli BL21(DE3) cells with the expression plasmid (e.g., pET22b carrying the gene of interest) and the chaperone plasmid pKJE7. Plate onto LB agar containing appropriate antibiotics (e.g., ampicillin and chloramphenicol).
- Culture and Growth: Inoculate a single positive colony into LB broth with antibiotics. Incubate at 37°C with shaking until the culture reaches the mid-logarithmic phase (OD600 ≈ 0.5).
- Chaperone Induction: Add L-arabinose to a final concentration of 0.5 mg/mL to induce chaperone expression. Continue incubation for 30 minutes at the production temperature.
- Target Protein Induction: Add IPTG to a final concentration of 0.5 mM to induce the expression of the target recombinant protein. The optimal temperature for this step varies; for anti-HER2 scFv, 30°C for 6 hours was effective [61].
- Harvesting and Analysis: Collect cells by centrifugation. Resuspend the cell pellet in a suitable lysis buffer and disrupt the cells by sonication. Separate the soluble and insoluble fractions by centrifugation. Analyze the supernatant for the presence of the soluble target protein using SDS-PAGE and subsequent purification via Ni-NTA affinity chromatography.
The following workflow diagram visualizes this multi-stage experimental process.
Protocol 2: Mild Solubilization and Refolding of Inclusion Bodies
This protocol emphasizes mild, detergent-free strategies for recovering active proteins, leveraging the finding that proteins in IBs can have native-like structure [59] [58].
Research Reagent Solutions:
- Lysis Buffer: Typically 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, possibly with protease inhibitors [65].
- Wash Buffer: Lysis buffer with 0.15 M NaCl or low concentrations of denaturants (e.g., 2 M Urea) to remove impurities [65] [58].
- Solubilization Buffer: A mild, detergent-free buffer tailored to the protein (e.g., 10 mM KPi buffer, PBS, or 0.01% acetic acid) [59].
- Refolding Additives: Arginine, glycerol, or redox couples (GSH/GSSG) to suppress aggregation and promote correct disulfide bond formation [62].
Methodology:
- IB Isolation and Washing: Harvest bacterial cells by centrifugation. Resuspend the cell pellet in lysis buffer and disrupt cells by sonication. Centrifuge the lysate at high speed (e.g., 8,000 × g for 10 minutes) to pellet the IBs. Wash the IB pellet thoroughly with wash buffer to remove cell debris and impurities.
- Spontaneous Solubilization: Resuspend the purified IBs in a suitable mild solubilization buffer. The choice of buffer (e.g., phosphate buffer or dilute acid) should be optimized for the target protein. Incubate the suspension with gentle agitation at a defined temperature (e.g., 37°C) for a period ranging from 16 to 48 hours [59].
- Recovery of Solubilized Protein: Remove the insoluble material by centrifugation. The active, solubilized protein will be present in the supernatant.
- Activity Monitoring: The success of solubilization should be monitored by assaying the biological activity of the supernatant (e.g., antimicrobial assays, enzymatic activity, or fluorescence), as this is a more sensitive indicator of correct folding than mere solubility [59].
- Refolding (if required): For proteins that are not fully active after spontaneous solubilization, a refolding step may be necessary. This can be achieved by dilution or dialysis against a refolding buffer containing additives like arginine and glycerol to prevent aggregation and facilitate correct folding [62].
The Broader Context: E. coli in the Landscape of Expression Hosts
The strategies discussed herein for E. coli must be evaluated within the broader thesis of host selection for heterologous protein production. While E. coli excels in simplicity and yield for many proteins, alternative hosts offer distinct advantages. Yeasts such as Komagataella phaffii and Kluyveromyces lactis are Crabtree-negative, enabling high biomass yields under respiratory conditions, which can translate to higher recombinant protein titers [12]. Furthermore, yeasts provide a eukaryotic folding environment capable of performing essential post-translational modifications, such as glycosylation, which are often required for the biological activity and stability of therapeutic proteins like antibodies and hormones [12]. Mammalian cells offer the most complex and human-like PTM machinery but at a significantly higher cost and with greater technical challenges. Therefore, the choice to use E. coli and combat its tendency to form IBs is often a calculated decision favoring speed and economy, suitable for proteins that do not require eukaryotic-specific modifications or when active protein can be successfully recovered from aggregates.
The choice between chaperone co-expression and refolding protocols is not mutually exclusive and should be guided by the specific protein and project goals. Chaperone co-expression is a powerful preemptive strategy that integrates well into high-throughput soluble expression pipelines and is ideal for proteins where correct folding in vivo is feasible. Refolding protocols, particularly mild solubilization methods, are essential rescue strategies for proteins that inevitably aggregate, offering a path to recover active protein from IBs.
For researchers, the following decision logic is recommended: Begin with expression condition optimization (e.g., lower temperature, reduced inducer concentration). If solubility remains low, implement chaperone co-expression. If IBs persist, employ mild, spontaneous solubilization screening. Traditional denaturation and refolding should be considered a last resort due to its empirical nature and potential for low yields. This multi-tiered approach maximizes the potential of E. coli as a robust and efficient host for recombinant protein production within the diverse toolkit of available expression systems.
The selection of a host organism is a foundational decision in heterologous expression, profoundly influencing the strategy and success of research in drug development and biotechnology. Escherichia coli, Saccharomyces cerevisiae, and Chinese Hamster Ovary (CHO) cells represent the three most prevalent host systems, each offering a unique balance of simplicity, protein processing capability, and translational relevance to human therapeutics [66] [12]. E. coli is prized for its rapid growth, well-understood genetics, and cost-effective cultivation, but it lacks the machinery for eukaryotic post-translational modifications [12] [67]. Yeasts, such as S. cerevisiae, bridge the gap, offering the simplicity of a unicellular organism with the ability to perform basic eukaryotic modifications, though their glycosylation patterns differ from humans [12]. Mammalian CHO cells provide the gold standard for producing complex biologics, including monoclonal antibodies, as they support human-like glycosylation and other complex modifications, albeit with higher costs and slower growth [66] [12]. This guide objectively compares the core engineering solutions—codon optimization, genomic integration, and CRISPR/Cas9 workflows—across these hosts, providing experimental data and protocols to inform research and development.
Codon Optimization: Enhancing Gene Expression for Protein Production
Codon optimization is a critical first step in synthetic biology, fine-tuning the nucleotide sequence of a foreign gene to match the translational machinery of the host organism without altering the amino acid sequence it encodes [66] [68]. This process overcomes the challenge of codon usage bias, where different species preferentially use specific synonymous codons for the same amino acid [66] [67]. The presence of rare codons in a heterologous gene can slow translation rates, cause errors, and drastically reduce protein yield [69] [67].
Key Parameters and Strategic Approaches
Successful codon optimization requires a multi-parameter approach beyond simply replacing rare codons. The key design criteria include:
- Codon Adaptation Index (CAI): This metric quantifies how well the codon usage of a gene matches the preferred codon usage of the host's highly expressed genes. A CAI value closer to 1.0 indicates a higher likelihood of strong expression [66] [69].
- GC Content: The percentage of Guanine and Cytosine nucleotides in the sequence must be optimized for the host. While increased GC content can enhance mRNA stability in E. coli, S. cerevisiae prefers A/T-rich codons, and CHO cells require a moderate, balanced GC content [66] [70].
- mRNA Secondary Structure: Stable secondary structures, especially near the 5' end, can hinder ribosome binding and translation initiation. The folding free energy (ΔG) is used to predict and minimize these structures [66] [68].
- Codon-Pair Bias (CPB): This refers to the non-random pairing of adjacent codons, which can also influence translational efficiency and accuracy [66] [68].
Different optimization tools employ varied strategies. Some, like JCat and OPTIMIZER, focus on strong alignment with host codon usage, while others, such as TISIGNER, employ different algorithms that can produce divergent results [66]. Emerging methods, including deep learning models, are being trained to capture the complex codon distribution patterns of host genomes, showing competitive performance in enhancing protein expression [69].
Comparative Performance Across Host Organisms
The effectiveness of codon optimization is best demonstrated through experimental case studies. The table below summarizes quantitative data from optimization campaigns in different host systems.
Table 1: Experimental Outcomes of Codon Optimization in Different Host Systems
| Host Organism | Target Protein | Key Optimization Parameter | Outcome: Before Optimization | Outcome: After Optimization | Fold Improvement | Source/Context |
|---|---|---|---|---|---|---|
| E. coli | SARS-CoV-2 RBD | CAI | CAI: 0.72 | CAI: 0.96 | - | [70] |
| S. cerevisiae | ROL (Lipase) | Protein Yield | 0.4 mg/mL | 2.7 mg/mL | 6.75x | [70] |
| S. cerevisiae | ROL (Lipase) | Enzyme Activity | 118.5 U/mL | 220.0 U/mL | 1.86x | [70] |
| S. cerevisiae | phyA (Phytase) | Protein Yield | 0.35 mg/mL | 2.2 mg/mL | 6.29x | [70] |
| S. cerevisiae | phyA (Phytase) | Enzyme Activity | 25.6 U/mL | 122 U/mL | 4.77x | [70] |
| Mammalian (HEK293) | Luciferase (LuxA) | Protein Expression (Bioluminescence) | 5x10⁵ RLU/mg | 2.7x10⁷ RLU/mg | 54x | [70] |
Experimental Protocol: A Step-by-Step Codon Optimization Workflow
The following protocol outlines a general workflow for designing and validating a codon-optimized gene for heterologous expression.
- Sequence Identification and Host Selection: Obtain the amino acid or nucleotide sequence of the target protein from a database like NCBI. Identify the expression host (e.g., E. coli, S. cerevisiae, CHO) based on project needs [70].
- In Silico Optimization:
- Tool Selection: Utilize a codon optimization tool (e.g., IDT's Codon Optimization Tool, GeneArt, JCat) and select the target host organism [66] [68].
- Parameter Setting: Set parameters for CAI, GC content, and avoid specific sequence motifs (e.g., restriction sites, cryptic splice sites for mammalian cells) [66] [70] [68].
- Sequence Analysis: Run the optimization algorithm to generate a synthetic DNA sequence. Analyze the output report for CAI, GC content, and other metrics [68].
- Gene Synthesis and Cloning: The optimized DNA sequence is synthesized de novo. The gene is then cloned into an appropriate expression vector containing host-specific regulatory elements (e.g., a T7 promoter for E. coli, an AOX1 promoter for Komagataella phaffii, or a CMV promoter for CHO cells) [12] [70] [68].
- Host Transformation and Cultivation: The recombinant vector is introduced into the host cells via transformation (for microbes) or transfection (for mammalian cells). Positive clones are selected and cultivated under conditions that induce protein expression [70].
- Validation and Effect Analysis:
- Molecular Verification: Confirm successful gene integration and sequence fidelity using PCR and sequencing [70].
- Expression Analysis: Analyze protein production through techniques like SDS-PAGE, Western Blot, or direct enzyme activity assays to compare yields and functionality against the non-optimized control [69] [70].
Genomic Integration and CRISPR/Cas9 Workflows for Stable Expression
While episomal plasmids are common for initial protein production, genomic integration provides a more stable and sustainable solution for long-term or industrial-scale expression, as it avoids issues of plasmid loss and metabolic burden [71]. CRISPR/Cas9 technology has revolutionized this field by enabling precise, programmable, and multiplexed integration of heterologous genes into the host genome [72] [73] [71].
The CRISPR/Cas9 System: Mechanism and Components
The CRISPR/Cas9 system is derived from a prokaryotic adaptive immune system and functions as a versatile genome engineering tool [72]. Its core components are:
- Cas9 Nuclease: An RNA-guided endonuclease that creates a double-strand break (DSB) in the DNA. It requires a specific Protospacer Adjacent Motif (PAM) sequence (e.g., 5'-NGG-3' for Streptococcus pyogenes Cas9) adjacent to the target site for recognition and cleavage [72] [71].
- Guide RNA (gRNA): A chimeric single guide RNA (sgRNA) that combines the functions of crRNA and tracrRNA. The 20-nucleotide spacer sequence at the 5' end of the sgRNA dictates the genomic target site by complementary base pairing [72] [71].
