Advanced Strategies for Tandem CRISPR sgRNA Array Construction: A Comprehensive Guide to Efficient Multiplexed Genome Editing
This article provides a comprehensive overview of modern approaches for constructing tandem CRISPR sgRNA arrays to achieve efficient multiplexed genome editing.
Advanced Strategies for Tandem CRISPR sgRNA Array Construction: A Comprehensive Guide to Efficient Multiplexed Genome Editing
Abstract
This article provides a comprehensive overview of modern approaches for constructing tandem CRISPR sgRNA arrays to achieve efficient multiplexed genome editing. It explores the foundational principles of multiplexed CRISPR technologies, detailing various genetic architectures for gRNA expression and processing. The content covers practical methodological strategies for array assembly and delivery across diverse systems, from mammalian cells to plants. It further addresses common troubleshooting challenges and optimization techniques, and concludes with rigorous validation frameworks and comparative analyses of different CRISPR systems. This resource is tailored for researchers, scientists, and drug development professionals seeking to implement sophisticated multiplexed editing approaches for functional genomics, metabolic engineering, and therapeutic development.
The Fundamentals of Multiplexed CRISPR: From Natural Systems to Synthetic Array Design
Multiplexed CRISPR technologies represent a transformative advancement in genetic engineering, enabling simultaneous modification of multiple genomic loci within a single experiment. Unlike single-guide CRISPR systems, multiplexed approaches employ numerous guide RNAs (gRNAs) or Cas enzymes expressed concurrently, vastly enhancing the scope and efficiency of genetic editing and transcriptional regulation. These technologies have become indispensable tools for functional genomics, complex disease modeling, metabolic engineering, and synthetic biology applications, allowing researchers to address biological questions with unprecedented scale and precision. The core innovation lies in the ability to express and process multiple gRNAs from engineered arrays, leveraging both natural CRISPR system components and synthetic biology approaches to achieve coordinated genetic perturbations.
Core Technologies and gRNA Expression Architectures
The foundation of multiplexed CRISPR editing lies in strategies for expressing and processing multiple gRNAs within target cells. Three primary genetic architectures have been developed for this purpose, each with distinct advantages and applications.
Individual Promoter Systems
The most straightforward approach involves expressing each gRNA under the control of an individual promoter, typically Pol III U6 promoters in mammalian cells and Pol III tRNA promoters in yeast and plants [1]. This method provides independent transcriptional control of each gRNA but becomes technically challenging with increasing numbers of guides due to vector size constraints and potential promoter interference.
Endogenous Processing Systems
Native CRISPR systems inherently process multiple guides from single transcripts, and this natural capacity has been engineered for synthetic systems. The Cas12a nuclease possesses intrinsic abilitiy to process pre-crRNA via recognition of hairpin structures formed within spacer repeats [1]. Similarly, arrays can be processed by RNase III in a tracrRNA-dependent manner, mimicking the natural processing mechanism of Type II CRISPR systems [1]. These approaches leverage evolved biological processing mechanisms but may be limited by the specific requirements of the processing enzymes.
Synthetic Processing Systems
Engineered systems utilize exogenous RNA cleavage elements to process gRNA arrays from single transcripts. Common strategies include:
- Ribozyme-flanked gRNAs: Hammerhead and hepatitis delta virus ribozymes flank each gRNA, enabling self-cleavage from primary transcripts [1]
- Csy4 processing: The bacterial Csy4 RNase recognizes a 28-nt stem-loop sequence and cleaves after the 20th nucleotide, allowing precise excision of gRNAs [1]
- tRNA-based processing: Endogenous tRNA-processing machinery (RNases P and Z) cleaves at pre-tRNA sequences inserted between gRNAs [2] [3]
Table 1: Comparison of Multiplexed gRNA Expression Systems
| System Type | Processing Mechanism | Max gRNAs Demonstrated | Key Advantages | Limitations |
|---|---|---|---|---|
| Individual Promoters | Transcriptional initiation | Variable | Simple design, independent regulation | Size constraints, promoter interference |
| Cas12a Processing | Native Cas12a cleavage | 10+ [1] | No additional factors needed | Limited to Cas12a systems |
| tRNA Processing | Endogenous RNases P & Z | 10 [2] | Universal across organisms | tRNA sequences may affect gRNA function |
| Ribozyme-mediated | Self-cleaving RNAs | 7+ [1] | Inducible systems possible | Larger sequence requirements |
| Csy4 Processing | Heterologous RNase | 12 [1] | High precision | Requires Csy4 co-expression |
Tandem sgRNA Array Construction Methods
The construction of highly repetitive gRNA arrays presents significant technical challenges due to sequence homology. Several cloning strategies have been developed to address these challenges.
Golden Gate Assembly
Golden Gate assembly utilizes type IIS restriction enzymes that cleave outside their recognition sequences, creating unique overhangs that allow directional assembly of multiple gRNA units [4]. This method enabled construction of a single CRISPR-Cas9 cassette with seven gRNAs [4]. Advanced versions like "PCR-on-ligation" have further enhanced this approach, allowing modular assembly of up to 10 gRNAs in the HEK293T cell line [4].
Modular Vector Systems
For plant systems, researchers have developed specialized binary vectors incorporating tRNA-gRNA arrays expressed under strong promoters. In citrus, the ES8Z promoter from Arabidopsis demonstrated robust expression when driving tRNA-sgRNA arrays, enabling efficient multiplex editing of at least four genes simultaneously [3]. Optimization of both Cas9 expression (using UBQ10 or RPS5a promoters) and sgRNA array expression significantly improved editing efficiency across multiple targets [3].
Viral Delivery Systems
Plant virus-derived vectors enable high-efficiency delivery of sgRNA arrays without conventional transformation. Engineered Potato virus X (PVX) vectors successfully expressed unspaced sgRNA arrays in solanaceous plants, achieving highly efficient multiplex editing in adult plant tissues within days [2]. Surprisingly, PVX vectors expressing sgRNA arrays without processing spacers still induced efficient gene editing, suggesting potential processing through unknown mechanisms [2]. This virus-induced genome editing (VIGE) strategy achieved nearly 80% indels in Nicotiana benthamiana lines constitutively expressing Cas9 [2].
Tandem sgRNA Array Construction and Implementation Workflow
Applications in Biological Engineering
Functional Genomic Screening
Multiplexed CRISPR enables genome-wide functional screening with unprecedented depth. The CRISPR-based double-knockout (CDKO) system utilizes paired gRNAs to create large deletions, identifying synthetic lethal interactions in K562 cells from 490,000 gRNA pairs [4]. Similarly, Perturb-seq combines single-cell RNA-seq with CRISPR barcoding, allowing complex phenotypic assessment of multiple perturbations [5]. This approach successfully decoupled the three branches of the unfolded protein response (UPR) by combinatorially repressing IRE1α, PERK, and ATF6 sensor genes [5].
Genome Engineering and Structural Variation
Dual-target editing facilitates programmed structural variations including:
- Large deletions: Two simultaneous DSBs create defined chromosomal deletions [4]
- Inversions and translocations: Targeted cutting at two sites can invert or translocate chromosomal segments [4]
- Gene knockouts: Large deletions within genes ensure complete functional disruption [4]
Notably, CRISPR-induced structural variations have been applied in hematopoietic stem cells to model clonal hematopoiesis and myeloid neoplasia, demonstrating the clinical relevance of these approaches [6].
Noncoding Genome Characterization
Large-scale CRISPR interference (CRISPRi) screens have enabled systematic functional characterization of noncoding cis-regulatory elements (CREs). The ENCODE Consortium conducted 108 screens comprising >540,000 perturbations across 24.85 megabases of genome, identifying 865 distinct functional CREs [7]. These efforts established that 4.0% of perturbed bases displayed regulatory function, with CREs predominantly overlapping accessible chromatin regions marked by H3K27ac [7].
Table 2: Quantitative Performance of Multiplexed CRISPR Applications
| Application Domain | Scale Demonstrated | Efficiency Metrics | Key Findings |
|---|---|---|---|
| Functional Screening | 490,000 gRNA pairs [4] | Identification of synthetic lethal interactions | Revealed gene networks and genetic interactions |
| Noncoding Element Mapping | 540,000 perturbations [7] | 4.0% of perturbed bases functional | 97.6% of CREs overlap ENCODE cCREs |
| Plant Genome Engineering | 4 simultaneous genes [3] | High-efficiency biallelic mutations | Virus-free edited progeny obtainable |
| Structural Variation | Large deletions, inversions, translocations [4] | Precise chromosomal rearrangements | Disease modeling in hematopoietic cells [6] |
Essential Reagents and Research Tools
Table 3: Research Reagent Solutions for Multiplexed CRISPR
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Cas Effectors | SpCas9, Cas12a [1] | DNA recognition and cleavage | Cas12a enables inherent array processing |
| gRNA Expression Promoters | U6, tRNA, ES8Z, UBQ10 [3] | Drive gRNA transcription | ES8Z effective in plants; U6 standard in mammals |
| Array Processing Elements | tRNAGly, Csy4, ribozymes [1] [2] | Excise individual gRNAs from arrays | tRNA systems use endogenous RNases |
| Delivery Vectors | Lentiviral, PVX-based [5] [2] | Introduce editing components | PVX vectors enable rapid plant editing |
| Assembly Systems | Golden Gate, MoClo toolkit [3] | Construct repetitive arrays | Type IIS enzymes enable modular cloning |
Multiplex CRISPR Reagent Systems and Applications
Detailed Experimental Protocol: tRNA-gRNA Array Implementation
Array Design and Synthesis
- Target Selection: Identify 20-nt protospacer sequences adjacent to PAM sites (5'-NGG-3' for SpCas9) for each target locus
- tRNA Selection: Incorporate Arabidopsis thaliana tRNAGly (GCC anticodon) sequences between each sgRNA unit [3]
- Array Synthesis: Synthesize the complete tRNA-sgRNA array as a gBlock fragment (Gene Universal Inc.) with appropriate flanking restriction sites (PacI-MluI) for cloning [3]
Vector Assembly
- Backbone Preparation: Digest binary vector (pYAO::hSpCas9 or similar) with PacI and MluI restriction enzymes
- Ligation: Insert the synthesized tRNA-gRNA array into the vector backbone using T4 DNA ligase
- Promoter Cloning: Clone the ES8Z or UBQ10 promoter upstream of the tRNA-gRNA array using SpeI and SbfI restriction sites [3]
- Transformation: Transform ligation reaction into competent E. coli cells and select on appropriate antibiotics
Plant Transformation and Analysis (Citrus Protocol)
- Explant Preparation: Germinate Carrizo citrange seeds in vitro on MS medium with vitamins, 30 g/L sucrose, and 2.5 g/L Phytagel (pH 5.8) for 4-6 weeks in darkness [3]
- Agrobacterium Transformation: Incubate 75-100 epicotyl explants with Agrobacterium tumefaciens strain EHA105 harboring the binary vector for 15 minutes [3]
- Selection and Regeneration: Transfer explants to selection media containing appropriate antibiotics and regenerate plants under 16-h-light/8-h-dark photoperiod at 28°C [3]
- Editing Efficiency Assessment: Extract genomic DNA from regenerated plants and perform PCR amplification of target loci followed by sequencing to quantify indel frequencies
Technical Considerations and Optimization Strategies
Enhancing Editing Efficiency
- Promoter Selection: Strong constitutive promoters (UBQ10, RPS5a) driving Cas9 expression significantly improve editing rates [3]
- Chromatin Accessibility: Exposure to heat stress can render chromatin more accessible to Cas9-sgRNA complexes [3]
- Array Configuration: Unspaced sgRNA arrays in PVX vectors unexpectedly achieved efficient editing, suggesting alternative processing mechanisms [2]
Specificity and Safety
- Nickase Systems: Paired Cas9 nickases targeting opposite DNA strands reduce off-target effects while maintaining on-target efficiency [4]
- Multiple Nicking: Increasing the number of nicks enhances homologous recombination efficiency with minimal indel generation [4]
Analytical Validation
- Multiplex Perturbation Validation: For combinatorial screens, ensure uniform perturbation efficiency across all targets by quantifying repression/editing of each individual target [5]
- Single-Cell Phenotyping: Integrate single-cell RNA sequencing with perturbation barcoding to resolve complex phenotypic outcomes [5]
The continued refinement of multiplexed CRISPR technologies promises to further expand capabilities in synthetic biology, therapeutic development, and functional genomics, establishing these approaches as cornerstone methodologies in biological engineering.
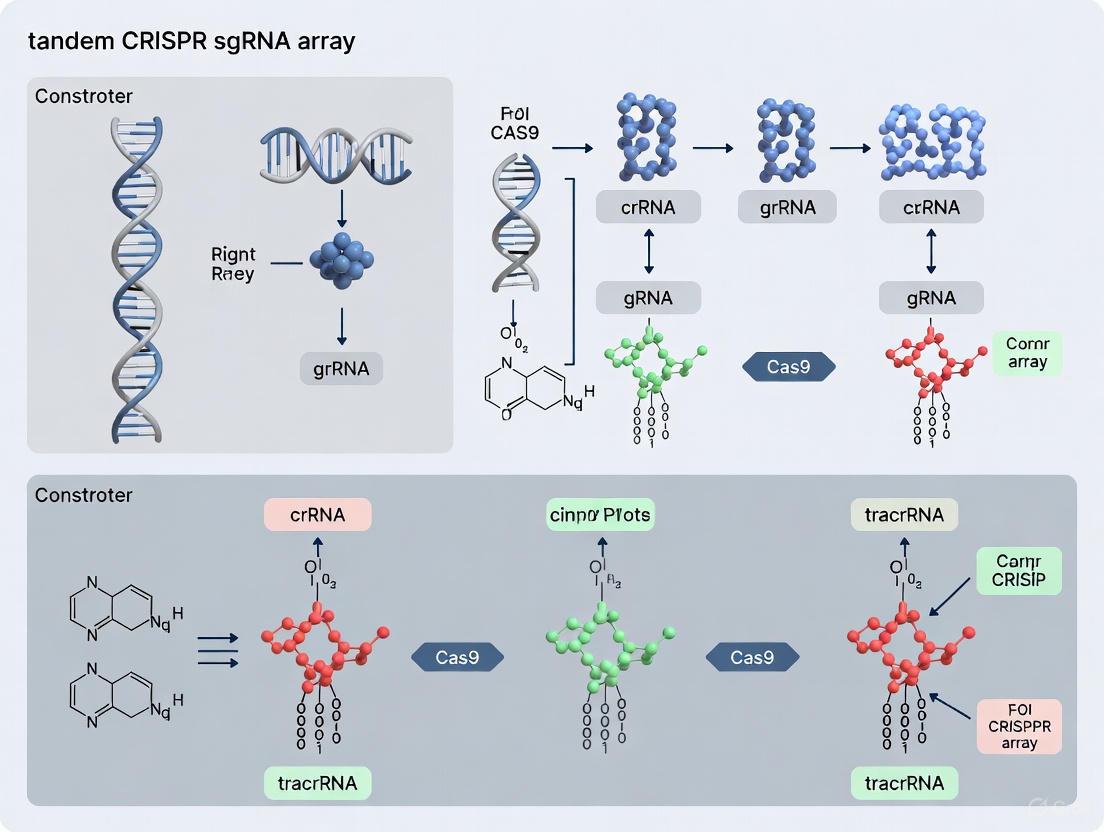
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) proteins constitute an adaptive immune system in bacteria and archaea. The heart of this system lies in the generation of mature CRISPR RNAs (crRNAs) from a long precursor transcript (pre-crRNA), which guide Cas proteins to cleave complementary invading nucleic acids [8]. This application note details the fundamental crRNA biogenesis pathways in native CRISPR-Cas systems, framing them as blueprints for constructing synthetic tandem guide RNA arrays. We provide a comparative analysis of processing mechanisms, structured protocols for leveraging these principles, and visualization of the core concepts to empower research in multiplexed genome editing.
In CRISPR-Cas adaptive immunity, a key step is the processing of a long primary transcript, the precursor crRNA (pre-crRNA), into short, mature guide crRNAs. The pre-crRNA is transcribed from a CRISPR array, which consists of a series of repeats interspaced by unique spacer sequences acquired from mobile genetic elements [8]. These mature crRNAs, each containing a spacer sequence, then guide one or more Cas proteins to recognize and destroy cognate invading genomes [8]. The molecular mechanisms behind pre-crRNA recognition and cleavage have evolved differently across CRISPR-Cas types, providing a rich toolkit of natural RNA-processing strategies [8]. Understanding these native mechanisms is crucial for repurposing them to express multiple guide RNAs (gRNAs) simultaneously—a capability highly sought after for multiplexed genetic perturbation studies [1].
Comparative Analysis of Native crRNA Processing Pathways
CRISPR-Cas systems are broadly divided into two classes based on their effector modules. Class 1 (Types I, III, IV) utilizes multi-protein effector complexes, while Class 2 (Types II, V, VI) employs single-protein effectors [9]. This classification underpins the distinct crRNA biogenesis pathways summarized in the table below.
Table 1: Comparative crRNA Biogenesis Pathways in Major CRISPR-Cas Types
| CRISPR-Cas Type | Processing Enzyme/Component | Key RNA Component | Cleavage Mechanism & Site | Resulting crRNA Structure |
|---|---|---|---|---|
| Type I | Cas6 (or Cas5d in I-C) [8] | pre-crRNA alone [8] | Metal-independent endoribonuclease cleavage within the repeat sequence, typically 8 nt upstream of the repeat-spacer boundary [8]. | Entire spacer flanked by partial repeat sequences; often contains a stable hairpin [8]. |
| Type II | RNase III + Cas9 [8] | pre-crRNA + trans-activating crRNA (tracrRNA) [8] | Dual-RNA structure formed by pre-crRNA:tracrRNA base-pairing is cleaved by housekeeping RNase III [8]. | Fused crRNA:tracrRNA (single-guide RNA, sgRNA) in engineered systems [1]. |
| Type III | Cas6 [8] | pre-crRNA alone [8] | Standalone Cas6 cleaves within repeats; often requires additional trimming for maturation [8]. | Mature crRNA used by complex; associated with cOA signaling for collateral activity [9]. |
| Type V (e.g., Cas12a) | Cas protein itself (e.g., Cas12a) [1] | pre-crRNA alone [1] | The nuclease (e.g., Cas12a) recognizes and cleaves its own pre-crRNA within the repeat [1]. | Mature crRNA ready for interference without further processing. |
The following diagram illustrates the logical relationships and key differences between these primary biogenesis pathways.
Native Processing Mechanisms as Blueprints for Synthetic sgRNA Arrays
The inherent ability of native CRISPR systems to process multiple spacers from a single transcript makes them ideal inspirations for synthetic multiplexed editing tools. Researchers have engineered various genetic architectures that mimic these natural strategies to express numerous gRNAs from a single Pol II or Pol III promoter [1].
The core principle involves constructing a synthetic array where individual gRNA units are separated by specific processing sequences. The choice of separator dictates the required processing machinery, each with distinct advantages. The workflow below outlines the primary methods for expressing and processing such synthetic gRNA arrays.
Table 2: Synthetic gRNA Array Architectures Inspired by Native Systems
| Array Architecture | Processing Mechanism | Key Feature | Example Application |
|---|---|---|---|
| Direct Cas Processing | The Cas protein itself (e.g., Cas12a) processes the array [1]. | Simple system; does not require additional processing factors. | Simultaneous cleavage of 5 targets in human cells [1]. |
| tRNA-spacer Array | Endogenous RNase P and RNase Z cleave flanking tRNA sequences [1]. | Ubiquitous cellular machinery; works across diverse organisms [1]. | Processing of tRNA–gRNA arrays from a single Pol II promoter [1]. |
| Ribozyme-flanked gRNAs | Self-cleaving ribozymes (e.g., Hammerhead, HDV) flank each gRNA [1]. | High modularity; does not rely on cellular proteins. | Expression of multiple gRNAs in yeast and plants [1]. |
| Csy4-dependent Array | The Cas6-family enzyme Csy4 cleaves a specific 28-nt RNA sequence [1]. | Highly specific and efficient processing. | Expression and processing of 12 sgRNAs in S. cerevisiae [1]. |
| Unspaced sgRNA Arrays | Not yet fully characterized; may leverage viral replication or endogenous nucleases [2]. | Simplified cloning; no spacers needed. | Efficient multiplex editing in N. benthamiana using a PVX vector [2]. |
Experimental Protocols
Protocol: Implementing a Cas12a-based gRNA Array for Multiplexed Knockout
This protocol leverages the intrinsic pre-crRNA processing capability of Cas12a (a Type V effector) to simultaneously target multiple genomic loci in mammalian cells [1].
Reagents:
- CasPEDIA entry for AsCas12a or LbCas12a
- Mammalian expression vector with a Pol II promoter (e.g., EF1a, CAG)
- Custom DNA fragment encoding the crRNA array (see Table 2)
- Target cells (e.g., HEK293T)
- Transfection reagent
Procedure:
- Array Design: Design a crRNA array where each 20-nt spacer targeting a specific genomic locus is separated by the native Cas12a direct repeat sequence (typically 19-23 nt).
- Vector Construction: Synthesize the crRNA array as a gBlock and clone it downstream of the Pol II promoter in the expression vector. On the same vector, ensure Cas12a is expressed from a separate promoter.
- Delivery: Transfect the constructed plasmid into target cells using a standard transfection protocol.
- Validation: Harvest cells 72-96 hours post-transfection. Assess editing efficiency at each target locus by tracking indels by decomposition (TIDE) or next-generation sequencing (NGS).
Protocol: Building a tRNA-gRNA Array for Multiplexed CRISPRi/a
This method uses endogenous tRNA-processing machinery to excise multiple gRNAs from a single transcript and is suitable for both CRISPR knockout and CRISPR interference/activation (CRISPRi/a) [1].
Reagents:
- dCas9-KRAB (for CRISPRi) or dCas9-VPR (for CRISPRa) expression vector
- tRNA-gRNA array cloned into a U6 or Pol II expression vector
- Target cells
- Appropriate transfection reagent
Procedure:
- Array Design: Design a DNA sequence where each sgRNA is flanked 5' and 3' by a 77-nt long pre-tRNA sequence. The tRNA sequence will be recognized by endogenous RNases P and Z.
- Cloning: Assemble the tRNA-gRNA array using Golden Gate or Gibson Assembly. Clone the final array into the chosen expression vector.
- Co-delivery: Co-transfect the tRNA-gRNA array plasmid with the dCas9-effector plasmid into the target cells.
- Phenotypic Analysis: After 4-5 days, assay for transcriptional repression (CRISPRi) or activation (CRISPRa) using qRT-PCR for target genes or a relevant phenotypic assay (e.g., fluorescence if targeting a reporter).
Table 3: Key Research Reagent Solutions for crRNA Processing and Multiplexed Screening
| Reagent / Resource | Function / Application | Source / Example |
|---|---|---|
| Cas Protein Effector Database (CasPEDIA) | Classification and functional assessment of Class 2 Cas enzymes for target selection [9]. | Publicly accessible database [9]. |
| MAGeCK Computational Tool | A bioinformatic workflow designed for the analysis of CRISPR screen data to identify enriched/depleted gRNAs [10] [11]. | Open-source software [10]. |
| CRISPR Cloud2 | A web-based platform for the analysis of CRISPR screen data, facilitating quality control and hit identification [10]. | Publicly accessible web tool [10]. |
| Addgene Repository | A non-profit source for CRISPR plasmids, including genome-wide gRNA libraries and Cas expression vectors [12]. | Addgene.org |
| Csy4 Nuclease | A specific Cas6-family endoribonuclease used for processing synthetic gRNA arrays flanked by its recognition sequence [1]. | Commercial vendors / Addgene. |
| Brunello CRISPR Knockout Library | A genome-wide human sgRNA library for pooled knockout screens [11]. | Addgene (Pooled Library #73179). |
The advent of CRISPR–Cas technology has revolutionized genome engineering, with its application extending from basic research to therapeutic development. A critical factor influencing the efficiency and success of CRISPR-based interventions is the strategy employed for guide RNA (gRNA) expression. For multiplexed genome editing—the simultaneous targeting of multiple genomic sites—the choice of genetic architecture for gRNA expression is paramount. These architectures primarily fall into two categories: the use of individual promoters for each gRNA and the use of single transcriptional units that produce a polycistronic array processed into individual gRNAs in vivo. The decision between these strategies impacts editing efficiency, stoichiometry of gRNA components, vector size, and overall experimental feasibility. This application note delineates the key genetic architectures for gRNA expression, providing a comparative analysis and detailed protocols to guide researchers in selecting and implementing the optimal system for their multiplexed editing objectives.
Comparative Analysis of gRNA Expression Architectures
The two predominant genetic architectures for expressing multiple gRNAs leverage distinct transcriptional and post-transcriptional mechanisms. Table 1 provides a systematic comparison of their core features.
Table 1: Comparison of Individual Promoter and Array-Based gRNA Expression Systems
| Feature | Individual Promoter System | Array-Based System |
|---|---|---|
| Genetic Design | Multiple, independent promoter-gRNA-terminator cassettes [13] [1] | Single promoter drives a transcript of tandem gRNAs separated by processing sites [1] |
| Transcriptional Control | Typically uses RNA Pol III promoters (e.g., U6, H1) for each gRNA [13] | Can use Pol II or Pol III promoters; Pol II allows inducible/tissue-specific control [13] [1] |
| gRNA Processing | Not required; each transcript is a mature gRNA | Requires enzymatic cleavage (e.g., Csy4, tRNA, Ribozyme) [1] [14] |
| Key Advantage | Simplicity of design; predictable expression | Compact vector size; defined gRNA stoichiometry from a single transcript [1] [14] |
| Key Limitation | Large plasmid size; potential for promoter cross-talk & recombination [13] | Requires co-expression of processing enzyme (for Csy4); processing efficiency varies [1] |
| Ideal Use Case | Expressing a small number (e.g., 2-4) of gRNAs | High-order multiplexing (e.g., >5 gRNAs); applications with strict size limits (e.g., AAV delivery) [14] |
The Individual Promoter Architecture
This conventional approach involves constructing a plasmid where each gRNA is expressed from its own dedicated promoter and terminator sequence [13]. In mammalian cells, the U6 small nuclear RNA polymerase III (Pol III) promoter is most commonly used due to its consistent expression and precise transcription initiation and termination [13] [14]. A significant consideration in this system is the risk of promoter cross-talk and transcriptional interference when multiple identical Pol III promoters are placed in close proximity, which can lead to reduced gRNA expression and transgene silencing, particularly in plants [13]. Furthermore, assembling plasmids with multiple repetitive cassettes can be technically challenging due to recombination in bacterial hosts. Despite these limitations, this architecture remains a robust and straightforward choice for experiments involving a limited number of gRNAs.
The Array-Based Architecture
Array-based systems consolidate multiple gRNA sequences into a single transcriptional unit. The nascent polycistronic RNA is subsequently processed into individual, functional gRNAs by co-expressed cellular or exogenous machinery [1]. This approach offers a more compact genetic design, which is crucial for viral vector applications with limited packaging capacity. Several highly effective processing mechanisms have been established:
- tRNA-based Processing: gRNAs are flanked by endogenous tRNA sequences, which are recognized and cleaved by ubiquitous cellular ribonucleases P and Z. This system is highly efficient and functions across diverse organisms without needing exogenous enzymes [1] [14].
- Ribozyme-based Systems: gRNAs are flanked by self-cleaving hammerhead (HH) and hepatitis delta virus (HDV) ribozymes. Upon transcription, the ribozymes catalyze their own excision, releasing the mature gRNA. This method is also enzyme-independent but can result in larger transcript sizes [13] [1].
- Csy4-based Processing: The bacterial endoribonuclease Csy4 cleaves with high specificity at a 28-base recognition sequence. When this sequence is placed between gRNAs in an array, co-expressed Csy4 releases mature gRNAs. A key consideration is potential cytotoxicity from high levels of Csy4 expression [1] [14].
- Cas12a-mediated Processing: The Cas12a (Cpf1) nuclease itself can process its own crRNA arrays, making it a native multiplexing system. A single transcript containing direct repeats flanking individual spacer sequences is cleaved by Cas12a to generate mature crRNAs [1].
Detailed Protocol: Golden Gate Assembly of Multiplexed gRNA Arrays
The following protocol, adapted from a peer-reviewed Bio-Protocol, details the assembly of multiplexed gRNA arrays using Golden Gate cloning, a highly efficient one-pot restriction-ligation method [15]. This protocol is designed to assemble up to 30 gRNA expression cassettes into a single vector.
Materials and Reagents
Table 2: Key Research Reagent Solutions
| Reagent/Plasmid | Function/Description | Source |
|---|---|---|
| pMA-SpCas9-g1 to g10 | Modular single gRNA expression vectors for initial oligo cloning | Addgene (#80784-80793) [15] |
| pMA-MsgRNA-EGFP | Destination array plasmid for final assembly of 11-30 gRNAs | Addgene (#80794) [15] |
| pFUS-A, pFUS-B1-B10 | Intermediate "acceptor" vectors for array construction | Golden Gate TALEN Kit (Addgene #1000000024) [15] |
| BsaI (BpiI), BsmBI (Esp3I) | Type IIS restriction enzymes for Golden Gate assembly | Thermo Fisher Scientific (FD0293, FD0454) |
| T4 DNA Ligase | Ligase for fragment joining during assembly | Thermo Fisher Scientific (EL0014) |
| Plasmid-Safe DNase | Digests linear DNA post-assembly to reduce background | Epicentre (E3101K) |
Step-by-Step Procedure
Step 1: gRNA Oligonucleotide Design and Preparation
- Design 20-nucleotide target sequences using established online tools (e.g., CRISPRscan, Benchling). A critical specificity check is required to minimize off-target effects.
