A Complete Guide to Golden Gate Assembly for Complex Multigene Constructs
This article provides a comprehensive resource for researchers and drug development professionals on employing Golden Gate Assembly for constructing complex multigene systems.
A Complete Guide to Golden Gate Assembly for Complex Multigene Constructs
Abstract
This article provides a comprehensive resource for researchers and drug development professionals on employing Golden Gate Assembly for constructing complex multigene systems. It covers foundational principles, detailed step-by-step protocols for modular cloning (MoClo) and high-fidelity assembly, and advanced troubleshooting strategies to overcome common challenges. Furthermore, it offers a comparative analysis with other DNA assembly methods and discusses validation techniques to ensure construct accuracy, empowering scientists to reliably build sophisticated genetic circuits for biomedical and clinical applications.
Understanding Golden Gate Assembly: Principles and Advantages for Synthetic Biology
Golden Gate Assembly represents a pivotal advancement in molecular cloning techniques, enabling the seamless, scarless assembly of multiple DNA fragments in a single reaction. This methodology leverages the unique properties of Type IIS restriction enzymes, which cleave DNA outside of their recognition sites, thereby allowing for the precise excision of DNA fragments with custom overhangs that facilitate ordered, scarless ligation. This application note delineates the core mechanism of Type IIS enzymes, provides detailed protocols for their use in constructing multigene vectors, and discusses their critical applications in synthetic biology and therapeutic development, framed within the context of advanced genetic construct research.
Restriction enzymes are fundamental tools in molecular biology, traditionally used for their ability to cleave DNA at specific palindromic sequences. However, Type IIS restriction enzymes constitute a distinct subclass characterized by their separation of recognition and cleavage functions. These enzymes recognize asymmetric DNA sequences and cleave at a defined distance outside of their recognition site, typically within 1 to 20 nucleotides [1] [2]. This functional dichotomy is enabled by a modular protein structure where the DNA recognition domain and the catalytic cleavage domain are physically distinct and connected by a short polypeptide linker [3] [4]. For instance, the well-studied FokI enzyme recognizes the sequence 5′-GGATG-3′ and cleaves the top and bottom strands 9 and 13 bases downstream, respectively, generating a four-base 5′ overhang [4]. This mechanism stands in stark contrast to traditional Type IIP enzymes, which cleave within their palindromic recognition sequences and often leave behind "scar" sequences in the final construct.
The Core Mechanism of Scarless Assembly
The scarless nature of Golden Gate Assembly is a direct consequence of the biochemical properties of Type IIS enzymes. The process involves a single-tube digestion-ligation reaction where the Type IIS enzyme and a DNA ligase are active simultaneously.
Biochemical Principles
- Precise Overhang Generation: Type IIS enzymes cleave at fixed positions outside their recognition sites, producing fragments with user-defined, single-stranded overhangs. The sequence of these overhangs is not dictated by the enzyme's recognition site but is instead encoded by the adjacent DNA sequence, allowing for the design of unique, complementary ends for each fragment [1] [5]. For example, enzymes like BsaI (recognition site: GGTCTC) generate 4-base 5′ overhangs, enabling the creation of 256 possible unique end combinations [5].
- Elimination of Recognition Sites: The recognition sites for the Type IIS enzyme are positioned on the primers or vector such that upon cleavage, they are physically separated from the DNA fragments of interest. Following ligation of the complementary overhangs, the recognition sites are absent from the final assembled construct. This ensures the product is immune to redigestion, allowing the reaction to proceed to completion in a single pot [1] [5].
- Simultaneous Digestion and Ligation: The assembly reaction is typically cycled between the restriction enzyme's optimal digestion temperature (e.g., 37°C for BsaI) and the ligase's optimal temperature (e.g., 16°C). This cycling drives the reaction equilibrium towards complete assembly by continuously cleaving incorrectly ligated products and favoring the formation of the correct, stable construct [6].