Upon DSB formation, the cell activates its DNA repair machinery. For targeted gene integration, the Homology-Directed Repair (HDR) pathway is harnessed. A donor DNA template, containing the gene of interest flanked by homology arms that match the sequences around the cut site, is used by the cell to repair the break, thereby seamlessly integrating the new genetic material [72] [73].
Host-Specific CRISPR/Cas9 Implementation
The application and efficiency of CRISPR/Cas9 vary significantly across host systems, reflecting their unique biology.
- In E. coli: CRISPR/Cas9 enables precise multiplex genome editing, allowing for targeted gene deletions (e.g., ldhA, pta) and the integration of metabolic pathways to overproduce platform chemicals like succinate and isobutanol [73].
- In S. cerevisiae: Yeast's highly efficient HDR pathway makes it exceptionally suited for CRISPR/Cas9. This allows for efficient multiplex editing, where multiple gRNAs are used simultaneously to integrate several genes or pathway modules in a single transformation, drastically accelerating the reconstruction of complex metabolic pathways, such as those for terpenoids and flavonoids [73] [71].
- In Mammalian Cells (e.g., CHO): CRISPR/Cas9 is used for advanced cell line engineering. Applications include knock-in of therapeutic protein genes, and gene knockout to alter glycosylation patterns for optimizing drug efficacy (glyco-engineering) [73].
Table 2: Comparison of CRISPR/Cas9 Applications Across Microbial and Mammalian Hosts
| Feature | E. coli | S. cerevisiae | CHO Cells |
|---|---|---|---|
| Primary Application | Pathway engineering for chemical production [73] | Multiplexed pathway reconstruction & metabolic engineering [73] [71] | Biopharmaceutical production & cell line development [73] |
| Editing Efficiency | High | Very High (due to efficient HDR) [71] | Moderate to High |
| Key Advantage | Rapid strain construction for industrial biotechnology [73] | One-step integration of multiple genes [71] | Human-like post-translational modifications [73] |
| Example Outcome | Succinate titers >80 g/L [73] | Chromosomal insertion of entire biosynthetic clusters [73] | Production of monoclonal antibodies with humanized glycans [73] |
Experimental Protocol: A Workflow for Multiplex Gene Integration in Yeast
The following protocol details a standard method for integrating multiple gene expression cassettes into the genome of S. cerevisiae using CRISPR/Cas9 [71].
- Target Selection and gRNA Design: Select specific genomic loci (e.g., "safe harbor" sites or non-essential genes) for integration. Design 20-nt sgRNA spacer sequences targeting these sites, ensuring they are unique in the genome and are immediately followed by a PAM (5'-NGG-3') [71].
- Donor DNA Construction: For each target locus, design a linear donor DNA fragment containing:
- 5' Homology Arm (300-500 bp) homologous to the genomic region upstream of the DSB.
- Gene Expression Cassette: The gene of interest under the control of a yeast promoter (e.g., TEF1) and terminator (e.g., CYC1).
- 3' Homology Arm (300-500 bp) homologous to the genomic region downstream of the DSB [71].
- CRISPR/Cas9 System Delivery: Co-transform the yeast cells with:
- A plasmid expressing the Cas9 nuclease (often codon-optimized for yeast) and a selectable marker.
- Plasmids or PCR fragments expressing the multiple sgRNAs.
- The linear donor DNA fragments for each integration site [71].
- Selection and Screening: Plate the transformed cells on selective media. Screen surviving colonies for correct integration using colony PCR, which amplifies the junction sequences between the genome and the integrated DNA [71].
- Strain Validation and Phenotypic Analysis: Sequence the PCR products to confirm error-free integration. Finally, cultivate the validated strain and measure the desired output, such as the titer of the target metabolite or recombinant protein [71].
Workflow Diagram: CRISPR/Cas9 Mediated Gene Integration
The following diagram visualizes the key steps in the CRISPR/Cas9 mechanism for gene integration.
Diagram Title: CRISPR/Cas9 Gene Integration Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
This section catalogs key reagents and materials required for executing the experimental workflows described in this guide.
Table 3: Essential Reagents for Heterologous Expression and Genome Engineering
| Reagent / Solution | Function | Host-Specific Examples & Notes |
|---|---|---|
| Codon Optimization Tool | In silico design of optimized DNA sequences for enhanced expression. | IDT Codon Optimization Tool [68], JCat [66], OPTIMIZER [66], Deep learning models [69]. |
| Expression Vector | Plasmid carrying the gene of interest and regulatory elements for replication and expression in the host. | pET series (E. coli) [70], pPICZ (Komagataella phaffii) [12], YEp plasmid (S. cerevisiae) [12], pcDNA3 (Mammalian cells). |
| Cas9 Nuclease | Engineered version of the Cas9 protein for targeted DNA cleavage. | Human-codon-optimized Cas9 with NLS for mammalian cells [72], Yeast-codon-optimized Cas9 for S. cerevisiae [71]. |
| Guide RNA (gRNA) | Synthetic RNA molecule that directs Cas9 to a specific genomic locus. | Can be expressed from a plasmid (e.g., under a U6 promoter) or synthesized as a crRNA-tracrRNA duplex [72] [71]. |
| Donor DNA Template | Linear DNA fragment containing the gene to be integrated, flanked by homology arms for HDR. | Can be a PCR product or a double-stranded DNA fragment. Homology arm length is critical for efficiency (e.g., 300-500 bp for yeast) [71]. |
| Host Strain | The engineered organism used for heterologous expression. | E. coli BL21(DE3) for protein expression [70], S. cerevisiae S288C for pathway engineering [66], CHO-K1 for biopharmaceutical production [66]. |
The production of recombinant proteins is a cornerstone of the modern biopharmaceutical industry, with applications ranging from therapeutic monoclonal antibodies to industrial enzymes. The selection of an appropriate host organism is a critical first step, as it directly influences the yield, quality, and cost of the final product. The three primary host systems—bacteria, yeast, and mammalian cells—offer a spectrum of capabilities, particularly in their handling of the secretory pathway, the complex cellular process responsible for synthesizing, folding, modifying, and exporting proteins. Bacterial systems like E. coli are prized for their simplicity and high growth rates but often struggle with the proper folding and post-translational modification of complex eukaryotic proteins [74]. This guide focuses on comparing the two leading eukaryotic systems: yeast and mammalian cells. We will objectively compare their performance in secreting recombinant proteins, supported by recent experimental data, and provide detailed methodologies for enhancing secretory yield, all within the context of selecting the optimal host for heterologous expression research.
Host System Comparison: Yeast vs. Mammalian Cells
The choice between yeast and mammalian cells involves balancing factors such as cost, growth speed, and the ability to produce complex, biologically active proteins. The table below provides a structured comparison of these two systems based on key performance metrics.
Table 1: Comparative Analysis of Yeast and Mammalian Host Systems for Recombinant Protein Secretion
| Feature | Yeast Systems (e.g., S. cerevisiae, K. phaffii) | Mammalian Systems (e.g., CHO, HEK-293) |
|---|---|---|
| Typical Titers | Varies by protein; examples include Transferrin at 2.33 g/L and Lipase at 11,000 U/L in fed-batch processes [49]. | Varies significantly; many therapeutic proteins produced at commercial scale (1-5 g/L and above). |
| Glycosylation Pattern | High-mannose type; can be engineered towards human-like patterns [49]. | Innately human-like or compatible glycosylation [74]. |
| Growth Rate & Cost | Rapid growth, low-cost media, high cell-density fermentation possible [49]. | Slower growth, complex and expensive media requirements [75]. |
| Secretion Efficiency | Highly efficient secretion machinery; MFα signal peptide is a robust tool [76]. | Efficient but can be a bottleneck for difficult-to-express proteins [77]. |
| Key Advantage | GRAS status, well-established synthetic biology tools, scalable fermentation [49]. | Gold standard for complex therapeutics requiring authentic PTMs [74]. |
| Primary Challenge | Non-human glycosylation can be immunogenic; requires engineering for humanized PTMs [49] [74]. | High metabolic cost of production; lower yields for some "difficult-to-express" proteins [77] [75]. |
| Ideal For | Industrial enzymes, non-glycosylated proteins, vaccines, scaffolded antibody fragments [49] [74]. | Full-length, complex therapeutic proteins like monoclonal antibodies and blood factors [74] [75]. |
Key Determinants of Secretory Yield
Understanding the factors that limit the secretory pathway is essential for developing strategies to enhance yield. Recent large-scale studies have moved beyond the assumption that high mRNA levels guarantee high protein output, revealing a more complex picture.
Protein-Specific Features
In mammalian cells, a systematic analysis of 2135 human secretome proteins expressed in CHO cells found that mRNA abundance of the transgene explained less than 1% of the observed variation in secretion titers [77]. Instead, machine learning models identified intrinsic protein features that account for approximately 15% of the secretion variability. The following table summarizes these key determinants.
Table 2: Key Protein Features Correlating with Secretion Efficiency in CHO Cells [77]
| Feature Category | Specific Feature | Correlation with Secretion |
|---|---|---|
| Biophysical Properties | Molecular Weight (MW) | Strong negative correlation (higher MW, lower titer) |
| Amino Acid Composition | Cysteine Content | Negative correlation (increased cysteine, lower titer) |
| Post-Translational Modifications | N-linked Glycosylation | Emerging as a key predictor |
| Structural Features | Disulfide Bonds | Negative correlation (more bonds, lower titer) |
These findings indicate that difficult-to-express proteins are often characterized by large size, high cysteine content, and complex disulfide bonding, which can challenge the folding capacity and quality control systems of the endoplasmic reticulum (ER) [77].
Host Cell Physiology and Metabolic Burden
The host cell's physiological state is a major determinant of success. Genome-scale metabolic models of CHO cells have been developed to compute the energetic costs and machinery demands of secreting a single protein molecule, which can require thousands of ATP equivalents [75]. For example, Factor VIII, a notoriously difficult-to-express protein, requires an estimated 9,488 ATP molecules per molecule produced, creating a significant metabolic burden [75].
Transcriptomic analyses reveal distinct physiological signatures between high- and low-producing cells:
- Low-producing cells show enrichment in pathways for ubiquitin-mediated proteasomal degradation and ER-associated degradation (ERAD), indicating unsuccessful protein folding and clearance [77].
- High-producing cells upregulate pathways for lipid metabolism and the oxidative stress response, suggesting these processes are critical for supporting successful recombinant protein production [77].
Furthermore, highly secretory cells appear to adapt by suppressing the expression of endogenous proteins that are metabolically expensive to synthesize and secrete, allowing for a more efficient allocation of nutrients [75].
Engineering Strategies and Experimental Protocols
Engineering the Yeast Secretory Pathway
1. Signal Sequence Engineering The MFα signal sequence from S. cerevisiae is the most widely used and optimized signal peptide for recombinant protein secretion in yeast, including K. phaffii [76]. It directs proteins into the post-translational translocation pathway.
- Optimization Strategies: Several engineered variants have been developed to enhance performance, including:
- Protocol – Testing Signal Peptide Efficiency:
- Clone your gene of interest downstream of the native MFα signal sequence and its optimized variants in a suitable expression plasmid (e.g., pPpT4AlphaS for K. phaffii).
- Transform the constructs into your yeast host strain.
- Culture transformants in a deep-well plate or small shake flasks under inducing conditions.
- Harvest cells and separate the supernatant via centrifugation.
- Quantify extracellular protein titer using a method like SDS-PAGE densitometry or a specific activity assay, and compare yields across the different signal sequence variants.
2. Uncoupling Production from Growth Decoupling protein production from rapid cell growth can significantly improve product yield on substrate. A 2025 study demonstrated that the optimal strategy differs for intracellular and secreted proteins in S. cerevisiae [78].
- For Secreted Proteins: The strong, constitutive PTEF1 promoter led to increased protein secretion rates and higher extracellular titers at lower specific growth rates controlled by nutrient limitation [78].
- Protocol – Fed-Batch for Secretion:
- Use a strain with your secretion construct under the control of the PTEF1 promoter.