- Oligo Design Rules: To ensure compatibility with the human U6 promoter in this system, the first base of the gRNA target site should be a 'G'. Furthermore, the target sequence must not contain internal BbsI, BsaI, or BsmBI restriction sites, as these would interfere with the cloning process [15].
- Order oligonucleotides with the appropriate overhangs. For targets starting with 'G':
- Sense oligo:
5′- CACC(N20) -3′ - Antisense oligo:
5′- AAAC(N20) -3′For targets not starting with 'G', add an extra 'G': - Sense oligo:
5′- CACCG(N20) -3′ - Antisense oligo:
5′- AAAC(N20)C -3′
- Sense oligo:
- Resuspend oligonucleotides to 100 µM in nuclease-free water. Anneal pairs by mixing 1 µL of each sense and antisense oligo with 2 µL of 10x NEB Buffer 2 and water to 20 µL. Heat the mixture to 95°C for 5 minutes and then allow it to cool slowly to room temperature (1-2 hours) [15].
Step 2: Cloning Individual gRNAs into Modular Vectors
- Digest 100-200 ng of a modular vector (e.g., pMA-SpCas9-g1) with BbsI (FastDigest) in a 20 µL reaction for 5-10 minutes at 37°C.
- Ligate the annealed oligo duplex into the digested vector using T4 DNA Ligase. A typical reaction uses a 1:50 molar ratio of vector to insert in a 20 µL volume.
- Transform the ligation product into competent E. coli, plate on LB-ampicillin plates, and incubate overnight at 37°C. Screen colonies by colony PCR or restriction digest to identify positive clones, which are then sequence-verified.
Step 3: Golden Gate Assembly into Multiplex Arrays
This step involves assembling the individual gRNA cassettes into intermediate or final array vectors. The overhangs created by Type IIS enzymes (BsaI or BsmBI) drive the ordered assembly.
Diagram 1: Workflow for Golden Gate Assembly of Multiplexed gRNA Arrays. The process involves two main phases: preparation of single gRNA vectors and the one-pot Golden Gate reaction to assemble them into a multiplex array.
- For 2-10 gRNAs: Combine 50-100 ng of each sequence-verified single gRNA vector (from Step 2) in a single tube. Set up a Golden Gate reaction mix containing:
- 100-200 ng of the pooled plasmid mix.
- 1x T4 DNA Ligase Buffer.
- 1 µL (10 U) of BsaI (FastDigest).
- 1 µL (5 U) of T4 DNA Ligase.
- Nuclease-free water to 20 µL.
- Run the following program in a thermal cycler:
- 10 cycles: (5 minutes at 37°C + 10 minutes at 16°C)
- 5 minutes at 50°C
- 5 minutes at 80°C
- Hold at 4°C.
- To reduce non-recombinant background, treat 5 µL of the Golden Gate product with 1 µL of Plasmid-Safe DNase and 1 µL of ATP for 30 minutes at 37°C [15].
- Transform 5 µL of the DNase-treated product into competent E. coli and plate on selective media (Spectinomycin for the described system). Screen resulting colonies using the universal primers (U6 Forward and Scr Reverse) listed in Table 1 of the search results [15].
Step 4: Validation of Assembled Arrays
- Confirm the assembly by analyzing the colony PCR products on an agarose gel. The correct clone should yield a single, sharp band of the expected size.
- Perform Sanger sequencing of the entire multiplex array using the universal primers to ensure the correct order and sequence of all gRNA cassettes. This is a critical quality control step before using the vector in functional experiments.
Application Notes and Troubleshooting
- Enhancing Knockout Efficiency with Dual gRNAs: For highly efficient gene knockout, consider using a dual-targeting strategy where two gRNAs are designed to target the same gene in close genomic proximity (e.g., 40-300 bp apart). This approach synergistically increases the generation of indel mutations and the probability of producing a frameshift or large deletion, leading to more consistent protein ablation, as demonstrated in human cell lines and primary T cells [16].
- Considerations for Plant Systems: When applying these architectures in plants, strong Pol III promoters like U3 and U6 are effective but can lead to gRNA expression variation and potential transgene silencing when multiple identical promoters are used [13]. The tRNA-processing system has proven particularly effective in plants, with protocols available for assembling arrays expressing up to 8 gRNAs for Agrobacterium-mediated transformation [14].
- Troubleshooting Poor Assembly Efficiency: If the Golden Gate assembly yields an insufficient number of correct clones, verify the purity and concentration of the input plasmids. Ensure that no gRNA target sequences contain internal BsaI or BsmBI sites. Increasing the number of thermal cycles (e.g., from 10 to 20) can also improve the yield of the final product.
The choice between individual promoter and array-based gRNA expression architectures is fundamental to the success of multiplexed CRISPR–Cas experiments. The individual promoter system offers simplicity and reliability for lower-level multiplexing, while array-based systems provide a compact and stoichiometrically defined solution for higher-order multiplexing and size-constrained applications. The Golden Gate assembly protocol detailed herein offers a robust, scalable, and cost-effective method for constructing complex multiplexed gRNA vectors, empowering researchers to fully harness the potential of CRISPR technologies in advanced genome engineering projects.
Multiplexed CRISPR technologies, which enable simultaneous editing or regulation of multiple genetic loci, have vastly enhanced the scope and efficiency of genetic engineering. The choice of CRISPR-associated (Cas) nuclease is a critical determinant for the success of such multiplexed applications. This application note provides a comparative analysis of three prominent systems—Streptococcus pyogenes Cas9 (SpCas9), Acidaminococcus sp. Cas12a (AsCas12a), and orthogonal systems combining multiple nucleases—focusing on their performance, practical protocols, and implementation within the context of tandem sgRNA array construction for multiplexed editing research. The content is structured to serve researchers, scientists, and drug development professionals by providing actionable methodologies and quantitative comparisons to guide experimental design.
Performance Comparison of Cas Enzymes for Multiplexing
The efficiency of Cas enzymes in multiplexed editing varies significantly. A systematic, side-by-side comparison of SpCas9, the enhanced version of AsCas12a (enAsCas12a), and the orthogonal SpCas9-enAsCas12a system (CHyMErA) in hTERT-immortalized retinal pigment epithelial (RPE1) cells revealed critical performance differences [17]. The study utilized combinatorial libraries targeting 10 core-essential and 10 tumor suppressor genes, with the following key quantitative outcomes:
Table 1: Quantitative Performance Metrics of Combinatorial CRISPR Systems in RPE1 Cells [17]
| CRISPR System | Effect Size Range (gRNAs) | Effect Size Range (Genes) | AU-ROC (Core-Essential Genes) | AU-ROC (Tumor Suppressor Genes) | Mean LFC (Core-Essential) | Mean LFC (Tumor Suppressor) |
|---|---|---|---|---|---|---|
| SpCas9 | 11.39 | 8.59 | 0.84 | 0.96 | -4.29 | 2.29 |
| enAsCas12a | 10.46 | 6.66 | 0.81 | 0.95 | -3.56 | 1.64 |
| CHyMErA | 6.47 | 3.19 | 0.82 | 0.92 | -1.15 | 0.65 |
Abbreviations: LFC, Log2-Fold Change; AU-ROC, Area Under the Receiver Operating Characteristic Curve.
The data identifies SpCas9 as the top performer in combinatorial screens, yielding the largest effect size range and the strongest separation of fitness phenotypes [17]. While enAsCas12a showed robust activity, its requirement for crRNA processing can delay the induction of phenotypic effects. The CHyMErA orthogonal system, while functional, demonstrated a substantially reduced effect size under the tested conditions.
Key Methodologies and Experimental Protocols
Golden Gate Assembly for gRNA Expression Arrays
The construction of tandem guide RNA arrays is a foundational technique for multiplexed editing. The Golden Gate Assembly method, which uses type IIS restriction enzymes (e.g., BsaI) to assemble multiple gRNA units in a single reaction, is a well-established and efficient protocol [18].
Detailed Protocol:
- Cloning of Individual gRNA Sequences: Clone each gRNA target sequence (T1 to Tn) into individual, modular gRNA expression vectors (e.g., pMA1 to pMAn). These vectors typically contain a U6 promoter, a BbsI cloning site for the gRNA insert, the SpCas9 gRNA scaffold, and a U6 terminator [18].
- Golden Gate Assembly into Array Vectors: Perform a single BsaI-based Golden Gate Assembly reaction to excise and ligate up to ten gRNA expression cassettes in a predefined order into a recipient "array" backbone plasmid (e.g., pFUS-B1 to pFUS-B10 from the TALEN assembly kit) [18].
- Large-Scale Array Construction (Optional): For arrays exceeding ten gRNAs, a third assembly step can be performed. Using another type IIS enzyme like BsmBI, multiple 10-gRNA arrays can be assembled into a final destination vector, such as pMsgRNA-EGFP, enabling the construction of arrays with up to 30 gRNA expression cassettes [18].
- Validation: Verify correct assembly using a two-pronged PCR screening strategy:
- Universal PCR: Use a U6 promoter-forward primer and a gRNA scaffold-reverse primer. A successful assembly produces a ladder-like pattern with bands increasing by ~392 bp for each additional gRNA cassette [18].
- Guide-Specific PCR: Use overlapping PCRs with primers specific to adjacent gRNA oligonucleotides to confirm the sequence and order of individual guides [18].
Implementing Orthogonal CRISPR Systems
Orthogonal systems, which employ two distinct Cas nucleases simultaneously, allow for complex genetic perturbations, such as concurrent gene knockout and activation. The multiSPAS (multiplex SpCas9-enAsCas12a) approach is an engineered system that avoids the need for hybrid gRNAs and enhances performance [17].
Detailed Protocol:
- Cell Line Engineering: Generate a stable cell line constitutively expressing both SpCas9 and enAsCas12a. Validate comparable nuclease activity and DNA damage response, for instance, by assessing proliferation rates after targeting a gene like TP53 with both nucleases [17].
- Library Design and Cloning: Design a dual-nuclease library where SpCas9 sgRNAs and enAsCas12a crRNAs are expressed from separate RNA Polymerase III promoters (e.g., hU6 or h7SK) within a single vector. Ensure the library is uniform (distribution skew < 2.5) to improve scalability and feasibility [17].
- Screening Workflow:
- Transduction: Transduce the engineered cell line with the lentiviral library at a low multiplicity of infection (MOI ~0.5) to ensure most cells receive only one gRNA combination. Maintain cells at a high coverage (e.g., 1000x per gRNA) throughout the screen [17].
- Phenotype Induction: Culture cells for a sufficient duration (e.g., 14 days) to allow for the manifestation of combinatorial phenotypes, such as fitness drops in essential gene knockouts.
- Harvest and Sequencing: Harvest genomic DNA from the final cell population and the initial plasmid library. Amplify the integrated gRNA sequences by PCR and subject them to next-generation sequencing.
- Data Analysis: Map sequencing reads to the library and calculate log2-fold changes (LFCs) in gRNA abundance. Use gene-level LFCs and ROC analysis to identify hits, such as core-essential genes or synthetic lethal interactions [17].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of multiplexed and orthogonal CRISPR screens relies on a core set of reagents and tools. The following table details essential components and their functions.
Table 2: Key Research Reagent Solutions for Multiplexed CRISPR Editing
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| Golden Gate Assembly Kit | Modular plasmid system with type IIS enzymes (BsaI) for ordered assembly of gRNA arrays [18]. | Construction of a 10-plex gRNA expression array for simultaneous gene knockout. |
| Orthogonal Cas Cell Line | Stable cell line expressing multiple Cas nucleases (e.g., SpCas9 + enAsCas12a) [17]. | Enabling orthogonal screens with CHyMErA or multiSPAS for combined knockout and transcriptional regulation. |
| Polymerase II/III Promoters | Promoters for gRNA expression; Pol III (U6) for short RNAs, Pol II for inducible/long arrays [1] [19]. | Driving expression of long crRNA arrays for Cas12a; tunable expression with inducible Pol II systems. |
| crRNA Processing Enzymes | Endogenous (Cas12a) or exogenous (Csy4) ribonucleases that process long transcripts into individual gRNAs [1]. | Processing a single transcript from a Pol II promoter into multiple functional crRNAs for Cas12a. |
| Lentiviral gRNA Libraries | Uniformly designed pools of vectors for delivering gRNA combinations into target cells [17]. | Conducting genome-wide combinatorial dropout screens to identify genetic interactions. |
Workflow and Architecture Visualization
The following diagrams illustrate the core experimental workflow for an orthogonal CRISPR screen and the primary genetic architectures for expressing multiplexed gRNAs in vivo.
Orthogonal CRISPR Screen Workflow
Multiplexed gRNA Expression Architectures
The strategic selection of Cas enzymes is paramount for successful multiplexed genome editing. Quantitative evidence strongly supports SpCas9 as the most robust nuclease for standard combinatorial knockout screens due to its large effect size and high efficiency [17]. However, the unique crRNA processing capability of Cas12a offers a distinct advantage for expressing long gRNA arrays from a single transcript, which can be beneficial for certain experimental designs, such as those using inducible Polymerase II promoters [1] [19]. For the most advanced applications requiring simultaneous and distinct genomic perturbations, such as knockout combined with transcriptional activation, orthogonal systems like multiSPAS represent the cutting edge, provided they are implemented with optimized reagents and protocols to mitigate their historically smaller effect sizes [20] [17]. By leveraging the protocols, reagents, and comparative data outlined in this application note, researchers can make informed decisions to effectively harness multiplexed CRISPR technologies for their specific research goals.
Application Notes
Multiplexed CRISPR technologies, which enable the simultaneous expression of numerous guide RNAs (gRNAs) or Cas enzymes, have dramatically expanded the scope and efficiency of genetic engineering. By moving beyond single-target editing, researchers can now address complex biological questions and engineering challenges, from recording cellular events and constructing synthetic genetic circuits to rewiring entire metabolic pathways [1]. The core of this capability lies in the design of tandem gRNA arrays, which allow for the coordinated expression of multiple guides from a single genetic construct.
The following table summarizes the core architectures for expressing these gRNA arrays, a foundational element for the applications discussed in this note.
Table 1: Genetic Architectures for Multiplexed gRNA Expression
| Architecture | Transcriptional Control & Processing | Key Features | Example Applications |
|---|---|---|---|
| Individual Promoters | Multiple Pol III promoters (e.g., U6) and terminators [1]. | High fidelity; avoids processing requirements; can be limited by promoter availability and size [1]. | CRISPR knockout screens [4]. |
| Native CRISPR Array | Single promoter with gRNAs flanked by direct repeats; processed by Cas proteins (e.g., Cas12a) or endogenous nucleases [1]. | Compact design; leverages natural processing mechanisms; high modularity [1]. | Simultaneous gene activation and repression in human cells [1]. |
| Ribozyme-Flanked | Pol II or Pol III promoter; gRNAs flanked by self-cleaving ribozymes (e.g., Hammerhead, HDV) [1]. | Amenable to inducible promoters; allows for tissue-specific expression [1]. | Multiplexed editing in plants and mammalian cells [1]. |
| tRNA–gRNA Array | Pol III promoter; gRNAs flanked by tRNA sequences; processed by endogenous RNase P and Z [1]. | Highly efficient processing; utilizes ubiquitous cellular machinery; compatible with high-throughput cloning [1]. | Processing of up to 16 gRNAs in yeast and plants [1] [21]. |
| Csy4 Processing | Single promoter; gRNAs flanked by 28-nt Csy4 recognition sequence; requires co-expression of Csy4 endoribonuclease [1]. | Precise cleavage; minimal gRNA scarring; but Csy4 cytotoxicity can be a concern at high levels [1]. | Expression and processing of 12 sgRNAs in S. cerevisiae [1]. |
Cellular Recorders
The ability to use CRISPR systems to record transient molecular events into durable DNA changes is a breakthrough in cellular monitoring. These "cellular recorders" convert dynamic biological signals, such as pathogen exposure or inflammatory responses, into stable, sequence-level genomic memories that can be read later via sequencing [1].
A primary strategy involves engineering CRISPR spacer acquisition systems from Type I CRISPR loci (e.g., from E. coli) into eukaryotic cells. These systems can capture and integrate synthetic "trigger" DNA sequences into a CRISPR array as new spacers upon activation of a specific signaling pathway. The sequential order of spacer acquisitions provides a temporal record of events [1].
Key Application: Recording inflammatory signaling events. A recorder can be designed where the activation of an inflammation-related transcription factor (e.g., NF-κB) drives the expression of the CRISPR acquisition machinery and a trigger DNA sequence. Each inflammatory pulse results in a new spacer acquisition, creating a historical log of inflammation within the cell's genome [1].
Genetic Circuits
Multiplexed CRISPR-Cas systems, particularly nuclease-null variants (dCas9, dCas12a), are powerful platforms for constructing sophisticated genetic circuits. By fusing dCas proteins to transcriptional repressors (CRISPRi) or activators (CRISPRa), researchers can create multi-layered logic gates and regulatory networks that control cellular behavior [1].
These circuits function by using multiple gRNAs to target dCas-effector fusions to specific promoters, thereby regulating the expression of downstream genes, which can themselves be other dCas proteins or gRNAs. This enables the creation of complex feedback loops, oscillators, and Boolean logic gates (AND, OR, NOT) [1].
Key Application: A two-layer AND gate circuit. In this design, the output gene (e.g., a fluorescent reporter) is under the control of a promoter that requires simultaneous activation by two different dCas9-activator complexes. The first input signal induces the expression of gRNA-A, which directs activator-A to the output promoter. The second input signal induces the expression of gRNA-B, which directs activator-B to the same promoter. Only when both inputs are present is the output gene robustly expressed, creating a logical AND gate [1].
Metabolic Pathway Engineering
A major application of multiplexed editing is the rewiring of metabolic pathways in microbial hosts for the overproduction of biofuels, pharmaceuticals, and commodity chemicals. This often requires the simultaneous knockout of competing pathways, fine-tuning the expression of multiple enzymes in a biosynthetic route, and introducing entirely new metabolic modules [1].
Multiplexed CRISPR editing allows for the rapid, one-step implementation of these complex genetic changes without the need for iterative rounds of engineering. This is crucial for balancing metabolic flux and minimizing the accumulation of intermediate metabolites that can be toxic or reduce yield [1].
Key Application: Engineering yeast for high-yield production of a target molecule, such as an alkaloid precursor. A multiplexed strategy could involve:
- Knockout: Simultaneous disruption of three genes (g1, g2, g3) in a competing pathway using Cas9 nuclease and a tRNA-gRNA array.
- Activation & Repression: Using dCas9-based CRISPRa to upregulate four key biosynthetic enzymes (genA, genB, genC, genD) and CRISPRi to downregulate a regulatory gene that inhibits the pathway (genR). This coordinated approach addresses multiple engineering bottlenecks at once, dramatically accelerating the strain development process [1].
Protocols
Protocol 1: Construction of a tRNA-gRNA Array for Multiplexed Knockout
This protocol describes the assembly of a multiplex tRNA-gRNA array using Golden Gate assembly, a method highly effective for cloning repetitive sequences [4] [1]. The resulting construct can be used for simultaneous knockout of up to seven genes in a single transformation.
Research Reagent Solutions:
- BsaI-HF v2 Restriction Enzyme: A type IIS enzyme for Golden Gate assembly.
- T4 DNA Ligase: For ligation of digested DNA fragments.
- pCAS9-TR plasmid: Destination vector containing a Cas9 expression cassette and a tRNA scaffold.
- pGRNA-tRNA modules: Library of entry clones with gRNA sequences flanked by tRNA parts.
Table 2: Key Reagents for tRNA-gRNA Array Construction
| Reagent | Function | Source/Example |
|---|---|---|
| BsaI-HF v2 | Type IIS restriction enzyme that cleaves outside its recognition site, creating unique overhangs for seamless assembly. | New England Biolabs |
| pCAS9-TR Vector | Destination vector with bacterial origin of replication, antibiotic resistance, Cas9 gene, and tRNA scaffold for gRNA integration. | Addgene (various) |
| gRNA-tRNA Modules | Pre-validated DNA fragments containing the target-specific gRNA sequence (20 nt) embedded within a tRNA architecture. | Synthesized as gBlocks (Integrated DNA Technologies) |
| Stbl3 E. coli | Recombinant-deficient E. coli strain used to propagate repetitive DNA sequences with high stability. | Thermo Fisher Scientific |
Detailed Procedure:
- gRNA Design: Design 20-nt gRNA sequences for each target gene using bioinformatics tools like CHOPCHOP or CRISPRscan to maximize on-target efficiency and minimize off-target effects [22].
- Module Preparation: Obtain DNA fragments for each gRNA-tRNA unit, either by synthesizing them as gBlocks or by PCR amplification from existing modules. Each unit must contain the BsaI recognition sites with overhangs compatible for assembly.
- Golden Gate Assembly:
- Set up a reaction mixture containing: 50 ng of pCAS9-TR vector, a 2:1 molar ratio of each gRNA-tRNA module, 1× T4 DNA Ligase Buffer, 10 units of BsaI-HF v2, and 400 units of T4 DNA Ligase.
- Run the following thermocycler program: 25 cycles of (37°C for 2 minutes + 16°C for 5 minutes), followed by a final digestion at 37°C for 15 minutes and heat inactivation at 80°C for 15 minutes.
- Transformation and Validation:
- Transform the assembled reaction into Stbl3 chemically competent E. coli cells to minimize recombination.
- Screen colonies by colony PCR and Sanger sequencing using primers that flank the integration site to confirm the correct assembly and order of all gRNA units.
The following workflow diagram illustrates the key experimental steps for constructing and implementing a multiplexed editing system.
Protocol 2: Implementing a dCas9-Based AND Gate Genetic Circuit
This protocol outlines the creation of a two-input AND gate in human HEK293T cells using dCas9 transcriptional activators.
Research Reagent Solutions:
- Lentiviral Vectors pLV-dCas9-VPR & pLV-gRNA: For stable integration of circuit components.
- dCas9-VPR Fusion Protein: A potent synthetic transcriptional activator (dCas9 fused to VP64, p65, and Rta).
- Lipofectamine 3000: For transient transfection of circuit components.
Detailed Procedure:
- Circuit Component Cloning:
- Clone the dCas9-VPR gene into a lentiviral vector under a constitutive promoter (e.g., EF1α).
- Clone two gRNA expression cassettes (gRNA-A and gRNA-B) targeting the promoter of your output gene into a separate lentiviral vector. Use a multiplexing architecture (e.g., Csy4 or tRNA) if they are on the same transcript.
- Place the output gene (e.g., GFP) under a minimal promoter containing the target sites for gRNA-A and gRNA-B.
- Lentivirus Production:
- Produce lentiviral particles for the dCas9-VPR vector and the gRNA-output vector in HEK293T cells using standard third-generation packaging systems.
- Cell Line Generation:
- Transduce HEK293T cells first with the dCas9-VPR lentivirus and select with the appropriate antibiotic (e.g., blasticidin) to create a stable cell line.
- Subsequently, transduce the dCas9-VPR-expressing cells with the gRNA-output reporter lentivirus and select with a second antibiotic (e.g., puromycin).
- Circuit Validation:
- The AND gate logic is inherent in the design. To test, measure the output signal (e.g., GFP fluorescence via flow cytometry) in the presence and absence of the inducers for gRNA-A and gRNA-B. High output should be observed only when both inducers are present.
The following diagram illustrates the logical structure and component relationships of the dCas9-based AND gate.
The Scientist's Toolkit
Successful implementation of multiplexed editing requires a suite of specialized reagents and tools. The following table details essential solutions for constructing and deploying tandem sgRNA arrays.
Table 3: Essential Research Reagent Solutions for Multiplexed Editing
| Tool Category | Specific Product/Resource | Explanation and Function |
|---|---|---|
| Bioinformatics Tools | CHOPCHOP, CRISPRscan [22] | Web-based platforms for designing highly efficient and specific gRNA sequences, critical for minimizing off-target effects in multiplexed experiments. |
| Assembly Systems | Golden Gate Assembly Kit (BsaI-HFv2) [4] [1] | A standardized molecular cloning system using Type IIS restriction enzymes to seamlessly assemble multiple gRNA units into a single vector. |
| Delivery Vectors | Lentiviral pLV-U6-gRNA-Ef1a-Puro [4] | A ready-to-use lentiviral vector for delivering gRNA arrays; allows for stable integration and selection in hard-to-transfect cells. |
| Cas Enzymes | High-Fidelity Cas9 (e.g., SpCas9-HF1) [4] | Engineered Cas9 variant with reduced off-target activity, essential for maintaining specificity when multiple gRNAs are expressed simultaneously. |
| Activation/Repression | dCas9-VPR / dCas9-KRAB [1] | Catalytically dead Cas9 (dCas9) fused to strong transcriptional activation (VPR) or repression (KRAB) domains for multiplexed gene regulation (CRISPRa/i). |
| Array Processing | Csy4 Endoribonuclease [1] | A highly specific RNA cleavage enzyme used to process a long transcript into individual functional gRNAs from a single array. |
| Validation (NGS) | CRISPResso2 [22] | A bioinformatics software package specifically designed to analyze next-generation sequencing data and quantify CRISPR editing efficiency and outcomes. |
Practical Implementation: Array Assembly Methods and Delivery Systems Across Biological Platforms
High-Accuracy crRNA Array Assembly Strategies for Streamlined Multiplex Construct Generation
Multiplex CRISPR technology, which enables the simultaneous targeting of multiple genomic loci, has revolutionized functional genomics and therapeutic development. The core of this technology lies in the efficient assembly of CRISPR RNA (crRNA) arrays—synthetic constructs that encode multiple guide RNAs within a single transcript. Unlike systems that rely on expressing each guide RNA from its own promoter, crRNA arrays are transcribed as a single unit and then processed into individual, functional guide RNAs by endogenous cellular machinery or the Cas protein itself [1]. This approach is indispensable for overcoming genetic redundancy, engineering polygenic traits, and performing complex genome-wide perturbations [21] [4]. However, the repetitive nature of the sequences within these arrays has historically made them difficult and time-consuming to construct with high accuracy. This application note details streamlined, high-accuracy strategies for crRNA array assembly, providing validated protocols and resources to empower researchers in the rapid generation of multiplex CRISPR constructs.
The foundation of any crRNA array is the repeat-spacer subunit, where the "spacer" is the target-specific guide sequence and the "repeat" is a conserved sequence that facilitates processing. Different CRISPR systems, such as those employing Cas12a or Cas13, inherently process these arrays by recognizing hairpin structures within the repeats [1]. The primary technical challenge in building arrays is assembling these highly repetitive subunits without recombination or errors. Traditional methods, such as Golden Gate Assembly, have been adapted for this purpose, but newer strategies offer significant improvements in speed, accuracy, and capacity.
A pivotal innovation is the CRATES (CRISPR Assembly through Trimmed Ends of Spacers) method. CRATES is a modular, one-pot assembly scheme that introduces defined, unique 4-base pair overhangs within the portion of the spacer sequence that is trimmed during natural crRNA biogenesis [23]. This design decouples the assembly junction from the functional targeting portion of the spacer and the conserved repeat, preventing misassembly and allowing for the highly efficient construction of arrays containing up to seven spacers in a single reaction [23]. Demonstrating the scalability of modern methods, a recent study utilized a novel high-accuracy strategy to successfully assemble arrays containing 12 crRNAs for AsCas12a and 15 crRNAs for RfxCas13d in a single reaction [24] [19].
The table below summarizes the key features of contemporary array assembly strategies.
Table 1: Key High-Accuracy crRNA Array Assembly Strategies
| Strategy Name | Core Principle | Key Advantage | Demonstrated Capacity (Spacers) | Compatible Cas Proteins |
|---|---|---|---|---|
| CRATES [23] | Introduction of unique 4-bp overhangs in the trimmed spacer region for one-pot assembly. | Modularity; decouples assembly junctions from functional sequences; enables library generation. | 7 | Cas12a, Cas9, Cas13a |
| High-Accuracy Strategy [24] [19] | Optimized, streamlined cloning strategy for large-scale array construction. | High-yield, cost- and time-saving assembly of very long arrays. | 15 (for RfxCas13d) | AsCas12a, RfxCas13d |
| tRNA-sgRNA Arrays [3] | Flanking sgRNAs with tRNA sequences, processed by endogenous RNase P and Z. | Leverages ubiquitous host enzymes; effective in plants and eukaryotes. | 4+ (validated in plants) | Cas9 |
Detailed Experimental Protocol: CRATES Assembly for Cas12a Arrays
This protocol provides a step-by-step guide for constructing a crRNA array for the Cas12a nuclease using the CRATES methodology [23].
Research Reagent Solutions
Table 2: Essential Reagents for CRATES Assembly
| Reagent / Material | Function / Explanation |
|---|---|
| Base Vector/Backbone | Contains constitutive promoter, 5' and 3' terminal repeats, a reporter gene (e.g., GFP) flanked by Type IIS sites, and a transcriptional terminator. |
| Repeat-Spacer Oligonucleotides | Complementary ~66-nt oligos that, when annealed, form a double-stranded repeat-spacer subunit with the required 4-nt overhangs. |
| Type IIS Restriction Enzyme (e.g., BsaI) | Cleaves the base vector and excises the reporter gene; creates compatible overhangs for ligation. |
| T4 DNA Ligase | Joins the assembled repeat-spacer subunits into the prepared backbone. |
| Thermocycler | Precisely controls temperature cycling for the one-pot digestion and ligation reaction. |
Step-by-Step Procedure
Vector Backbone Preparation: Clone the necessary regulatory elements into your base vector: a promoter (e.g., a Pol II or Pol III promoter), a 5' terminal repeat, a cassette containing a GFP or other reporter gene flanked by two Type IIS restriction sites (e.g., BsaI sites in opposite orientations), a 3' terminal repeat, and a terminator [23].
Repeat-Spacer Subunit Design and Annealing:
- For each spacer, design two complementary single-stranded oligonucleotides that, when annealed, form a double-stranded DNA fragment with the structure:
[3' overhang from previous subunit]-[Repeat]-[Spacer]-[5' overhang to next subunit]. - The 4-nt overhangs should be chosen from a validated set of non-palindromic, unique sequences to ensure proper directional assembly [23].