Visualizing the Workflow
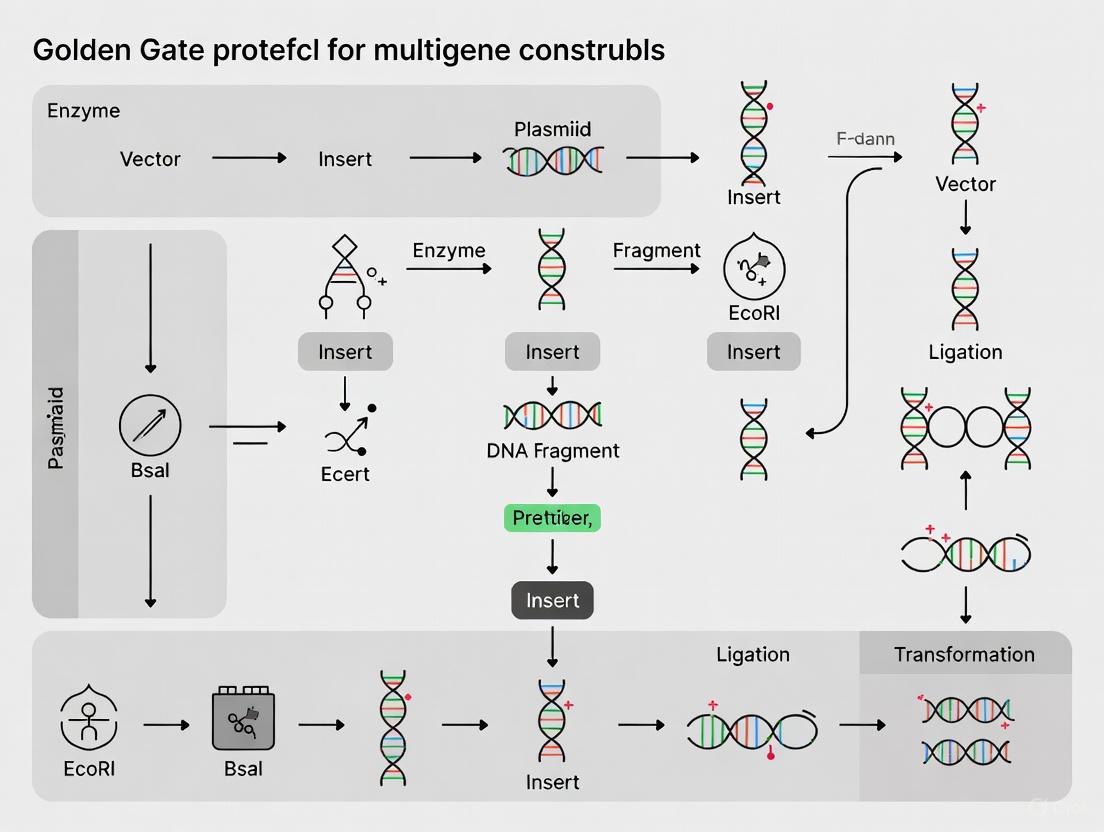
The following diagram illustrates the core mechanism of a Golden Gate Assembly reaction for assembling two DNA fragments into a vector backbone.
Key Type IIS Enzymes for Golden Gate Assembly
Several Type IIS enzymes are commonly employed in Golden Gate Assembly, each with distinct recognition sequences and cleavage characteristics. The choice of enzyme can influence assembly efficiency, particularly with complex or repetitive sequences.
Table 1: Commonly Used Type IIS Restriction Enzymes in Golden Gate Assembly
| Enzyme | Recognition Sequence (5′→3′) | Cleavage Pattern | Overhang Length | Common Applications |
|---|---|---|---|---|
| BsaI (Eco31I) | GGTCTC | (1/5) | 4 bp | Most common; standard MoClo systems [1] [7] |
| BsmBI-v2 | CGTCTC | (1/5) | 4 bp | Alternative to BsaI; higher temperature optimum (55°C) [8] [7] |
| BbsI (BpiI) | GAAGAC | (2/6) | 4 bp | Golden Gate assembly; TALEN construction [1] [7] |
| Aa | CACCTGC | (4/8) | 4 bp | Larger recognition site reduces internal site frequency [1] [7] |
| SapI (BspQI) | GCTCTTC | (1/4) | 3 bp | Creates 3-base overhangs; useful for specific toolkits [8] [7] |
| FokI | GGATG | (9/13) | 4 bp | Primarily used in engineered nucleases (e.g., TALENs) [3] [4] |
Protocol: MoClo for Multigene Construct Assembly
The Modular Cloning (MoClo) system is a hierarchical, standardized framework that leverages Golden Gate Assembly for building complex multigene constructs [9]. The following protocol details a standard MoClo workflow.