- Initiate a fed-batch fermentation with an initial batch phase for growth.
- Transition to a controlled feed of growth-limiting nutrient (e.g., carbon source) to gradually reduce the specific growth rate (µ) to a target range (e.g., 0.02 - 0.1 h⁻¹).
- Monitor cell density and extracellular product titer throughout the process. The highest secretory titers are expected during the slow-growth phase [78].
The diagram below illustrates the yeast protein secretion pathway and key engineering targets.
Engineering the Mammalian Secretory Pathway
1. Addressing Difficult-to-Express Proteins For proteins identified as difficult-to-express due to features like high molecular weight and cysteine content (see Table 2), rational engineering of the protein itself can be effective [77].
- Strategy: Use machine learning models trained on the 218-feature dataset to predict the expressibility of a target protein and identify problematic domains. Follow with site-directed mutagenesis to reduce molecular complexity without compromising function, for example, by mutating non-essential cysteine residues or simplifying glycosylation patterns.
2. Host Cell Engineering and Small Molecule Enhancement Engineering the host cell to alleviate metabolic and secretory bottlenecks is a powerful approach.
- Transcriptomic-Guided Engineering: Analysis of high-producing CHO cells suggests that engineering efforts should focus on enhancing lipid metabolism and the oxidative stress response, while simultaneously downregulating ubiquitin-proteasome system components to reduce degradation of the recombinant product [77].
- Small Molecule Intervention: A 2025 study used a computational method (DECCODE) to match the transcriptomic signature of high-producing mammalian cells to drug-induced profiles. This identified several small molecules, including Filgotinib and Ruxolitinib, that boost transgene expression.
- Protocol – Small Molecule Treatment:
- Culture engineered mammalian cells (e.g., H1299, CHO) and transfert with your genetic payload.
- At 4 hours post-transfection, add the identified small molecule (e.g., 1-10 µM Filgotinib) to the culture medium.
- Continue culture for 24-72 hours and analyze transgene expression (e.g., via fluorescence or ELISA) compared to an untreated control. This treatment can enhance expression by 10-50% [79].
- Protocol – Small Molecule Treatment:
The diagram below outlines the mammalian secretion pathway and its key bottlenecks.
The Scientist's Toolkit: Essential Research Reagents
This section details key reagents and tools used in the featured studies to engineer and optimize the secretory pathway.
Table 3: Key Research Reagent Solutions for Secretory Pathway Engineering
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| MFα Signal Sequence & Variants | Directs recombinant proteins into the yeast secretory pathway. | The primary signal peptide for secreting proteins in K. phaffii and S. cerevisiae; optimized variants improve titer and quality [76]. |
| Constitutive & Inducible Promoters (e.g., PTEF1, PHSP12) | Controls the timing and level of gene expression. | PTEF1 for stable secretion during slow growth; PHSP12 for stress-induced, growth-uncoupled intracellular production [78]. |
| Genome-Scale Metabolic Models (e.g., iCHO2048s) | Computational models that predict metabolic costs and bottlenecks. | Used to calculate ATP demand of secreting a specific protein and identify targets for host cell engineering [75]. |
| DECCODE Computational Tool | Matches transcriptomic signatures to drug-induced profiles to identify productivity-enhancing molecules. | Identified Filgotinib and Ruxolitinib as small molecules that boost transgene expression in mammalian cells [79]. |
| CRISPR/Cas9 Systems | Enables precise genome editing for host cell engineering. | Knocking out competing host cell proteins or inserting optimized genetic circuits to re-direct cellular resources [49] [75]. |
Mastering the secretory pathway in yeast and mammalian systems requires a holistic understanding of both the intrinsic properties of the target protein and the physiological state of the host cell. While mammalian cells like CHO remain the gold standard for producing the most complex biologics, advanced engineering in yeast is making it an increasingly powerful and cost-effective alternative. The future of heterologous protein production lies in the integrated application of protein engineering, host cell tailoring, and bioprocess optimization, including novel strategies like small molecule enhancement. By leveraging the comparative data, engineering strategies, and experimental protocols outlined in this guide, researchers can make informed decisions and develop robust, high-yielding production systems for their specific recombinant protein targets.
Overcoming Cellular Toxicity and Metabolic Burden Through Inducible Systems and Medium Design
The production of recombinant proteins is a cornerstone of modern biotechnology, driving advancements in biopharmaceuticals, industrial enzymes, and basic research [4] [80]. A fundamental challenge in this field involves balancing the high-level production of target proteins against the physiological health of the host cells. Introducing and expressing foreign genes places a substantial metabolic burden on host organisms, diverting precious cellular resources—such as energy, nucleotides, amino acids, and ribosomes—away from essential growth and maintenance functions [81] [82]. This burden often manifests as reduced cell growth, decreased viability, and ironically, lower overall protein yields. Furthermore, the expression of foreign pathways can lead to the accumulation of toxic intermediates, exacerbating cellular stress and limiting production efficiency [82]. To mitigate these interconnected issues, researchers have developed sophisticated strategies centered on inducible expression systems and refined medium design. This guide objectively compares how these strategies are applied across the three primary host systems—bacterial, yeast, and mammalian cells—providing a framework for selecting the optimal platform for specific research or production goals.
Host System Comparison: Strengths, Weaknesses, and Ideal Applications
The choice of host organism is a critical first step in designing a recombinant protein expression experiment. Each system offers a unique set of advantages and limitations, largely defined by its cellular machinery and metabolic capabilities. The table below provides a detailed comparison of the three main host systems.
Table 1: Comparison of Major Heterologous Protein Expression Systems
| Feature | Bacterial Systems (E. coli) | Yeast Systems (S. cerevisiae, K. phaffii) | Mammalian Cells (CHO, HEK293) |
|---|---|---|---|
| Typical Hosts | Escherichia coli, Bacillus subtilis [4] | Saccharomyces cerevisiae, Komagataella phaffii [4] [21] | CHO, HEK293 [80] |
| Cost & Technical Barrier | Low cost, minimal technical requirements [4] | Low to moderate cost, easy to manipulate [4] [21] | High cost, complex culture requirements [80] |
| Growth Speed | Very fast (short doubling time) [4] | Rapid growth rate [4] [21] | Slow growth, laborious scale-up [4] |
| Post-Translational Modifications | Limited; unable to perform most eukaryotic PTMs (e.g., complex glycosylation) [4] | Capable of many PTMs (e.g., glycosylation), but patterns differ from humans (hypermannosylation) [4] [21] | Full range of human-like PTMs (e.g., complex glycosylation), ensuring protein activity [4] [80] |
| Ideal Protein Types | Non-glycosylated proteins, enzymes for industrial applications [4] | Secreted eukaryotic proteins, vaccines, some therapeutic proteins [4] [21] | Complex therapeutic proteins (e.g., monoclonal antibodies, cytokines) [80] |
| Key Challenges | Formation of inclusion bodies, metabolic burden, lack of PTMs [4] [82] | Hyperglycosylation, proteolytic degradation, metabolic burden [4] [21] | Viral contamination susceptibility, high cost, low protein output [4] [80] |
Quantitative Analysis of Metabolic Burden and Toxicity
The metabolic burden is not merely a theoretical concern; it has quantifiable impacts on cell physiology and production metrics. Experimental data helps illustrate the severity of this burden and the efficacy of mitigation strategies.
Table 2: Experimental Data on Metabolic Burden and Inducible System Performance
| Experimental Context | Key Findings | Impact of Burden/Toxicity | Citation |
|---|---|---|---|
| E. coli with synthetic TCP biodegradation pathway | Metabolic burden and toxicity exacerbation observed on single cell and population levels. | Cell growth and productivity are significantly hampered by the burden of heterologous protein expression and toxic intermediate accumulation. | [82] |
| In silico model of multicellular control architecture | Distributing control functions across different cell populations mitigates metabolic burden effects. | Limited ribosome availability is a key factor; distributed architectures enhance circuit reliability and performance compared to single-cell implementations. | [81] |
| K. phaffii with engineered inducible promoter (DAPG-iSynP) | > 1000-fold induction of gene expression with minimal leakiness achieved through promoter insulation and operator mutagenesis. | Leaky expression from non-optimized promoters constitutively drains cellular resources. Tightly controlled induction decouples growth and production phases, boosting yield. | [83] |
| S. cerevisiae engineering for protein production | Heterologous proteins can reach up to 49.3% (w/w) of the yeast's own protein content. | Despite high potential yield, metabolic burden and inefficient secretion often keep yields below theoretical maxima, requiring systematic engineering. | [21] |
Mechanisms and Mitigation: Inducible Systems and Medium Design
The Role of Inducible Expression Systems
Inducible systems are favored over stable, constitutive expression because they offer temporal control, allowing researchers to separate the cell growth phase from the protein production phase. This decoupling is one of the most effective ways to reduce metabolic burden [83] [84]. The following diagram illustrates the core concept of metabolic burden resulting from resource competition.
The most advanced inducible systems address a key flaw: leakiness, or unwanted expression before induction. As identified in yeast, leakiness is often caused by cryptic transcriptional activation from upstream sequences. This can be mitigated by inserting >1-kbp insulator sequences and directly fusing operator repeats upstream of the TATA-box [83]. The following workflow outlines the strategic process for implementing an optimized inducible system.
Table 3: Commonly Used Inducible Gene Expression Systems
| System Name | Origin | Inducer Molecule | Mechanism of Action | Key Features |
|---|---|---|---|---|
| Tetracycline (Tet)-On/Off | E. coli Tn10 operon [84] | Doxycycline (a tetracycline derivative) [84] | In Tet-On, reverse Tet transactivator (rtTA) binds operator and activates transcription ONLY in the presence of doxycycline [84]. | High induction (>1000-fold), low background; requires tetracycline-free serum [83] [84]. |
| Cumate | Pseudomonas putida [84] | Cumate [84] | In reverse activator configuration, mutant cTA (rcTA) binds operator upon cumate addition, triggering expression [84]. | Can be combined with Tet system for multi-gene control; low leakiness [84]. |
| DAPG-iSynP | Synthetic (based on E. coli PhlF) [83] | 2,4-diacetylphloroglucinol (DAPG) [83] | DAPG-responsive synthetic transcription activator (rPhlTA) binds operator (phlO) to activate transcription [83]. | >10³-fold induction demonstrated in yeasts; minimal toxicity [83]. |
| Lac/IPTG | E. coli lac operon [82] | Isopropyl β-D-1-thiogalactopyranoside (IPTG) [82] | IPTG binds to Lac repressor (LacI), causing it to dissociate from the operator and allow transcription [84]. | Can contribute to metabolic burden; less efficient in mammalian cells [84] [82]. |
The Role of Medium Design and Cultivation Strategies
The design of the growth medium and fermentation process is inextricably linked to managing metabolic burden. Key strategies include:
- Feedstock Selection: Using sustainable, non-food-competing feedstocks like methanol and formate (C1 compounds) is a growing trend in microbial fermentation to reduce costs and environmental impact [85]. The choice of substrate influences carbon efficiency and can minimize the formation of toxic by-products.
- Nutrient Balancing: Ensuring an optimal balance of carbon, nitrogen, and other essential nutrients prevents the premature cessation of cell growth and supports protein production during the induction phase.
- Induction Timing and Conditions: For inducible systems, the optical density or growth phase at which the inducer is added is critical. Induction during mid-to-late exponential phase often allows for a robust biomass base before diverting resources to protein production. The concentration of the inducer (e.g., IPTG, doxycycline) also requires optimization to maximize yield while minimizing toxicity and burden [82].
The Scientist's Toolkit: Essential Reagents and Solutions
The experimental strategies discussed rely on a set of core reagents and molecular biology tools. The following table details these essential components.