- The spacer sequence should include the 20-nt target-specific region, with the 4-nt assembly junction placed in the 3' region that will be trimmed during crRNA maturation.
- Anneal the oligonucleotides in a thermocycler by mixing equimolar amounts, heating to 95°C for 3 minutes, and cooling slowly to 25°C.
- For each spacer, design two complementary single-stranded oligonucleotides that, when annealed, form a double-stranded DNA fragment with the structure:
One-Pot Golden Gate Assembly:
- Set up a reaction mixture containing:
- 50-100 ng of the prepared base vector.
- Equimolar amounts of each annealed repeat-spacer subunit.
- 1× T4 DNA Ligase Buffer.
- 10 U of Type IIS Restriction Enzyme (e.g., BsaI-HFv2).
- 400 U of T4 DNA Ligase.
- Nuclease-free water to a total volume of 20 µL.
- Run the following program in a thermocycler [23]:
- Cycle 1: 37°C for 5 minutes (digestion), 16°C for 5 minutes (ligation).
- Repeat Cycle 1 25-50 times.
- Final Step: 60°C for 10 minutes (enzyme inactivation), 80°C for 10 minutes.
- Set up a reaction mixture containing:
Transformation and Screening:
- Transform 2-5 µL of the assembly reaction into competent E. coli cells.
- Plate on selective media and screen colonies. Successful assembly will result in the excision of the GFP reporter, producing white/colorless colonies, while unsuccessful clones retain GFP, producing fluorescent green colonies [23].
- Pick non-fluorescent colonies and verify correct assembly by colony PCR and Sanger sequencing.
Diagram 1: CRATES One-Pot Assembly Workflow
Expression and Processing of Assembled Arrays
Once assembled, the crRNA array must be transcribed and processed correctly within the target cell. The choice of promoter is critical and depends on the desired application.
- Polymerase II (Pol II) Promoters: These are often inducible and can be tissue-specific. Arrays expressed by Pol II promoters produce a long transcript that requires efficient processing (e.g., via the embedded Cas12a processing signal) to release functional crRNAs. These promoters can drive complex expression patterns distinct from Pol III promoters [24] [19].
- Polymerase III (Pol III) Promoters: Promoters like U6 are commonly used as they produce high yields of short, non-coding RNAs with defined start and end sites, making them ideal for crRNA expression. They have been shown to be highly effective in driving tRNA–sgRNA arrays for Cas9 in plants [3].
Processing of the primary transcript is handled by different mechanisms, as illustrated below.
Diagram 2: crRNA Array Transcription & Processing
Results and Validation
Robust validation is essential to confirm the functionality of assembled crRNA arrays. The high-accuracy strategy mentioned enabled the assembly of arrays with up to 15 spacers [24] [19]. When testing arrays, it is critical to measure editing efficiency at each target locus, typically using next-generation sequencing or T7 Endonuclease I assays.
Table 3: Expected Outcomes from a High-Accuracy Assembly
| Validation Metric | Expected Outcome | Method of Assessment |
|---|---|---|
| Assembly Success Rate | >95% for arrays with ≤5 spacers [23]. | Colony PCR, sequencing. |
| Array Length | Successful construction of arrays with 12-15 spacers [24]. | Gel electrophoresis, sequencing. |
| Multiplex Editing Efficiency | High, target-dependent efficiency; simultaneous knockout of multiple genes (e.g., 3-4 genes in plants) [21] [3]. | NGS of target loci, phenotypic analysis. |
| Processing Fidelity | Clean production of mature crRNAs without extraneous intermediates. | Northern Blot, RNA-seq. |
Troubleshooting and Technical Notes
- Low Assembly Yield: Ensure the repeat-spacer subunits are annealed correctly and are added in equimolar amounts. Increase the number of thermocycler cycles for assemblies with more than five spacers.
- Incorrect Arrays: Verify the design of the 4-nt overhangs to ensure they are unique and non-palindromic. Re-check the sequence of synthesized oligonucleotides.
- Poor Editing Efficiency: Optimize the promoter driving the array (test Pol II vs. Pol III). For Cas12a arrays, ensure the final array is flanked by terminal repeats, as this has been shown to prevent the generation of an non-functional, extraneous crRNA from the 3' end [23]. Also, be aware that the spacer sequence itself and the global secondary structure of the pre-crRNA can impact crRNA biogenesis and nuclease activity [23].
- Application in Plants: For plant systems, the use of tRNA–sgRNA arrays processed by endogenous RNases has proven highly effective for multiplex editing with Cas9. Promoters such as Arabidopsis UBQ10 and RPS5a have been shown to drive high expression of Cas9, while U6 Pol III promoters or the ES8Z Pol II promoter are effective for the sgRNA array [3].
The advent of multiplexed CRISPR technologies has revolutionized genetic engineering by enabling simultaneous targeting of multiple genomic loci. A critical advancement underpinning this capability is the development of engineered guide RNA (gRNA) arrays, where multiple gRNAs are transcribed as a single polycistronic transcript and subsequently processed into individual, functional gRNAs. Four principal mechanisms have emerged as the most efficient and widely adopted for this processing: tRNA-based processing, ribozyme-mediated cleavage, Csy4 ribonuclease processing, and Cas12a intrinsic self-processing. These systems bypass the need for multiple repetitive promoters, thereby enhancing vector compactness, maintaining gRNA stoichiometry, and improving overall editing efficiency. The strategic selection of an appropriate processing mechanism is paramount for successful experimental outcomes in multiplexed genome editing, transcriptional regulation, and metabolic pathway engineering.
Table 1: Overview of gRNA Array Processing Mechanisms
| Processing Mechanism | Core Principle | Key Components | Primary Organism Demonstrations |
|---|---|---|---|
| tRNA | Exploits endogenous tRNA processing machinery | tRNA-gRNA array, RNase P, RNase Z | Plants, yeast, human cells, Drosophila |
| Ribozyme | Utilizes self-cleaving catalytic RNA motifs | Hammerhead (HH) and Hepatitis Delta Virus (HDV) ribozymes | Plants, mammalian cells, yeast |
| Csy4 | Employs a sequence-specific bacterial endoribonuclease | Csy4 enzyme, Csy4 recognition sequence | Yeast, mammalian cells, bacteria |
| Cas12a Self-processing | Leverages the innate RNase activity of the Cas12a protein | Cas12a nuclease, CRISPR array with direct repeats | Human cells, plants, yeast, porcine embryos |
Comparative Performance Analysis
Quantitative assessments across diverse organisms reveal distinct performance characteristics for each processing system. The choice of mechanism significantly impacts editing efficiency, multiplexing capacity, and practical implementation.
Table 2: Comparative Performance of gRNA Processing Systems
| Processing Mechanism | Reported Editing Efficiency | Multiplexing Capacity (Number of gRNAs) | Notable Advantages | Key Limitations |
|---|---|---|---|---|
| tRNA (PTG) | 87% for 8 genes in yeast [25]; Robust in rice [26] | Up to 8 in a single transcript [25] | High efficiency; uses endogenous enzymes; no co-factor needed in many systems [25] [27] | gRNA sequence context can influence processing efficiency |
| Ribozyme (STU-RZ) | Less robust than tRNA or Csy4 in rice [26] | Demonstrated for multiple targets [26] | Self-cleaving; no protein co-factors required [28] | Potentially low in vivo processing activity [26] |
| Csy4 | More robust than RZ system in rice [26] | Up to 12 sgRNAs in S. cerevisiae [28] | High precision; clean release of gRNAs [28] [29] | Requires constitutive co-expression of Csy4; cytotoxicity at high levels [28] |
| Cas12a Self-processing | Efficient C-to-T conversion on targets with TTTV PAM (e.g., 69.9%) [30]; Up to 15 targets in human cells [30] | Up to 15 target sites in human cells [30] | Most compact system; no additional processing factors needed [28] [30] | Compromised efficiencies and restricted PAM for wild-type; requires engineered variants [31] |
Detailed Methodologies and Protocols
tRNA-gRNA Array (Polycistronic-tRNA-gRNA - PTG)
The tRNA system leverages the highly conserved endogenous tRNA processing machinery, specifically ribonucleases P and Z, which recognize the cloverleaf secondary structure of tRNA precursors and cleave at their 5' and 3' ends, respectively [28] [27]. This mechanism allows for the precise release of multiple gRNAs from a single transcript.
Protocol: Implementation of PTG System in Yeast [25]
- gRNA-TRNA Array Design: Design a synthetic gene where each gRNA is flanked by identical tRNA sequences (e.g., tRNA-Gly). The array structure is:
Promoter - [tRNA - gRNA] - [tRNA - gRNA] - ... - Terminator. - Vector Construction: Clone the synthesized PTG array into an expression vector downstream of a strong Pol III promoter, such as the SNR52 promoter.
- Donor Template Preparation: For gene knock-outs, generate 100 bp double-stranded DNA donor templates via PCR. These donors should introduce frame-shifting mutations (e.g., 8-bp deletions including the PAM sequence).
- Transformation: Co-transform the PTG expression plasmid and the donor DNA fragments into Saccharomyces cerevisiae using a standard lithium acetate protocol.
- Screening and Validation: After outgrowth, screen colonies for successful multiplexed edits using PCR genotyping and subsequent DNA sequencing.
Csy4 Ribonuclease System
Csy4, an endoribonuclease from Pseudomonas aeruginosa, provides a highly specific processing mechanism. It binds to a 28-nucleotide sequence in the CRISPR repeats and cleaves the immediate downstream nucleotides, offering clean and precise excision of gRNAs [28] [29].
Protocol: Csy4-Mediated Processing for Multiplexed Editing [26] [28]
- Array Construction: Assemble a gRNA array where each gRNA is flanked by the 28-nt Csy4 recognition sequence.
- Vector Assembly: Clone this array into a single expression cassette, which can be driven by either a Pol II or Pol III promoter.
- Csy4 Co-expression: Ensure constitutive expression of the Csy4 gene from the same or a separate vector. The Csy4 must be co-expressed with the gRNA array to function.
- Delivery and Expression: Deliver the constructed vector(s) into the target cells (e.g., rice protoplasts or stable transformants).
- Efficiency Assessment: Analyze genome editing efficiency via restriction fragment length polymorphism (RFLP) assay or deep sequencing of the target sites.
Ribozyme-Based Systems (Hammerhead and HDV)
Ribozyme systems use cis-acting catalytic RNA motifs, such as Hammerhead (HH) and Hepatitis Delta Virus (HDV) ribozymes, which flank the gRNA sequence and self-cleave during transcription to release the mature gRNA, without requiring any protein co-factors [26] [28].
Protocol: Single Transcript Unit (STU) with Ribozymes [26]
- STU Construct Design: Design a construct where the Cas9 protein coding sequence and the sgRNA(s) are combined into a single open reading frame, linked by a polyA sequence. The sgRNA component must be flanked by HH ribozyme at the 5' end and HDV ribozyme at the 3' end.
- Vector Construction: Clone the STU construct under the control of a single Pol II promoter.
- Expression and Processing: Upon transcription in the host system (e.g., rice), the ribozymes undergo self-catalyzed cleavage, releasing the functional sgRNA.
- Evaluation: Compare editing robustness against dual-promoter systems and other STU systems (e.g., Csy4, tRNA) via transient expression in protoplasts or stable transformation.
Cas12a Self-Processing System
Cas12a (formerly Cpf1) possesses intrinsic RNase activity that allows it to process its own CRISPR RNA (crRNA) precursor. A single transcript containing a CRISPR array with direct repeats separating the crRNA spacers is cleaved by Cas12a into mature crRNAs, each guiding the nuclease to a specific DNA target [28] [30] [31]. This makes it the most streamlined system for multiplexing.
Protocol: Multiplexed Base Editing with Cas12a [30]
- Base Editor Selection: Choose an optimized Cas12a-derived base editor. Recent studies have identified LbCas12a variants (e.g., BEACON1, BEACON2, enAsBE1.1) fused with highly active deaminases like hA3A (human APOBEC3A) as particularly effective [30] [31].
- crRNA Array Design: Design a crRNA array where individual spacer sequences are separated by the native Cas12a direct repeat sequence.
- Delivery into Human Cells:
- Plasmid Transfection: Co-transfect HEK293 cells with two plasmids: one expressing the Cas12a-base editor and another (e.g., a hU6 promoter-driven plasmid) expressing the crRNA array.
- Selection and Outgrowth: Treat transfected cells with puromycin (2 µg/mL) for selection. Allow an outgrowth phase of 7 days post-transfection to enrich for edited cells.
- Assessment of Editing: Harvest genomic DNA and use high-throughput amplicon sequencing to quantify base conversion efficiency and purity at all target sites.
The Scientist's Toolkit: Essential Reagents
Table 3: Key Research Reagent Solutions
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| LbCas12a-/dCas12a-BE variants | Engineered base editors for multiplexed C-to-T or A-to-G editing with expanded PAM recognition (e.g., NTTN, TYCN) [30] [31]. | Precise installation of multiple point mutations in human cell lines. |
| Polymerase III Promoters (U6, SNR52) | Drive high-level expression of short, non-coding RNAs like gRNAs and PTG arrays [25] [29]. | Constitutive expression of gRNA arrays in yeast (SNR52) and mammalian cells (hU6). |
| hA3A (human APOBEC3A) deaminase | A highly active cytidine deaminase domain used in advanced CBEs to boost editing efficiency [30] [31]. | Improving the performance of Cas12a-derived CBEs in human cells and embryos. |
| Golden Gate Assembly | A modular DNA assembly technique ideal for cloning repetitive gRNA arrays [25] [1]. | Rapid, one-pot construction of plasmids containing multiple gRNA expression cassettes. |
| tRNA-Gly | A short (71 bp) tRNA sequence used as a processing element in PTG arrays [25]. | Serving as a highly efficient and compact processing site for multiplex gRNA expression in yeast and plants. |
The application of tandem CRISPR sgRNA arrays for multiplexed genome editing represents a transformative approach in functional genomics and therapeutic development. A critical hurdle in leveraging this technology is the efficient delivery of these large, repetitive genetic constructs into target cells. Delivery systems are broadly categorized into viral and non-viral approaches, each with distinct trade-offs between efficiency, cargo capacity, safety, and immunogenicity. The choice of delivery vector is paramount, as it must not only transport the CRISPR machinery but also accommodate the substantial size and complex architecture of sgRNA arrays designed for simultaneous targeting of multiple genomic loci. This document outlines detailed application notes and protocols for the most prominent delivery systems, providing a framework for researchers to select and implement the optimal strategy for their multiplexed editing goals.
Core Concepts and Cargo Formats
The effectiveness of a delivery system is intrinsically linked to the format of the CRISPR cargo. For multiplexed editing involving sgRNA arrays, the cargo is typically the array construct itself, which can be delivered in different overarching formats alongside the Cas nuclease.
- Plasmid DNA (pDNA): This format involves a DNA plasmid encoding the Cas nuclease and the tandem sgRNA array. While stable and simple to produce, prolonged expression from plasmids can increase the risk of off-target effects and immune responses [32] [33].
- mRNA/sgRNA: In this approach, in vitro transcribed mRNA encoding the Cas protein is co-delivered with the in vitro transcribed sgRNA array. This method offers transient expression, reducing off-target risks, but the RNA molecules are fragile and require protection during delivery [32] [33].
- Ribonucleoprotein (RNP): The Cas protein is pre-complexed with the in vitro transcribed sgRNA array to form an RNP complex. This method is the most transient, leading to rapid editing and the highest reported fidelity with minimal off-target effects, making it ideal for clinical applications [32] [33]. However, production of large quantities of pure protein and RNA can be challenging.
The following diagram illustrates the decision-making workflow for selecting an appropriate delivery system based on key experimental parameters.
Viral Vector Delivery Systems
Viral vectors are engineered viruses that exploit natural viral transduction mechanisms to deliver genetic cargo with high efficiency. They are particularly useful for hard-to-transfect cells and for applications requiring persistent expression.
Table 1: Comparison of Viral Delivery Systems for CRISPR
| Vector | Adeno-Associated Virus (AAV) | Lentivirus (LV) | Adenovirus (AdV) |
|---|---|---|---|
| Payload Capacity | < 4.7 kb [32] | ~8 kb [32] | Up to 36 kb [32] |
| Integration Profile | Non-integrating (episomal) [32] | Integrates into host genome [32] | Non-integrating (episomal) [32] |
| Advantages | Low immunogenicity; FDA-approved variants; high tissue specificity [32] | Infects dividing & non-dividing cells; stable long-term expression [32] | Very high cargo capacity; high titer production [32] |
| Disadvantages | Very limited cargo size; potential pre-existing immunity [34] [32] | Risk of insertional mutagenesis; complex safety handling [34] [32] | Can trigger strong immune responses [34] [32] |
| Best for Multiplexed Editing | Delivering sgRNA arrays to cells expressing stable Cas9; use of smaller Cas orthologs [32] | Delivery of large tandem sgRNA arrays where long-term expression is desired [32] | Delivery of very large CRISPR cargo, including full Cas9 and extensive sgRNA arrays [32] |
Protocol: Lentiviral Production and Transduction for sgRNA Array Delivery
This protocol is designed for the delivery of a tandem sgRNA array construct via lentiviral vectors, which is suitable for large arrays and provides sustained expression.
Materials:
- Research Reagents: HEK293T cells, lentiviral transfer plasmid (containing sgRNA array and Cas9), lentiviral packaging plasmids (psPAX2, pMD2.G), polyethylenimine (PEI), culture medium, puromycin.
- Equipment: Tissue culture hood, CO₂ incubator, ultracentrifuge, biosafety level 2 (BSL-2) facility.
Method:
- Day 1: Cell Seeding: Seed HEK293T cells in a 10 cm tissue culture dish to reach 70-80% confluency within 24 hours.
- Day 2: Transfection: Co-transfect the HEK293T cells with the lentiviral transfer plasmid and the packaging plasmids (psPAX2 and pMD2.G) using PEI. For one dish, use a 1:1:1 mass ratio (e.g., 10 µg transfer plasmid : 10 µg psPAX2 : 10 µg pMD2.G).
- Day 3: Media Change: Replace the transfection medium with fresh culture medium.
- Day 4 & 5: Harvesting: Collect the viral supernatant 48 and 72 hours post-transfection. Pool the supernatants and filter through a 0.45 µm filter to remove cell debris.
- Virus Concentration (Optional): Concentrate the virus by ultracentrifugation at 50,000 × g for 2 hours. Resuspend the viral pellet in a small volume of PBS or serum-free medium.
- Day 6: Transduction: Incubate your target cells with the lentiviral supernatant in the presence of a transduction enhancer (e.g., polybrene). Centrifuge the plate (spinoculation) to enhance infection efficiency.
- Selection: 48-72 hours post-transduction, begin selecting transduced cells with an appropriate antibiotic (e.g., 1-2 µg/mL puromycin) for 5-7 days.
Non-Viral Delivery Systems
Non-viral methods offer advantages in safety, scalability, and reduced immunogenicity. They are ideal for transient CRISPR expression, which is well-suited for RNP delivery and can minimize off-target effects.
Table 2: Comparison of Non-Viral Delivery Systems for CRISPR
| Method | Electroporation/ Nucleofection | Lipid Nanoparticles (LNPs) | Cell-Penetrating Peptides (CPPs) |
|---|---|---|---|
| Cargo Format | RNP, mRNA, pDNA [33] | RNP, mRNA, pDNA [32] | RNP, Protein [32] |
| Mechanism | Electrical pulses create pores in cell membrane [33] | Lipid vesicles fuse with cell membrane and release cargo [32] | Peptides facilitate translocation across membrane [32] |
| Advantages | High efficiency for hard-to-transfect cells (e.g., HSCs) [33] | Low immunogenicity; clinically validated; potential for organ targeting [32] | Low cytotoxicity; high specificity through peptide engineering [32] |
| Disadvantages | Can cause significant cell death; requires specialized equipment [33] | Endosomal entrapment can limit efficiency; variable optimization [32] | Lower editing efficiency compared to physical methods [32] |
| Best for Multiplexed Editing | Gold standard for RNP delivery to primary and stem cells [33] | In vivo delivery of CRISPR components, including sgRNA arrays as mRNA [32] | Delivery of pre-assembled RNPs for various in vitro applications [32] |
Protocol: RNP Delivery via Nucleofection for Hematopoietic Stem Cells
This protocol is optimized for delivering pre-assembled Cas9 RNP complexes with in vitro transcribed sgRNA arrays into sensitive hematopoietic stem cells (HSCs), a method renowned for high efficiency and minimal off-target effects.
Materials:
- Research Reagents: Recombinant Cas9 protein, in vitro transcribed tandem sgRNA array, Nucleofector Kit for HSCs (e.g., Lonza), pre-warmed culture medium, cytokines (SCF, TPO, FLT3-L).
- Equipment: Nucleofector device, biosafety cabinet, water bath, centrifuge.
Method:
- RNP Complex Assembly: Complex the purified Cas9 protein with the sgRNA array at a molar ratio of 1:2 (e.g., 10 µg Cas9 : 3.5 µg sgRNA for a 100-nt array) in a low-salt buffer. Incubate at room temperature for 10-20 minutes to form the RNP.
- Cell Preparation: Harvest and count HSCs. Centrifuge 1 × 10⁵ to 5 × 10⁵ cells and carefully aspirate the supernatant.
- Nucleofection Preparation: Resuspend the cell pellet in 20 µL of Nucleofector Solution from the kit. Add the pre-assembled RNP complex directly to the cell suspension and mix gently.
- Nucleofection: Transfer the cell-RNP mixture into a certified cuvette. Select the appropriate program on the Nucleofector device (e.g., "DS-138" for human HSCs) and run the program.
- Recovery: Immediately after nucleofection, add 80 µL of pre-warmed culture medium to the cuvette. Gently transfer the cells to a culture plate containing pre-warmed, cytokine-supplemented medium.
- Analysis: Assess editing efficiency 48-72 hours post-nucleofection via genomic DNA extraction, PCR amplification of the target loci, and next-generation sequencing (e.g., T7E1 assay or NGS).
The Scientist's Toolkit: Essential Reagents for sgRNA Array Construction and Delivery
Table 3: Key Research Reagent Solutions
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| Cas9 Expression Plasmid | Source of Cas9 nuclease when using DNA-based delivery. | pX330 (Addgene), p201N-Cas9 (Addgene) [35] [36]. |
| sgRNA Cloning Vector | Backbone for inserting and expressing sgRNA spacer sequences. | pUC-gRNA (Addgene) [36]. |
| High-Fidelity DNA Assembly Mix | Cloning of repetitive sgRNA arrays with high efficiency. | Gibson Assembly Master Mix, Golden Gate Assembly kits [1]. |
| IVT sgRNA Kit | Generation of sgRNA arrays as RNA for RNP or mRNA delivery. | HiScribe T7 Quick High Yield RNA Synthesis Kit. |
| Cationic Lipids / Polymers | Formulation of LNPs and polyplexes for nucleic acid or RNP delivery. | Lipofectamine CRISPRMAX, jetOPTIMUS. |
| Nucleofection Kits | Specialized reagents for electroporation of primary cells. | Lonza P3 Primary Cell 4D-Nucleofector Kit for HSCs [33]. |
| Viral Packaging System | Production of lentiviral or AAV particles. | 2nd/3rd generation lentiviral packaging systems (psPAX2, pMD2.G). |
| Selective Antibiotics | Selection of successfully transduced/transfected cells. | Puromycin, Blasticidin, G418. |
Visualization of Tandem sgRNA Array Architectures
A key challenge in multiplexed editing is the construction and processing of tandem sgRNA arrays. The following diagram illustrates the primary genetic architectures used for expressing multiple gRNAs from a single transcript and their respective processing mechanisms.
Multiplexed CRISPR editing represents a significant leap forward in genetic engineering, enabling researchers to simultaneously modify multiple genomic loci within a single cell. This capability is crucial for addressing polygenic traits, dissecting complex genetic pathways, and engineering sophisticated cellular functions. A key technological innovation driving this progress is the development of tandem guide RNA (gRNA) array systems, which allow for the coordinated expression of multiple guide RNAs from a single transcriptional unit. These systems overcome the limitations of traditional single-guide approaches and open new possibilities for comprehensive genome manipulation. This application note presents three detailed case studies demonstrating successful implementations of multiplexed editing across diverse biological systems—mammalian cells, citrus plants, and primary immune cells—highlighting the protocols, reagents, and quantitative outcomes that researchers can leverage for their own investigations.
The efficiency of multiplexed editing hinges on the strategic design of gRNA expression systems. Different CRISPR nucleases and expression architectures offer distinct advantages for various applications. The table below summarizes the primary technologies employed in multiplexed editing workflows.
Table 1: Key Technologies for Multiplexed Genome Editing
| Technology | Mechanism for Multiplexing | Key Advantages | Reported Editing Efficiency Range | Ideal Applications |
|---|---|---|---|---|
| Cas9-based Systems | Multiple individual gRNA expression cassettes or tRNA-gRNA arrays | High efficiency, well-characterized, extensive toolkits | Varies by target; up to 93% in plants [21] | High-efficiency knockout, base editing |
| Cas12a-based Systems | Single transcript with self-processing crRNA arrays | Native array processing, simplified delivery | 29.5% to 69.9% in human cells [30] | High-level multiplexing (5-15 targets) |
| Base Editing (BE) | Fusion of deaminase to nuclease; uses gRNA arrays | No double-strand breaks; precise nucleotide conversion | Up to 39% in human cells [30]; near 100% in NK cells [37] | Point mutation introduction, therapeutic editing |
| Prime Editing (PE) | pegRNA guides reverse transcriptase template | Versatile all possible base-to-base conversions | Lower than BE; requires optimization [38] | Precise sequence writing without donors |
The following diagram illustrates the fundamental workflow and gRNA processing mechanisms for implementing multiplexed CRISPR editing across different systems, highlighting the critical decision points for array design and delivery.
Case Study 1: Multiplex Base Editing in Human Cells Using Cas12a
Recent research has demonstrated the application of Cas12a-derived base editors for precision multiplexed editing in human cells. The study aimed to overcome key limitations in mammalian genome engineering, particularly the limited ability to simultaneously edit multiple loci with base-pair level precision, which has hindered the generation of polygenic phenotypes. Researchers developed and optimized six different Cas12a-derived base editing systems, focusing on their ability to process multiple gRNAs from a single transcript for simultaneous editing at multiple genomic sites [30].
Key Protocols and Workflow
Cell Culture and Transfection:
- Maintain HEK293 cells in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C with 5% CO₂.
- Seed cells at 70-80% confluence in 24-well plates one day prior to transfection.
- Transfect cells using polyethylenimine (PEI) reagent at a 3:1 PEI:DNA ratio.
- Use 500 ng of base editor plasmid and 500 ng of gRNA expression plasmid per well.
gRNA Array Design and Cloning:
- Design gRNA arrays with direct repeat sequences separating individual spacer sequences.
- Clone arrays under the human U6 (hU6) promoter in expression plasmids.
- For multiplexing, construct arrays containing 2-15 distinct gRNAs targeting genes such as RUNX1, DNMT1, and EMX1 for cytosine base editing (CBE) and CDKN2A, VEGFA, and DYRK1A for adenine base editing (ABE).
Selection and Outgrowth:
- At 24 hours post-transfection, begin selection with 2 μg/mL puromycin.
- Maintain selection for 48 hours, then replace with fresh medium without puromycin.
- Allow cells to recover and outgrow for 7 days post-transfection before analysis.
- Harvest cells for genomic DNA extraction and editing efficiency analysis.
Quantitative Results and Outcomes
Table 2: Performance of Cas12a-Derived Base Editors in Human Cells
| Base Editor Type | Number of Targets | Editing Efficiency Range | Key Observations | Bystander Mutation Reduction |
|---|---|---|---|---|
| dLbCas12a-CBE (BEACON1/2) | 3 | Up to 39% ± 5% | Robust editing across all targets | Achieved with truncated gRNAs |
| dLbCas12a-ABE (LbABE8e) | 3 | Variable by target | Position-dependent effects in arrays | Not specifically reported |
| dAsCas12a-CBE (enAsBE1.1/1.2) | 3 | Modest at limited sites | Lower efficiency than LbCas12a systems | Not specifically reported |
| dAsCas12a-ABE (enAsABE8e) | 3 | Modest at limited sites | Inconsistent editing across array | Not specifically reported |
The study achieved a landmark three-fold improvement in multiplex editing capacity, successfully targeting up to 15 distinct genomic sites simultaneously in human cell lines. Editing efficiency was significantly influenced by protocol optimization, with the highest efficiencies achieved using 2 μg/mL puromycin selection followed by a 7-day outgrowth phase. Researchers also developed a Cas12a gRNA engineering approach using truncated gRNAs to direct editing outcomes toward single base-pair conversions, effectively reducing bystander mutations [30].
Critical Reagents and Solutions
Table 3: Essential Research Reagents for Cas12a Multiplex Editing
| Reagent/Solution | Function | Specifications/Alternatives |
|---|---|---|
| LbCas12a- and AsCas12a-derived BEs | Core editing machinery | BEACON1, BEACON2, enAsBE1.1, enAsBE1.2, LbABE8e, enAsABE8e |
| hU6 promoter plasmid | gRNA array expression | Enables high-level Pol III transcription |
| PEI transfection reagent | Plasmid delivery | Cost-effective alternative to commercial reagents |
| Puromycin dihydrochloride | Selection of transfected cells | 2 μg/mL concentration, 48-hour treatment |
| Direct repeat spacers | gRNA processing in arrays | Native Cas12a processing elements |
Case Study 2: Multiplex Gene Editing in Citrus Using tRNA-gRNA Arrays
Citrus species present significant challenges for genetic engineering due to long reproductive cycles, high heterozygosity, and complex regeneration processes. Researchers developed an efficient multiplex CRISPR/Cas9 approach for citrus gene editing using tRNA-based sgRNA arrays to overcome these limitations and enable simultaneous targeting of multiple genes. The study focused on optimizing both Cas9 and sgRNA array expression to achieve high-efficiency editing in the Carrizo citrange cultivar, with the goal of disrupting multiple genes simultaneously to address disease susceptibility and other agronomic traits [3].