Experimental Workflow
The hierarchical nature of the MoClo system is visualized in the workflow below, progressing from basic parts to a complete multigene construct.
Step-by-Step Procedure
Step 1: Preparation of Level 0 Modules (Basic Parts)
- Fragment Generation: Amplify or synthesize DNA sequences for basic biological parts (e.g., promoters, coding sequences (CDS), terminators) using primers that append the appropriate Type IIS recognition sites (e.g., BsaI sites for MoClo) and the required 4-bp fusion overhangs. The sites must be oriented to face inward, ensuring they are removed upon cleavage [9] [5].
- Domestication: Screen all parts for internal recognition sites for the Type IIS enzyme used. Remove any internal sites via silent mutagenesis (for CDS) or other methods to prevent internal cleavage [5].
- Cloning into Entry Vectors: Perform a Golden Gate reaction to clone each domesticated part into a Level 0 acceptor vector.
- Reaction Setup:
- 50 ng Level 0 destination vector (containing a negative selection marker like ccdB)
- 20 fmol of each purified PCR fragment or 10-50 ng of each plasmid-derived part
- 1 µL BsaI-HFv2 (or equivalent Type IIS enzyme)
- 1 µL T4 DNA Ligase (400 U/µL)
- 1X T4 DNA Ligase Buffer
- Nuclease-free water to 20 µL
- Thermocycling Conditions:
- Reaction Setup:
- Sequence Verification: Transform the reaction into competent E. coli, select positive clones, and verify the sequence of all Level 0 modules.
Step 2: Assembly of Level 1 Transcription Units
- Design: Combine Level 0 parts (e.g., promoter, CDS, terminator) in the desired order to form a functional transcription unit (TU). The 4-bp overhangs between parts must be designed to be specific and complementary in the intended order [9].
- Golden Gate Assembly:
- Reaction Setup: Mix equimolar amounts (typically 50-100 ng each) of the required Level 0 plasmids (promoter, CDS, terminator) and a Level 1 acceptor vector. Use the same enzyme and ligase concentrations as in Step 1.
- Use the same thermocycling profile as for Level 0 assembly.
- Analysis: Verify correct assembly of Level 1 TUs by colony PCR, diagnostic restriction digest, and/or sequencing.
Step 3: Assembly of Level 2 Multigene Constructs
- Design: Combine multiple Level 1 TU plasmids in the desired order and orientation within a final destination vector. This step often uses a different Type IIS enzyme (e.g., BsmBI or Aa) to avoid internal sites within the Level 1 modules [9].
- Final Golden Gate Assembly:
- Reaction Setup: Mix equimolar amounts of the Level 1 plasmids and the Level 2 destination vector. Use the appropriate Type IIS enzyme for this final assembly (e.g., BsmBI-v2 with incubation at 55°C).
- Thermocycling for BsmBI-v2: 30 cycles of: 55°C for 2 minutes → 16°C for 5 minutes; followed by 55°C for 5 minutes and 80°C for 10 minutes.
- Validation: Validate the final multigene construct using analytical techniques such as long-range sequencing or restriction fingerprinting.
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of Golden Gate Assembly requires a set of core reagents and materials.