Table 4: Key Research Reagent Solutions for Mitigating Toxicity and Burden
| Reagent / Tool | Function | Application Examples |
|---|---|---|
| Synthetic Inducible Promoters | Enables tight, temporal control of gene expression with high induction and minimal leakiness. | DAPG-iSynP in K. phaffii [83]; Tet-On system in mammalian cells [84]. |
| Codon-Optimized Genes | Maximizes translation efficiency by matching the host's codon usage bias, improving protein yield and solubility. | Production of Talaromyces emersonii enzymes in S. cerevisiae [21]. |
| CRISPR/Cas9 Systems | Allows precise genome editing for knocking out proteases, integrating genes at high-expression loci, and engineering host chassis. | Creating protease-deficient A. niger strain [8]; genome editing in S. cerevisiae [21]. |
| Insulator DNA Sequences | Prevents cryptic transcriptional activation from upstream sequences, a major cause of promoter leakiness. | >1-kbp KpARG4 sequence used to insulate yeast iSynPs [83]. |
| Chemical Inducers | Small molecules that trigger the expression from inducible promoters. | Doxycycline (for Tet systems), DAPG, Cumate, IPTG [83] [84] [82]. |
| Specialized Growth Media | Supports high cell density and provides essential nutrients while avoiding components that interfere with inducers. | Tetracycline-free fetal bovine serum for Tet systems [84]; Synthetic Mineral Medium for E. coli [82]. |
Effectively overcoming cellular toxicity and metabolic burden is paramount to achieving high yields of functional recombinant proteins. While the fundamental challenge of resource competition is universal, the optimal solution is host-dependent. Bacterial systems benefit most from the simple decoupling of growth and production via strong, tight inducible promoters like T7/lac. Yeast systems leverage their secretory capacity and GRAS status, requiring engineering of both hyper-expression promoters and humanized glycosylation pathways. Mammalian cells, as the most complex hosts, are indispensable for producing sophisticated biologics, where the high cost of inducible expression is justified by the need for authentic post-translational modifications.
The future of the field lies in the intelligent integration of strategies. This includes combining advanced inducible systems with genome-scale metabolic models to predict and preempt bottlenecks, and employing synthetic biology tools like CRISPR to create next-generation chassis cells inherently resistant to burden and toxicity. By carefully matching the expression strategy to the target protein and host system, researchers can maximize productivity while maintaining cell viability.
Head-to-Head System Comparison: Data-Driven Selection for Your Project
This guide provides a direct comparison of the three predominant heterologous protein expression systems: bacterial, yeast, and mammalian cells. The selection of an appropriate host is a critical first step in recombinant protein production, impacting not only the yield and cost but also the biological activity and therapeutic efficacy of the final product. The data summarized herein are compiled from recent scientific literature to offer researchers a foundational resource for project planning and system selection.
Table 1: Core Comparison of Heterologous Expression Systems
| Parameter | Bacterial (E. coli) | Yeast (S. cerevisiae / K. phaffii) | Mammalian (CHO / HEK293) |
|---|---|---|---|
| Typical Yield | High (mg/L to g/L) for soluble, non-glycosylated proteins [11] | High, can reach up to 49.3% (w/w) of cellular protein for S. cerevisiae [21] | Varies; often lower than microbial systems, but suitable for therapeutics [11] |
| Cost | Low (simple media, high cell density) [10] | Low to Moderate [10] | High (complex media, expensive infrastructure) [11] [10] |
| Timeline | Short (hours to days) [10] | Short (days) [10] | Long (weeks to months) [10] |
| Post-Translational Modifications (PTMs) | Limited; lacks eukaryotic glycosylation, disulfide bond formation in periplasm [10] [86] | Basic PTMs (e.g., high-mannose glycosylation); can be engineered for human-like patterns [10] [21] | Most complex; produces proteins with human-like glycosylation and other PTMs [10] |
| Best For | Simple, non-glycosylated proteins; research proteins; industrial enzymes [10] [87] | Proteins requiring basic eukaryotic folding/secretion; some therapeutics (e.g., insulin) [12] [21] | Complex proteins requiring authentic human PTMs (e.g., monoclonal antibodies, receptors) [10] [88] |
| Key Challenge | Formation of inclusion bodies; absence of complex PTMs [11] [86] | Hyper-mannosylation can be immunogenic; secretion efficiency can vary [10] [21] | High cost, technical complexity, and longer production timelines [11] [10] |
Experimental Protocols for Key Analyses
The following section details standard methodologies used to generate the comparative data presented in this guide.
Protocol for Evaluating Recombinant Protein Yield
Objective: To quantify the volumetric and specific yield of a recombinant protein produced in different host systems.
Materials:
- Shake flasks or bioreactors
- Host-specific growth media
- Induction agent (if using inducible promoters)
- Centrifuges and cell disruption equipment (for intracellular proteins)
- Purification system (e.g., FPLC, affinity resins)
- SDS-PAGE equipment
- Spectrophotometer or ELISA plate reader
Method:
- Strain Transformation & Culture: Transform the expression vector containing the gene of interest (GOI) into the respective host (E. coli, yeast, mammalian cells). Inoculate a starter culture and grow overnight under selective conditions [11] [21].
- Induction/Expression: Dilute the culture to the recommended optical density (OD). For inducible systems, add the appropriate inducer (e.g., IPTG for E. coli, methanol for K. phaffii). Continue incubation for the host-optimized duration and temperature [86] [87].
- Harvesting:
- Purification: Purify the protein using a tag-based affinity method (e.g., Ni-NTA for His-tag) or other chromatography techniques [12].
- Quantification:
- Total Protein: Use the Bradford or BCA assay.
- Target Protein: Analyze purified fractions by SDS-PAGE with densitometry, or use a specific functional assay (e.g., ELISA, enzymatic activity) [21].
- Calculation:
- Volumetric Yield (mg/L) = (Mass of purified protein (mg)) / (Culture volume (L))
- Specific Yield (mg/gDCW) = (Mass of purified protein (mg)) / (Dry Cell Weight (g))
Protocol for Analyzing Glycosylation Patterns
Objective: To characterize the N-linked glycosylation profile of a recombinant glycoprotein, a key differentiator between eukaryotic systems.
Materials:
- Purified glycoprotein
- Denaturing buffer
- PNGase F enzyme
- C18 solid-phase extraction tips
- Mass Spectrometry (MS) system (e.g., MALDI-TOF/TOF or LC-ESI-MS)
- Data analysis software
Method:
- Enzymatic Deglycosylation: Denature the purified glycoprotein. Incubate with PNGase F to release N-glycans from the polypeptide backbone [10].
- Glycan Cleanup: Desalt and purify the released glycans using C18 tips or graphitized carbon cartridges.
- MS Analysis:
- Spot the purified glycans onto a MALDI target plate with a suitable matrix.
- Acquire mass spectra in positive or negative ion mode.
- For detailed structural analysis, use LC-ESI-MS/MS.
- Data Interpretation: Assign glycan structures based on mass-to-charge (m/z) ratios and fragmentation patterns. Compare the profiles to known standards [10].
Glycosylation Analysis Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Heterologous Protein Expression
| Reagent / Solution | Function | Host Application |
|---|---|---|
| Codon-Optimized Gene | Synthetic gene sequence tailored to the host's codon usage bias to maximize translation efficiency [66] [21]. | All |
| Expression Vector | Plasmid containing host-specific promoter (e.g., T7, AOX1, CMV), origin of replication, and selectable marker [11] [89]. | All |
| Affinity Tags | Peptides (e.g., His-tag, GST, MBP) fused to the target protein to facilitate purification and sometimes enhance solubility [11] [87]. | All |
| Specialized Growth Media | Chemically defined or complex media formulated to support high-density growth and recombinant protein production (e.g., LB for E. coli, YPD for yeast, DMEM for mammalian cells) [87]. | All |
| Induction Agents | Chemicals to trigger expression from inducible promoters (e.g., IPTG for E. coli, Methanol for K. phaffii, Tetracycline for mammalian cells) [21]. | All |
| Lysis Buffers | Solutions for breaking open cells to extract intracellular proteins; composition varies with host cell wall/membrane structure [11]. | E. coli, Yeast |
| Affinity Resins | Chromatography media (e.g., Ni-NTA, Protein A/G) for purifying tagged or native proteins [12]. | All |
| PNGase F | Enzyme used to release and analyze N-linked glycan chains from glycoproteins [10]. | Yeast, Mammalian |
In heterologous expression research, the choice of a host organism is a critical determinant of the structural and functional fidelity of the recombinant protein produced. One of the most significant factors in this regard is protein glycosylation, a post-translational modification where sugar chains are attached to specific amino acid residues. This modification profoundly influences the stability, solubility, immunogenicity, and biological activity of therapeutic proteins [90]. The glycosylation machinery of bacteria, yeast, and mammalian cells differs vastly, leading to distinct glycan profiles. This guide provides a detailed, objective comparison of these glycosylation patterns, underpinned by experimental data, to inform the selection of an appropriate expression system for research and drug development.
Glycosylation is a complex enzymatic process that occurs in the secretory pathway, primarily within the endoplasmic reticulum and Golgi apparatus. The nature of the glycans attached to a protein is determined by the host cell's unique repertoire of glycosyltransferases and glycosidases [91]. The following diagram illustrates the fundamental differences in the N-glycosylation pathways of yeast and mammalian cells, which are absent in bacteria.
Comparative Analysis of Glycosylation Profiles
The core structure for all N-glycans is conserved (Asn-GlcNAc₂Man₃), but its extension and modification differ dramatically between hosts [90]. The table below provides a structured, quantitative comparison of the key glycosylation characteristics across bacterial, yeast, and mammalian expression systems.
Table 1: Glycosylation Profile Comparison Across Expression Systems
| Feature | Bacterial Systems | Yeast Systems | Mammalian Systems |
|---|---|---|---|
| N-linked Glycosylation | Absent [92] | Present; High-mannose type (Man8-14GlcNAc2 to Man>50GlcNAc2) [93] [94] | Present; Complex type [92] |
| Common N-glycan Structures | Not applicable | Man8-14GlcNAc2 (upon OCH1 deletion) [93] | Biantennary complex (e.g., G0, G1, G2 with Fuc, GlcNAc) [95] |
| O-linked Glycosylation | Present (e.g., on pili, flagella) [96] [97] | Present; Mannose-based chains [98] [93] | Present; Mucin-type (initiated with GalNAc) [90] [93] |
| Key Monosaccharides | Unique sugars (e.g., Pse, Leg, Bacillosamine) [96] [97] | Predominantly Mannose [93] | Galactose, Sialic Acid, Fucose, GlcNAc [93] [95] |
| Typical Expression Hosts | E. coli | S. cerevisiae, P. pastoris | CHO, HEK293 |
| Impact on Therapeutic Proteins | Non-glycosylated products may have short half-life [93] | Hypermannosylation causes rapid clearance & immunogenicity [93] [95] | Human-like glycosylation; optimal pharmacokinetics [92] |
Detailed Experimental Analysis of Glycans
Understanding the specific composition and structure of glycans requires specialized experimental protocols. The following section details key methodologies used to characterize and modify the O-glycans of a model fungal glycoprotein, providing a template for similar analyses.
Experimental Protocol: O-Glycan Analysis of a Fungal Cellobiohydrolase
This protocol, adapted from studies on Trichoderma reesei cellobiohydrolase I (TrCel7A) expressed in Aspergillus oryzae, outlines the steps for mapping O-glycan structures [98].
Objective: To determine the extent, composition, and linkage of O-glycans in the linker region of the TrCel7A glycoprotein.
Workflow Diagram:
Methodology Details:
- Protein Expression and Purification: The TrCel7A wild-type and a variant with N-glycosylation sites knocked out (TrCel7A ΔN-glyc) are expressed in Aspergillus oryzae and purified using standard chromatographic techniques [98].
- O-glycan Release via Reductive β-elimination: O-linked glycans are chemically released from the protein backbone using sodium hydroxide (NaOH). To prevent degradation of the released glycans via "peeling" reactions, the reducing ends are stabilized by the addition of a reducing agent, sodium borohydride (NaBH₄) [98].
- Glycan Composition Analysis: The released glycans are analyzed to determine their monosaccharide constituents. In the case of TrCel7A, this revealed the presence of only hexose (Hex) residues, ranging from Hex₁ to Hex₁₄, with Hex₂ being the most prevalent. Further monosaccharide analysis showed the hexoses were mannose (Man) and galactose (Gal) in a ratio of 1.3:1 [98].