Key Protocols and Workflow
Vector Design and Assembly:
- Utilize Golden Gate assembly for modular construction of multigene targeting vectors.
- Employ the pYAO::hSpCas9 backbone with optimized promoters for Cas9 expression.
- Test various promoters for sgRNA array expression, including Pol III promoters (U6) and Pol II promoters (UBQ10, ES8Z).
- Design tRNA-gRNA arrays with Arabidopsis thaliana tRNA-Gly sequences separating individual sgRNAs.
- Synthesize array fragments commercially and clone into destination vectors.
Citrus Transformation:
- Surface-sterilize Carrizo citrange seeds and germinate in vitro on MS medium with vitamins.
- Use etiolated seedling epicotyls as explants for transformation.
- Prepare Agrobacterium tumefaciens strain EHA105 harboring the binary vector.
- Infect explants by incubation with Agrobacterium suspension for 15 minutes.
- Co-cultivate on agar media containing MS salts, sucrose, and benzyladenine (BA).
- Transfer to selection media containing appropriate antibiotics to select transformed tissue.
- Regenerate shoots on media with cytokinins, then root on media with auxins.
Molecular Analysis:
- Extract genomic DNA from regenerated plantlets using CTAB method.
- PCR-amplify target regions and sequence using Sanger or next-generation sequencing.
- Analyze editing efficiency by tracking indels at each target site.
Quantitative Results and Outcomes
Table 4: Multiplex Editing Efficiency in Citrus Using Optimized Systems
| Promoter for Cas9 | Promoter for gRNA Array | Number of Targets | Editing Efficiency | Key Findings |
|---|---|---|---|---|
| UBQ10 | AtU6-26 | 4 | Significantly improved | Strong Pol III promoters perform well |
| RPS5a | ES8Z | 4 | Robust editing | ES8Z effective as Pol II alternative |
| UBQ10 | tRNA with Pol II promoter | 4 | High efficiency | tRNA processing enables multiplexing |
| Conventional 35S | Conventional U6 | 4 | Lower efficiency | Benchmark for comparison |
The optimized system achieved efficient multiplex editing, enabling the simultaneous disruption of at least four genes in citrus. Promoter choice significantly influenced editing efficiency, with the Arabidopsis UBQ10 and RPS5a promoters driving high levels of Cas9 expression, and Pol III promoters (U6) generally outperforming other options for gRNA expression, though the ES8Z Pol II promoter also showed effectiveness. The tRNA-processing system efficiently liberated individual gRNAs from the polycistronic transcript, enabling functional multiplex editing. This represented a significant advancement for citrus biotechnology, moving multiplex editing from proof-of-concept to practical application [3].
Critical Reagents and Solutions
Table 5: Essential Research Reagents for Citrus Multiplex Editing
| Reagent/Solution | Function | Specifications/Alternatives |
|---|---|---|
| pYAO::hSpCas9 backbone | Cas9 expression | Human codon-optimized SpCas9 |
| Arabidopsis U6-26 promoter | gRNA expression | High-activity Pol III promoter |
| tRNA-Gly sequences | gRNA processing | Arabidopsis thaliana, GCC anticodon |
| Agrobacterium EHA105 | Plant transformation | Virulent strain for citrus |
| MS medium with vitamins | Plant tissue culture | Standard citrus regeneration |
Case Study 3: Multiplex Base Editing in Primary NK Cells for Cancer Immunotherapy
Natural killer (NK) cells have emerged as promising candidates for allogeneic "off-the-shelf" cancer immunotherapy, but their efficacy can be limited by various immunosuppressive mechanisms. Researchers developed a non-viral platform for multiplex base editing of primary human NK cells to enhance their antitumor functionality while maintaining their favorable safety profile. The study aimed to simultaneously disrupt multiple immune checkpoint genes in CAR-NK cells without inducing double-strand breaks, thereby creating potent effector cells resistant to tumor-mediated suppression [37].
Key Protocols and Workflow
NK Cell Isolation and Culture:
- Isolate primary human NK cells from healthy donor peripheral blood using negative selection kits.
- Culture cells in NK-specific medium (e.g., RPMI-1640 with 10% FBS) supplemented with IL-2 (100-200 U/mL) or IL-15 (10-20 ng/mL).
- Maintain cells at 1-2×10⁶ cells/mL in a 37°C, 5% CO₂ humidified incubator.
- Activate cells with cytokines (IL-2/IL-15) or activating beads for 24-48 hours before editing.
Base Editor Delivery and Multiplex Editing:
- Use ABE8e adenine base editor for precise A•T to G•C conversions.
- Design sgRNAs to target splice sites or introduce premature stop codons in immune checkpoint genes (AHR, CISH, TIGIT, PDCD1).
- Combine up to six sgRNAs simultaneously in a single electroporation.
- Electroporate NK cells using a specialized protocol: 1-5 million cells per reaction, 2-5 μg of base editor mRNA, 1-2 μg of total sgRNA.
- Use optimized pulse parameters for primary NK cells (e.g., 1500V, 20ms, 1 pulse).
- Immediately transfer electroporated cells to pre-warmed complete medium.
CAR Integration and Functional Assays:
- Combine base editing with non-viral CAR integration using TcBuster transposon system.
- Electroporate CAR transposon plasmid together with base editor components.
- Expand edited NK cells for 7-14 days with appropriate cytokines.
- Assess editing efficiency by targeted amplicon sequencing.
- Evaluate functionality through in vitro cytotoxicity assays against tumor cell lines.
- Test in vivo efficacy and safety using xenograft mouse models.
Quantitative Results and Outcomes
Table 6: Multiplex Base Editing Outcomes in Primary Human NK Cells
| Target Genes | Number of Edits | Editing Efficiency | Functional Outcome | Toxicity Observations |
|---|---|---|---|---|
| Single gene targets | 1 | Up to 100% knockout | Improved function for AHR, CISH, TIGIT, PDCD1 | No significant toxicity |
| TIGIT, PDCD1, CISH (TPCko) | 3 | Highly efficient | Strongest in vitro improvement | Minimal genotoxicity |
| Six simultaneous targets | 6 | Efficient editing | Maintained cytotoxicity | Low translocation rates |
| TPCko + IL-15 CAR | 3 + CAR | High efficiency | Superior in vitro and in vivo function | Systemic toxicity in some models |
The study demonstrated that base editors could achieve highly efficient multiplex editing in primary NK cells, with up to 100% knockout efficiency for single genes and maintained high efficiency when combining up to six sgRNAs simultaneously. The triple knockout of TIGIT, PDCD1, and CISH (TPCko) showed the most significant functional improvement, enhancing NK cell cytotoxicity against tumor targets. When combined with IL-15-expressing CAR, the triple-edited NK cells showed improved persistence in vivo but also exhibited some systemic toxicity in xenograft models, highlighting the need for careful safety assessment. Importantly, base editing resulted in minimal genotoxicity, with low rates of translocations and only one identified off-target site in non-coding regions [37].
Critical Reagents and Solutions
Table 7: Essential Research Reagents for NK Cell Multiplex Editing
| Reagent/Solution | Function | Specifications/Alternatives |
|---|---|---|
| ABE8e base editor | Precise A•T to G•C conversion | Eighth generation adenine base editor |
| NK cell isolation kit | Primary cell purification | Negative selection from PBMCs |
| IL-2/IL-15 cytokines | NK cell activation and expansion | Critical for post-editing recovery |
| Electroporation system | Component delivery | Specialized protocols for sensitive NK cells |
| TcBuster transposon | Non-viral CAR integration | Enables combined editing and CAR delivery |
Technical Considerations and Troubleshooting
gRNA Array Design Optimization
The success of multiplexed editing experiments heavily depends on optimal gRNA array design. For Cas12a systems, ensure proper direct repeat sequences between crRNAs and consider position effects, as editing efficiency can vary based on a gRNA's location within the array [30]. For Cas9 systems using tRNA-processing, select appropriate tRNA species (e.g., Arabidopsis tRNA-Gly for citrus) and verify processing efficiency through RNA analysis [3]. When designing base editing experiments, consider the editing window and potential bystander mutations, which can be mitigated through gRNA truncation strategies [30].
Delivery Method Optimization
Different cell types require specialized delivery approaches. For difficult-to-transfect primary cells like NK cells, optimized electroporation protocols are essential, balancing efficiency with cell viability [37]. For plant systems, Agrobacterium-mediated transformation remains the gold standard, but strain selection and co-cultivation conditions must be optimized for each species [3]. In mammalian cell lines, chemical transfection methods can be effective, but may require optimization of DNA:reagent ratios and timing [30].
Analysis of Complex Editing Outcomes
Multiplexed editing generates complex mutation patterns that require sophisticated analysis methods. For base editing applications, deep sequencing of target loci is necessary to quantify efficiency and assess editing purity [30]. In plant systems, careful molecular screening is required to identify transgene-free edited events, particularly when using co-editing strategies with selection markers [39]. For therapeutic applications, comprehensive off-target assessment using methods like rhAmpSeq is recommended, though current prediction tools for base editor off-targets remain limited [37].
The case studies presented here demonstrate the remarkable progress in multiplexed genome editing across diverse biological systems. From achieving 15-plex base editing in human cells using Cas12a arrays [30], to simultaneous four-gene editing in citrus using tRNA-processed gRNAs [3], to therapeutic engineering of primary NK cells through multiplex base editing [37], these approaches are pushing the boundaries of genetic engineering. The continued refinement of gRNA array designs, delivery methods, and analysis techniques will further expand the capabilities of multiplexed editing, enabling more sophisticated manipulation of complex biological systems for both basic research and therapeutic applications.
Dual-sgRNA Library Designs for Enhanced Knockdown Efficacy in CRISPRi Applications
CRISPR interference (CRISPRi) has emerged as a powerful tool for programmable gene repression in genetic research and drug development. A significant advancement in this field is the development of dual-sgRNA library designs, which substantially enhance knockdown efficacy compared to conventional single-guide approaches. Unlike CRISPR knockout systems that create permanent DNA breaks, CRISPRi utilizes a catalytically dead Cas9 (dCas9) fused to repressor domains to block transcription, enabling reversible and titratable gene repression without genotoxic stress [40] [41]. This technical note details the implementation of dual-sgRNA libraries for improved genetic screening outcomes.
Dual-sgRNA libraries represent a strategic evolution in CRISPRi technology by targeting each gene with two distinct sgRNAs expressed from a tandem cassette. This design achieves stronger phenotypic effects while maintaining an ultra-compact library size – typically 1-3 elements per gene compared to 5 or more in traditional libraries [40]. The compact nature of these libraries reduces screening costs and enables applications in cell types where large library sizes are prohibitive, such as primary cells and stem cell-derived models [42] [43].
Performance Advantages of Dual-sgRNA Designs
Quantitative Efficacy Comparisons
Empirical studies directly comparing single and dual-sgRNA libraries demonstrate clear advantages of the dual-guide approach. In genome-wide growth screens conducted in K562 cells, dual-sgRNA libraries produced significantly stronger growth phenotypes for essential genes previously identified by the Cancer Dependency Map (DepMap).
Table 1: Performance Comparison of Single vs. Dual-sgRNA Libraries in Genetic Screens
| Parameter | Single-sgRNA Library | Dual-sgRNA Library |
|---|---|---|
| Library size (elements per gene) | 1 | 1 (containing 2 sgRNAs) |
| Mean growth rate (γ) for essential genes | -0.20 | -0.26 |
| Statistical significance | Reference | p-value = 6 × 10−15 |
| Correlation with published CRISPRi screens | r = 0.82 | r = 0.83 |
| Essential gene detection (AUC) | >0.98 | >0.98 |
| Phenotypic strength | Baseline | 29% decrease in growth rate |
Data adapted from Replogle et al. [40]
The dual-sgRNA approach demonstrates particular utility for challenging screening scenarios, including identification of synthetic-lethal interactions, mapping genetic modifiers, and probing essential gene functions [42]. The enhanced efficacy stems from simultaneous targeting of multiple sites within a gene promoter, leading to more complete transcriptional repression.
Applications in Genetic Modifier Screening
Dual-sgRNA libraries enable systematic mapping of epistatic relationships through a flexible design that incorporates a fixed "anchor point" sgRNA paired with a genome-wide library of second guides delivered in a single vector [42]. This platform allows researchers to:
- Identify functional redundancy by simultaneously targeting genes with overlapping functions
- Assign factors to parallel pathways through synthetic genetic interactions
- Reveal genetic interactions on a genome-wide scale for essential biological processes
- Utilize sensitive phenotypic readouts compatible with fluorescent reporter systems
This approach is particularly valuable for dissecting complex biological systems containing partially redundant pathways, such as tail-anchored protein insertion into the endoplasmic reticulum mediated by parallel GET and EMC pathways [42].
Experimental Protocols
Dual-sgRNA Library Construction
The following protocol details the construction of a dual-sgRNA library for genome-wide CRISPRi screening:
Materials:
- CRISPRi-v2 library backbone (Addgene)
- Oligonucleotides for fixed sgRNA sequence
- Restriction enzymes (appropriate for cloning strategy)
- T4 DNA ligase
- Competent E. coli cells
- Lentiviral packaging plasmids
Method:
Clone fixed sgRNA: Introduce the predetermined anchor guide sequence into the hU6-CR3 cassette using standard restriction enzyme cloning with complementary DNA oligos [42].
Assemble dual-guide construct: Ligate the hU6-CR3 cassette containing the fixed sgRNA into the CRISPRi-v2 library backbone using compatible restriction sites, creating an mU6-CR1-hU6-CR3 architecture.
Verify library quality: Sequence the resulting library using barcoded 5' CRISPRi-v2 index primers paired with a reverse primer complementary to the hU6 region to ensure presence of both guide RNAs.
Package lentivirus: Produce lentiviral particles using standard packaging cell lines (e.g., HEK293T) and concentration methods.
Titer virus: Determine viral titer to achieve optimal multiplicity of infection (MOI ~0.3) for screening applications.
Critical considerations:
- The fixed sgRNA should be pre-validated for efficient target gene knockdown
- Account for minimal guide loss (~1%) due to restriction sites within the library
- Include appropriate non-targeting control guides
- Validate library representation by next-generation sequencing
Cell Line Engineering and Screening
Stable dCas9 cell line generation:
Select an appropriate CRISPRi effector. Recent comparisons indicate that Zim3-dCas9 provides an optimal balance between strong on-target knockdown and minimal non-specific effects on cell growth or transcriptome [40].
Introduce dCas9-repressor fusion (e.g., dCas9-Zim3 or dCas9-SALL1-SDS3) via lentiviral transduction or stable integration.
Select and validate clones for consistent dCas9 expression and functionality using positive control sgRNAs.
Genetic screening workflow:
Transduce target cells with the dual-sgRNA library at low MOI to ensure single integration events.
Apply selection (e.g., puromycin) 24-48 hours post-transduction to eliminate untransduced cells.
Harvest reference sample (T0) at day 4-6 post-selection for baseline sgRNA representation.
Apply relevant phenotypic selection or sorting based on experimental design:
- For growth screens: culture cells for 14-21 days and harvest final population
- For FACS-based screens: sort cells based on fluorescent reporter expression [42]
Extract genomic DNA from reference and selected populations.
Amplify sgRNA cassettes using optimized PCR protocols to minimize bias.
Sequence amplified products and quantify sgRNA abundance changes to calculate phenotypic effects.
Optimized Reagent Solutions
Table 2: Essential Research Reagents for Dual-sgRNA CRISPRi Applications
| Reagent Category | Specific Examples | Function & Application Notes |
|---|---|---|
| CRISPRi Effectors | Zim3-dCas9, dCas9-KRAB, dCas9-SALL1-SDS3 | Transcriptional repressors; Zim3-dCas9 shows optimal balance of efficacy and minimal non-specific effects [40] |
| Library Backbones | CRISPRi-v2, Dual-sgRNA modified vectors | Foundation for library construction; contain marker genes (BFP, puromycin resistance) for selection [42] |
| sgRNA Design Tools | CRISPRi v2.1 algorithm | Machine learning-based guide prediction using FANTOM and Ensembl TSS data [41] |
| Delivery Systems | Lentiviral vectors, Synthetic sgRNA + dCas9 mRNA | Lentiviral for stable integration; synthetic for rapid, transient repression (24-144 hours) [41] |
| Validation Assays | RT-qPCR, Western blot, FACS reporters | Confirm repression efficiency; RT-qPCR most common for transcriptional assessment [42] [41] |
| Cell Lines | K562, RPE1, Jurkat, HT29, iPSCs | Validated models for CRISPRi screening; available with stable dCas9 expression [40] |
Technical Considerations and Troubleshooting
Implementation Challenges
While dual-sgRNA libraries offer significant advantages, several technical aspects require attention:
- Library amplification bias: Use optimized PCR conditions with minimal cycles to maintain representation
- Vector recombination: The lentiviral reverse transcriptase can undergo template switching between identical repeats in dual-sgRNA cassettes [40]
- Effector selection: Different dCas9 repressor domains vary in their knockdown efficiency and non-specific effects; Zim3-dCas9 currently represents an optimal choice [40]
- sgRNA design: Target regions 0-300 base pairs downstream of the transcription start site (TSS) for optimal repression [41]
Advanced Applications
The dual-sgRNA approach enables sophisticated screening applications:
- Multiplexed gene knockdown: Pooling multiple sgRNAs enables simultaneous repression of several genes, useful for studying redundant pathways or gene families [41]
- CRISPRgenee system: Combining CRISPRi with CRISPR knockout in a single system using Zim3-Cas9 fusion and dual guides for enhanced loss-of-function effects [43]
- Genetic interaction mapping: Fixed anchor guides combined with random second guides enable systematic epistasis analysis [42]
Dual-sgRNA library designs represent a significant advancement in CRISPRi technology, offering enhanced knockdown efficacy in a compact format ideal for sophisticated genetic screens. By implementing the protocols and considerations outlined in this application note, researchers can leverage these systems to address complex biological questions with improved sensitivity and reliability. The continued optimization of effector domains, delivery methods, and library designs will further expand the utility of dual-sgRNA approaches in basic research and drug development.
Overcoming Technical Challenges: Optimization Strategies for Enhanced Editing Efficiency and Specificity
In tandem CRISPR sgRNA array construction for multiplexed genome engineering, achieving high editing efficiency at all intended target sites is a fundamental challenge. Low efficiency can stem from multiple factors, including suboptimal guide RNA (gRNA) design, inefficient delivery of editing components, and inadequate Cas9 expression levels. The simultaneous targeting required for multiplexed editing, especially in complex research applications such as gene network analysis or polygenic trait engineering, exacerbates these challenges. When using sgRNA arrays, the editing efficiency can vary significantly between different guides within the same array due to differences in their individual targeting efficiency and the structural properties of the array itself. This protocol provides a comprehensive, experimentally-validated framework to systematically address these limitations, enabling researchers to achieve more consistent and efficient multiplex editing outcomes. By implementing robust strategies for gRNA design, optimizing delivery methods, and fine-tuning Cas9 expression, researchers can significantly enhance the performance of their tandem CRISPR systems for advanced genome engineering applications.
AI-Enhanced gRNA Design for Improved On-Target Activity
The design of guide RNAs is the most critical determinant of CRISPR editing efficiency. Traditional rule-based design methods often fail to account for the complex sequence and epigenetic features that influence gRNA activity. Artificial intelligence (AI) models, particularly deep learning frameworks, now enable more accurate prediction of gRNA efficacy by integrating multiple contextual factors.
State-of-the-Art AI Tools for gRNA Design
Table 1: Bioinformatics Tools for Enhanced gRNA Design
| Tool/Model | Key Features | Applicable Systems | Performance Advantages |
|---|---|---|---|
| CRISPRon | Integrates sequence features with epigenomic information (chromatin accessibility) | Cas9 variants | More accurate efficiency ranking compared to sequence-only predictors [44] |
| Kim et al. Model | Predicts activity of SpCas9 variants (xCas9, Cas9-NG) | SpCas9 variants with altered PAM specificities | Optimized guide selection for non-NGG PAM targets [44] |
| CRISPR-Net | Combines CNN and bi-directional GRU to analyze guides with mismatches/indels | Cas9, Cas12a | Analyzes guides with up to four mismatches or indels relative to targets [44] |
| Croton | Variant-aware indel prediction pipeline | CRISPR-Cas9 | Accounts for nearby genetic variants that might alter editing outcomes [44] |
| CHOPCHOP | User-friendly web interface, off-target predictions | Multiple nucleases | Widely adopted, commonly highlighted in tool analyses [35] [22] |
Experimental Protocol: gRNA Design and Validation Workflow
Step 1: Target Identification and gRNA Selection
- Input your target genomic sequence into multiple AI-based design tools (CRISPRon, CHOPCHOP)
- For multiplexed arrays, select 3-5 candidate gRNAs per target locus with highest predicted efficiency scores
- Prioritize gRNAs with minimal off-target potential using integrated prediction algorithms
- For tandem arrays, avoid gRNAs with significant complementarity that may cause array instability
Step 2: gRNA Cloning and Array Construction
- Clone individual gRNAs into expression vectors using BsaI restriction sites following Golden Gate assembly [4]
- For tandem arrays, employ tRNA or ribozyme-mediated processing systems to ensure proper individual guide expression [21] [45]
- Verify array integrity by Sanger sequencing across all junction sites
Step 3: In Vitro Validation of gRNA Activity
- Transcribe gRNAs in vitro using T7 RNA polymerase (protocol 1.4) [35]
- Incubate gRNAs with Cas9 protein and target DNA fragments containing the intended target sites
- Analyze cleavage efficiency by gel electrophoresis or fragment analysis
- Select the most active gRNAs for each target before proceeding to cellular experiments
gRNA Design and Validation Workflow
Delivery System Optimization for Multiplex Editing
Efficient delivery of CRISPR components is particularly challenging for multiplex editing due to the larger size of sgRNA arrays and the need for coordinated expression of all components. The choice of delivery method significantly impacts editing efficiency, especially in difficult-to-transfect cells.
Quantitative Comparison of Delivery Methods
Table 2: Delivery Methods for CRISPR Components in Multiplex Editing
| Delivery Method | Max Carrying Capacity | Typical Efficiency | Advantages | Limitations |
|---|---|---|---|---|
| Lentiviral Vectors | ~8kb insert size | Variable (10-80% depending on cell type) | Stable integration, broad tropism | Insert size limitations, random integration [4] |
| Lipid Nanoparticles (LNPs) | ~10kb mRNA | High in susceptible cells (50-90%) | Low immunogenicity, clinical relevance | Cytotoxicity at high concentrations [45] |
| Electroporation | No practical limit | High in immune cells, stem cells (30-95%) | Applicable to wide range of macromolecules | Specialized equipment required, cell toxicity [35] |
| Virus-Like Particles (VLPs) | Limited cargo space | Moderate (20-60%) | High specificity, minimal off-target effects | Complex production, limited cargo capacity [45] |
| Polyethylenimine (PEI) | No practical limit | Variable (10-70%) | Cost-effective, simple use | Cytotoxicity, lower efficiency in some cell types [35] |
Experimental Protocol: Delivery Optimization for sgRNA Arrays
Step 1: Delivery Method Selection and Preparation
- For plasmid-based delivery of sgRNA arrays, use high-purity endotoxin-free plasmid preparations
- For ribonucleoprotein (RNP) delivery, complex purified Cas9 protein with in vitro transcribed sgRNAs at 1:2 molar ratio and incubate for 10 minutes at room temperature before delivery
- For viral delivery, package sgRNA arrays in lentiviral or AAV vectors, considering the cargo size limitations of AAV (~4.7kb)
Step 2: Delivery Condition Optimization
- Perform dose-response experiments with varying amounts of CRISPR components (e.g., 0.5-5μg plasmid DNA, 10-100pmol RNP per 10^5 cells)
- Optimize delivery parameters specific to your cell type:
- Electroporation: Voltage, pulse length, cell density
- Lipofection: Lipid:DNA ratio, incubation time
- Viral transduction: MOI, spinoculation parameters, polybrene concentration
- Include appropriate controls (empty vector, non-targeting gRNA)
Step 3: Post-Delivery Processing and Analysis
- Allow 48-72 hours for expression and editing after delivery before initial assessment
- For RNP delivery, analyze editing efficiency at 24-48 hours post-delivery
- Use a portion of cells to assess delivery efficiency via included fluorescent markers or immunostaining
- Harvest cells for genomic DNA extraction and editing efficiency analysis
Cas9 Expression Tuning and Validation
The expression level of Cas9 nuclease significantly impacts both editing efficiency and specificity. Excessive Cas9 expression can increase off-target effects, while insufficient expression reduces on-target editing. For multiplex editing applications, maintaining optimal Cas9 levels is particularly important when targeting multiple loci simultaneously.
Cas9 Expression Systems and Characterization
Constitutive vs. Inducible Expression Systems
- Use strong constitutive promoters (EF1α, CAG, UBC) for maximum expression across cell types [35]
- Employ inducible systems (doxycycline, cumate) for temporal control of Cas9 expression to minimize off-target effects
- Consider self-inactivating systems that reduce Cas9 exposure after editing occurs
Cas9 Variant Selection
- For multiplex editing with extended target range, use Cas9 variants with relaxed PAM requirements (xCas9, SpCas9-NG) [44]
- For improved specificity, especially with multiple gRNAs, use high-fidelity variants (eSpCas9, SpCas9-HF1)
- Consider smaller Cas variants (CasMINI, Cas12f1) when delivery efficiency is limiting [46] [45]
Experimental Protocol: Cas9 Expression Optimization
Step 1: Quantitative Assessment of Cas9 Expression
- Transfert cells with Cas9 expression constructs and analyze expression 48 hours post-transfection
- For protein-level detection, use Western blotting with anti-Cas9 antibodies
- For mRNA-level detection, use RT-qPCR with Cas9-specific primers
- Correlate expression levels with editing efficiency across a range of expression vectors
Step 2: Editing Efficiency Validation
- Extract genomic DNA 72-96 hours post-transfection using silica column-based methods [35]
- Amplify target regions by PCR using primers flanking each target site
- Assess editing efficiency using one of these methods:
- T7 Endonuclease I or Surveyor assay for initial efficiency estimation
- Sanger sequencing with ICE analysis for quantitative indel assessment [47]
- Next-generation amplicon sequencing for comprehensive editing characterization
Step 3: Specificity Validation
- Perform off-target prediction for all gRNAs in the array using tools like Cas-OFFinder [22]
- Amplify and sequence top potential off-target sites (typically 3-5 per gRNA)
- For comprehensive off-target assessment, use GUIDE-seq or CIRCLE-seq methods
Cas9 Expression Optimization Workflow
Integrated Workflow for Enhanced Multiplex Editing
Combining optimized gRNA design, efficient delivery, and tuned Cas9 expression creates a synergistic effect that significantly enhances multiplex editing efficiency. This integrated approach is particularly valuable for complex applications such as gene network dissection and combinatorial trait engineering.
Comprehensive Experimental Protocol: Tandem sgRNA Array Workflow
Step 1: Design and Construction Phase
- Identify all target loci and design 3-5 gRNAs per locus using AI-based tools (Section 2.1)
- Select top 2 gRNAs per locus based on predicted efficiency and specificity scores
- Design tandem sgRNA array with selected gRNAs separated by appropriate processing elements (tRNA, ribozymes)
- Include a tracking mechanism (e.g., barcode sequence) to identify successful delivery
- Clone the complete expression cassette into appropriate delivery vector
Step 2: Delivery and Expression Phase
- Prepare high-quality vector DNA or RNP complexes as appropriate for your cell type
- Transduce/transfect cells using optimized parameters (Section 3.2)
- For stable expression systems, apply appropriate selection 24 hours post-delivery
- For inducible systems, induce Cas9 expression at optimal cell density
Step 3: Analysis and Validation Phase
- Harvest cells at 72-96 hours post-editing for initial efficiency assessment
- Extract genomic DNA and amplify all target loci
- Use ICE analysis for rapid quantification of editing efficiency [47]
- For comprehensive analysis, perform NGS amplicon sequencing of all target sites
- Expand edited cells and isolate single-cell clones if needed
- Validate phenotypic consequences of multiplex editing through functional assays
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Enhanced Multiplex Editing
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Cas9 Expression Systems | pX330, lentiCas9-Blast, pCW-Cas9 | Provides nuclease function | Choose based on need for constitutive, inducible, or transient expression [35] |
| gRNA Cloning Vectors | pU6-sgRNA, LRG2.1, pRG2 | gRNA expression and array assembly | tRNA-gRNA vectors enable efficient processing of tandem arrays [21] |
| Delivery Reagents | Lipofectamine CRISPRMAX, Neon Electroporation System, PEI MAX | Component delivery | Optimize reagent:DNA ratio for each cell type [35] |
| Efficiency Assessment | T7 Endonuclease I, ICE Analysis Software, NGS kits | Editing quantification | ICE provides NGS-quality data from Sanger sequencing [47] |
| AI Design Tools | CRISPRon, CHOPCHOP, DeepCRISPR | gRNA selection and optimization | Access through web interfaces or local installation [44] [22] |
Implementing the comprehensive strategies outlined in this protocol—from AI-enhanced gRNA design to delivery optimization and Cas9 expression tuning—enables researchers to significantly improve editing efficiency in tandem CRISPR sgRNA arrays for multiplexed genome engineering. The quantitative frameworks and step-by-step protocols provide a systematic approach to address the key bottlenecks in multiplex editing efficiency. As CRISPR technology continues to evolve, with emerging developments in AI-guided design and novel delivery platforms, these foundational principles will remain essential for achieving robust and efficient multiplex editing across diverse research applications.