Table 2: Essential Research Reagent Solutions for Golden Gate Assembly
| Reagent/Material | Function | Example Products & Notes |
|---|---|---|
| Type IIS Restriction Enzyme | Digests DNA at specific positions to generate custom overhangs. | BsaI-HFv2 (NEB), FastDigest Eco31I (Thermo Fisher). High-Fidelity (HF) versions reduce star activity [8] [7]. |
| T4 DNA Ligase | Joins DNA fragments via complementary overhangs. | T4 DNA Ligase (NEB, Thermo Fisher). Requires ATP. Often provided with reaction buffer [5]. |
| Entry/Destination Vectors | Plasmid backbones for storing parts and final assembly. | Vectors with ccdB negative selection marker (e.g., MoClo toolkit vectors) to minimize background from empty vectors [9] [6]. |
| High-Quality DNA Parts | Inserts for assembly. | PCR-amplified fragments or pre-cloned sequences in entry vectors. Must be domestication-checked [5]. |
| Competent E. coli | For transformation post-assembly. | High-efficiency chemically competent cells (>10⁷ CFU/µg) for optimal results with complex assemblies. |
Applications in Multigene Construct Research
The precision and efficiency of Type IIS enzyme-based assembly have made it a cornerstone technology in modern biological engineering.
- Synthetic Biology and Metabolic Engineering: Golden Gate Assembly allows researchers to construct entire synthetic metabolic pathways by seamlessly assembling multiple genes and regulatory elements into a single operon or multigene construct, enabling the production of complex biomolecules or the introduction of novel metabolic functions into host organisms [1].
- Viral Vector and Vaccine Development: The system is instrumental in constructing complex viral genomes for gene therapy and vaccine development. For instance, a plasmid-based reverse genetics system using seven vectors with Type IIS sites was developed to seamlessly assemble the full-length ~30,000 nucleotide SARS-CoV-2 genome cDNA, facilitating the study of viral mutations and the development of countermeasures [1].
- Advanced Plant Biotechnology: Golden Gate toolkits like MoClo and GoldenBraid are extensively used in plant genetic engineering to build complex multigene constructs for crop improvement, enabling the stacking of multiple traits such as disease resistance and drought tolerance in a precise, predictable manner [1] [9].
- CRISPR-Cas9 and Genome Editing: Golden Gate Assembly is the method of choice for constructing CRISPR-Cas9 vectors by efficiently assembling multiple guide RNA expression cassettes and the Cas9 nuclease coding sequence into a single vector, streamlining the creation of sophisticated genome editing tools [1].
The core mechanism of Type IIS restriction enzymes, which separates DNA recognition from catalytic cleavage, is the fundamental innovation enabling the seamless and scarless assembly characteristic of the Golden Gate method. This application note has detailed the biochemical principles, provided a comparative analysis of key enzymes, and outlined a robust protocol for assembling multigene constructs via the MoClo system. As the demand for complex genetic engineering in therapeutic development and synthetic biology grows, mastery of Golden Gate Assembly and its underlying mechanisms becomes increasingly critical for researchers aiming to engineer biological systems with high precision and efficiency.
The advent of Golden Gate assembly has revolutionized the construction of complex multigene DNA constructs, becoming an indispensable tool in synthetic biology and pharmaceutical development. This protocol employs Type IIS restriction enzymes, which cleave DNA outside their recognition sequences, enabling the seamless, one-pot assembly of multiple DNA fragments [10] [5]. For research on multigene constructs—such as those for metabolic engineering, recombinant protein production, and synthetic gene circuits—the method offers three paramount advantages: exceptional scalability for assembling dozens of fragments, precise directionality to control the order and orientation of parts, and unparalleled reusability of standardized DNA parts [11] [12]. These features collectively address the pressing need for high-throughput, reliable, and flexible cloning strategies in modern drug development and basic research. This application note details the underlying mechanisms and provides robust protocols to leverage these benefits effectively.
The Golden Gate Mechanism: A Foundation for Advanced Cloning
The core principle of Golden Gate assembly relies on the unique properties of Type IIS restriction enzymes (e.g., BsaI, BsmBI). Unlike traditional restriction enzymes, Type IIS enzymes bind to a specific DNA recognition site but cleave the DNA strand at a predetermined distance away from this site [10] [13]. This cleavage produces user-defined, non-palindromic overhangs, often 4 bases in length, which are independent of the enzyme's recognition sequence.