- Glycosyl Linkage Analysis: This technique involves permethylation of the glycans followed by acid hydrolysis and gas chromatography-mass spectrometry (GC-MS) to determine how the monosaccharides are linked together. For TrCel7A, the main linkages identified were terminal mannopyranose (45%), 2-linked mannopyranose at the reducing end (31%), and 4-linked galactopyranose (12%) [98].
- Enzymatic Trimming (Glycoengineering): An enzymatic toolbox is used to controllably trim the O-glycans. This includes:
- NnGH92: A glycoside hydrolase family 92 (GH92) α-1,2-mannosidase from Neobacillus novalis that trims mannose residues.
- AaGH2: A GH2 β-galactofuranosidase from Amnesia atrobrunnea that removes terminal galactofuranose residues.
- Jack Bean α-Mannosidase (JBM): A broad-specificity mannosidase targeting α-1,2/α-1,3/α-1,6 linkages [98].
- Functional Consequence Assay: The impact of O-glycosylation on protein function is evaluated by comparing the enzymatic activity (kinetic parameters) of the native and deglycosylated forms of TrCel7A [98].
The Scientist's Toolkit: Key Research Reagents
The following table lists essential reagents used in the aforementioned experiments, along with their specific functions in glycosylation analysis.
Table 2: Key Reagents for Glycosylation Analysis and Engineering
| Reagent | Function/Application |
|---|---|
| Sodium Borohydride (NaBH₄) | Reducing agent used in reductive β-elimination to stabilize released O-glycans by preventing "peeling" reactions [98]. |
| GH92 α-1,2-mannosidase (NnGH92) | Exoglycosidase that specifically trims α-1,2-linked mannose residues from fungal O-glycans [98]. |
| Jack Bean α-Mannosidase (JBM) | A broad-specificity exoglycosidase used to trim a variety of α-linked mannosyl residues (α-1,2/α-1,3/α-1/6) from glycans [98]. |
| Endoglycosidase H (Endo H) | An endoglycosidase that hydrolyzes the chitobiose core of high-mannose and hybrid-type N-glycans, commonly used for deglycosylation [98]. |
| Peptide-N-glycosidase F (PNGase F) | An amidase that removes almost all types of N-glycans from glycoproteins by cleaving the bond between the innermost GlcNAc and asparagine residue [98]. |
Glycoengineering Strategies for Humanization
The inherent glycosylation patterns of microbial hosts often necessitate engineering to make them suitable for producing human therapeutic proteins. The diagram below summarizes the primary strategies used to "humanize" glycosylation in yeast.
Key Strategies Explained:
- Gene Deletion: The most common approach involves knocking out genes encoding enzymes responsible for non-human glycosylation. For example, deleting the OCH1 gene (an α-1,6-mannosyltransferase) in yeasts like Pichia pastoris and Saccharomyces cerevisiae* is a primary step to eliminate hypermannosylation [93]. This can result in glycoproteins with shorter Man3-14GlcNAc2 glycans instead of the typical Man>50GlcNAc2 [93].
- Heterologous Enzyme Expression: To produce complex human-like glycans, yeast systems are engineered to express functional mammalian glycosyltransferases and glycosidases. This includes enzymes like β-1,4-galactosyltransferase and sialyltransferases, which are necessary to add terminal galactose and sialic acid residues, respectively [94]. Advanced tools like the CRISPR-Cas9 system are now employed for precise multiplexed gene editing to implement these complex pathway changes [93].
- System Selection: Some non-conventional yeasts, such as Pichia pastoris, are naturally less prone to hyperglycosylation and are therefore considered more suitable starting platforms for humanization efforts [93].
Impact on Therapeutic Protein Efficacy
The glycosylation profile of a therapeutic protein, particularly monoclonal antibodies (mAbs), directly dictates its safety and efficacy through several critical mechanisms.
Table 3: Functional Impact of Key Glycan Features on Therapeutic Antibodies
| Glycan Feature | Impact on Therapeutic Monoclonal Antibodies (mAbs) |
|---|---|
| Core Fucose | Decreases Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) by reducing binding to Fcγ receptors [95]. |
| Terminal Galactose | Enhances Complement-Dependent Cytotoxicity (CDC) by improving binding to the C1q complex [95]. |
| Bisecting GlcNAc | Increases ADCC by enhancing affinity for Fcγ receptors [95]. |
| High-Mannose | Reduces serum half-life and can increase immunogenicity due to clearance by mannose receptors [95]. |
| Sialic Acid | Can influence anti-inflammatory activity [95]. |
The selection of a host system for heterologous protein expression is a fundamental decision that directly determines the glycosylation profile and, consequently, the biological activity of the product. Bacterial systems are incapable of eukaryotic N-glycosylation, limiting their use for proteins where glycans are essential. Yeast systems produce high-mannose glycans, which often lead to rapid clearance and immunogenicity in humans, though significant progress in glycoengineering has made the production of humanized glycans a reality. Mammalian cells, notably CHO cells, remain the gold standard for producing complex, human-like glycans required for the optimal efficacy and safety of most therapeutic glycoproteins, including monoclonal antibodies. Researchers must weigh these distinct glycosylation outcomes, along with factors like cost, yield, and scalability, to align their host system choice with the specific application of the recombinant protein.
The selection of an appropriate host organism is a foundational decision in the development of any bioprocess for heterologous protein production. This choice critically influences both the economic viability and technical scalability of the entire production pipeline, from initial gene expression to final protein purification. The three dominant host systems—bacterial, yeast, and mammalian cells—each possess a distinct profile of advantages and limitations, governed by their inherent biological capabilities. Key differentiators include the ability to perform complex post-translational modifications (PTMs), achieve high protein yields, and the associated cost structures of cell culture and media [35] [99] [80]. This guide provides an objective, data-driven comparison of these platforms, focusing on the critical upstream and downstream processing considerations that inform process development and scale-up within the pharmaceutical and biotechnology industries.
Systematic Comparison of Expression Hosts
A comprehensive evaluation of host systems requires a multi-faceted analysis of performance metrics, cost drivers, and typical applications. The following tables summarize the core characteristics and economic considerations for bacterial, yeast, and mammalian cell platforms.
Table 1: Core Characteristics and Performance Metrics of Major Expression Hosts
| Parameter | Bacterial (E. coli) | Yeast (S. cerevisiae / K. phaffii) | Mammalian (CHO / HEK293) |
|---|---|---|---|
| Typical Expression Timeline | 2–3 weeks [99] | 2–4 weeks [12] | 4–6 weeks (stable lines) [99] |
| Post-Translational Modification Capability | None or limited [99] [80] | Simple glycosylation, disulfide bonds [49] [12] | Complex, human-like PTMs (glycosylation, phosphorylation) [35] [80] |
| Typical Yield (Therapeutic Proteins) | High for simple proteins | High; e.g., Transferrin at 2.33 g/L [49] | High for complex proteins; lower volumetric yield than microbes but higher functionality |
| Secretion Efficiency | Often forms inclusion bodies [99] | Generally efficient secretion [49] [100] | Efficient secretion into culture medium [80] |
| Genetic Manipulation Complexity | Low; extensive toolkit available | Moderate; tools highly developed for S. cerevisiae [49] [12] | High; more complex and time-consuming [79] |
| Representative Proteins | Non-glycosylated cytokines, enzymes [99] | Insulin, hepatitis vaccine, albumin [12] | Monoclonal antibodies, complex glycoproteins [35] [80] |
Table 2: Economic and Scalability Assessment
| Consideration | Bacterial (E. coli) | Yeast (S. cerevisiae / K. phaffii) | Mammalian (CHO / HEK293) |
|---|---|---|---|
| Upstream Cost Drivers | Inexpensive culture media [24] | Inexpensive defined media, high cell-density fermentation [49] | High-cost media (up to 80% of direct cost), slow growth rates [24] |
| Downstream Cost Drivers | Often required refolding from inclusion bodies, increasing step count [99] | Simplified purification due to secretion; may require glycoform separation | Complex purification; stringent validation for therapeutics |
| Scalability | Excellent; facile scale-up to very large volumes | Excellent; well-established industrial fermentation [49] | Moderate; requires sophisticated bioreactor control and monitoring |
| Process Development Time | Short | Short to moderate | Lengthy, particularly for stable cell line generation |
| Relative Cost Estimate | Low [99] | Low to Medium [99] | High [99] |
Experimental Protocols for Host Evaluation
To generate comparative data like that presented in this guide, researchers employ standardized experimental workflows to assess protein expression and quality across different host systems.
Protocol for Parallel Protein Expression and Titer Analysis
Objective: To compare the yield and quality of a target protein expressed in E. coli, S. cerevisiae, and HEK293 cells.
Methodology:
- Gene and Vector Construction: The gene of interest is codon-optimized for each host and cloned into appropriate expression vectors: a T7-promoter vector for E. coli (e.g., pET), a galactose-inducible vector for S. cerevisiae (e.g., pYES2), and a CMV-promoter vector for HEK293 cells (e.g., pcDNA3.1) [100].
- Host Transformation/Transfection: E. coli and S. cerevisiae are transformed via heat shock and lithium acetate methods, respectively. HEK293 cells are transfected using polyethylenimine (PEI) [100] [79].
- Cell Culture and Induction: Cultures are grown in shake flasks or bioreactors under optimal conditions. Protein expression is induced at mid-log phase using IPTG (E. coli), galactose (S. cerevisiae), or via continuous promotion (HEK293).
- Harvest and Lysis: Cells are harvested by centrifugation. Microbial cells are lysed enzymatically or mechanically, while mammalian cells often secrete the protein, allowing collection of the supernatant [80].
- Titer Analysis: The concentration of the target protein in the lysate or supernatant is quantified via ELISA or by SDS-PAGE with densitometry analysis.
Protocol for Glycosylation Analysis via Lectin Blotting
Objective: To characterize the glycosylation patterns of the recombinant protein produced in different eukaryotic hosts.
Methodology:
- Protein Purification and Separation: The target protein is partially purified from each host system and separated by SDS-PAGE.
- Western Blotting: Proteins are transferred from the gel onto a nitrocellulose or PVDF membrane.
- Lectin Staining: The membrane is probed with digoxigenin-labeled lectins (e.g., ConA for mannose-type glycans, SNA for sialic acid). A secondary anti-digoxigenin antibody conjugated to alkaline phosphatase is then applied.
- Detection: Glycosylation profiles are visualized using a chromogenic or chemiluminescent substrate. The resulting banding patterns reveal host-specific glycosylation, such as the high-mannose glycans typical of S. cerevisiae versus the complex, sialylated glycans of CHO cells [12] [80].
Key Signaling Pathways and Cellular Engineering
A host cell's capacity for protein production is governed by its metabolic and regulatory networks. Engineering these pathways is key to enhancing yield and quality.
The Unfolded Protein Response (UPR) in Eukaryotic Secretion
The UPR is a critical signaling pathway in eukaryotic cells that is activated upon the accumulation of unfolded proteins in the endoplasmic reticulum (ER). For secretory proteins, a robust UPR is essential to maintain ER homeostasis and ensure correct protein folding.
Engineering a Synthetic Protein Hyperexpression System
Advanced metabolic engineering in yeast, such as S. cerevisiae, involves the coordinated optimization of multiple genetic elements to create a hyperexpression host. This systems-level approach goes beyond simple gene insertion.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful host evaluation and engineering rely on a suite of specialized reagents and tools.