Minimizing Off-Target Effects Through High-Fidelity Cas Variants and Specific gRNA Design
The application of CRISPR-based gene editing technologies, from basic research to therapeutic development, is substantially hampered by off-target effects. These effects occur when the CRISPR machinery, particularly the Cas nuclease, acts on untargeted genomic sites with sequence similarity to the intended target, leading to unintended alterations that can confound experimental results and pose significant safety risks in clinical applications [48] [49]. In the specific context of tandem CRISPR sgRNA array construction for multiplexed editing, where multiple guide RNAs are expressed simultaneously to target several genomic loci, the risk of off-target activity is magnified due to the increased complexity of the system. This application note provides a detailed framework for minimizing off-target effects through the combined use of high-fidelity Cas variants and strategically designed guide RNAs, with specific consideration for multiplexed editing applications.
High-Fidelity Cas Variants: Mechanisms and Performance Data
Wild-type Cas nucleases, such as the commonly used Streptococcus pyogenes Cas9 (SpCas9), possess a reasonable tolerance for mismatches between the guide RNA and target DNA, enabling them to cleave sequences with up to 3-5 base pair mismatches [48]. To address this limitation, protein engineering approaches have produced several high-fidelity Cas9 variants with reduced off-target activity while maintaining robust on-target editing.
Engineered High-Fidelity Cas9 Variants
These variants employ distinct molecular strategies to enhance specificity, primarily by reducing the nuclease's affinity for non-target DNA strands or introducing conformational checks.
Table 1: Characteristics of High-Fidelity SpCas9 Variants
| Variant | Key Mutations | Mechanism of Fidelity Enhancement | Reported On-Target Efficiency |
|---|---|---|---|
| eSpCas9(1.1) | K848A, K1003A, R1060A | Reduces protein affinity for the non-target DNA strand, decreasing stability of mismatch-containing helices [50]. | Severely compromised or absent in base editing configurations [50]. |
| SpCas9-HF2 | N497A, R661A, Q695A, Q926A, D1135E | Decreases energetics of the Cas9-sgRNA complex to diminish cleavage at mismatched sites [50]. | ~50% reduction compared to SpCas9-pBE at multiple targets [50]. |
| HypaCas9 | N692A, M694A, Q695A, H698A | Stringently traps the HNH domain at a conformational checkpoint in the presence of mismatches [50]. | Comparable to wild-type SpCas9 (e.g., 53% vs. 53% at an ALS target) [50]. |
| OpenCRISPR-1 | AI-generated (∼400 mutations from SpCas9) | An artificial-intelligence-generated editor designed de novo for optimal properties [51]. | Comparable or improved activity relative to SpCas9 [51]. |
Performance Considerations for Multiplexed Editing
When deploying these high-fidelity variants in multiplexed systems, their performance must be carefully evaluated. Research in rice has demonstrated that high-fidelity base editors (HF-pBEs) derived from these variants exhibit distinct multiplex-genome editing efficiencies [50]. For instance, the editing efficiency of HypaCas9-pBE was comparable to SpCas9-pBE, whereas SpCas9-HF2-pBE showed reduced efficiency, and eSpCas9(1.1)-pBE showed no base-editing ability at the tested targets [50]. This underscores the necessity of empirically validating each variant for a given application. Furthermore, the use of a tandemly arrayed tRNA-sgRNA architecture has been shown to enhance the base-editing efficiencies of high-fidelity Cas9s, rescuing the activity of some variants by up to 25.5-fold [50].
Strategic gRNA Design for Enhanced Specificity
The guide RNA's spacer sequence, which determines target recognition through Watson-Crick base pairing, is a critical determinant of specificity. Rational gRNA design can significantly reduce the likelihood of off-target binding and cleavage.
Core Principles of Specific gRNA Design
- Leverage Mismatch-Sensitive "Seed" Regions: The PAM-distal region of the gRNA spacer (approximately 10-12 nucleotides) forms a "seed" sequence where mismatches are least tolerated [52]. Designing gRNAs such that the target site's unique nucleotides fall within this seed region can dramatically enhance specificity. If the single-nucleotide variant (SNV) of interest lies outside this region, one strategy is to use mutagenic primers to relocate the PAM, thereby positioning the SNV within the seed for detection [52].
- Introduce Synthetic Mismatches: A counter-intuitive yet effective strategy involves intentionally introducing a synthetic mismatch into the gRNA at a specific position relative to the SNV. This can increase the penalty score for binding to the off-target sequence, making it less likely to reach the cleavage threshold. This approach has been successfully applied in Cas13a-based diagnostics (SHERLOCK) and for other effectors like Cas12 and Cas9 [52].
- Optimize gRNA Length and Composition: Shorter gRNAs (17-18 nucleotides instead of 20) can reduce off-target activity by decreasing the stability of the DNA:RNA duplex at imperfectly matched sites [48]. Furthermore, gRNAs with higher GC content tend to form more stable duplexes at the on-target site, which can improve specificity [48].
- Chemical Modifications: Synthetic gRNAs can be modified with molecules such as 2'-O-methyl analogs (2'-O-Me) and 3' phosphorothioate bonds (PS) to reduce off-target edits while potentially increasing on-target efficiency [48].
gRNA Design for Tandem Arrays
In multiplexed editing systems, the principles above must be applied to each individual gRNA within the array. This necessitates careful in silico analysis of every proposed gRNA sequence to predict and minimize potential off-targets across the genome. Tools like CRISPOR and others implement algorithms that provide off-target scores, helping researchers select guides with an optimal on-target to off-target activity ratio [48].
Table 2: Strategies for High-Specificity gRNA Design and Validation
| Strategy | Methodological Approach | Key Considerations |
|---|---|---|
| In Silico Prediction | Use tools like Cas-OFFinder, FlashFry, or DeepCRISPR to nominate potential off-target sites based on sequence similarity [49]. | Algorithms primarily consider sequence complementarity and may not fully account for chromatin environment. Essential for pre-screening gRNA designs. |
| Seed Region Targeting | Design gRNA so that unique bases in the target site are located in the PAM-distal seed region (positions ~1-12) [52]. | Highly effective for discriminating against sequences with single-nucleotide differences. |
| Synthetic Mismatch | Deliberately introduce a mismatched base in the gRNA sequence to destabilize binding to off-targets [52]. | Effectiveness is context-dependent and requires empirical testing to find the optimal position and base change. |
| gRNA Truncation | Shorten the gRNA spacer sequence to 17-18 nucleotides (truncated guides, or "tru-gRNAs") [48]. | Can reduce off-target activity but may also impair on-target efficiency, requiring a balance. |
Integrated Experimental Protocol for Off-Target Assessment
The following protocol provides a step-by-step guide for designing a high-specificity multiplexed editing experiment and rigorously assessing its off-target profile.
Protocol: Design, Delivery, and Validation of High-Fidelity Multiplexed Editing
Part 1: gRNA Selection and Array Construction
- Target Identification: Define all genomic loci to be targeted in the multiplexed experiment.
- gRNA Design: For each target locus, design 3-5 candidate gRNAs using a design tool (e.g., CRISPOR).
- Prioritize gRNAs with high predicted on-target scores and low off-target scores.
- Apply the principles in Section 3.1, ensuring unique bases fall within the seed region.
- Use in silico tools (e.g., Cas-OFFinder) to generate a list of potential off-target sites for each candidate gRNA for later validation [49].
- Array Assembly: Assemble the selected gRNAs into a single transcriptional unit using a method suitable for your experimental system. Robust options include:
- tRNA-based processing: Flank each gRNA with endogenous tRNA sequences, which are processed by cellular RNases P and Z [1]. This system is highly modular and functions in a wide range of organisms.
- Cas12a-based crRNA arrays: Utilize the inherent processing capability of Cas12a, which cleaves its own pre-crRNA transcript at hairpin structures formed within the direct repeats [1].
Part 2: Delivery and Expression
- Vector Construction: Clone the gRNA array into an expression vector alongside a gene encoding a high-fidelity Cas variant (e.g., HypaCas9, OpenCRISPR-1). Use a promoter system that enables controlled, short-term expression (e.g., inducible promoter) to limit the window for off-target activity [48].
- Cell Delivery: Transfect or transduce the target cells with the constructed vector. The choice of delivery vehicle (e.g., virus, nanoparticle) can impact the duration of Cas/gRNA expression and thus off-target risk [48].
Part 3: On-Target and Off-Target Analysis
- On-Target Efficiency Validation:
- After sufficient time for editing, harvest genomic DNA.
- Amplify the targeted loci by PCR and analyze editing efficiency using Sanger sequencing (analyzed with tools like Inference of CRISPR Edits - ICE) or next-generation sequencing (NGS) [48].
- Comprehensive Off-Target Assessment:
- Targeted Sequencing: Sequence the potential off-target sites identified in silico during Step 2. This is a cost-effective method for validating high-probability off-targets [48] [49].
- Unbiased Genome-Wide Screening: For therapeutic applications or critical experiments, employ an unbiased method to discover unanticipated off-targets.
- GUIDE-seq: Integrates double-stranded oligodeoxynucleotides (dsODNs) into double-strand breaks (DSBs) in cells, followed by NGS to map integration sites genome-wide. It is highly sensitive and has a low false-positive rate [49].
- CIRCLE-seq: A cell-free method that circularizes sheared genomic DNA, incubates it with Cas9-gRNA ribonucleoprotein (RNP) complexes, and sequences the linearized cleavage products. It is highly sensitive and does not require a reference genome [49].
The following workflow summarizes the integrated experimental protocol:
Table 3: Key Research Reagent Solutions for High-Fidelity Multiplexed Editing
| Reagent / Tool Category | Specific Examples | Function & Application Note |
|---|---|---|
| High-Fidelity Cas Effectors | HypaCas9, SpCas9-HF2, eSpCas9(1.1), OpenCRISPR-1 [51] [50]. | Engineered or AI-generated nucleases with reduced non-specific DNA binding; foundational for reducing off-target cleavage in multiplexed setups. |
| gRNA Array Scaffolds | tRNA-gRNA, Cas12a direct repeat array, Csy4 recognition site array [1]. | Genetic architectures for expressing multiple gRNAs from a single transcript; choice affects processing efficiency and final gRNA stoichiometry. |
| In Silico Prediction Tools | Cas-OFFinder, FlashFry, DeepCRISPR, CRISPOR [49]. | Computational tools for nominating potential off-target sites and scoring gRNAs during the design phase. |
| Off-Target Detection Kits | Commercial GUIDE-seq or CIRCLE-seq kits [49]. | Experimentally validated systems for genome-wide, unbiased identification of nuclease off-target sites; critical for pre-clinical safety assessment. |
| Chemically Modified gRNAs | 2'-O-Me/PS modified synthetic gRNAs [48]. | Enhanced stability and specificity guides; particularly useful for RNP delivery in therapeutic contexts. |
Minimizing off-target effects is not a single-step solution but a comprehensive strategy integral to the experimental design process. For researchers constructing tandem CRISPR sgRNA arrays for multiplexed editing, this involves the synergistic application of high-fidelity Cas variants like HypaCas9 or AI-designed OpenCRISPR-1, coupled with rigorously designed gRNAs that leverage seed region targeting and computational prediction. The workflow must be capped with empirical, sensitive off-target detection methods such as GUIDE-seq or CIRCLE-seq to obtain a full picture of editing specificity. By adhering to the protocols and principles outlined in this application note, scientists can significantly enhance the precision and safety of their multiplexed genome engineering efforts, accelerating the transition of CRISPR technologies from bench to bedside.
The construction of tandem CRISPR sgRNA arrays is a foundational technique for advanced multiplexed genome engineering. Unlike pooled libraries where guides are processed in mixed populations, arrayed libraries allow for the perturbation of genes individually in distinct wells, making them applicable to virtually all screenable cell phenotypes, including non-selectable ones [53]. The primary technical challenge in building these multi-guide constructs stems from the repetitive nature of the sequences involved. The repeating scaffolds, identical promoters, and other homologous elements create a genetic environment prone to inappropriate recombination events in both bacterial cloning intermediates (such as E. coli and Agrobacterium) and the final plant or mammalian hosts [21]. This recombination leads to plasmid instability, sequence deletions, and rearrangements, ultimately resulting in incomplete or dysfunctional CRISPR libraries with heterogeneous and unpredictable gene-perturbation efficacy [53] [21].
This Application Note details proven methodologies to overcome these challenges, enabling the reliable generation of high-complexity, highly active arrayed libraries for multiplexed editing research. The strategies discussed below are critical for researchers aiming to perform precise combinatorial gene knockouts, transcriptional activation, or epigenetic silencing screens.
Key Challenges and Strategic Solutions
The table below summarizes the major challenges in sgRNA array assembly and the corresponding strategic solutions developed to address them.
Table 1: Key Challenges and Strategic Solutions for sgRNA Array Assembly
| Challenge | Impact on Assembly | Proposed Solution |
|---|---|---|
| Repetitive Promoters | Homologous recombination leading to plasmid deletion and rearrangement [21]. | Use of diverse, non-identical Pol-III promoters (e.g., hU6, mU6, hH1, h7SK) to drive each sgRNA [53]. |
| Repetitive sgRNA Scaffolds | Recombination causing guide loss and functional failure [53]. | Employing distinct tracrRNA variants for each sgRNA in the array [53]. |
| High-Throughput Cloning Bottleneck | Traditional cloning (gel purification, colony picking) is slow, laborious, and not easily automatable [53]. | Implementation of automated, high-throughput liquid-phase assembly methods like ALPA cloning [53]. |
| Low Editing Efficiency & Heterogeneity | Single sgRNAs often yield low and variable perturbation, reducing screen robustness [53]. | Targeting each gene with a quadruple-sgRNA (qgRNA) array to ensure high, consistent efficacy [53]. |
Experimental Protocols and Workflows
ALPA Cloning for High-Throughput qgRNA Assembly
The Automated Liquid-Phase Assembly (ALPA) cloning method is designed for the one-pot, high-throughput construction of quadruple-sgRNA (qgRNA) plasmids, effectively bypassing the need for single-colony picking and minimizing recombination [53].
Materials & Reagents
- Precursor Vector: pYJA5, containing puromycin and TagBFP selection markers, and flanked by lentiviral LTRs and PiggyBac transposon elements for flexible delivery. The vector contains a removable ampicillin resistance (AmpR) gene [53].
- Oligonucleotides: 59-mer primers for each sgRNA, comprising the 20-nt protospacer and constant regions with amplification primer annealing sites.
- Constant-Fragment Templates: DNA templates for PCR amplification of the sgRNA expression units.
- Enzymes: BbsI restriction enzyme, Gibson assembly mix, and high-fidelity DNA polymerase.
- Competent Cells: Recombination-deficient chemically competent E. coli.
- Selection Antibiotics: Ampicillin and Trimethoprim.
Procedure
- Vector Digestion: Digest the pYJA5 precursor vector with BbsI to remove the AmpR cassette flanked by BbsI sites [53].
- sgRNA Amplicon Generation: In three separate PCRs, mix the sgRNA oligonucleotide primers with their corresponding constant-fragment templates to generate three individual amplicons. These amplicons and the digested vector are designed with distinct, non-homologous overlapping ends (∼20 nt) for subsequent Gibson assembly [53].
- Gibson Assembly: Assemble the three amplicons and the digested pYJA5 vector using Gibson assembly. The first amplicon incorporates a trimethoprim-resistant dihydrofolate reductase gene (TmpR), enabling a selection switch from ampicillin to trimethoprim [53].
- Transformation and Selection: Transform the Gibson assembly reaction directly into recombination-deficient chemically competent E. coli. Plate the bacteria on trimethoprim-containing media. The dual antibiotic selection (AmpR in the precursor, TmpR in the final plasmid) selectively enriches for correctly assembled plasmids without the need for single-colony isolation [53].
- High-Throughput Processing: Perform all steps, including transformation and subsequent magnetic bead-based plasmid minipreps, in 384-well and deep-96-well plates using custom-made equipment for automation. This pipeline enables the construction of over 2,000 plasmids per week [53].
Validation
- PCR Screening: Perform colony PCR with primers flanking the qgRNA insert. Correct assembly is indicated by a product of the expected size (e.g., 2.2 kb) [53].
- Sequencing Verification: Sanger sequence colonies from independent cloning procedures. The ALPA method typically yields 83–93% of colonies with correct qgRNA sequences, with low rates of recombination (0–10%) and point mutations (3–14%) [53].
Alternative Method: Golden Gate Assembly for Multiplex Arrays
For assembly workflows not requiring the specific ALPA selection system, Golden Gate assembly is a widely adopted and effective strategy for combining multiple gRNA units.
Materials & Reagents
- Type IIS Restriction Enzymes: Such as BsaI or BbsI, which cut outside their recognition sequence, creating unique overhangs.
- Destination Vector: Contains the Cas9 expression cassette and necessary backbone elements.
- gRNA Modules: Individual gRNA expression units flanked by Type IIS recognition sites, designed with unique overhangs for ordered assembly.
Procedure
- Design: Design each gRNA module with flanking Type IIS sites so that digestion generates unique, non-palindromic 4-bp overhangs that dictate the assembly order.
- Digestion and Ligation: In a single reaction tube, combine the destination vector and all gRNA modules with the Type IIS restriction enzyme and DNA ligase. The reaction cycles between digestion and ligation, seamlessly assembling the fragments in the predefined order [4].
- Transformation: Transform the final reaction product into competent E. coli and select with the appropriate antibiotic.
This method has been successfully used to construct CRISPR-Cas9 cassettes with up to seven gRNAs [4]. An advanced "PCR-on-ligation" variation of this technique has enabled 10-plex gene editing in human cell lines [4].
Research Reagent Solutions
The table below lists essential reagents and their functions for successful array assembly and implementation.
Table 2: Key Research Reagents for Arrayed CRISPR Library Construction
| Reagent / Tool | Function / Application | Key Features / Examples |
|---|---|---|
| Diverse Pol-III Promoters | Drives sgRNA expression while minimizing sequence homology. | Human U6, Mouse U6, Human H1, Human 7SK [53]. |
| Distinct tracrRNA Variants | Forms the functional sgRNA complex while reducing scaffold repetition. | Different sequences yielding the same secondary structure [53]. |
| Precursor Vector (e.g., pYJA5) | Backbone for sgRNA array assembly and delivery. | Contains Puromycin & TagBFP for selection; Lentiviral LTRs & PiggyBac for delivery; AmpR/TmpR selection switch [53]. |
| Type IIS Restriction Enzymes | Enzymatic digestion for Golden Gate assembly. | BsaI, BbsI; creates unique overhangs for seamless, ordered assembly [4]. |
| Recombination-Deficient E. coli | Bacterial host for plasmid propagation. | Reduces plasmid recombination during amplification (e.g., Stbl3, SURE cells) [53]. |
| Small Molecule Enhancers | Increases editing efficiency post-assembly. | Repsox (TGF-β inhibitor): shown to increase NHEJ efficiency 3.16-fold in porcine cells [54]. |
Validation and Functional Assessment
After successful array assembly, rigorous validation of its functional efficacy is crucial.
Functional Testing in Cell Lines
- CRISPRa (Activation): Co-transfect HEK293 cells with the qgRNA plasmid and a CRISPR activator (e.g., dCas9-VPR). Measure target gene expression via qRT-PCR or surface marker expression (e.g., CD2, CD4, CD200) using flow cytometry. qgRNA vectors have been shown to massively increase target gene activation compared to single sgRNAs, with substantially reduced cell-to-cell heterogeneity [53].
- CRISPRko (Knockout): Transduce the cell line of interest with the lentiviral qgRNA library and select with puromycin. Assess indel formation at the target loci using T7E1 assay or TIDE sequencing. Quadruple-sgRNA libraries have demonstrated high perturbation efficacy, with reported deletion efficiencies of 75–99% [53].
Application in Genetic Screens The practical utility of arrayed qgRNA libraries is demonstrated in large-scale screens. For example, an arrayed activation screen of 1,634 human transcription factors successfully identified 11 novel regulators of the cellular prion protein PrPC [53]. Similarly, pooled versions of ablation libraries have uncovered novel modifiers of autophagy that were previously undetected with less robust tools [53].
The challenges of repetitive sequences and recombination in tandem CRISPR array assembly can be effectively managed through strategic molecular design and modern cloning techniques. The core principles involve replacing repetitive elements with diverse counterparts, employing high-throughput, selection-based cloning methods like ALPA, and functionally validating the libraries using robust biological assays. Adherence to these protocols enables researchers to reliably construct high-quality, genome-wide arrayed libraries, thereby unlocking the full potential of multiplexed CRISPR editing for sophisticated functional genomics and drug discovery.
Optimizing gRNA Stoichiometry and Positional Effects in Tandem Arrays
Within the context of tandem CRISPR sgRNA array construction for multiplexed editing research, optimizing the physical arrangement and relative abundance of guide RNAs (gRNAs) is paramount to experimental success. Tandem gRNA arrays, in which multiple gRNA sequences are transcribed as a single transcript and subsequently processed into individual functional guides, enable simultaneous targeting of multiple genomic loci [1]. The efficiency of such multiplexed editing is heavily influenced by two critical factors: gRNA stoichiometry (the relative abundance of each gRNA) and positional effects (the impact of a gRNA's location within the array on its functionality) [1]. This Application Note provides a comprehensive framework for optimizing these parameters, supported by quantitative data and detailed protocols, to enhance the reliability of multiplexed CRISPR experiments for researchers and drug development professionals.
The fundamental challenge in tandem array design stems from the observation that not all gRNAs within an array are expressed or function with equal efficiency [1]. Position-dependent effects can lead to inconsistent editing outcomes, while imbalances in gRNA stoichiometry may result in incomplete perturbation of intended targets. By systematically addressing these variables through the strategies outlined herein, researchers can achieve more predictable and efficient multiplexed editing, thereby improving the quality of functional genomics studies and therapeutic development workflows.
Tandem gRNA Array Architectures and Processing Mechanisms
Tandem gRNA arrays can be implemented using several distinct genetic architectures, each with unique processing mechanisms that influence overall performance [1]. The choice of architecture determines the processing method, which in turn affects gRNA stoichiometry and the impact of positional effects.
Table 1: Comparison of Tandem gRNA Array Architectures
| Architecture Type | Processing Mechanism | Key Features | Typical gRNA Capacity | Organisms Demonstrated |
|---|---|---|---|---|
| Cas12a-based Array | Self-processing by Cas12a nuclease | Utilizes native crRNA processing; simple design | 5+ gRNAs | Human cells, plants, yeast, bacteria |
| Ribozyme-flanked Array | Self-cleaving hammerhead and HDV ribozymes | Compatible with Pol II/III transcription; precise excision | 2-7 gRNAs | Mammalian cells, yeast |
| tRNA-gRNA Array | Endogenous tRNA processing machinery | Uses RNase P and Z cleavage; highly efficient | 12+ gRNAs | Yeast, plants, mammalian cells |
| Csy4-based Array | Heterologous RNase (Csy4) | High processing efficiency; requires co-expression of processing enzyme | 12+ gRNAs | Mammalian cells, yeast, bacteria |
The Cas12a (Cpf1) system leverages the inherent processing capability of the Cas12a protein itself, which cleaves pre-crRNA transcripts based on hairpin structures formed within spacer repeats [1]. This system enables concurrent expression of both the effector protein and gRNA array from a single polymerase II (Pol II) promoter, as demonstrated by the simultaneous targeting of five loci for cleavage and ten for transcriptional regulation in human cells [1].
Alternative architectures employ exogenous processing elements. The ribozyme-based approach flanks each gRNA with self-cleaving hammerhead and hepatitis delta virus (HDV) ribozymes, which autocatalytically excise the gRNAs without requiring protein cofactors [1]. The tRNA-gRNA system exploits endogenous cellular machinery, where RNases P and Z recognize and cleave at tRNA sequences inserted between gRNAs [55]. Similarly, the Csy4 system uses a heterologous bacterial endonuclease that recognizes specific 28-nucleotide RNA sequences flanking each gRNA, though this requires co-expression of the Csy4 protein and may introduce cytotoxicity at high concentrations [1].
Figure 1: Tandem gRNA Array Architecture Processing Pathways. Different array architectures utilize distinct processing mechanisms to liberate individual gRNAs from a single transcript.
Quantitative Analysis of gRNA Spacing and Positional Effects
Optimal gRNA Spacing in Tandem Systems
The genomic distance between adjacent gRNA target sites significantly influences editing efficiency, particularly when using dual gRNA approaches for enhanced knockout efficiency. Research demonstrates that positioning two gRNAs at close genomic proximity (40-300 bp) creates a synergistic effect that dramatically improves editing efficiency compared to single gRNA approaches [16].
Table 2: Editing Efficiency Based on Inter-guide Distance
| Genomic Distance Between gRNAs | Editing Efficiency | Synergistic Benefit* | Key Observations |
|---|---|---|---|
| < 40 bp | Variable | Low | Steric hindrance may reduce Cas9 binding |
| 40-300 bp | High (near 100% allelic editing) | Significant | Optimal range for synergistic effect |
| > 300 bp | Moderate | Reduced | Approaches additive effect of independent gRNAs |
*Synergistic benefit calculated as: %GE(T) - %GE(A), where %GE(T) is the actual tandem editing efficiency and %GE(A) is the calculated additive effect [16].
This synergistic effect is quantified as the difference between the observed tandem editing efficiency and the calculated additive effect of the two gRNAs working independently [16]. The calculated additive effect is derived as: %GE(A) = %GE(D) + [100 - %GE(D)] × [%GE(H)/100], where %GE(D) represents the gene editing percentage of the driver gRNA and %GE(H) represents the helper gRNA efficiency [16].
Positional Effects in gRNA Arrays
The position of a gRNA within a tandem array significantly affects its abundance and activity. gRNAs located closer to the 5' end of the transcript often exhibit higher expression levels due to transcriptional priority, while those at the 3' end may suffer from reduced abundance [1]. This positional bias can be mitigated through strategic array design and the use of specific processing systems.
Table 3: Positional Effects Across Different Array Types
| Array Architecture | 5' End gRNA Efficiency | Middle gRNA Efficiency | 3' End gRNA Efficiency | Recommended Mitigation Strategies |
|---|---|---|---|---|
| Cas12a-based | High | Moderate | Moderate | Balance array order across experiments |
| Ribozyme-flanked | High | Moderate | Low to moderate | Use bidirectional promoters |
| tRNA-gRNA | Moderate | Moderate | Moderate | Most consistent across positions |
| Csy4-based | High | High | Moderate | Requires Csy4 co-expression |
The tRNA-gRNA array architecture generally demonstrates the most consistent performance across array positions, as the endogenous tRNA processing machinery efficiently recognizes and processes tRNA sequences regardless of their location within the transcript [1] [55]. This system has been successfully implemented in diverse organisms including yeast, plants, and mammalian cells, with demonstrations of up to 12 gRNAs processed from a single array [1].
Experimental Protocol for Tandem gRNA Array Implementation
Design and Cloning of Tandem gRNA Arrays
This protocol outlines the steps for designing and constructing a tandem gRNA array using the tRNA-gRNA architecture, which provides high processing efficiency and relatively uniform gRNA expression [1] [55].
Materials Required:
- Target sequence information for genes of interest
- Benchling or similar gRNA design tool (www.benchling.com)
- DNA oligonucleotides for gRNA sequences
- Cloning vector with tRNA array backbone
- Restriction enzymes and ligase or Gibson Assembly reagents
- Competent E. coli cells for transformation
Step-by-Step Procedure:
gRNA Design and Selection (Timing: 15-30 minutes)
- Import the fully annotated gene of interest into a gRNA design tool such as Benchling [56].
- For each target gene, generate 20 bp gRNA sequences using the tool's algorithm, selecting those with high on-target and low off-target scores [56].
- Verify that selected gRNAs contain a synonymous change that disrupts the Protospacer Adjacent Motif (PAM) site to prevent Cas9 recutting after editing [56].
- Check SpliceAI prediction (https://spliceailookup.broadinstitute.org/) to ensure PAM modifications do not impact splicing, using a threshold score of >0.2 as potentially affecting splicing [56].
Array Architecture Assembly (Timing: 2-3 days)
- Design the tandem array with the structure: Promoter-tRNA-gRNA1-tRNA-gRNA2-tRNA-gRNA3-terminator.
- For tRNA-gRNA arrays, flank each gRNA with 77-nt long pre-tRNA genes that will be processed by RNases P and Z [1].
- Assemble the array using Golden Gate or Gibson Assembly methods, which are particularly suited for handling repetitive sequences [1].
- Transform the assembled construct into competent E. coli cells and plate on selective media.
- Verify correct assembly by colony PCR and Sanger sequencing of the entire array.
Figure 2: Tandem gRNA Array Construction Workflow. Key steps in designing, assembling, and verifying tandem gRNA arrays for multiplexed genome editing.
Cell Transfection and Editing Validation
Materials Required:
- Appropriate cell line (e.g., HEK293T, U2OS, or custom cell models)
- Cas9 source (mRNA, protein, or stable expression)
- Transfection reagent (e.g., DharmaFECT Duo for synthetic crRNAs)
- Tissue culture media and supplements
- PCR reagents for genomic amplification
- Sequencing primers for edited loci
Procedure:
Cell Preparation and Transfection (Timing: 3-4 days)
Editing Efficiency Analysis (Timing: 2-3 days)
- Harvest cells 72 hours post-transfection for initial efficiency assessment [57].
- Extract genomic DNA using standard protocols.
- Amplify target regions by PCR with primers flanking the edited sites.
- Analyze editing efficiency using TIDE (Tracking of Indels by Decomposition) or ICE (Inference of CRISPR Edits) software [16].
- For population-level editing, confirm protein knockdown via Western blot or functional assays [57].