- One-Pot Reaction: The assembly is performed in a single-tube reaction containing the Type IIS enzyme, DNA ligase (commonly T4 DNA ligase), the destination vector, and the DNA fragments to be assembled. The reaction is typically cycled between the restriction enzyme's optimal temperature (e.g., 37°C) and the ligase's optimal temperature (e.g., 16°C). This cycling drives the reaction forward; any incorrectly ligated products that retain the Type IIS recognition site are re-digested, while the correct final product, lacking the recognition site, is stable and accumulates [10] [5] [12].
- Seamlessness: Because the enzyme recognition sites are positioned distal to the cleavage site, they are removed from the assembly during digestion. The resulting fragments are ligated via their complementary overhangs, leaving no extra nucleotides ("scars") between the assembled parts [5] [13]. This scarless ligation is critical for maintaining open reading frames and generating native-like genetic constructs.
The following diagram illustrates the core workflow of a Golden Gate assembly reaction, from initial digestion to the final seamless product.
Key Advantages and Quantitative Assessment
Scalability: Hierarchical Assembly of Complex Constructs
Scalability refers to the ability to assemble a large number of DNA fragments into a single construct efficiently. Golden Gate achieves this through hierarchical assembly strategies such as the Modular Cloning (MoClo) system [12]. In this framework, basic genetic elements (promoters, coding sequences, terminers) are first cloned into Level 0 vectors. These are then assembled into transcription units (Level 1), which can be further combined into multigene constructs (Level 2 and beyond) [11] [12]. This modular approach breaks down the assembly of highly complex constructs into manageable, standardized steps.
Research demonstrates the remarkable scalability of this method. The original MoClo study successfully assembled a 33 kb construct containing 11 transcription units from 44 individual basic modules in just three sequential cloning steps [12]. Advances in understanding ligase fidelity and reaction optimization have further pushed these limits, enabling the assembly of more than 50 DNA fragments in a single reaction [10].
Table 1: Quantitative Performance of Golden Gate Assembly
| Metric | Performance Data | Context / Citation |
|---|---|---|
| Fragments in Single Reaction | >50 fragments | Optimized protocol [10] |
| Assembly Efficiency | 95-100% correct colonies | For assemblies of up to 10 fragments [12] |
| Construct Size Demonstrated | 33 kilobases (kb) | 11 transcription units [12] |
| Standard Overhang Length | 4 bases | Allows for 256 (4^4) possible unique sequences [10] [5] |
Directionality: Precursive Control over Assembly Order
Directionality ensures that DNA fragments assemble in a predefined order and orientation. This is a fundamental advantage over traditional cloning and is critical for building functional genetic devices. In Golden Gate assembly, directionality is encoded by the unique 4-base overhangs created by the Type IIS enzyme [10] [5]. Each overhang sequence is designed to be complementary only to the overhang of its intended neighbor. This design prevents incorrect assemblies and ensures the orderly and oriented ligation of multiple fragments in a single reaction [13]. The specificity of these interactions allows researchers to pre-determine the exact architecture of the final multigene construct.
Reusability: Standardized Parts for High-Throughput Workflows
Reusability is a cornerstone of synthetic biology, and Golden Gate cloning excels through the creation of standardized, reusable part libraries. DNA parts—such as promoters, coding sequences, and terminators—are individually cloned into standardized entry vectors (e.g., Level 0 modules in MoClo) [11] [12]. Once a part is cloned, sequenced, and validated, it becomes a permanent resource that can be easily shared and reused in countless assembly reactions without the need for re-cloning [11] [6]. This "clone once, use forever" paradigm drastically reduces labor, time, and cost, and promotes reproducibility and collaboration across the scientific community. Numerous standardized toolkits for various host organisms are available from repositories like Addgene [11].
The diagram below illustrates how these three advantages interconnect within a hierarchical assembly workflow, enabling the reuse of standardized parts to build ever-larger constructs with precise directionality.
Application Notes for Multigene Construct Research
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of Golden Gate assembly relies on a set of core reagents. The table below details essential materials and their functions.