Table 3: Essential Research Reagents for Heterologous Expression
| Reagent / Material | Function | Host Application |
|---|---|---|
| pET / pYES2 / pcDNA3.1 Vectors | Standardized expression plasmids with inducible promoters for each host system. | All (Bacterial, Yeast, Mammalian) [100] |
| Polyethylenimine (PEI) | A synthetic polymer used for transient transfection of mammalian cells, facilitating DNA uptake. | Mammalian Cells [79] |
| Hygromycin B | An antibiotic used as a selection marker to maintain plasmids or select for genomically integrated genes in eukaryotic cells. | Yeast, Mammalian Cells [100] |
| Phusion High-Fidelity DNA Polymerase | A PCR enzyme used for accurate amplification of gene inserts and genetic parts with low error rates. | All (Cloning) [100] |
| T4 DNA Ligase | Enzyme that catalyzes the joining of DNA fragments, essential for most cloning workflows. | All (Cloning) [100] |
| Filgotinib / Ruxolitinib | Small molecule drugs identified via computational screening to boost recombinant protein production in engineered mammalian cells. | Mammalian Cells [79] |
The economic and scalability assessment of bacterial, yeast, and mammalian host systems reveals a clear trade-off between simplicity/cost and processing complexity/functionality. Bacterial systems offer the most cost-effective and scalable solution for proteins that do not require eukaryotic PTMs. Yeasts, particularly non-conventional species like K. phaffii, present a balanced platform with good scalability, lower costs, and eukaryotic secretion machinery, albeit with simplified glycosylation. Mammalian cells remain the indispensable choice for producing the most complex therapeutic proteins, such as monoclonal antibodies, where authentic glycosylation is critical for biological activity and safety, despite higher upstream costs and longer process development times [99] [80]. The decision matrix for host selection thus ultimately depends on the specific protein target, its structural and functional requirements, and the intended application in research or medicine.
Market Share and Industry Adoption Trends for Biopharmaceutical Production
The global biopharmaceutical market is experiencing robust growth, driven by the increasing prevalence of chronic diseases and advancements in biotechnology. This guide provides an objective comparison of the three primary heterologous expression systems—bacterial, yeast, and mammalian cells—evaluating their performance based on current market data and scientific research. For researchers and drug development professionals, understanding the adoption trends, technical capabilities, and limitations of each system is crucial for selecting the appropriate platform for therapeutic protein production. The analysis reveals that while mammalian systems dominate the market for complex biologics, advanced yeast and bacterial platforms are gaining traction for specific applications through continuous engineering improvements, creating a diversified and competitive production landscape.
The biopharmaceutical market has demonstrated significant expansion and is projected to continue this trajectory over the next decade. The market encompasses biologic medicines derived from living organisms, including monoclonal antibodies, vaccines, gene therapies, and biosimilars.
Table 1: Global Biopharmaceutical Market Size and Projections
| Metric | 2024 Value | 2034 Projected Value | CAGR (2025-2034) |
|---|---|---|---|
| Market Size (Source 1) | USD 469.47 Billion | USD 1,796.21 Billion | 14.36% [101] |
| Market Size (Source 7) | USD 422.5 Billion | USD 921.5 Billion | 8.2% [102] |
Note: Variances in projections are due to different methodological assumptions and market segment definitions.
This growth is fueled by the rising demand for targeted therapies and the high prevalence of chronic diseases. According to the World Health Organization (WHO), cancer caused nearly 10 million deaths in 2023, with cases expected to rise from 20 million in 2024 to 30 million by 2040. Additionally, over 537 million adults live with diabetes globally, and autoimmune diseases impact nearly 10% of the global population [102].
Host System Performance Comparison
The choice of host system is a fundamental decision in biopharmaceutical development, with bacterial, yeast, and mammalian cells offering distinct advantages and limitations.
Table 2: Host System Comparison for Heterologous Protein Production
| Parameter | Bacterial Systems (E. coli) | Yeast Systems (S. cerevisiae, K. phaffii) | Mammalian Systems (CHO, HEK293) |
|---|---|---|---|
| Market Position | Established for simple proteins | Mature platform for vaccines, insulins; evolving for complex proteins [12] | Dominant for complex molecules (mAbs, advanced therapies) [101] |
| Typical Yield | High for simple proteins (g/L) | Variable; can be high with engineered strains [21] | Lower titer but high activity for complex proteins [103] |
| Production Timeline | Rapid (hours) | Rapid (days) | Slow (weeks) [12] |
| Cost & Scalability | Low cost, highly scalable | Low cost, highly scalable | Very high cost, complex scale-up [103] |
| Key Strength | Simplicity, high yield of simple proteins | Eukaryotic secretion, GRAS status, genetic tractability [21] | Full human-like PTMs (e.g., complex glycosylation), essential for many therapeutics [103] |
| Key Limitation | Lack of eukaryotic PTMs, intracellular aggregation [12] | Non-human, hypermannosylation glycosylation pattern; burden on host resources [103] [12] | |
| Ideal Application | Non-glycosylated proteins, peptides, antibiotics | Secreted enzymes, vaccines, generic peptides, engineered human glycoproteins [12] [21] | Monoclonal antibodies, complex fusion proteins, blood factors [101] [102] |
Experimental Protocols for Host Evaluation
Protocol: Assessing Cellular Burden in Heterologous Expression
A critical factor in host performance is the metabolic burden imposed by recombinant protein production, which competes for essential cellular resources [103].
Objective: To quantify the resource load imposed by a heterologous genetic construct on a host cell factory. Materials: Host cells (e.g., HEK293T, CHO-K1, S. cerevisiae), test plasmid with gene of interest, capacity monitor plasmid (constitutively expressing a fluorescent protein like mKATE), transfection reagents, flow cytometer or microplate reader. Methodology:
- Co-transfection: Co-transfect host cells with a fixed amount of the capacity monitor plasmid and the test plasmid.
- Fluorescence Measurement: After a defined period, measure the fluorescence intensity of both the protein of interest and the fluorescent protein from the capacity monitor.
- Data Analysis: A decrease in the monitor's fluorescence compared to a control (co-transfected with an empty vector) is inversely proportional to the resource load imposed by the test construct. This allows for the quantification of burden caused by different genetic parts (promoters, polyA signals, etc.) [104].
Protocol: Heterologous Expression of Natural Products in Streptomyces
For the discovery and production of bacterial natural products, heterologous expression in optimized Streptomyces strains is a key strategy [105].
Objective: To express a cryptic Biosynthetic Gene Cluster (BGC) in a engineered Streptomyces chassis to discover or overproduce a natural product. Materials:
- Microbial Heterologous Expression Platform (Micro-HEP): Includes specialized E. coli strains (e.g., with rhamnose-inducible Redα/β/γ recombinase system) and a chassis S. coelicolor strain (e.g., A3(2)-2023 with endogenous BGCs deleted and multiple RMCE sites) [105].
- Cloning Vectors: Modular plasmids with sites for recombinases (Cre-lox, Vika-vox, etc.). Methodology:
- BGC Capture: Identify and clone the target BGC from the native bacterium into a shuttle vector using bioinformatics and cloning techniques like TAR or ExoCET [106] [105].
- Vector Engineering: Use Red recombineering in the E. coli component of Micro-HEP to insert an RMCE cassette (containing oriT and RTS) into the BGC-containing plasmid.
- Conjugative Transfer: Transfer the engineered plasmid from E. coli to the Streptomyces chassis via bacterial conjugation.
- Genomic Integration: The BGC is integrated into the chromosome of the chassis strain via RMCE, which avoids plasmid backbone integration and allows for multi-copy integration.
- Fermentation & Analysis: Cultivate the engineered Streptomyces and analyze the metabolite profile (e.g., via HPLC-MS) to detect new or increased product yields [105].
Visualizing Workflows and Challenges
Heterologous Expression Workflow
Diagram 1: Generalized workflow for heterologous protein production, highlighting key challenges across all host systems.
Resource Competition in Expression Hosts
Diagram 2: Resource competition is a universal challenge where heterologous expression diverts cellular machinery, creating burden and reducing yield [103] [104].
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Heterologous Expression Research
| Reagent / Tool | Function & Application | Examples / Notes |
|---|---|---|
| Optimized Expression Vectors | Plasmids designed for specific hosts with strong promoters and selection markers. | YEp, YCp for S. cerevisiae; Modular systems (GoldenPiCS) for K. phaffii; RMCE cassettes for Streptomyces [12] [105]. |
| Specialized Chassis Strains | Engineered host cells with enhanced production capabilities or simplified backgrounds. | S. coelicolor A3(2)-2023 (BGC-deleted); S. cerevisiae with humanized glycosylation; E. coli with T7 RNA polymerase [105] [21]. |
| Capacity Monitor Plasmids | Quantify the metabolic burden and resource load of genetic constructs [104]. | Plasmids with constitutive fluorescent reporters (e.g., mKATE). A decrease in signal indicates high resource competition. |
| Recombineering Systems | Enable precise genetic modifications in hard-to-engineer hosts. | Red α/β/γ system in E. coli for cloning and modifying large BGCs [105]. |
| Conjugative Transfer Strains | Facilitate the transfer of large DNA constructs from E. coli to other hosts (e.g., Streptomyces). | E. coli ET12567(pUZ8002) or improved Micro-HEP E. coli strains for enhanced stability [105]. |
The biopharmaceutical production landscape is dynamic, with mammalian cell culture maintaining its dominance for the most complex therapeutics, a fact reflected in its substantial market share. However, bacterial and yeast systems remain indispensable and are continuously being improved through advanced engineering strategies aimed at overcoming their inherent limitations, such as metabolic burden and non-human post-translational modifications. The choice of host is ultimately dictated by the target molecule's complexity, required volume, and cost constraints. Future growth will be fueled by the convergence of synthetic biology, artificial intelligence, and innovative engineering approaches across all host platforms, further blurring the lines of their traditional applications and enabling the next generation of biologic medicines.
The selection of an appropriate host organism is a critical first step in the successful production of recombinant proteins, with profound implications for both research outcomes and biomanufacturing efficiency. The global market for recombinant proteins, expected to reach $2850.5 million by 2022, underscores the economic and scientific importance of this decision [107]. Researchers and drug development professionals must navigate a complex landscape of host options, primarily categorized into bacterial, yeast, and mammalian systems, each with distinct advantages and limitations. This guide provides a structured, evidence-based framework for selecting the optimal expression host based on protein characteristics and intended application, supported by comparative experimental data and practical methodologies.
Each major host system occupies a specific niche in the recombinant protein production ecosystem, balancing factors such as post-translational modification capability, scalability, and cost.
Bacterial Systems (primarily E. coli) represent the most established and economically efficient platform for producing simple, non-glycosylated proteins. Their rapid growth, well-characterized genetics, and inexpensive cultivation make them ideal for research-scale protein production and industrial enzymes that tolerate prokaryotic expression environments [108] [1].
Yeast Systems including Saccharomyces cerevisiae, Komagataella phaffii (formerly Pichia pastoris), and others offer a compelling compromise between prokaryotic simplicity and eukaryotic complexity. These unicellular fungi perform basic post-translational modifications while maintaining the scalability and cost-effectiveness of microbial fermentation [107]. K. phaffii has gained particular prominence for therapeutic protein production, with commercial products including human insulin, serum albumin, and hepatitis B vaccine [107].
Mammalian Systems (especially CHO and HEK293 cells) represent the gold standard for producing complex therapeutic proteins requiring human-like glycosylation patterns. Despite higher costs and technical complexity, their ability to correctly fold, assemble, and modify sophisticated biologics makes them indispensable for biopharmaceutical manufacturing [109] [108]. CHO cells alone account for approximately 89% of therapeutic proteins produced in mammalian systems [110].
Table 1: Core Characteristics of Major Expression Systems
| Parameter | Bacterial (E. coli) | Yeast (K. phaffii) | Mammalian (CHO) |
|---|---|---|---|
| Growth Rate | Very fast (20-30 min doubling) [1] | Fast (90 min doubling for S. cerevisiae) [1] | Slow (12-24 hour doubling) [108] |
| Cost | Low | Moderate | High |
| Post-Translational Modifications | Limited or none [108] | Basic glycosylation, disulfide bonds [107] | Human-like complex glycosylation [108] |
| Typical Yield Range | Up to several g/L [108] | Up to 10 g/L for some proteins [108] | 1-5 g/L (up to 10 g/L optimized) [108] |
| Membrane Protein Expression | Generally poor [37] | Good for many eukaryotic membrane proteins [37] | Excellent, native folding environment [37] |
| Key Advantage | Speed, cost, scalability | Balance of cost and eukaryotic processing | Authentic protein processing and modification |
| Primary Limitation | Lack of PTMs, inclusion bodies | Hypermannosylation, simpler glycosylation | Cost, complexity, technical requirements |
Decision Framework: Selecting Your Host System
The following decision pathway provides a systematic approach to host selection based on protein characteristics and application requirements. This framework integrates empirical findings from comparative studies to guide researchers through critical decision points.