Clonal Isolation and Validation (Timing: 2-3 weeks)
- For single-cell cloning, isolate cells 72 hours post-transfection using dilution method or flow sorting [57].
- Plate cells at low density in 96-well plates and culture for 10-14 days until visible colonies form [16] [57].
- Isplicate individual clones and expand for genomic analysis.
- Sequence each target locus in clonal lines using Sanger sequencing.
- Analyze sequencing data using deconvolution tools such as CRISP-ID to determine editing patterns [57].
Research Reagent Solutions
Table 4: Essential Reagents for Tandem gRNA Array Experiments
| Reagent Category | Specific Examples | Function/Application | Commercial Sources |
|---|---|---|---|
| gRNA Design Tools | Benchling, CRISPOR, CHOPCHOP | gRNA sequence design with on-target/off-target scoring | Web-based platforms |
| Cas9 Enzymes | Wild-type SpCas9, HiFi Cas9 variants, Cas12a | Target DNA cleavage with varying specificities | Addgene, commercial suppliers |
| Array Cloning Systems | tRNA-gRNA backbone, Csy4 processing system, Ribozyme vectors | Tandem gRNA array assembly | Addgene, academic donations |
| Synthetic gRNAs | Edit-R crRNAs, tracrRNA | Synthetic RNA for rapid testing without cloning | Horizon Discovery |
| Delivery Tools | DharmaFECT Duo, Electroporation systems | Introduction of CRISPR components into cells | Commercial suppliers |
| Analysis Software | TIDE, ICE, CRISP-ID | Quantification of editing efficiency and pattern analysis | Web-based tools |
The Edit-R system from Horizon Discovery provides predesigned crRNAs that are algorithm-optimized for genome-wide coverage of human, mouse, and rat genes, with modifications for nuclease resistance that improve DNA-free editing efficiency [57]. These synthetic RNAs can be complexed with tracrRNA and delivered alongside Cas9 mRNA or protein for rapid testing of gRNA efficiency before committing to array construction.
For array assembly, the tRNA-gRNA system offers particular advantages for marker-free multiple gene editing, as demonstrated in Trichoderma reesei for enhanced cellobiose production [55]. This approach can be adapted for various host organisms by utilizing species-specific tRNA sequences.
Optimizing gRNA stoichiometry and positional effects in tandem arrays is essential for successful multiplexed genome editing. Key considerations include selecting the appropriate array architecture for the specific application, maintaining optimal spacing between gRNA target sites (40-300 bp for synergistic effects), and implementing strategies to mitigate positional biases within arrays [16] [1]. The tRNA-gRNA array system generally provides the most consistent performance across array positions, while Cas12a-based arrays offer simplified design and processing [1] [55].
By following the detailed protocols and leveraging the reagent solutions outlined in this Application Note, researchers can significantly improve the efficiency and reliability of their multiplexed editing experiments. These optimized approaches facilitate more complex genetic engineering applications, including combinatorial gene knockout studies, metabolic pathway engineering, and the generation of sophisticated disease models for drug development. As CRISPR technology continues to evolve, further refinements in array design and processing will undoubtedly enhance our ability to precisely manipulate multiple genetic elements simultaneously.
{Application Notes and Protocols}
Reducing Cellular Toxicity and Mosaicism Through Timing and Inducible Systems
The construction of tandem single-guide RNA (sgRNA) arrays is a powerful strategy for multiplexed CRISPR-Cas9 genome editing, enabling the simultaneous perturbation of multiple genetic targets. However, two significant challenges often compromise experimental outcomes and therapeutic potential: cellular toxicity and mosaicism. Toxicity can arise from the simultaneous generation of multiple double-strand breaks (DSBs), which can overwhelm DNA repair mechanisms and trigger apoptosis or p53-mediated stress responses [58]. Mosaicism—the presence of a mixture of edited and unedited cells in a population—occurs when editing is incomplete or asynchronous, a particular concern in embryonic editing and clinical applications where uniform genotype is critical [59]. This Application Note details protocols leveraging inducible systems and temporal control to mitigate these issues, providing a robust framework for advanced multiplexed editing research.
Quantitative Analysis of Toxicity and Mosaicism Challenges
The tables below summarize the core problems and the strategic solutions explored in this document.
Table 1: Key Challenges in Multiplexed CRISPR Editing
| Challenge | Primary Cause | Impact on Experimental/Clinical Outcomes |
|---|---|---|
| Cellular Toxicity | Simultaneous generation of multiple DSBs, leading to sustained DNA damage response and p53 activation [58]. | Reduced cell viability, clonal outgrowth of p53-deficient cells, skewed experimental results, and compromised therapeutic efficacy. |
| Mosaicism | Incomplete or asynchronous editing events, particularly in post-mitotic cells or during early embryonic development [59]. | Heterogeneous phenotype within a cell population, unpredictable developmental outcomes, and potential long-term tissue instability [59]. |
| Genomic Instability | Error-prone repair of multiple DSBs via Non-Homologous End Joining (NHEJ), leading to large deletions and complex rearrangements [59]. | Scars and structural variants (SVs) in the genome; studies report ~5-6 SVs in edited trisomic clones [59]. |
Table 2: Strategic Solutions and Their Mechanisms of Action
| Solution Strategy | Mechanism | Key Advantage |
|---|---|---|
| Inducible Cas9 Systems | Decouples the delivery of CRISPR components from the timing of active nuclease function, allowing cells to recover from transfection/transduction stress before editing is triggered. | Enables temporal control; editing can be initiated at an optimal cell density or developmental stage. |
| Tandem tRNA-sgRNA Arrays | Utilizes endogenous tRNA processing machinery to cleave a polycistronic transcript into multiple functional sgRNAs, minimizing the delivery of repetitive sequences that can trigger recombination [60]. | Increases co-expression efficiency of multiple sgRNAs from a single transcript while reducing genomic instability [60]. |
| Non-Repetitive sgRNA Mutants | Employs high-throughput screening to identify functional sgRNA sequences with minimal homology, preventing homologous recombination between identical sequences in tandem arrays [61]. | Enables multiplex editing systems that can delete up to four genes with ~30% efficiency without genotype loss [61]. |
| Pharmacologic Inhibition of NHEJ | Uses small molecules to transiently inhibit the dominant NHEJ pathway, thereby favoring the more precise Homology-Directed Repair (HDR) when a template is present [62]. | Can improve the ratio of precise edits to error-prone indels, reducing genomic scarring. |
Protocol: Implementing an Inducible, Multi-sgRNA System for Reduced Toxicity
This protocol outlines the steps for constructing and utilizing a tRNA-sgRNA array expressed from an inducible promoter to achieve efficient, low-toxicity, multiplexed editing.
I. Molecular Cloning of the Inducible tRNA-sgRNA Array
Research Reagent Solutions:
- Vector Backbone: A plasmid containing an inducible promoter (e.g., tetracycline- or cumate-inducible), a puromycin resistance gene, and the hCas9 gene expressed from a separate, constitutive promoter.
- tRNA-sgRNA Cassette: A synthesized DNA fragment containing the 5S rRNA promoter (for strong Pol III-driven expression), followed by the tandemly arrayed tRNA-sgRNA architecture, and a T6 terminator [60].
- tRNA-sgRNA Architecture: For a 3-gRNA array, the structure is:
[5S rRNA Promoter]-(sgRNA1 scaffold)-(tRNAGly)-(sgRNA2 scaffold)-(tRNAGly)-(sgRNA3 scaffold)-[T6 Terminator]. The endogenous RNase P and RNase Z enzymes will recognize and cleave the tRNA, releasing the individual sgRNAs [60].
Procedure:
- Design sgRNAs and Array: Using established design rules [63] [64], select three 20-nucleotide sgRNA spacer sequences with high on-target and low off-target scores.
- Acquire Cassette: Synthesize the full tRNA-sgRNA expression cassette as a gBlock gene fragment with appropriate overhangs for your inducible vector.
- Golden Gate Assembly: Digest the vector backbone and the gBlock fragment with BsaI-HFv2. Perform a ligation reaction to insert the cassette downstream of the inducible promoter in the vector.
- Transform and Verify: Transform the assembled plasmid into a high-efficiency E. coli strain (e.g., JM109). Isolve plasmids from resulting colonies and verify the correct sequence of the entire array via Sanger sequencing.
II. Cell Culture, Transfection, and Inducible Editing
Procedure:
- Cell Seeding: Seed HEK293T or HCT116 cells in a 12-well plate to reach 70-80% confluence at the time of transfection.
- Transfection: Transfect 1 µg of the verified plasmid using a lipofection reagent (e.g., Lipofectamine LTX) according to the manufacturer's instructions.
- Post-Transfection Recovery: 6 hours post-transfection, replace the transfection medium with fresh, complete growth medium. This recovery period is critical for reducing toxicity associated with the transfection process itself.
- Selection: 24 hours post-transfection, add puromycin (1-2 µg/mL, concentration to be determined by a kill curve) to the medium to select for successfully transfected cells. Maintain selection for 48 hours.
- Induction of Editing: After the 48-hour selection, add the inducer molecule (e.g., 1 µg/mL doxycycline) to the culture medium. This activates the promoter and initiates the expression of the tRNA-sgRNA array, leading to the production of Cas9-sgRNA complexes and the onset of genome editing.
- Harvest and Analysis: Harvest cells 72-96 hours after induction. Extract genomic DNA and assess editing efficiency at all target loci using the qEva-CRISPR method [65] or next-generation sequencing.
The Scientist's Toolkit: Essential Reagents
Table 3: Key Research Reagents for Implementing Inducible, Low-Toxicity Multiplex Editing
| Reagent / Solution | Function / Explanation | Example (if specified in search results) |
|---|---|---|
| tRNA-sgRNA Array Cassette | A DNA construct where multiple sgRNA sequences are separated by tRNA (e.g., tRNAGly). The cell's endogenous enzymes process this into individual, functional sgRNAs, improving co-expression and reducing toxicity from repetitive sequences [60]. | Glycine tRNA (tRNAGly) from T. reesei [60]. |
| Inducible Expression System | A genetic system where the expression of the sgRNA array (or Cas9) is controlled by an external stimulus (e.g., a small molecule). This allows temporal control over the onset of editing. | Tetracycline- or cumate-inducible promoters. |
| qEva-CRISPR Assay | A quantitative, ligation-based method for evaluating on-target and off-target editing efficiency. It is highly sensitive, can detect all mutation types (including large deletions), and is suitable for multiplex analysis [65]. | Used for analyzing edits in TP53, VEGFA, and HTT genes [65]. |
| Non-Repetitive sgRNA Mutants | sgRNA sequences that are functionally identical but have different nucleotide compositions, preventing homologous recombination when expressed in tandem, thereby maintaining genomic stability [61]. | Identified via high-throughput screening platform [61]. |
| Lipid Nanoparticles (LNPs) | A non-viral delivery vehicle for in vivo delivery of CRISPR components (e.g., Cas9 mRNA and sgRNA). They are less immunogenic than viral vectors and allow for potential re-dosing [66]. | Used in clinical trials for hATTR and the first personalized in vivo CRISPR therapy [66]. |
Workflow and Pathway Visualization
The following diagram illustrates the logical workflow and key biological pathways involved in the protocol for reducing toxicity and mosaicism.
Titled: Workflow for Toxicity Reduction
The synergistic application of inducible systems and advanced sgRNA array architectures provides a powerful and necessary methodology for overcoming the critical bottlenecks of cellular toxicity and mosaicism in multiplexed CRISPR editing. By granting researchers precise temporal control and minimizing genotoxic stress, these protocols enable more reliable functional genomics screens, the creation of more accurate disease models, and pave the way for safer therapeutic genome engineering. The tools and strategies outlined here are essential for any research program aiming to harness the full potential of tandem CRISPR sgRNA arrays.
Improving Knockout Homogeneity with Synergistic Tandem gRNA Strategies
In CRISPR-Cas9-based gene knockout (KO) experiments, a common challenge is the phenotypic heterogeneity that arises from incomplete and variable editing outcomes. This heterogeneity persists even when using isolated KO subclones, complicating the interpretation of functional data [16]. Achieving a uniform, biallelic knockout in a pool of cells is critically dependent on the efficiency of the guide RNA (gRNA) but is often hampered by the cell's error-prone non-homologous end joining (NHEJ) repair pathway, which generates a spectrum of insertions and deletions (indels) [67] [68].
A powerful strategy to overcome this limitation is the use of synergistic tandem gRNAs. This approach involves the simultaneous use of two gRNAs targeting the same genomic locus in close proximity (40–300 bp apart). The co-expression of these "driver" and "helper" gRNAs synergizes to edit nearly 100% of targeted alleles, resulting in a predictable, large deletion between the cut sites. This significantly reduces allelic heterogeneity and residual protein expression, enabling the generation of high-confidence molecular omics data from edited cell pools without the need for lengthy subcloning procedures [16].
This Application Note details the implementation of this strategy, providing validated protocols and resources to enhance the homogeneity and efficiency of your gene knockout projects.
Key Principles and Quantitative Evidence
The tandem gRNA strategy leverages the simultaneous introduction of two double-strand breaks (DSBs) within a single gene. The repair of these breaks often results in the deletion of the intervening genomic segment. This large deletion is more likely to disrupt the open reading frame and is less susceptible to rescue mechanisms such as exon skipping or the use of alternative translation start sites, which can occur with single, small indels [16] [4].
Quantitative data from the seminal study demonstrates the superior performance of this method compared to single gRNAs or additive combinations [16].
Table 1: Quantitative Benefits of Synergistic Tandem gRNA Strategy
| Metric | Single gRNA (Driver) | Calculated Additive Effect | Synergistic Tandem gRNAs | Key Improvement |
|---|---|---|---|---|
| Alleles with Indels | Up to ~80% | Calculated as: %GE(D) + [100 - %GE(D)] × [%GE(H)/100] | Close to 100% [16] | Near-complete allele coverage |
| Residual Protein | Often detectable by MS | N/A | Low or undetectable by MS3 mass spectrometry [16] | Confident functional knockout |
| Allelic Heterogeneity | High (diverse indels) [16] | N/A | Low (predictable large deletion) [16] | Uniform phenotypic output |
| Model System Compatibility | Limited by subcloning | N/A | Broad (cell lines, iPSCs, primary/non-dividing cells) [16] | Versatile application |
The synergistic benefit is calculated as the difference between the observed gene editing percentage with the tandem combination and the calculated additive effect [%GE(T) - %GE(A)], which can be substantial [16].
Experimental Protocol
The following protocol is adapted for a typical experiment in human cell lines (e.g., HepG2) using ribonucleoprotein (RNP) electroporation, but can be modified for other delivery methods.
Tandem gRNA Design and Assembly
gRNA Design:
- Using a design tool (e.g., Benchling), select two target sites within the same exon of your gene of interest.
- Genomic Proximity: The Cas9 cut sites should be 40–300 base pairs apart [16]. A shorter distance increases the likelihood of a single PCR product for analysis and a definitive knockout.
- Specificity: Design gRNAs with at least three mismatches to any other NGG PAM site in the genome to minimize off-target effects [16].
- Synthesis: gRNAs can be synthesized in vitro as single-guide RNAs (sgRNAs) from DNA oligonucleotides using a transcription kit or purchased as synthetic sgRNAs or crRNA:tracrRNA duplexes from commercial suppliers [16].
Multiplex Expression (Optional): For stable expression, the two gRNAs can be cloned into a single plasmid. Several systems facilitate this:
- Dual-Promoter Vectors: Use a plasmid with two different RNA polymerase III promoters (e.g., human U6 and mouse U6) to drive the expression of each gRNA [4] [14].
- Golden Gate Assembly: This method uses Type IIS restriction enzymes (e.g., BsmBI, BsaI) to clone multiple gRNA expression cassettes into a destination vector in a defined order. Kits are available to express up to 7 gRNAs from a single plasmid [4] [14] [69].
- Polycistronic Transcripts: gRNAs can be expressed from a single promoter and flanked by self-cleaving RNA elements (e.g., Csy4 ribonuclease sites or tRNA) that process the long transcript into individual gRNAs. This is advantageous for viral delivery with limited cargo capacity [14].
Cell Electroporation and Editing
RNP Complex Formation: For highest efficiency and reduced off-target effects, use the RNP complex.
- Complex recombinant Cas9 protein with the two sgRNAs at a molar ratio of 1:2:2 (Cas9:gRNA1:gRNA2) in a suitable buffer. Incubate at room temperature for 10-20 minutes to form the RNP.
Cell Preparation and Electroporation:
- Culture and passage HepG2 cells using standard methods. For primary cells like human CD4+ T cells, activate them 3 days prior to editing with anti-CD3/anti-CD28 and interleukin-2 [16].
- Detach adherent cells and resuspend them in the appropriate electroporation buffer (e.g., SF buffer for HepG2, P3 for primary T cells).
- Mix the cell suspension with the pre-formed RNP complex and electroporate using a 4D-Nucleofector system (or equivalent). Use manufacturer-recommended programs (e.g., EH-100 for HepG2, EH-115 for T cells) [16].
- Include controls: a "mock" electroporation (no RNP) and single gRNA electroporations.
Post-Electroporation Recovery:
- Immediately transfer the electroporated cells to pre-warmed culture medium.
- For sensitive cells, consider using conditioned medium or recovery supplements [16].
- Analyze editing efficiency 2-4 days post-electroporation.
Validation and Analysis
Genomic DNA Analysis:
- PCR Amplification: Design PCR primers that flank the two target sites. The amplicon should be small enough that the deleted fragment results in a visibly smaller band on a gel.
- Editing Efficiency: Purify the PCR product and analyze by Sanger sequencing. Use web tools like TIDE or ICE (Synthego) to quantify the overall indel percentage and deconvolute the mixture of sequences [16].
- The successful tandem knockout will show a high proportion of a single, clean sequence trace corresponding to the large deletion.
Functional Validation (Protein Detection):
- Mass Spectrometry: The gold-standard for detecting residual protein. MS3 mass spectrometry can reliably quantify the absence of the target protein, which often persists in single-gRNA KOs despite the presence of indels [16].
- Immunoblotting/Flow Cytometry: If a high-quality antibody is available, these methods can provide a semi-quantitative assessment of protein depletion. However, they may miss truncated protein variants.
The Scientist's Toolkit
Table 2: Essential Research Reagents for Tandem gRNA Knockouts
| Reagent / Solution | Function / Explanation | Example Products / Kits |
|---|---|---|
| gRNA Design Tool | In silico design of gRNAs with specificity checks and off-target scoring. | Benchling, IDT Alt-R Custom Cas9 crRNA |
| In vitro Transcription Kit | Synthesis of high-quality sgRNAs from DNA templates. | TranscriptAid Kit (Thermo) |
| Synthetic gRNAs | Chemically modified gRNAs for enhanced stability and editing efficiency. | IDT Alt-R CRISPR-Cas9 sgRNA |
| Recombinant Cas9 Protein | High-purity Cas9 for RNP complex formation. | IDT Alt-R S.p. Cas9 Nuclease V3 |
| Electroporation System | Hardware for efficient RNP delivery into hard-to-transfect cells. | Lonza 4D-Nucleofector System |
| Multiplex gRNA Cloning Kit | Modular systems for assembling multiple gRNAs into a single vector. | Yamamoto Lab Multiplex CRISPR/Cas9 Assembly Kit, Gersbach Lab plasmids [14] |
| Editing Analysis Software | Web-based tools to quantify indel efficiency from Sanger sequencing data. | TIDE, ICE (Synthego) |
Workflow and Strategic Application
The following diagram illustrates the critical steps and decision points in the synergistic tandem gRNA strategy.
Diagram: Tandem gRNA knockout workflow. MS: Mass Spectrometry.
The synergistic tandem gRNA strategy represents a significant advancement in CRISPR-Cas9 gene editing by directly addressing the critical issue of phenotypic heterogeneity. By implementing the detailed protocols and utilizing the recommended toolkit outlined in this Application Note, researchers can achieve more predictable, complete, and homogeneous gene knockouts. This method is particularly valuable for functional genomics, proteomic studies, and research involving difficult-to-culture primary cells, where subcloning is impractical or impossible, thereby accelerating the generation of robust and interpretable biological data.
Performance Assessment: Benchmarking Array Systems and Validation Frameworks
{#abstract} Abstract
Combinatorial CRISPR screening represents a transformative approach for probing genetic interactions and dependencies, particularly for redundant gene pairs that are missed in single-gene knockout studies. This application note provides a comparative performance analysis of three major combinatorial CRISPR systems—dual spCas9 with alternative tracrRNAs, orthogonal spCas9-saCas9, and enCas12a—based on a systematic empirical benchmark. We summarize key quantitative data in structured tables and provide detailed protocols for implementing these systems, specifically framed within the context of constructing tandem sgRNA arrays for multiplexed editing research. The findings indicate that a combination of specific alternative tracrRNAs for spCas9 (VCR1-WCR3) consistently outperforms other systems in single-gene knockout efficacy, positional balance of sgRNAs, and reduced recombination rates, offering researchers a robust framework for selecting and optimizing combinatorial genome-editing tools. {#abstract}
The next frontier in functional genomics involves identifying combinatorial genetic interactions, such as context-specific dependencies on redundant paralogous genes, which are frequently overlooked in conventional single-gene knockout studies [70]. Combinatorial CRISPR technology, which enables the simultaneous knockout of two or more genes, has emerged as a powerful method to address this challenge. Several platforms have been developed for multiplexed editing, including dual spCas9 systems, orthogonal spCas9-saCas9 systems, and enCas12a (Cpf1) systems [70] [1]. However, a direct, comparative evaluation of these systems has been historically difficult due to the use of different gene sets, sgRNA designs, and cell lines across independent studies [70].
This application note synthesizes findings from a controlled, comparative benchmark of ten distinct combinatorial CRISPR libraries targeting paralog pairs. The analysis is presented with a focus on their application in tandem sgRNA array construction, providing researchers with clear performance metrics and practical protocols to guide experimental design in multiplexed editing research.
Results and Performance Data {#1-results-and-performance-data}
The following table summarizes the core performance characteristics of the three major combinatorial CRISPR systems, as benchmarked in a pooled screen targeting 616 genes and 454 paralog pairs [70].
| CRISPR System | PAM Sequence | Single-gene KO Efficiency (ROC-AUC) | Positional Balance (Correlation, r) | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Dual spCas9 (VCR1-WCR3) | NGG | ~0.95 (Highest) | 0.91 (Highest) | Superior single-gene knockout; Excellent sgRNA balance; Low recombination rate [70]. | Requires careful tracrRNA pairing to avoid recombination [70]. |
| Orthogonal spCas9-saCas9 | spCas9: NGGsaCas9: NNGRRT | ~0.85 | Moderate | Orthogonal systems minimize recombination; Broad PAM recognition [70] [71]. | Lower single-gene knockout efficacy compared to best dual-spCas9 systems [70]. |
| enCas12a (Cpf1) | TTTV | ~0.80 | Moderate | Native processing of crRNA arrays; Simplified multiplexing [1] [72]. | Lower single-gene knockout efficacy; Performance is highly PAM- and sgRNA design-dependent [70]. |
Abbreviations: KO, Knockout; ROC-AUC, Receiver Operating Characteristic - Area Under the Curve; PAM, Protospacer Adjacent Motif.
In-depth Performance Analysis {#1-2-in-depth-performance-analysis}
Single-gene Knockout Efficacy: The benchmark evaluated performance using ROC-AUC and null-normalized mean difference (NNMD) for a predefined set of core essential and nonessential genes. Libraries utilizing specific alternative
spCas9tracrRNA combinations (e.g.,VCR1-WCR3,WCR3-VCR1) consistently outperformed both the orthogonalspCas9-saCas9system and theenCas12asystem across multiple cell lines (IPC298, MELJUSO, PK1). These top-performing tracrRNA pairs demonstrated superior separation of essential and nonessential genes and showed stronger depletion of pan-essential genes than even the genome-wide Avana library [70].sgRNA Positional Balance and Promoter Effects: A critical challenge in combinatorial screens is ensuring balanced knockout efficiency between the two sgRNAs. The
VCR1-WCR3library exhibited the highest correlation (r = 0.91) between the left and right sgRNAs and the highest percentage (82.7%) of pan-essential genes being effectively targeted by both sgRNAs. Flipping the tracrRNA positions (VCR1-WCR3vs.WCR3-VCR1) revealed minimal positional bias from the tracrRNAs themselves. However, a consistent and stronger depletion was observed for sgRNAs driven by the U6 promoter compared to the H1 promoter, irrespective of the linked tracrRNA [70].Recombination Rate and Library Design: Homology between tracrRNA sequences was identified as a major factor leading to recombination and reduced library quality. The
WCR2-WCR3library, which uses two more homologous tracrRNAs, showed a detectable increase in recombination compared to the less homologousVCR1-WCR3pair. This higher recombination rate directly contributed to a decrease in observed knockout performance. When the analysis accounted for recombined sequences, the performance of theWCR2-WCR3library significantly improved, highlighting the importance of selecting heterologous tracrRNA pairs and the potential for sequencing artifacts to impact results [70].Optimizing enCas12a and spCas9 sgRNA Design: The performance of the
enCas12asystem was notably improved when using sgRNAs with the canonical TTTV PAM, compared to non-canonical PAMs [70]. Furthermore, forspCas9, sgRNAs that were "pre-validated" from the Avana library and exhibited high agreement across hundreds of cell lines showed significantly superior performance compared to sgRNAs designed using Rule Set2 alone. This underscores the critical importance of leveraging empirically validated sgRNAs for library construction [70].
{#fig1} Workflow for a combinatorial CRISPR screen.
The Scientist's Toolkit {#2-the-scientists-toolkit}
The following table lists essential reagents and their functions for establishing combinatorial CRISPR screens, as featured in the benchmark studies and related resources.
| Research Reagent / Tool | Function / Application | Notes |
|---|---|---|
| Alternative TracrRNAs (VCR1, WCR3) | Engineered tracrRNA sequences for dual-spCas9 systems. | Minimize recombination and improve sgRNA balance [70]. |
| enCas12a (enPAM+GB) | Enhanced Cas12a nuclease with expanded PAM recognition. | Used for combinatorial screens; prefers canonical TTTV PAM [70]. |
| SpCas9 Nuclease (NEB #M0386) | Wild-type Streptococcus pyogenes Cas9 protein. | Used for in vitro validation and RNP delivery [73]. |
| T7 Endonuclease I / Authenticase | Enzymes for detecting indel mutations post-editing. | Cleave heteroduplex DNA at mismatch sites; measure editing efficiency [73]. |
| NEBNext Ultra II DNA Library Prep Kits | Kits for preparing sequencing libraries from edited genomes. | For NGS-based analysis of on- and off-target editing events [73]. |
| Golden Gate Assembly (BsaI-HF v2) | Modular cloning system for constructing sgRNA arrays. | Enables seamless assembly of repetitive sgRNA sequences into vectors [72]. |
| PVX-based Viral Vector | Plant virus vector for delivering sgRNA arrays in plants. | Enables highly efficient, multiplexed editing in solanaceous species (VIGE) [2]. |
Experimental Protocols {#3-experimental-protocols}
Protocol: Library Construction for Dual-spCas9 Screens {#3-1-protocol-library-construction-for-dual-spcas9-screens}
This protocol outlines the steps for constructing a pooled combinatorial CRISPR library using the high-performing VCR1-WCR3 tracrRNA system [70].
sgRNA Selection and Array Design:
- Select six pre-validated sgRNAs per target gene from established libraries (e.g., Avana library) [70].
- For each dual-gene target, design a library construct expressing two sgRNAs. Place one sgRNA coupled with the
VCR1tracrRNA under a U6 promoter and the second sgRNA coupled with theWCR3tracrRNA under an H1 promoter. - Ensure the two tracrRNA sequences have low homology to minimize recombination during library amplification [70].
Oligonucleotide Library Synthesis:
- Synthesize the oligonucleotide library encoding the sgRNA-tracrRNA cassettes. Use pooled oligo synthesis to generate the entire library in a single tube.
Golden Gate Assembly:
- Clone the oligonucleotide pool into the lentiviral backbone vector using Golden Gate Assembly with BsaI-HF v2 [72].
- The destination vector should contain the necessary elements for replication in E. coli and subsequent viral production.
Library Validation and Quality Control:
- Transform the assembled library into a high-efficiency E. coli strain and perform a large-scale plasmid preparation.
- Validate the library complexity and distribution by next-generation sequencing (NGS) of the plasmid DNA.
- Check for low recombination rates by gel electrophoresis or by analyzing the NGS reads spanning the tracrRNA regions [70].
Protocol: Functional Screen in a Cas9-Expressing Cell Line {#3-2-protocol-functional-screen-in-a-cas9-expressing-cell-line}
Lentivirus Production and Titration:
- Produce lentivirus by co-transfecting the sgRNA library plasmid with packaging plasmids (e.g., psPAX2, pMD2.G) into HEK293T cells.
- Harvest the viral supernatant, concentrate if necessary, and determine the viral titer by transducing a target cell line and measuring the percentage of puromycin-resistant cells (or other relevant selection markers).
Cell Line and Transduction:
- Use a cell line that constitutively expresses SpCas9 (e.g., IPC298 melanoma cells) [70].
- Transduce the cells at a low Multiplicity of Infection (MOI ~0.3) to ensure most cells receive a single viral integrant. Include a non-targeting control sgRNA library.
- Culture the transduced cells under appropriate selection for at least 5-7 days to ensure effective knockout before initiating the screen.
Pooled Screening and Sample Collection:
- Passage the cells for a minimum of 14 population doublings to allow for phenotypic depletion.
- Collect a minimum of 50 million cells per replicate at the endpoint to maintain library representation. Also, collect genomic DNA from the initial transduced cell population (T0) as a reference.
Next-Generation Sequencing and Data Analysis:
- Amplify the integrated sgRNA sequences from the genomic DNA via PCR using primers compatible with your NGS platform [73].
- Sequence the amplicons to a depth that ensures >500x coverage per sgRNA.