Table 2: Key Research Reagent Solutions for Golden Gate Assembly
| Reagent / Material | Function & Importance | Examples & Notes |
|---|---|---|
| Type IIS Restriction Enzyme | Catalyzes digestion, generating defined overhangs. | BsaI-HFv2, BsmBI-v2; High-fidelity (HF) versions reduce star activity [10]. |
| T4 DNA Ligase | Joins DNA fragments via complementary overhangs. | Requires ATP; often provided in specialized master mixes [10] [14]. |
| Entry/Level 0 Vectors | Backbones for hosting and storing standardized DNA parts. | Vectors contain antibiotic resistance and a cloning cassette with Type IIS sites [11] [12]. |
| Destination Vectors | Accept assembled transcription units or multigene constructs. | Designed with outward-facing Type IIS sites for final assembly [5] [12]. |
| Standardized Part Libraries | Collections of pre-cloned genetic elements. | Available from Addgene (e.g., MoClo Toolkit, GoldenBraid Kit) for various organisms [11]. |
| ccdB Negative Selection | Counterselection marker to reduce background from empty vectors. | Toxin gene replaced during cloning, eliminating non-recombinant clones [6]. |
Standard Protocol for Multigene Assembly
This protocol is adapted for assembling multiple DNA fragments into a destination vector, suitable for creating multigene constructs for expression in various host systems [10] [14] [13].
Materials:
- Purified plasmid DNA: Entry clones (e.g., Level 0 modules) and destination vector.
- Type IIS Restriction Enzyme (e.g., BsaI-HFv2).
- T4 DNA Ligase and corresponding reaction buffer (or commercial Golden Gate assembly mix).
- Thermocycler.
- Competent E. coli cells.
Method:
- Reaction Setup: In a single tube, combine the following:
- 50-100 ng of destination vector.
- Molar equivalent of each insert fragment (a typical fragment:vector ratio is 2:1).
- 1 µL of Type IIS restriction enzyme (e.g., BsaI-HFv2).
- 1 µL of T4 DNA Ligase (or follow kit instructions if using a master mix).
- 1X T4 DNA Ligase Reaction Buffer.
- Nuclease-free water to a final volume of 20 µL.
- Thermocycling: Place the reaction tube in a thermocycler and run the following program:
- Cycle (repeat 25-50 times):
- 37°C for 2-5 minutes (digestion)
- 16°C for 2-5 minutes (ligation)
- Final Digestion: 60°C for 5-10 minutes (optional, to digest any residual misassembled products).
- Hold: 4°C or 80°C for enzyme heat inactivation.
- Cycle (repeat 25-50 times):
- Transformation and Screening:
- Transform 2-5 µL of the final reaction into competent E. coli cells.
- Plate onto LB agar plates containing the appropriate antibiotic for the destination vector.
- Screen resulting colonies by colony PCR, restriction digest, or sequencing.
Advanced Technique: Golden EGG for Simplified Workflow
The Golden EGG (Entry for Golden Gate cloning) system is a recent innovation that simplifies the initial creation of entry clones. It uses a single universal entry vector and a single Type IIS enzyme (e.g., BsaI) for both creating entry clones and performing the final assembly [6].
Key Modifications to the Standard Protocol:
- Primer Design: PCR primers are designed with a specific 5' extension (
NGGTCTCHGTCTCNn1n2n3n4) that, after cloning and digestion, allows the release of the insert with any desired overhang sequence. - Cloning Site: The pEGG entry vector contains a cassette with a ccdB negative selection marker, flanked by outward-directed BsaI sites.
- Temperature Profile: A critical "cold treatment" step (incubation at 4°C) is incorporated to shift the reaction kinetics towards ligation, maximizing the yield of correct entry clones despite the presence of internal BsaI sites in the ligation product [6]. This eliminates the need for strict domestication of internal sites and streamlines the entire process.
Golden Gate assembly provides a powerful and efficient framework for constructing complex multigene DNA molecules. Its core advantages—scalability through hierarchical design, directionality via programmable overhangs, and reusability of standardized parts—make it an superior choice for high-throughput research and development in synthetic biology and drug discovery. The provided protocols and toolkit offer a clear roadmap for scientists to implement this technology, enabling the rapid and reliable engineering of biological systems for advanced therapeutic applications.