Framework Application Guidance:
The decision pathway begins with the most critical differentiator: glycosylation requirements. Proteins requiring complex, human-like glycosylation patterns (such as many therapeutic antibodies) typically necessitate mammalian hosts, as neither bacterial nor yeast systems can replicate these modifications authentically [107] [108]. For membrane proteins, mammalian systems generally provide superior results due to their compatible lipid environment and associated folding machinery, though some plant membrane transporters have been successfully expressed in yeast [37].
For non-glycosylated or simply glycosylated proteins, the decision shifts to economic and scale considerations. Bacterial systems provide maximum cost efficiency for simple proteins at any scale, while yeast systems offer the best balance of eukaryotic processing capability and scalability for industrial production [107] [108]. K. phaffii specifically can achieve protein yields exceeding 10 g/L under optimized conditions, rivaling bacterial systems for many applications while providing superior processing for eukaryotic proteins [108].
Experimental Data and Performance Comparison
Recent studies provide quantitative comparisons of host system performance across multiple parameters, enabling evidence-based decision-making.
Yield Comparison Across Host Systems
Table 2: Typical Protein Yields by Host System and Application
| Host System | Typical Yield Range | Therapeutic Examples | Key Limitations |
|---|---|---|---|
| E. coli | Several g/L for simple proteins [108] | Insulin, growth hormone [107] | No glycosylation, inclusion body formation [107] |
| S. cerevisiae | Variable, generally lower than K. phaffii [107] | Insulin, glucagon, hepatitis B vaccine [107] | Hypermannosylation, lower titers than K. phaffii [107] |
| K. phaffii | Up to 10 g/L for optimized proteins [108] | Human serum albumin, interferon-alpha 2b [107] | Still simpler glycosylation than mammalian systems [107] |
| Insect Cells | 100 mg/L - 1 g/L [108] | Various viral vaccines | More complex culture than microbial systems |
| CHO Cells | 1-5 g/L (up to 10-15 g/L optimized) [108] [111] | Monoclonal antibodies, complex therapeutics | High cost, technical complexity, longer timelines [108] |
Case Study: Recombinant Gelatin Production in K. phaffii
A 2025 study demonstrated a systematic approach to optimizing recombinant human-like gelatin (hlrGEL) production in K. phaffii, illustrating key optimization principles applicable across host systems [112]. Researchers employed post-transformational vector amplification (PTVA) by screening with increasing Zeocin concentrations (200, 400, 800, and 1,200 µg/mL) to select for transformants with elevated gene copy numbers [112].
The experimental outcomes demonstrated a direct correlation between gene copy number and protein expression, up to an optimal threshold:
- Copy number: 4.29×10³/DNA (ng) → Expression: 0.21 mg/mL
- Copy number: 5.66×10³/DNA (ng) → Expression: 0.31 mg/mL
- Copy number: 6.01×10³/DNA (ng) → Expression: 0.36 mg/mL
- Copy number: 6.29×10³/DNA (ng) → Expression: 0.19 mg/mL [112]
Notably, expression declined at the highest copy number, indicating that excessive gene dosage can be counterproductive—an important consideration for expression optimization [112].
The study also implemented single-cell laser Raman spectroscopy (SCLRS) as a rapid, non-destructive screening method for identifying high-producing strains, detecting characteristic peaks at 1447 cm⁻¹, 1658 cm⁻¹, and 2929-2943 cm⁻¹ that correlated with expression levels [112]. This approach enabled high-throughput screening without cell disruption or staining, significantly accelerating strain development.
Case Study: Mammalian Host Engineering for Enhanced Production
A 2024 CHO cell study demonstrated how genetic engineering can overcome inherent limitations of host systems [109]. Researchers optimized expression vectors by incorporating Kozak sequences (GCCGCCRCC) and leader peptides upstream of target genes, resulting in significant yield improvements:
- Enhanced GFP expression increased 1.26-fold with Kozak sequence alone and 2.2-fold with combined Kozak and leader sequences [109]
- Secreted alkaline phosphatase (SEAP) production increased 1.37-1.55-fold across transient and stable expression systems [109]
Additionally, CRISPR/Cas9-mediated knockout of the Apaf1 gene (a key regulator of mitochondrial apoptosis pathway) enhanced recombinant protein production by reducing apoptosis, particularly under culture stress [109]. This strategic host cell engineering addressed a fundamental limitation in mammalian cell culture—viability maintenance in high-density production bioreactors.
Essential Methodologies and Workflows
Standardized Experimental Protocols
Protocol 1: High-Copy Strain Selection in K. phaffii
- Linearization: Digest expression vector with appropriate restriction enzyme to target integration into the AOX1 genomic locus [112]
- Transformation: Introduce linearized vector into competent GS115 cells via electroporation [112]
- Selection Plate Preparation: Prepare YPDZ plates (Yeast Extract-Peptone-Dextrose + Zeocin) with antibiotic concentrations ranging from 200-1200 µg/mL [112]
- Copy Number Amplification: Use the Post-Transformational Vector Amplification (PTVA) method by plating on increasing Zeocin concentrations to select for multi-copy integrants [112]
- Screening: Employ single-cell laser Raman spectroscopy (SCLRS) or traditional methods (SDS-PAGE, Western blot) to identify high-expressing clones [112]
- Validation: Quantify exact gene copy number via real-time quantitative PCR (qPCR) [112]
Protocol 2: Mammalian Expression Vector Optimization
- Vector Design: Incorporate strong constitutive or inducible promoter (CMV, EF-1α, CR5) with Kozak sequence (GCCGCCRCC) immediately upstream of start codon [109] [110]
- Regulatory Elements: Add chromatin-opening elements (UCOE, MAR) to maintain epigenetic activation of integrated vectors [110]
- Signal Peptide Selection: Include appropriate secretion signal (native or optimized) for extracellular production [110]
- Transfection: Use polyethylenimine (PEI) or lipofection methods for vector delivery to suspension-adapted CHO or HEK293 cells [110]
- Culture Optimization: Maintain cultures in serum-free media with nutrient feeding for 7-14 days, monitoring viability and productivity [109]
Advanced Engineering Strategies
Recent advances have expanded the toolbox for host system optimization:
Yeast Engineering: CRISPR/Cas9 has been successfully employed in K. phaffii to create protease-deficient strains by knocking out yapsin (YPS) genes, reducing proteolytic degradation of target proteins and increasing bovine intestinal alkaline phosphatase (BIAP II) yield by 2.5-fold [113]. A novel dual-color qPCR (DC-qPCR) method enables precise determination of target gene dosage, enhancing screening efficiency [113].
Mammalian Cell Engineering: Beyond Apaf1 knockout, strategies include overexpression of anti-apoptotic factors (Bcl-2, Bcl-xL) and engineering of unfolded protein response pathways to enhance secretion capacity [110]. Binary systems like the cumate gene switch enable inducible expression with 3-4 fold higher yields compared to constitutive promoters [110].
Bacterial Engineering: While not covered in detail in the current search results, E. coli strains have been engineered with disulfide isomerases and chaperones to improve folding of complex proteins, and with orthogonal translation systems for incorporation of non-natural amino acids.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Host System Implementation and Optimization
| Reagent/Cell Line | Function/Application | Key Characteristics |
|---|---|---|
| K. phaffii GS115 | Methanol-utilizing expression host | HIS4 mutant, allows selection of AOX1-integrated transformants [112] |
| CHO-S Cells | Mammalian suspension culture host | Adapted to serum-free suspension culture, suitable for large-scale production [109] |
| HEK293-6E Cells | High-density mammalian expression | Expresses truncated EBNA1, enables high-level transient expression [110] |
| pPICZα Vector | K. phaffii expression vector | Contains AOX1 promoter, Zeocin resistance, α-factor secretion signal [112] |
| Zeocin | Selection antibiotic | Selects for Shble resistance marker in yeast and mammalian systems [112] |
| PEI Transfection Reagent | Polyethylenimine-based DNA delivery | Cost-effective transfection for suspension cultures, suitable for large-scale TGE [110] |
| Single-Cell Laser Raman Spectroscopy | Non-destructive screening | Identifies high-producing clones without cell disruption [112] |
Comparative Analysis and Future Directions
Each host system continues to evolve through genetic engineering and process optimization. Bacterial systems are being engineered for improved disulfide bond formation and folding of eukaryotic proteins, expanding their utility beyond simple polypeptides. Yeast systems, particularly K. phaffii, are undergoing humanization of glycosylation pathways to produce proteins with more mammalian-like N-glycans, potentially bridging the gap between microbial and mammalian production capabilities [107]. Mammalian systems are benefiting from extensive host cell engineering to enhance productivity, product quality, and process robustness—with recent perfusion bioreactor technologies achieving cell densities of 150×10⁶ cells/mL and extended production durations [111].
Emerging technologies such as artificial intelligence-assisted sequence design, advanced CRISPR-based genome editing, and high-throughput screening methodologies are accelerating host optimization across all platforms [114]. The integration of multi-omics analyses and computational modeling promises more predictive and rational host selection in the future.
The selection of an appropriate expression host remains a multidimensional decision balancing protein characteristics, production requirements, and economic constraints. Bacterial systems excel for simple, non-glycosylated proteins where cost and speed are paramount. Yeast platforms, particularly K. phaffii, offer an optimal balance for many eukaryotic proteins requiring basic post-translational modifications at industrial scale. Mammalian systems remain essential for complex biologics requiring authentic human-like glycosylation. By applying the systematic framework and experimental approaches outlined in this guide, researchers can make informed, evidence-based decisions that maximize the success of their recombinant protein production initiatives.
Conclusion
The choice between bacterial, yeast, and mammalian expression systems is not a one-size-fits-all decision but a strategic trade-off. Bacteria offer unmatched speed and cost-efficiency for simple proteins, yeast provides a powerful balance for many eukaryotic proteins requiring basic folding and secretion, and mammalian cells remain indispensable for the production of complex, glycosylated therapeutics. The future of heterologous expression lies in the continued engineering of these hosts—through synthetic biology, CRISPR, and AI-driven design—to create next-generation cell factories that blur the lines between these traditional categories. By applying the comparative framework and optimization strategies outlined, researchers can make informed decisions that de-risk projects and accelerate the development of vital recombinant proteins for biomedical research and clinical applications.