- Map the sequencing reads to the reference library and calculate log2 fold-changes (LFC) for each sgRNA pair between T0 and the endpoint using established analysis pipelines (e.g., MAGeCK).
- Identify hit genes or gene pairs based on significant depletion or enrichment.
{#fig2} Decision guide for selecting a CRISPR system.
This systematic comparison establishes that the combinatorial CRISPR landscape has a clear front-runner for pooled, dual-gene knockout screens in terms of efficacy and robustness: the dual spCas9 system utilizing specific alternative tracrRNA pairs like VCR1-WCR3. Its superior performance in single-gene knockout, excellent positional balance, and low recombination rate make it highly suitable for constructing complex tandem sgRNA arrays. The orthogonal spCas9-saCas9 system remains a valuable tool, particularly when its distinct PAM requirements are necessary, though it may require deeper screening to achieve the same level of confidence as the best dual-spCas9 systems. The enCas12a system, while offering the native advantage of processing crRNA arrays from a single transcript, currently requires more stringent sgRNA selection and optimization to match the knockout efficacy of the Cas9-based systems [70].
For researchers engaged in multiplexed editing, the key takeaways are:
- Library Design is Critical: The choice of tracrRNA pairs for dual-
spCas9systems directly impacts performance and recombination rates. Using highly heterologous pairs likeVCR1-WCR3is recommended. - Leverage Validated Reagents: Whenever possible, use pre-validated sgRNAs and promoters with known performance characteristics to maximize screen quality.
- Context Matters: The optimal system may still depend on the specific experimental context, such as the need for a particular PAM or delivery vehicle (e.g.,
saCas9for AAV delivery).
In conclusion, this application note provides a validated roadmap for implementing high-performance combinatorial CRISPR screens, emphasizing the critical parameters in system selection and library construction to ensure robust and interpretable results in functional genomics research.
Benchmarking Alternative TracrRNA Combinations for Balanced and Efficient Dual Knockouts
Combinatorial CRISPR knockout screens are transformative tools for probing genetic interactions and identifying synthetic lethal pairs for therapeutic targeting. A significant challenge in dual-knockout screens is achieving balanced and efficient cutting from both single-guide RNAs (sgRNAs) within a single cell. Traditional approaches using identical or highly similar tracrRNA sequences are prone to homologous recombination, leading to vector instability and imbalanced knockout efficacy. This application note benchmarks alternative tracrRNA combinations, a strategy that utilizes distinct tracrRNA variants to mitigate recombination and enhance performance. Framed within the broader objective of optimizing tandem sgRNA arrays for multiplexed editing, we present a validated protocol and quantitative analysis to identify the most robust system for dual-genome editing.
Results and Benchmarking Data
Systematic evaluation of ten distinct combinatorial CRISPR libraries revealed that the choice of tracrRNA combination critically impacts both single-guide knockout efficiency and the balance of dual-knockout efficacy.
Table 1: Performance Metrics of Alternative TracrRNA Combinations [70]
| TracrRNA Combination (5' - 3') | Single-Gene KO Efficiency (ROC-AUC) | Left/Right sgRNA Correlation (r) | Recombination Rate | Key Findings |
|---|---|---|---|---|
| VCR1 - WCR3 | High | 0.91 | Low | Most balanced and efficacious; 82.7% of pan-essential genes had LFC < -1 for both sgRNAs. |
| WCR3 - VCR1 | High | High | Low | Performance was not detectably altered from VCR1-WCR3, indicating minimal positional bias. |
| WCR2 - WCR3 | Intermediate | High | High | Decreased performance linked to higher recombination between homologous tracrRNAs. |
| SCR27 - SCR43 | Low | Low | Not Reported | Poor separation of essential and non-essential genes; tracrRNAs were likely inert. |
Key Insights:
- Optimal Performance: The VCR1-WCR3 pair emerged as the superior combination, demonstrating robust single-gene knockout and the highest correlation (r=0.91) between the log-fold changes (LFC) of the left and right sgRNAs, indicating a balanced dual knockout [70].
- Impact of Recombination: Libraries using more homologous tracrRNA sequences (e.g., WCR2-WCR3) suffered from higher recombination rates, which was directly linked to a decrease in functional knockout performance. Calculating LFC based on sequenced crRNAs alone underestimated the efficacy; analysis of full sgRNA sequences revealed significantly improved performance [70].
- Promoter Effect: A consistent, slight bias towards stronger depletion was observed for sgRNAs driven by the U6 promoter compared to the H1 promoter, regardless of the tracrRNA variant used [70].
Experimental Workflow for Benchmarking TracrRNAs
Experimental Protocol
This protocol details the steps for conducting a benchmark screen to evaluate tracrRNA combinations, as derived from the cited studies [70].
1. Library Design and Cloning
- sgRNA Selection: Select 4-6 sgRNAs per gene from a pre-validated library (e.g., the Avana library) for high confidence in on-target activity [70].
- TracrRNA Variants: Design dual-gRNA vectors where the two sgRNA expression cassettes utilize distinct tracrRNA backbones. The recommended combination is VCR1 for the first sgRNA and WCR3 for the second [70].
- Promoter Choice: Use consistent, strong promoters (e.g., U6) for both sgRNAs. Note that U6 may yield slightly stronger depletion than H1 [70].
- Cloning and QC: Assemble the library using Golden Gate or Gibson Assembly. Validate the final library by deep sequencing to confirm representation and assess recombination rates in the plasmid DNA by analyzing reads that span the tracrRNA regions [70].
2. Cell Line Preparation and Screening
- Cell Line: Use a cell line stably expressing Streptococcus pyogenes Cas9 (SpCas9). The screen cited was performed in IPC298 (meloma) cells [70].
- Viral Transduction: Transduce the pooled library into the cell line at a low Multiplicity of Infection (MOI ~0.3) to ensure most cells receive a single vector. Maintain a 500x library coverage throughout.
- Selection and Passaging: Apply puromycin selection (or other appropriate selection) 24 hours post-transduction for 3-5 days. After selection, split cells into replicate control and experimental arms. Passage cells continuously, maintaining coverage, for approximately 12-14 population doublings.
3. Sequencing and Data Analysis
- Genomic DNA (gDNA) Extraction: Harvest cells at the initial timepoint (post-selection) and the final timepoint. Extract gDNA from all samples.
- Amplification and Sequencing: PCR-amplify the sgRNA regions from the gDNA. For critical assessment, use sequencing primers that extend through the tracrRNA sequences to directly measure recombination [70].
- Bioinformatic Analysis:
- Read Alignment: Map sequences to the reference library.
- Fitness Calculation: Calculate log-fold changes (LFC) for each sgRNA pair between the initial and final timepoints using a tool like MAGeCK [74].
- Efficacy Assessment: Generate Receiver Operating Characteristic (ROC) curves and calculate Area Under the Curve (AUC) using a predefined set of core essential and non-essential genes.
- Balance Assessment: Calculate the correlation coefficient (r) between the LFCs of the left- and right-positioned sgRNAs targeting the same essential genes.
- Recombination Analysis: Compare the ratio of reads mapping to the full sgRNA (crRNA + tracrRNA) versus those mapping to the crRNA only in both plasmid and genomic DNA samples [70].
The Scientist's Toolkit
Table 2: Essential Research Reagents and Tools [74] [70]
| Item | Function/Description | Example/Source |
|---|---|---|
| Alternative TracrRNAs | Engineered variants of the native tracrRNA that reduce homologous recombination in dual-gRNA vectors. | VCR1, WCR3 [70] |
| Validated sgRNA Library | A curated set of sgRNAs with high on-target efficacy, used as the source for crRNA sequences. | Avana library, Brunello library [74] [70] |
| Dual-Guide Expression Vector | A lentiviral backbone containing two sgRNA expression cassettes with unique tracrRNAs. | Custom construct [70] |
| SpCas9-Expressing Cell Line | A mammalian cell line that constitutively expresses the Cas9 nuclease. | IPC298, HAP1, RPE1 [70] |
| Analysis Software | Computational tools for quantifying sgRNA abundance and calculating gene fitness effects. | MAGeCK, Chronos [74] |
Visualizing the Vector Architecture
Optimal Dual-sgRNA Vector Design
The advent of tandem CRISPR sgRNA arrays has revolutionized multiplexed genome editing, enabling the simultaneous perturbation of multiple genetic loci in a single experiment. This powerful approach facilitates comprehensive functional genomics studies, from investigating gene networks and synthetic lethality to engineering complex metabolic pathways [1] [45]. However, the increased complexity of multiplex editing necessitates equally sophisticated validation strategies to confirm intended genomic alterations, assess functional consequences, and rule out unintended off-target effects. This application note provides a detailed framework for researchers employing multiplex CRISPR systems, outlining integrated validation methodologies spanning genomics, proteomics, and functional assays to ensure robust and interpretable results in basic research and drug development applications.
Genomic Validation Through Next-Generation Sequencing
Sequencing-based validation provides the most direct evidence of successful genome editing by characterizing induced mutations at DNA level.
Next-Generation Sequencing Approaches
Table 1: NGS Methods for Validating Multiplex CRISPR Editing
| Method | Key Applications in Multiplex Editing | Key Advantages | Throughput | Quantitative Data |
|---|---|---|---|---|
| Amplicon Sequencing | Verification of indels at specific target sites; assessment of editing efficiency | High sensitivity; cost-effective for multiple targets; enables deep sequencing | Moderate to High | Editing efficiency per target; mutation spectrum |
| Whole Genome Sequencing (WGS) | Comprehensive off-target profiling; identification of large structural variations | Unbiased genome-wide coverage; detects unexpected rearrangements | Low | Off-target sites; chromosomal rearrangements |
| RNA-seq | Transcriptomic consequences of multiplexed perturbations; alternative splicing analysis | Captures genome-wide expression changes; identifies pathway alterations | Moderate to High | Differential expression; pathway enrichment |
For sgRNA arrays targeting multiple genomic loci, amplicon sequencing represents the most efficient and cost-effective validation method. This targeted approach involves PCR amplification of genomic regions flanking each target site, followed by NGS library preparation and deep sequencing to characterize mutation spectra [75]. The high sequencing depth (typically >1000× coverage) enables precise quantification of editing efficiencies across all targeted loci and detection of heterogeneous editing outcomes within cell populations.
Whole genome sequencing, while more resource-intensive, provides critical safety assessment by identifying potential off-target effects at unpredicted genomic locations. This is particularly important for therapeutic applications where comprehensive genomic integrity must be verified [76].
Protocol: Amplicon Sequencing for Multiplex Editing Validation
Day 1: Genomic DNA Isolation and Quality Control
- Isolate genomic DNA from edited cells (minimum 100 ng/µL concentration recommended) using silica-membrane column kits or magnetic bead-based methods.
- Verify DNA integrity and purity via agarose gel electrophoresis or fragment analyzer (DNA Integrity Number >7 recommended).
- Quantify DNA using fluorometric methods (e.g., Qubit) for accurate concentration measurement.
Day 2: Target Amplification and Library Preparation
- Design primer pairs flanking each sgRNA target site with Illumina adapter overhangs.
- Perform PCR amplification in 25 µL reactions: 2.5 µL 10× buffer, 0.5 µL dNTPs (10 mM), 0.5 µL each primer (10 µM), 0.125 µL polymerase, 20 ng gDNA, nuclease-free water to volume.
- Cycling conditions: 98°C for 30 s; 35 cycles of 98°C for 10 s, 60°C for 30 s, 72°C for 30 s; final extension 72°C for 5 min.
- Clean PCR products using magnetic beads (0.8× volume) and elute in 25 µL nuclease-free water.
Day 3: Indexing and Pooling
- Add dual indices and sequencing adapters via limited-cycle PCR (8 cycles).
- Quantify libraries using fluorometry and pool equimolar amounts (minimum 50 ng each library).
- Validate library quality using Bioanalyzer or TapeStation (expect single peak at appropriate size).
Day 4: Sequencing and Data Analysis
- Sequence on Illumina platform (MiSeq or MiniSeq recommended for rapid turnaround) with minimum 1000× coverage.
- Analyze data using CRISPR-specific tools (CRISPResso2, Cas-Analyzer, or custom pipelines):
- Align sequences to reference genome
- Quantify indel frequencies and spectra for each target
- Calculate statistical significance of editing efficiencies
Proteomic Validation of Editing Outcomes
While genomic validation confirms DNA-level alterations, proteomic analysis verifies functional consequences at protein level, essential for evaluating knockout efficiency, splice variants, and pathway perturbations.
Mass Spectrometry-Based Proteomics
Mass spectrometry provides unbiased, global protein quantification, making it ideal for validating multiplex editing where multiple gene products may be simultaneously affected. As demonstrated in CRISPR-Cas9 generated prion protein (PrPC) knockout models, quantitative proteomics can reveal unexpected functional relationships and validate editing efficacy through differential protein expression analysis [77].
Isobaric tagging approaches (TMT, iTRAQ) enable multiplexed comparative analyses, allowing simultaneous quantification of protein expression across multiple edited cell lines or conditions in a single MS run, significantly reducing technical variability [77].
Table 2: Proteomic Methods for Multiplex Editing Validation
| Method | Key Applications | Key Advantages | Limitations | Compatibility with Multiplexing |
|---|---|---|---|---|
| Western Blotting | Target protein knockout confirmation; partial fragment detection | High specificity; accessible | Low throughput; limited multiplexing | Moderate (sequential probing) |
| Multiplexed Immuno-fluorescence | Spatial protein expression in heterogeneous cell populations; co-localization studies | Single-cell resolution; spatial context | Antibody availability; semi-quantitative | High (multiple channels) |
| Mass Spectrometry-Based Proteomics | Global protein quantification; pathway analysis; post-translational modifications | Unbiased; highly multiplexable; quantitative | Instrument access; expertise required | Very High (isobaric tagging) |
Protocol: Quantitative Proteomics for Edited Cell Validation
Week 1: Sample Preparation and Protein Extraction
- Culture control and edited cells under identical conditions (minimum triplicate biological replicates).
- Harvest cells at 80-90% confluence, wash with cold PBS, and lyse using RIPA buffer with protease/phosphatase inhibitors.
- Quantify protein concentration using BCA assay, normalizing samples to equal concentration.
- Reduce proteins with 5 mM DTT (30 min, 56°C), alkylate with 15 mM iodoacetamide (30 min, dark), and digest with trypsin (1:50 ratio, 37°C, overnight).
Week 2: TMT Labeling and Fractionation
- Desalt peptides using C18 solid-phase extraction columns.
- Label peptides with TMT reagents (0.8 mg reagent per 100 µg peptide, 1 hour incubation).
- Quench reaction with 5% hydroxylamine (15 min).
- Combine labeled samples in equal ratios and desalt.
- Fractionate using high-pH reverse-phase chromatography (collect 12-24 fractions).
Week 3: LC-MS/MS Analysis and Data Processing
- Analyze fractions by LC-MS/MS using Orbitrap Fusion Lumos or similar high-resolution mass spectrometer.
- Gradient: 2-35% acetonitrile over 120 min, 300 nL/min flow rate.
- Data-dependent acquisition: MS1 at 120,000 resolution, MS2 at 50,000 resolution.
- Search data against appropriate database (UniProt) using Sequest HT or MaxQuant.
- Apply statistical analysis (t-tests, ANOVA) with multiple testing correction to identify significantly altered proteins.
- Perform pathway enrichment analysis using GO, KEGG, or Reactome databases.
Functional Validation Assays
Functional assays bridge the gap between genetic perturbation and phenotypic outcome, providing critical biological context for multiplex editing experiments.
High-Content Phenotypic Screening
High-content imaging enables multiparametric analysis of cellular phenotypes, making it particularly valuable for assessing complex outcomes of multiplex editing. The integration of fluorescent reporters with automated image analysis facilitates high-throughput functional validation, as demonstrated in multiplexed antiviral assays where multiple fluorescently-tagged viruses enabled simultaneous assessment of antiviral activity [78].
CRISPR-based genetic perturbations combined with single-cell RNA sequencing (Perturb-seq) represent a powerful functional validation approach, enabling high-resolution mapping of genotype-phenotype relationships across complex cell populations [79].
Protocol: High-Content Imaging for Functional Assessment
Day 1: Cell Seeding and Staining
- Seed edited and control cells in black-walled, clear-bottom 96-well or 384-well plates (2,000-5,000 cells/well for 384-well format).
- Culture for 24-48 hours to reach 60-70% confluence.
- Fix cells with 4% paraformaldehyde (15 min), permeabilize with 0.1% Triton X-100 (10 min), and block with 3% BSA (1 hour).
- Stain with primary antibodies targeting proteins of interest (diluted in blocking buffer, 2 hours room temperature or overnight at 4°C).
- Wash 3× with PBS, then incubate with fluorescent secondary antibodies and nuclear stain (Hoechst or DAPI, 1 hour).
- Wash 3× with PBS and add 100 µL PBS for imaging.
Day 2: Image Acquisition and Analysis
- Acquire images using high-content imaging system (ImageXpress Micro Confocal or similar) with 20× objective.
- Capture minimum 9 fields per well to ensure statistical power.
- For multiplexed assays, use appropriate filter sets to separate fluorescence signals (e.g., DAPI, FITC, TRITC, Cy5).
- Analyze images using CellProfiler or similar software:
- Identify nuclei using intensity thresholding
- Define cytoplasm using propagation or watershed algorithms
- Measure intensity, texture, and morphological features per cell
- Export single-cell data for statistical analysis
- Perform statistical analysis using R or Python:
- Compare edited vs. control populations using t-tests or Mann-Whitney tests
- Apply multiple testing correction for multiparametric data
- Use dimensionality reduction (t-SNE, UMAP) to visualize population differences
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Multiplex Editing Validation
| Category | Reagent/Solution | Function | Example Applications |
|---|---|---|---|
| NGS Library Prep | Amplicon PCR Master Mix | Amplifies target loci for sequencing | Targeted validation of editing efficiency |
| NGS Library Prep | Dual Indexing Kits | Enables sample multiplexing in sequencing | Pooling multiple samples/cell lines |
| Proteomics | TMT or iTRAQ Reagents | Multiplexed protein quantification | Comparative analysis of edited vs. control cells |
| Proteomics | Trypsin/Lys-C | Protein digestion for mass spectrometry | Sample preparation for bottom-up proteomics |
| Cell Staining | Multiplexed Antibody Panels | Simultaneous detection of multiple targets | High-content imaging and flow cytometry |
| Cell Staining | Cell Viability Dyes (PI, DAPI) | Distinguish live/dead cells | Cytotoxicity assessment post-editing |
| Cell Culture | Selection Antibiotics (Puromycin) | Enrich for successfully transfected cells | Selection following CRISPR delivery |
| Software | CRISPResso2, Cas-Analyzer | NGS data analysis for editing outcomes | Quantification of indel frequencies and spectra |
| Software | CellProfiler, ImageJ | Automated image analysis | High-content screening data extraction |
| Software | MaxQuant, Proteome Discoverer | Mass spectrometry data processing | Protein identification and quantification |
Comprehensive validation of multiplex CRISPR editing requires an integrated approach combining genomic, proteomic, and functional methodologies. Sequencing provides direct evidence of intended genetic alterations, proteomics confirms the functional consequences at protein level, and phenotypic assays bridge the gap to biological relevance. As multiplex editing technologies continue to evolve toward increasingly complex applications in functional genomics and therapeutic development, robust validation frameworks will remain essential for generating reliable, interpretable data. The protocols outlined herein provide a foundation for researchers to implement these critical validation strategies, ensuring the continued advancement of multiplex genome editing with appropriate scientific rigor.
Evaluating Positional Effects and Promoter Influence on sgRNA Efficacy in Arrays
The construction of tandem CRISPR sgRNA arrays has become a fundamental technique for conducting multiplexed genome editing experiments, enabling the simultaneous perturbation of multiple genetic targets. However, a significant challenge in implementing this approach is the frequent observation of variable editing efficiencies across different guide RNAs (gRNAs) within the same array. This variability often stems from positional effects within the array architecture and the choice of promoter systems driving gRNA expression. This application note provides a detailed experimental framework for systematically evaluating these factors to design more predictable and efficient multiplexed editing systems. The protocols are framed within the context of advanced CRISPR research, integrating recent findings on promoter and scaffold engineering to optimize array performance for both basic research and therapeutic development [80] [21].
Background and Significance
Multiplexed CRISPR screens have revolutionized functional genomics by enabling systematic interrogation of gene networks and polygenic traits. The simplicity of programming CRISPR systems with short guide RNAs makes them ideally suited for targeting multiple loci simultaneously [4]. However, when gRNAs are arranged in tandem arrays, their efficacy can be significantly influenced by their physical position within the array and the regulatory elements controlling their expression. Position-dependent effects can lead to inconsistent gRNA representation and activity, complicating the interpretation of screening results [80] [21].
Recent advances in synthetic biology have highlighted the limitations of relying on a minimal set of standardized parts, such as the ubiquitous U6 promoter for gRNA expression. Studies now demonstrate that sequence diversification of both promoters and gRNA scaffolds can dramatically improve the performance and reliability of multiplexed systems while reducing repetitive elements that compromise genetic stability [80]. Furthermore, the integration of computational tools for predicting gRNA efficiency and minimizing off-target effects remains crucial for experimental success [74] [22]. This document outlines standardized protocols to quantify these effects and provides actionable strategies for constructing optimized sgRNA arrays.
Experimental Design and Quantitative Benchmarking
Key Factors Affecting sgRNA Array Performance
Table 1: Critical Parameters Influencing sgRNA Array Efficacy
| Factor Category | Specific Parameter | Impact on Efficacy | Measurement Approach |
|---|---|---|---|
| Promoter Strength | Polymerse III promoter variant (U6, H1, 7SK) | Directly correlates with gRNA expression levels and editing efficiency [80] | Prime editing efficiency scores (edit scores) normalized to plasmid barcode frequency |
| Scaffold Design | gRNA sequence and structural stability | Affects Cas protein binding and complex formation [80] | Comparison of editing rates across scaffold variants |
| Positional Context | Linear position within array | Influences transcriptional termination and processing efficiency [80] [21] | Normalized read counts from sequencing of array transcripts |
| Sequence Diversity | Repetitive sequence content (Lmax) | Impacts genetic stability during synthesis and assembly; Lmax < 40 bp recommended [80] | Calculation of longest shared repeat between pairs of sequences |
Performance Metrics of Promoter and Scaffold Variants
Table 2: Experimentally Determined Performance of Diversified U6 Promoters and gRNA Scaffolds
| Component Type | Design Strategy | Number Tested | Activity Range | Notable Performers |
|---|---|---|---|---|
| Pol III Promoters | Evolutionary diversification | 97 | Several orders of magnitude | Ornithorhynchus anatinus (platypus) U6: 1.2-1.8× human U6-1 [80] |
| Pol III Promoters | Synthetic diversification | 112 | Several orders of magnitude | 28 promoters within 5-fold of standard human U6-1 activity [80] |
| gRNA Scaffolds | Mutagenesis and miniaturization | 154 | Up to 4.5-fold difference | H.sapiens–S.pyogenes chimera: comparable to standard scaffold [80] |
Experimental Workflow for Evaluating sgRNA Arrays
Detailed Protocols
Protocol 1: Library Design and Vector Construction for Assessing Positional Effects
Objective: To construct a diversified sgRNA array library enabling systematic evaluation of positional and promoter influences on editing efficacy.
Materials:
- Diversified Promoter Library: Evolutionarily diversified (89 vertebrate U6 orthologs) and synthetically diversified U6 promoters (112 variants) [80]
- Scaffold Variants: 154 sequence-diverse gRNA scaffolds with minimized repetitiveness [80]
- Assembly System: Golden Gate assembly components with type IIS restriction enzymes [4] [21]
- Vector Backbone: Lentiviral transfer plasmid compatible with mammalian systems
Procedure:
- Design sgRNA Array Architecture:
- Select 4-6 target genomic loci with validated sgRNA sequences
- For each target, design a corresponding barcode (5-bp insertional barcode recommended) [80]
- Arrange targets in different sequential positions across array designs
Promoter and Scaffold Selection:
Library Assembly:
- Use Golden Gate assembly with BsaI or similar type IIS enzymes to concatenate promoter-gRNA units [4]
- Assemble 12-24 array configurations with systematic position rotation for each gRNA
- Clone final arrays into lentiviral backbone with unique molecular identifiers for tracking
Quality Control:
- Verify assembly by Sanger sequencing across all junctions
- Quantify library diversity by next-generation sequencing of plasmid pool
- Confirm absence of recombination in E. coli and Agrobacterium (for plant systems) [21]
Protocol 2: Delivery and Functional Assessment in Cellular Systems
Objective: To quantitatively measure editing efficiencies of sgRNA array variants in relevant biological contexts.
Materials:
- Cell Lines: K562, HEK293T, and iPSCs for mammalian systems [80]
- Editing System: PEmax stable cell lines for prime editing assessments [80]
- Sequencing Reagents: Illumina-compatible library preparation kit
- Transfection Reagents: Lipofectamine 3000 or similar for mammalian cell delivery
Procedure:
- Library Delivery:
- For mammalian cells: Package lentiviral vectors at MOI < 0.3 to ensure single integration
- Transduce at least 1000x library coverage to maintain representation
- Include non-targeting control sgRNAs for background subtraction [74]
Harvesting and Nucleic Acid Extraction:
- Harvest cells 72-96 hours post-transduction for genomic DNA and RNA
- Extract gDNA using silica column-based methods
- Isolve RNA using TRIzol with DNase I treatment
Amplicon Library Preparation:
- Design primers to amplify edited genomic loci with Illumina adapters
- Include unique molecular identifiers to correct for PCR amplification bias
- For transcriptional assessment, reverse transcribe and amplify array-derived gRNAs
Sequencing and Primary Analysis:
- Sequence on Illumina platform to achieve >1000x coverage per sample
- Demultiplex samples based on experimental barcodes
- Align sequences to reference genome using BWA or Bowtie2
Protocol 3: Computational Analysis of Editing Efficiencies
Objective: To quantify positional and promoter effects from sequencing data and identify optimal array configurations.
Materials:
- Computational Resources: Linux workstation with ≥16GB RAM
- Software Tools: CRISPResso2, MAGeCK, or custom analysis pipelines [74] [22]
- Reference Files: Genome sequence for relevant species and annotation files
Procedure:
- Edit Score Calculation:
Positional Effect Analysis:
- Compare editing efficiencies for identical gRNAs in different array positions
- Fit linear mixed models with position as fixed effect and gRNA identity as random effect
- Calculate positional bias index as: (max efficiency - min efficiency) / max efficiency
Promoter Strength Correlation:
- Correlate promoter activity scores from massively parallel reporter assays with editing outcomes [80]
- Identify promoter features (TF binding sites, sequence motifs) associated with consistent performance
Data Visualization and Interpretation:
- Generate heatmaps of editing efficiency by position and promoter type
- Create scatter plots comparing promoter strength predictions with observed editing rates
- Perform cluster analysis to identify optimal promoter-gRNA pairings
Factors Affecting sgRNA Array Performance
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for sgRNA Array Engineering
| Tool Category | Specific Resource | Function and Application | Key Features |
|---|---|---|---|
| Promoter Libraries | Diversified U6 promoter sets [80] | Drive gRNA expression with varying strengths | 209 sequence-diverse variants with Lmax < 40 bp |
| Scaffold Variants | Engineered gRNA scaffolds [80] | Optimize Cas protein binding and stability | 154 designs with minimal repetitiveness |
| Assembly Systems | Golden Gate assembly [4] [21] | Construct multi-gRNA arrays efficiently | Type IIS enzyme compatibility (BsaI, BbsI) |
| Screening Libraries | Vienna-single/dual libraries [74] | Benchmark sgRNA performance | Minimal genome-wide libraries with top VBC-scored guides |
| Analysis Software | CRISPResso2 [22] | Quantify editing efficiencies from sequencing | Handles multiplexed editing outcomes |
| Design Tools | CHOPCHOP, Cas-OFFinder [22] | Design sgRNAs and predict off-target effects | Web-based or command-line interfaces |
Troubleshooting and Technical Considerations
Common Challenges and Solutions
Low Editing Efficiency Across Array: Replace low-activity promoters with higher-performing variants identified in Table 2. Consider the platypus U6 promoter, which outperforms human U6-1 by 1.2-1.8-fold [80].
Positional Bias: Implement promoter-gRNA combinations with demonstrated consistent performance across positions. Synthetic promoters often show more uniform behavior than evolutionarily diversified ones [80].
Genetic Instability During Assembly: rigorously monitor Lmax during design phase to ensure < 40 bp for all repetitive elements. This facilitates stable synthesis and yeast-based assembly of complex arrays [80].
Variable Dual-Targeting Efficiency: Note that dual-targeting strategies can enhance knockout efficiency but may trigger DNA damage response even in non-essential genes [74].
Advanced Applications
The principles outlined here extend beyond basic gene knockout to include:
- CRISPRactivation (CRISPRa) for gain-of-function studies using dCas9-transcriptional activator fusions [81]
- Multiplexed epigenetic editing for simultaneous regulation of multiple genes [4]
- Chromosomal engineering for large-scale genomic rearrangements [21]
Systematic evaluation of positional effects and promoter influence is essential for designing effective sgRNA arrays for multiplexed genome editing. By employing the detailed protocols and reference data provided herein, researchers can create more predictable and efficient multiplexed editing systems. The integration of diversified genetic parts, robust delivery systems, and comprehensive computational analysis represents the current state-of-the-art in complex genome engineering. As CRISPR technology continues to evolve, these foundational principles will enable increasingly sophisticated manipulations of biological systems for both basic research and therapeutic applications.
Assessment of Recombination Rates in Different Array Configurations and Designs
The construction of synthetic tandem guide RNA (gRNA) arrays is a foundational technique for multiplexed CRISPR-Cas applications, enabling simultaneous editing, activation, or repression of multiple genomic targets. The configuration of these arrays—encompassing their genetic architecture, processing mechanisms, and assembly strategy—profoundly influences critical performance parameters, with recombination rates being a primary concern for functional integrity and experimental reproducibility [1]. High recombination rates in repetitive DNA sequences can lead to array rearrangements, gRNA dropout, and inconsistent editing outcomes, ultimately compromising screening reliability and therapeutic efficacy.
This Application Note provides a systematic framework for assessing recombination rates across prevalent array designs. We detail standardized protocols for quantifying recombination events and present a comparative analysis of configuration performance to support robust, high-complexity multiplexed editing workflows.
Quantitative Comparison of Array Configuration Performance
The table below summarizes the key performance characteristics, including reported recombination rates, for different gRNA array configurations.
Table 1: Performance Characteristics of Different gRNA Array Configurations
| Array Configuration | Processing Mechanism | Reported Recombination Rate | Key Advantages | Documented Limitations |
|---|---|---|---|---|
| Quadruple-sgRNA (qgRNA) [53] | Four distinct Pol III promoters | 7–17% (during ALPA cloning) | Massive synergistic activation; reduced cell-to-cell heterogeneity | Requires high-throughput cloning (e.g., ALPA) |
| Cas12a crRNA Array [1] | Endogenous Cas12a nuclease | Not explicitly quantified | Simplified, nuclease-driven processing; high multiplexing capacity | Potential for pre-mature array processing |
| tRNA-gRNA Array [1] | Endogenous RNase P and Z | Not explicitly quantified | Ubiquitous processing across organisms; highly modular | Can be inefficient in some cell types |
| Csy4-gRNA Array [1] | Heterologous Csy4 endoribonuclease | Not explicitly quantified | High processing fidelity; precise gRNA stoichiometry | Cytotoxicity at high Csy4 concentrations |
| Ribozyme-gRNA Array [1] | Self-cleaving ribozymes (HH/HDV) | Not explicitly quantified | No co-expressed proteins needed; compatible with Pol II promoters | Larger construct size due to ribozyme sequences |
| CRISPR-StAR Array [82] | Cre-loxP/lox5171 recombination | Optimized to ~55% active / 45% inactive sgRNAs | Built-in internal control for complex screens; overcomes bottleneck effects | Requires Cre-ERT2 system; more complex vector design |
Experimental Protocols for Recombination Rate Assessment
Protocol 1: High-Throughput Cloning and Sequencing for qgRNA Arrays
This protocol uses the Automated Liquid-Phase Assembly (ALPA) method to construct and quality-control complex qgRNA arrays [53].
- Principle: The ALPA method assembles four distinct sgRNA expression cassettes into a single vector via Gibson assembly, with a selection switch from ampicillin to trimethoprim to enrich for correct constructs. Recombination rates are assessed by Sanger sequencing of bacterial colonies.
- Materials:
- Research Reagent Solutions:
- ALPA Precursor Vector (pYJA5): Contains ampicillin resistance (AmpR) flanked by BbsI sites and a landing pad for Gibson assembly.
- TMP-resistant Dihydrofolate Reductase (TmpR): Cloned into the first amplicon for post-assembly selection.
- Four Distinct Pol III Promoters: (e.g., human U6, mouse U6, human H1, human 7SK) to minimize homologous recombination.
- Gibson Assembly Master Mix: For isothermal assembly of multiple DNA fragments.
- Research Reagent Solutions:
- Procedure:
- Fragment Preparation: Digest the pYJA5 vector with BbsI to remove the AmpR cassette. Generate three sgRNA-containing amplicons via PCR using primers with 20-nt overlapping ends for Gibson assembly.
- Gibson Assembly: Mix the digested vector and the three amplicons in a 1:1 molar ratio with Gibson Assembly Master Mix. Incubate at 50°C for 1 hour.
- Transformation & Selection: Transform the assembly reaction into recombination-deficient chemically competent E. coli. Plate cells on agar containing trimethoprim to select for successfully assembled plasmids.
- Recombination Rate Quantification: Pick a statistically significant number of bacterial colonies (e.g., ≥22 per assembly reaction). Perform colony PCR with primers flanking the qgRNA insert. Purify plasmids from colonies with correct insert size and subject them to Sanger sequencing.
- Data Analysis: Analyze sequencing chromatograms. Classify colonies as:
- Correct: All four sgRNA sequences and promoters are intact.
- Recombined: Evidence of homologous recombination between repetitive elements (e.g., promoter regions).
- Mutated: Presence of point mutations, likely from oligo synthesis errors.
- Calculate the recombination rate as: (Number of recombined colonies / Total number of sequenced colonies) × 100.
- Technical Notes: Performing assembly and transformation in 384-well plates is recommended for high-throughput library generation. Using a recombination-deficient E. coli strain is critical to minimize bias.
Protocol 2: Single-Cell DNA Sequencing for Multiplexed Array Analysis
This protocol leverages droplet-based single-cell sequencing to detect recombination-derived editing patterns in a population of cells expressing a multiplexed gRNA array [83].
- Principle: Cells are transduced with a multiplexed gRNA array and then analyzed at the single-cell level to determine the co-occurrence of intended edits. A deviation from the expected combinatorial pattern indicates recombination or uneven processing within the array.
- Materials:
- Research Reagent Solutions:
- Tapestri Platform (Mission Bio) or equivalent: For droplet-based single-cell DNA sequencing.
- Custom Amplification Panel: A multiplex PCR panel designed with ~40 amplicons covering all on-target gRNA sites, predicted off-target sites, and positive control genomic loci.
- Cell Line: A Cas9-expressing cell line (e.g., murine Ba/F3 or HEK293) transduced with the multiplexed gRNA array library.
- Research Reagent Solutions:
- Procedure:
- Cell Preparation and Sequencing: Harvest cells at an appropriate time post-transduction (e.g., 10-14 days). Prepare a single-cell suspension and load onto the Tapestri platform according to manufacturer's instructions for generating single-cell barcoded amplicon libraries.
- NGS and Primary Analysis: Sequence the libraries. Use the instrument's software to demultiplex cell barcodes and align reads to each amplicon.
- Variant Calling and Zygosity Analysis: Use software like CRISPResso2 to quantify editing events in each cell for each target locus.
- Co-occurrence Analysis: For each cell barcode, determine the number and identity of successfully edited loci.
- Data Analysis: The final output is a matrix of cells (rows) by edited loci (columns).
- The percentage of cells containing the full, intended combination of edits reflects the array's functional stability.
- A high frequency of cells with partial edits (e.g., only 1 or 2 out of 4 expected edits) suggests array recombination or processing failure.
- The distribution of co-editing frequencies directly informs the effective recombination rate within the cell population.
Protocol 3: Internally Controlled Screening with CRISPR-StAR
This protocol uses the CRISPR-StAR system, which employs an inducible, dual-recombinase system to generate internal controls, allowing for precise quantification of editing efficiency and indirect assessment of array stability in complex models [82].
- Principle: The CRISPR-StAR vector contains a Cre-inducible sgRNA cassette with intercalated loxP and lox5171 sites. Cre activity stochastically generates either an active sgRNA or an inactive one, creating isogenic control and experimental populations within a single clone. This controls for heterogeneity and enables accurate measurement of sgRNA function post-bottleneck.
- Materials:
- Research Reagent Solutions:
- CRISPR-StAR Vector Backbone: Contains the inducible sgRNA expression cassette and a unique molecular identifier (UMI) for clonal tracking.
- Cre-ERT2-Expressing Cell Line: A Cas9-positive cell line expressing a tamoxifen-inducible Cre recombinase.
- 4-Hydroxytamoxifen (4-OHT): For induction of Cre-mediated recombination.
- Research Reagent Solutions:
- Procedure:
- Library Transduction and Bottleneck: Transduce the Cre-ERT2/Cas9 cell line with the CRISPR-StAR library at high coverage. Pass the cells through a stringent bottleneck (e.g., by limiting dilution or in vivo engraftment) to create single-cell-derived clones.
- Clonal Expansion and Induction: Expand the clones. Administer 4-OHT to induce recombination, which will generate a mixed population of active and inactive sgRNA-bearing cells within each UMI-marked clone.
- Phenotypic Readout and Sequencing: After a period of phenotypic selection (e.g., 14 days), harvest cells and perform genomic DNA sequencing to quantify the abundance of active vs. inactive sgRNA-bearing cells within each UMI clone.
- Data Analysis: For a given sgRNA, the functional effect is calculated by comparing the abundance of cells with the active conformation to the internal control (inactive conformation) within each UMI clone, then aggregating across all relevant clones.
- Technical Notes: The optimal active-to-inactive ratio for maximum dynamic range is approximately 55:45. The initial vector design may require optimization of lox site positioning and sequence context to achieve this balance. This method is particularly powerful for in vivo screens where engraftment bottlenecks are unavoidable.
Workflow Visualization
Figure 1. Decision Workflow for Selecting a Recombination Assessment Protocol. This diagram guides the selection of the most appropriate protocol based on the array configuration and the experimental model. Each path yields a specific metric for quantifying recombination or its functional consequences, leading to a final comparative analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Array Construction and Recombination Assessment
| Research Reagent | Function / Application | Example Use Case / Benefit |
|---|---|---|
| ALPA Cloning System [53] | High-throughput, liquid-phase plasmid assembly for multi-cassette vectors. | Enables construction of complex qgRNA arrays with a selection switch (AmpR to TmpR) to minimize false positives. |
| Distinct Pol III Promoters [53] | Drive expression of individual gRNAs in an array with minimal sequence homology. | Reduces homologous recombination in bacterial and mammalian cells (e.g., human U6, mouse U6, human H1, human 7SK). |
| Recombination-Deficient E. coli | A bacterial strain engineered to suppress homologous recombination. | Essential for stable propagation of repetitive gRNA arrays during plasmid amplification. |
| Csy4 Endoribonuclease [1] | Processes long gRNA array transcripts by cleaving at specific 28-nt recognition sites. | Provides high-fidelity, stoichiometric gRNA production; useful for defined array expression. |
| CRISPR-StAR Vector [82] | An inducible sgRNA system with Cre/lox logic for generating internal control cells. | Allows for precise, internally controlled functional genetics in heterogeneous systems and in vivo. |
| Droplet-Based Single-Cell Seq Platform [83] | Simultaneously detects multiple CRISPR-induced gene edits in thousands of single cells. | Directly quantifies co-editing patterns and zygosity, providing a direct readout of array stability and function. |
Long-term Stability and Heritability of Edits in Progeny and Cell Lines
The construction of tandem CRISPR sgRNA arrays has revolutionized multiplexed genome editing by enabling the simultaneous targeting of multiple genetic loci from a single transcript [1]. However, a significant challenge in employing this powerful technology, especially for long-term studies and therapeutic applications, is ensuring the stable persistence and reliable heritability of the introduced edits. While initial editing efficiencies can be high, the long-term fate of these edits in proliferating cell lines and their faithful transmission to progeny is not guaranteed. Factors such as cellular fitness costs, DNA repair pathway choice, and the potential for karyotypic instability can profoundly influence the outcome of multiplexed editing experiments [84] [85]. This Application Note details quantitative findings on edit stability, provides protocols for its assessment, and outlines strategies to enhance the heritability of edits, all within the framework of advanced tandem sgRNA array technology.
Quantitative Data on Editing Stability and Persistence
A systematic understanding of how edited cell populations evolve over time is critical for experimental design and data interpretation. The data summarized below reveal common trends in the persistence of edits.
Table 1: Persistence of CRISPR-Cas9 Edited Hematopoietic Stem and Progenitor Cells (HSPCs) in Animal Models This meta-analysis of 15 in vivo studies reveals a key challenge in maintaining edited cell populations over time [84].
| Time Point / Condition | Relative Engraftment/Persistence (vs. Control) | Notes |
|---|---|---|
| Early Engraftment | Equivalent | Edited cells initially engraft with similar efficiency to unedited controls. |
| Later Time Points | Reduced | A noticeable decline in the proportion of edited cells is observed over time. |
| Secondary Transplantation | Further Reduced | Persistence is more significantly challenged upon serial transplantation. |
| Targeting Hemoglobin Genes (HDR or NHEJ) | Significantly Reduced | Specific locus targeting impacts stability; no significant difference between repair pathways. |
Table 2: Factors Influencing Long-Term Stability of CRISPR Edits Multiple technical and biological factors contribute to the long-term stability of genomic edits [1] [84] [85].
| Factor | Impact on Stability | Experimental Evidence |
|---|---|---|
| Cellular Fitness Cost | High | Edited cells, especially those with large indels or multiple DSBs, may be outcompeted by unedited or healthier cells [84]. |
| Chromosomal Instability | High | CRISPR-Cas9 can cause large deletions and chromosomal rearrangements, leading to selective disadvantages [85]. |
| Multiplexing Strategy | Medium | Tandem arrays with multiple gRNAs per gene (e.g., qgRNAs) can enhance editing efficiency but require careful assembly to avoid toxicity [53]. |
| Targeted Locus | Variable | Genomic context, gene essentiality, and transcriptional activity can influence stability. |
| DNA Delivery Method | Variable | RNP delivery may reduce off-target effects and cytotoxicity compared to plasmid delivery, potentially improving stability [85]. |
Figure 1: The trajectory of edited cell populations over time, showing a common decline due to various cellular pressures. Achieving stable outcomes requires strategic intervention to counter these factors.
Protocols for Assessing Edit Stability and Karyotypic Integrity
Rigorous validation is paramount after performing multiplexed edits to ensure that the intended modifications are present and that no unintended, potentially destabilizing alterations have occurred.
Protocol: Longitudinal Tracking of Edited Cell Populations
This protocol is designed to monitor the proportion of edited cells over multiple generations.
- Initial Editing and Clonal Isolation: Transfert cells with your tandem sgRNA array and Cas9 (or use a stable Cas9 cell line). For bulk populations, proceed to step 2. For clonal analysis, seed cells at a density of 1 cell per well in a 96-well plate and expand for 2-3 weeks [85].
- Timepoint Sampling: Passage cells at a consistent dilution ratio. At each passage (e.g., every 3-4 days for immortalized lines), collect an aliquot of ~1x10^6 cells for genomic DNA (gDNA) extraction. Continue for at least 10-15 passages.
- gDNA Extraction and Amplification: Extract gDNA using a commercial kit. Amplify the target loci by PCR using high-fidelity DNA polymerase. Primer design is critical: ensure they flank the edited region(s) by at least 50-100 bp.
- Edit Quantification via Sanger Sequencing and ICE Analysis:
- Purify the PCR products and submit for Sanger sequencing.
- Upload the sequencing chromatogram files (.ab1) from both edited and a control (unedited) sample to the ICE (Inference of CRISPR Edits) webtool (Synthego).
- Input the gRNA target sequence(s) and select the correct nuclease.
- The tool will output an Indel Percentage (editing efficiency) and a Knockout Score (proportion of frameshift indels) for each sample [47].
- Tracking the Indel Percentage across timepoints will reveal the stability of the edited population.
Protocol: Detection of Large-Scale Genomic Rearrangements
Standard PCR-based screening can miss large deletions or translocations induced by multiple, concurrent double-strand breaks. This cytogenetic protocol provides a robust check for karyotypic integrity [85].
Metaphase Spread Preparation:
- Culture cells to ~70% confluency.
- Add Colcemid (final concentration 50 ng/mL) to the culture medium for 3 hours to arrest cells in metaphase.
- Detach cells, swell them in a pre-warmed hypotonic solution (75 mM KCl) for 20 minutes at 37°C, and fix them in fresh Carnoy's fixative (3:1 methanol:glacial acetic acid).
- Drop the fixed cell suspension onto clean microscope slides and air-dry overnight [85].
Fluorescence In Situ Hybridization (FISH):
- Probe Selection: Choose BAC (Bacterial Artificial Chromosome) probes that flank the target locus (e.g., one probe upstream and one downstream). Label probes with different fluorophores (e.g., SpectrumGreen and SpectrumRed) via nick translation.
- Hybridization: Denature the metaphase spread DNA and the labeled probe mix. Apply the denatured probes to the slides and hybridize overnight at 37°C in a humidified chamber.
- Washing and Detection: Perform post-hybridization washes in 0.1x SSC at 60°C. If using biotinylated probes, detect with streptavidin-Cy5. Mount slides in DAPI/Vectashield [85].
Analysis:
- Image at least 25 metaphase spreads per cell line using a fluorescence microscope equipped with appropriate filters.
- Normal Signal: Two pairs of co-localized or closely adjacent green and red signals.
- Large Deletion: Separation of one green and one red signal on one chromatid, indicating the loss of the intervening sequence.
Strategies to Enhance Stability and Heritability of Edits
To counter the observed decline in edit persistence, implement the following strategies in your experimental design.
Table 3: Reagent Solutions for Enhanced Multiplexed Editing
| Reagent / Tool | Function | Example & Application Notes |
|---|---|---|
| Stable Cas9 Cell Lines | Provides uniform, constitutive Cas9 expression, minimizing delivery variability. | GeneHero Cas9 cell lines (e.g., HEK293, HCT116) [86]. Ideal for high-throughput sgRNA screening. |
| Quadruple-guide RNA (qgRNA) Vectors | Expresses 4 distinct sgRNAs per gene, massively increasing editing efficiency and robustness [53]. | T.spiezzo/T.gonfio libraries; uses ALPA cloning. Reduces heterogeneity and escape from editing. |
| High-Fidelity Cas9 Variants | Engineered to reduce off-target cleavage, minimizing karyotypic instability. | eSpCas9(1.1), SpCas9-HF1, HypaCas9 [87]. Crucial for sensitive applications and reducing fitness costs. |
| Ribonucleoprotein (RNP) Delivery | Direct delivery of pre-complexed Cas9 protein and gRNA; transient activity, high efficiency, low toxicity. | Synthesized sgRNA complexed with Cas9 protein and electroporated [85]. Redces off-targets and DNA vector integration. |
| ICE Analysis Tool | Web tool for quantitative analysis of CRISPR editing efficiency and indel profiles from Sanger data. | Synthego ICE (free online tool) [47]. Provides indel % and KO score for stability tracking. |
Figure 2: Strategic solutions to counter the common causes of editing instability, leading to more reliable and heritable multiplexed editing outcomes.
Employ qgRNA Vectors for Robust Editing: When constructing tandem arrays, design them to include multiple, non-overlapping sgRNAs targeting a single gene. The synergy between these guides leads to higher knockout efficiency (75–99% ablation shown in one study) [53], ensuring a higher initial rate of bi-allelic editing and leaving fewer cells to outcompete the successfully edited ones over time.
Utilize Stable Cas9 Cell Lines: For long-term or sequential editing projects, using a clonal cell line with Cas9 stably integrated into a "safe harbor" locus (e.g., AAVS1) ensures consistent nuclease expression across the entire population and over countless passages, eliminating variability from transient delivery [86].
Prioritize High-Fidelity Cas9 Variants and RNP Delivery: To minimize the introduction of off-target mutations and chromosomal rearrangements that compromise cellular fitness, use high-fidelity Cas9 enzymes like SpCas9-HF1 or eSpCas9(1.1) [87]. Where possible, deliver the CRISPR machinery as a purified RNP complex via electroporation. This method is not only highly efficient but also minimizes the time the nuclease is active in the cell, reducing the window for off-target cleavage [85].
Implement Rigorous and Longitudinal Validation: Do not rely solely on initial PCR and Sanger sequencing of a small number of clones. Employ the protocols in Section 3 to track editing rates over multiple passages and to screen master cell banks for karyotypic abnormalities. This ensures that the cell lines used for downstream experiments and data generation are genetically stable and accurately represent the intended genotype.
Conclusion
Tandem CRISPR sgRNA arrays represent a powerful advancement in multiplexed genome editing, enabling sophisticated applications from basic research to therapeutic development. The successful implementation requires careful consideration of array design, processing mechanisms, and delivery systems tailored to specific biological contexts. Optimization of promoter systems, addressing technical challenges like recombination and off-target effects, and rigorous validation using comparative frameworks are essential for achieving high-efficiency, specific editing. Future directions will likely involve the integration of artificial intelligence for guide design and outcome prediction, the development of more compact and efficient array systems, and the expansion of these technologies into clinical applications for complex genetic diseases. As these technologies mature, they will continue to transform our ability to dissect complex biological networks and engineer novel therapeutic interventions.
References
- 1. Multiplexed CRISPR technologies for gene editing and ... [https://www.nature.com/articles/s41467-020-15053-x]
- 2. Efficient Cas9 multiplex editing using unspaced sgRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8251967/]
- 3. An efficient multiplex approach to CRISPR/Cas9 gene editing ... [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-024-01274-4]
- 4. Applications of multiplexed CRISPR–Cas for genome ... [https://www.nature.com/articles/s12276-025-01500-6]
- 5. A multiplexed single-cell CRISPR screening platform enables ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5315571/]
- 6. Multiplex CRISPR/Cas9-Based Genome Editing in Human ... [https://www.sciencedirect.com/science/article/pii/S1934590917303181]
- 7. Multicenter integrated analysis of noncoding CRISPRi ... [https://www.nature.com/articles/s41592-024-02216-7]
- 8. Biogenesis pathways of RNA guides in archaeal and bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5965381/]
- 9. An updated evolutionary classification of CRISPR–Cas ... [https://www.nature.com/articles/s41564-025-02180-8]
- 10. Bioinformatics approaches to analyzing CRISPR screen data [https://pmc.ncbi.nlm.nih.gov/articles/PMC10019185/]
- 11. Genome Annotation / CRISPR screen analysis / Hands-on [https://training.galaxyproject.org/training-material/topics/genome-annotation/tutorials/crispr-screen/tutorial.html]
- 12. High-content CRISPR screening - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10200264/]
- 13. Efficient expression of multiple guide RNAs for CRISPR/ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9590505/]
- 14. CRISPR 101: Multiplex Expression of gRNAs [https://blog.addgene.org/crispr-101-multiplex-expression-of-grnas]
- 15. A Golden Gate-based Protocol for Assembly of Multiplexed ... [https://bio-protocol.org/en/bpdetail?id=2059&type=0]
- 16. A Tandem Guide RNA-Based Strategy for Efficient CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7194318/]
- 17. Optimized metrics for orthogonal combinatorial CRISPR ... [https://www.nature.com/articles/s41598-023-34597-8]
- 18. Golden Gate Assembly of CRISPR gRNA expression array ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11108369/]
- 19. High-accuracy crRNA array assembly strategy for multiplex ... [https://www.sciencedirect.com/science/article/pii/S2162253124003159]
- 20. Orthogonal CRISPR systems for targeted integration and ... [https://www.sciencedirect.com/science/article/pii/S1525001625006628]
- 21. harnessing multiplex editing for polygenic trait engineering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12527382/]
- 22. CRISPR-Cas9 and Its Bioinformatics Tools: A Systematic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12109910/]
- 23. Modular one-pot assembly of CRISPR arrays enables ... [https://www.nature.com/articles/s41467-019-10747-3]
- 24. High-accuracy crRNA array assembly strategy for multiplex ... [https://pubmed.ncbi.nlm.nih.gov/39897580/]
- 25. A gRNA-tRNA array for CRISPR-Cas9 based rapid ... [https://www.nature.com/articles/s41467-019-09005-3]
- 26. Single transcript unit CRISPR 2.0 systems for robust Cas9 and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6576101/]
- 27. Polycistronic tRNA and CRISPR guide-RNA enables highly ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X16320034]
- 28. Multiplexed CRISPR technologies for gene editing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7062760/]
- 29. The Multiplexed CRISPR targeting platforms - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6699180/]
- 30. Precision multiplexed base editing in human cells using ... [https://www.nature.com/articles/s41467-025-59653-x]
- 31. Multiplexed base editing through Cas12a variant-mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9630288/]
- 32. CRISPR Delivery Methods: Cargo, Vehicles, and Challenges [https://www.synthego.com/blog/delivery-crispr-cas9/]
- 33. Exploring non-viral methods for the delivery of CRISPR-Cas ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-024-03848-4]
- 34. Non-viral delivery systems for CRISPR/Cas9-based genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5944364/]
- 35. Comprehensive protocols for CRISPR/Cas9-based gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4988528/]
- 36. Multiplex Editing of the Nucleoredoxin1 Tandem Array in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10460964/]
- 37. Next-generation multiplex-edited CAR-NK cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC12439146/]
- 38. Multiplex engineering and multifunction T cells for precise ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1680410/full]
- 39. Transgene-free genome editing in citrus and poplar trees ... [https://link.springer.com/article/10.1007/s00299-025-03627-2]
- 40. Maximizing CRISPRi efficacy and accessibility with dual- ... [https://elifesciences.org/articles/81856]
- 41. CRISPR interference [https://horizondiscovery.com/en/applications/crisprmod/crispri]
- 42. A dual sgRNA library design to probe genetic modifiers using ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09754-y]
- 43. CRISPR GENome and epigenome engineering improves loss ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12272254/]
- 44. Artificial Intelligence for CRISPR Guide RNA Design [https://arxiv.org/html/2508.20130v1]
- 45. Programmable Multiplex Genome Editing: Innovations in ... [https://www.sciepublish.com/article/pii/680]
- 46. Comparison of CRISPR-Cas9, CRISPR-Cas12f1, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12131857/]
- 47. How To Use ICE: A Detailed Guide for Analyzing CRISPR ... [https://www.synthego.com/guide/how-to-use-crispr/ice-analysis-guide/]
- 48. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 49. Off-target effects in CRISPR/Cas9 gene editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10034092/]
- 50. Multiplex nucleotide editing by high-fidelity Cas9 variants ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6873407/]
- 51. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 52. Recent advances in CRISPR-based single-nucleotide fidelity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12219407/]
- 53. Arrayed CRISPR libraries for the genome-wide activation ... [https://www.nature.com/articles/s41551-024-01278-4]
- 54. Improving the Efficiency of CRISPR/Cas9-Mediated Non ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12384319/]
- 55. A tRNA-gRNA array-based CRISPR system for efficient ... [https://www.sciencedirect.com/science/article/pii/S0926669025007150]
- 56. Protocol for the saturation and multiplexing of genetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10658368/]
- 57. Multiplex CRISPR-Cas9 [https://horizondiscovery.com/en/applications/gene-editing/multiplexed-gene-knockout]
- 58. Frontiers | CRISPR /Cas9 technology in tumor research and drug... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1552741/full]
- 59. Cutting to the core: Down syndrome, CRISPR, and the future ... [https://law.stanford.edu/2025/11/23/cutting-to-the-core-down-syndrome-crispr-and-the-future-of-human-diversity-part-ii/]
- 60. An efficient CRISPR /Cas9 genome editing system based on a multiple... [https://biotechnologyforbiofuels.biomedcentral.com/articles/10.1186/s13068-024-02468-7]
- 61. Protocol for identification of sgRNA mutants using high ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11981739/]
- 62. Resources for the design of CRISPR gene editing experiments [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0823-x]
- 63. Optimized sgRNA design to maximize activity and minimize off-target... [https://pubmed.ncbi.nlm.nih.gov/26780180/]
- 64. Compact and highly active next-generation libraries for... [https://elifesciences.org/articles/19760]
- 65. qEva-CRISPR: a method for quantitative evaluation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6158505/]
- 66. CRISPR Clinical Trials: A 2025 Update [https://innovativegenomics.org/news/crispr-clinical-trials-2025/]
- 67. Current Strategies for Increasing Knock-In Efficiency ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10931195/]
- 68. Optimizing CRISPR delivery for drug discovery [https://www.drugdiscoverynews.com/optimizing-crispr-delivery-for-drug-discovery-16487]
- 69. One-Day Construction of Multiplex Arrays to Harness ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8173669/]
- 70. Comparative optimization of combinatorial CRISPR screens [https://www.nature.com/articles/s41467-022-30196-9]
- 71. How to Choose the Right Cas Variant for Every CRISPR ... [https://www.synthego.com/guide/how-to-use-crispr/cas-nuclease-variants/]
- 72. Simple-to-use CRISPR-SpCas9/SaCas9/AsCas12a vector ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8664446/]
- 73. Validation of CRISPR-based Gene Editing [https://www.neb.com/en-us/products/crispr-cas-gene-editing/crispr-validation?srsltid=AfmBOorLF4RkYR_t0U78Tz5Zdi_I4vMfAqR03OCbVVzuVqdJgpity8vV]
- 74. A benchmark comparison of CRISPRn guide-RNA design ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11386-3]
- 75. Functional Genomics via CRISPR-Cas - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6309720/]
- 76. Comparing Gene Editing Platforms: CRISPR vs. Traditional ... [https://blog.crownbio.com/comparing-gene-editing-platforms-crispr-vs-traditional-methods]
- 77. Combining CRISPR-Cas9 Gene Editing With Functional ... [https://www.thermofisher.com/us/en/home/references/newsletters-and-journals/bioprobes-journal-of-cell-biology-applications/bioprobes-74/bioprobes-74-crispr-cas9-functional-proteomics.html]
- 78. Multiplexed multicolor antiviral assay amenable for high- ... [https://www.nature.com/articles/s41467-023-44339-z]
- 79. Unraveling the future of genomics: CRISPR, single-cell ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2025.1565387/full]
- 80. A parts list of promoters and gRNA scaffolds ... [https://www.nature.com/articles/s41587-025-02896-2]
- 81. CRISPR activation: identifying and using novel genes for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12450905/]
- 82. CRISPR-StAR enables high-resolution genetic screening ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12611787/]
- 83. High throughput single-cell detection of multiplex CRISPR ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02174-1]
- 84. Persistence of CRISPR/Cas9 gene edited hematopoietic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8235122/]
- 85. CRISPR-Cas9 Causes Chromosomal Instability and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6919265/]
- 86. GeneHero™ CRISPR-Cas9 stable cell lines [https://www.genecopoeia.com/product/cas9-cell-line/]
- 87. CRISPR Guide [https://www.addgene.org/guides/crispr/]