References
- 1. Heterologous expression [https://en.wikipedia.org/wiki/Heterologous_expression]
- 2. Comparison of bacterial, yeast, insect and mammalian ... [https://www.sciencedirect.com/science/article/abs/pii/S0022175998000775]
- 3. Overview of Protein Expression Systems [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-protein-expression-systems.html]
- 4. Popular Cell Expression Systems in the Biotechnology ... [https://www.chromatographyonline.com/view/popular-cell-expression-systems-biotechnology-industry]
- 5. Technological developments driving industrial natural ... [https://pubs.rsc.org/en/content/articlelanding/2025/np/d4np00072b]
- 6. Recent Advances in the Heterologous Expression of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9225448/]
- 7. The development of Burkholderia bacteria as heterologous ... [https://pubs.rsc.org/en/content/articlelanding/2025/np/d5np00024f]
- 8. Development of an efficient heterologous protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12236015/]
- 9. Heterologous Expression - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/heterologous-expression]
- 10. A concise guide to choosing suitable gene expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10643540/]
- 11. Factors involved in heterologous expression of proteins in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10148705/]
- 12. Comparison of Yeasts as Hosts for Recombinant Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6027275/]
- 13. Advances in Escherichia coli-Based Therapeutic Protein ... [https://www.mdpi.com/2673-8449/3/4/21]
- 14. Impact of the Expression System on Recombinant Protein ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2021.682001/full]
- 15. Heterologous Expression of Difficult to Produce Proteins in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10815505/]
- 16. Recent advances in recombinant production of soluble ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02646-8]
- 17. High‐Throughput Pipeline for Protein Expression and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12306927/]
- 18. Strategies to maximize heterologous protein expression in ... [https://www.sciencedirect.com/science/article/pii/S1046592806001951]
- 19. Pichia pastoris: A highly successful expression system for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7228273/]
- 20. Transforming non-conventional yeasts into key players in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12081427/]
- 21. Engineering strategies for enhanced heterologous protein ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02299-z]
- 22. Current advances of Pichia pastoris as cell factories for ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.1059777/full]
- 23. Engineering Pichia pastoris for the efficient production of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12278525/]
- 24. Smart culture medium optimization for recombinant protein ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002241]
- 25. Advantages & Disadvantages of Protein Expression Systems [https://www.betalifesci.com/blogs/articles/advantages-and-disadvantages-of-various-protein-expression-systems?srsltid=AfmBOoqa1KRMQtueWEuwvXzlidfnWjDmvoSMiXvF1OSK3JnkpBbqbf9j]
- 26. Expression of mammalian proteins for diagnostics and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9175168/]
- 27. Mammalian Cell–Based Protein Expression [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/protein-expression-handbook/pex-handbook-mammalian-cell-based-protein-expression.html]
- 28. A Review of Therapeutic Protein Expression By ... [https://www.bioprocessintl.com/expression-platforms/a-review-of-therapeutic-protein-expression-by-mammalian-cells]
- 29. Overview of Recombinant Protein Expression Systems [https://futurefields.io/blogs/flylab/overview-of-recombinant-protein-expression-systems?srsltid=AfmBOopmGOXfIw-npzf2Lzh3hT6PviMMgrF1jFK3DVVFdgedUdK95pgW]
- 30. Protein Expression Host Systems | Selection Guide ... [https://www.betalifesci.com/pages/protein-expression-host-selection?srsltid=AfmBOoqVytbg5l1VywRum-Uvr_Z001K7oJ4nX1ftp3PcnhrNPOFzJ674]
- 31. Cell Engineering for Increasing Production of Recombinant ... [https://pubmed.ncbi.nlm.nih.gov/41261329/]
- 32. Overview of Post-Translational Modifications (PTMs) [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-post-translational-modification.html]
- 33. Protein posttranslational modifications in health and diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC10152985/]
- 34. Multiple Post-translational Modifications Affect ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3411053/]
- 35. Protein Expression Systems, Which One Should I Choose? [https://www.biosynth.com/blog/protein-expression-systems]
- 36. Engineering PVX vectors harboring heterologous VSRs to ... [https://www.nature.com/articles/s41598-025-24470-1]
- 37. Yeast Heterologous Expression Systems for the Study of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10342074/]
- 38. Efficient size-independent chromosome delivery from yeast to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5397165/]
- 39. Gene Expression in Mammalian Cells and its Applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC3848218/]
- 40. Optimizing viral transduction in immune cell therapy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12046913/]
- 41. Efficient site-specific integration of large genes in ... [https://www.nature.com/articles/s41551-024-01227-1]
- 42. 5 reasons to move from shake flasks to a bioreactor [https://infors-ht.com/en/blog/5-reasons-to-move-from-shake-flasks-to-a-bioreactor]
- 43. CHO cell cultures in shake flasks and bioreactors present ... [https://www.sciencedirect.com/science/article/abs/pii/S1369703X19300464]
- 44. Experimental studies from shake flasks to 3 L stirred tank ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10127095/]
- 45. Comparing Shaker Flasks with a Single-Use Bioreactor for ... [https://www.bioprocessintl.com/microbial-cell-culture/comparing-shaker-flasks-with-a-single-use-bioreactor-for-growing-yeast-seed-cultures]
- 46. when shake flasks mimic 15000-L bioreactors better [https://pubmed.ncbi.nlm.nih.gov/26097232/]
- 47. The development of Burkholderia bacteria as heterologous ... [https://pubs.rsc.org/en/content/articlehtml/2025/np/d5np00024f]
- 48. A highly efficient heterologous expression platform to facilitate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12076808/]
- 49. Engineering strategies for enhanced heterologous protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10801990/]
- 50. A Comprehensive Review of the Protein Subunit Vaccines ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.927306/full]
- 51. Higher-Order Structure Comparability: Case Studies of ... [https://www.bioprocessintl.com/biosimilars/higher-order-structure-comparability-case-studies-of-biosimilar-monoclonal-antibodies]
- 52. 综述:细菌重组蛋白生产的进展:异源系统中的策略与挑战 [https://www.ebiotrade.com/newsf/2025-10/20251019082409578.htm]
- 53. 蛋白质表达系统 [https://www.sigmaaldrich.com/HK/zh/technical-documents/technical-article/protein-biology/protein-expression/protein-expression-systems?srsltid=AfmBOoqbCiUpPaNDy4QgQMvPo2F1v-YHmWxSDUk7yZizy7Um1vuvBpuY]
- 54. Developing Aspergillus as a host for heterologous ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975008000906]
- 55. 产几丁质酶侧孢短芽孢杆菌的筛选及其酶学性质研究 [https://html.rhhz.net/SWJSTB/html/2016-6-174.htm]
- 56. A new high-performance heterologous fungal expression ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2015.00184/full]
- 57. Assessing Brevibacillus sp. C17: An indigenous isolated ... [https://www.sciencedirect.com/science/article/abs/pii/S2214714417307316]
- 58. Protein recovery from inclusion bodies of Escherichia coli ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-015-0222-8]
- 59. Time to consider ruling out inclusion bodies denaturing ... [https://www.nature.com/articles/s41598-024-77899-1]
- 60. Challenges Associated With the Formation of Recombinant ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2021.630551/full]
- 61. Co-Expression of Chaperones for Improvement of Soluble ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9926028/]
- 62. Overview of refolding methods on misfolded recombinant ... [https://jabonline.in/abstract.php?article_id=937&sts=2]
- 63. Solubilization and refolding of inclusion bodies by detergents [https://www.sciencedirect.com/science/article/abs/pii/S1046592825001330]
- 64. Quality comparison of recombinant soluble proteins and ... [https://www.sciencedirect.com/science/article/pii/S187167842200053X]
- 65. Pre‐folding purification procedures for inclusion body ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12171399/]
- 66. Comparative Analysis of Codon Optimization Tools [https://pmc.ncbi.nlm.nih.gov/articles/PMC12010093/]
- 67. Strategies of codon optimization for high-level ... [https://www.sciencedirect.com/science/article/abs/pii/S2452014417300614]
- 68. Codon Optimization: Understanding the Basics | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/codon-optimization-the-basics-explained/]
- 69. Codon optimization with deep learning to enhance protein ... [https://www.nature.com/articles/s41598-020-74091-z]
- 70. Codon Optimization for Different Expression Systems [https://www.biosynsis.com/blog/codon-optimization-for-different-expression-systems-key-points-and-case-studies/]
- 71. Multiplex Genome Editing in Yeast by CRISPR/Cas9 [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2021.719148/full]
- 72. CRISPR/Cas9 System: A Bacterial Tailor for Genomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6167567/]
- 73. Microbial Genome Editing with CRISPR–Cas9 [https://www.mdpi.com/2311-5637/11/7/410]
- 74. Recombinant monoclonal antibody production in yeasts [https://www.sciencedirect.com/science/article/abs/pii/S0141813024021846]
- 75. Genome-scale reconstructions of the mammalian secretory ... [https://www.nature.com/articles/s41467-019-13867-y]
- 76. The MFα signal sequence in yeast-based protein secretion [https://pmc.ncbi.nlm.nih.gov/articles/PMC12141369/]
- 77. Deciphering the determinants of recombinant protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12541331/]
- 78. different strategies for intracellular and secreted proteins in yeast [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02848-0]
- 79. Computational identification of small molecules for ... [https://www.nature.com/articles/s41467-025-62529-9]
- 80. Recombinant Protein Expression in Mammalian Cells [https://www.evitria.com/journal/recombinant-antibodies/recombinant-protein-expression-mammalian-cells/]
- 81. In silico analysis of metabolic burden effects on a ... [https://www.sciencedirect.com/science/article/pii/S0947358025001517]
- 82. Computational Modelling of Metabolic Burden and ... [https://www.mdpi.com/2076-2607/7/11/553]
- 83. Designing strong inducible synthetic promoters in yeasts [https://www.nature.com/articles/s41467-024-54865-z]
- 84. How to Choose the Right Inducible Gene Expression System ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6721553/]
- 85. A blueprint for designing the next-generation of synthetic C ... [https://www.nature.com/articles/s41467-025-64483-y]
- 86. Folding of heterologous proteins in bacterial cell factories [https://www.sciencedirect.com/science/article/abs/pii/S0734975022001756]
- 87. Strategies for Enhancing Recombinant Protein Expression [https://www.procellsystem.com/resources/cell-culture-academy/strategies-for-enhancing-recombinant-protein-expression-core-techniques-for-efficient-and-reliable-results-2057]
- 88. Manipulating gene expression levels in mammalian cell ... [https://www.sciencedirect.com/science/article/pii/S1871678423000663]
- 89. Yeast Heterologous Expression Systems for the Study of ... [https://www.mdpi.com/1422-0067/24/13/10768]
- 90. Glycosylation: mechanisms, biological functions and ... [https://www.nature.com/articles/s41392-024-01886-1]
- 91. Mammalian glycosylation in immunity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2768770/]
- 92. Protein Expression Systems [https://www.antibodies-online.com/resources/17/5177/protein-expression-systems/]
- 93. Humanization of Yeasts for Glycan-Type End-Products [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.930658/full]
- 94. The humanization of N- glycosylation pathways in yeast [https://www.nature.com/articles/nrmicro1087?error=cookies_not_supported&code=9939351b-8e53-432c-bfac-f4ae853024b9]
- 95. Principles of N-Linked Glycosylation Variations of IgG-Based... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7345016/]
- 96. Commensal bacterial glycosylation at the interface of host ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355684/]
- 97. Bacterial glycosylation, it's complicated [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.1015771/full]
- 98. Analysis of fungal high-mannose structures using CAZymes [https://pmc.ncbi.nlm.nih.gov/articles/PMC8970417/]
- 99. Protein Expression Host Systems | Selection Guide ... [https://www.betalifesci.com/pages/protein-expression-host-selection?srsltid=AfmBOopTiJMJBkT2JLg_nMX2n24s9rgSpTqfFc0cnIc80P8MtO9RqLll]
- 100. ECO-Casein/iGEM NCHU 2025/methods [https://2025.igem.wiki/nchu/experiments/]
- 101. Biopharmaceuticals Market Size and Forecast 2025 to 2034 [https://www.precedenceresearch.com/biopharmaceutical-market]
- 102. Biopharmaceutical Market Size & Share Report, 2025 - 2034 [https://www.gminsights.com/industry-analysis/biopharmaceutical-market]
- 103. Burden Imposed by Heterologous Protein Production in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10512257/]
- 104. Resource-aware construct design in mammalian cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10275982/]
- 105. A highly efficient heterologous expression platform to facilitate ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02722-z]
- 106. Bacterial natural product discovery by heterologous expression [https://pmc.ncbi.nlm.nih.gov/articles/PMC10727000/]
- 107. Comparison of Yeasts as Hosts for Recombinant Protein Production - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6027275/]
- 108. Comparison of Protein Yield Between Different Expression ... [https://synapse.patsnap.com/article/comparison-of-protein-yield-between-different-expression-hosts]
- 109. Optimization of a novel expression system for recombinant ... [https://www.nature.com/articles/s41598-024-76995-6]
- 110. Improvement strategies for transient gene expression in ... [https://link.springer.com/article/10.1007/s00253-024-13315-y]
- 111. Proteomics Reveals Distinctive Host Cell Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11747259/]
- 112. Efficient extracellular expression and rapid screening of ... [https://www.nature.com/articles/s41598-025-14855-7]
- 113. Top 26 Yeast papers published in 2025 [https://scispace.com/journals/yeast-j2vudwwj/2025]
- 114. Engineering Genetic Elements for Microbial Protein ... [https://www.sciencedirect.com/science/article/pii/S2405805X2500170X]