The demand for complex DNA constructs in synthetic biology and drug development has driven the creation of standardized, high-throughput cloning methods. Among these, Modular Cloning (MoClo), GoldenBraid, and the iGEM BioBrick system represent prominent tiered assembly standards that leverage the power of Golden Gate assembly. These systems provide hierarchical, modular workflows for efficiently assembling multiple genetic parts into functional multigene constructs, significantly accelerating research in metabolic engineering, genetic circuit development, and therapeutic protein production [15] [16].
Golden Gate assembly itself is a pivotal method that exploits the properties of Type IIS restriction enzymes, which cut DNA outside their recognition sites, creating unique 4-base overhangs. This enables the seamless, one-pot, directional assembly of multiple DNA fragments without introducing scar sequences [17]. The standardization of parts and assembly rules across these systems allows for the creation of reusable part libraries and facilitates collaboration across research institutions by ensuring compatibility and reproducibility [16].
Core Principles of Golden Gate Assembly
Golden Gate assembly enables efficient, seamless DNA construction through the simultaneous activity of a Type IIS restriction enzyme and DNA ligase. Type IIS enzymes such as BsaI and BsmBI recognize asymmetric DNA sequences but cleave outside these sites, generating user-defined 4-base overhangs. This mechanism allows for the ordered assembly of multiple DNA fragments in a single reaction, as the original restriction sites are eliminated from the final construct, making the process irreversible [17].
The assembly relies on careful design of complementary overhangs between adjacent DNA fragments to ensure correct ordering. The use of T4 DNA ligase ensures covalent bonding after fragment annealing. This method has been optimized to assemble up to 52 DNA fragments in a single reaction, demonstrating remarkable capability for constructing complex genetic systems [17] [16]. Key advantages include:
- Seamless Assembly: No nucleotide scars remain between assembled fragments
- Directional Cloning: Proper overhang design ensures correct fragment orientation
- High Efficiency: Simultaneous digestion and ligation in one tube reduces hands-on time
- Modularity: Standardized parts can be reused in different combinations
Comparative Analysis of Cloning Standards
System Architectures and Methodologies
MoClo (Modular Cloning) The MoClo system employs a three-level hierarchical architecture (Level 0, 1, and 2) for constructing multigene constructs. Basic genetic elements (promoters, coding sequences, terminators) are first cloned as standardized Level 0 modules. These are assembled into Level 1 vectors to create complete transcriptional units. Finally, multiple Level 1 plasmids are combined into Level 2 vectors capable of holding up to six transcriptional units, with potential for further iteration to increase complexity [15] [18]. The system primarily uses BsaI for Level 0 to Level 1 assemblies and BpiI (also known as BbsI) for Level 1 to Level 2 assemblies, with careful domestication of parts to eliminate internal restriction sites [19].
GoldenBraid GoldenBraid employs a dual cassette system where transcriptional units are assembled in standard α and β vectors. These are then combined through a binary rotation strategy that allows for iterative assembly of increasingly complex constructs. The system is designed for infinite recursion, enabling the creation of very large DNA assemblies. GoldenBraid 2.0 includes specialized parts for plant synthetic biology and CRISPR/Cas9 applications [15] [18].
iGEM BioBrick System The iGEM standard, used by the international Genetically Engineered Machine competition, employs a simpler three-part assembly system based on standard prefixes and suffixes. While earlier versions used traditional restriction enzymes, more recent implementations have incorporated Golden Gate assembly methods. The system is designed for educational use and standardization across multiple laboratories, emphasizing part reuse and documentation [20].
Comparative Technical Specifications
Table 1: Technical comparison of cloning standards
| Feature | MoClo | GoldenBraid | iGEM BioBrick |
|---|---|---|---|
| Assembly Method | Golden Gate | Golden Gate | Traditional REST/Golden Gate |
| Hierarchical Levels | 3+ (Level 0, 1, 2...) | Binary recursion | Single/Two-level |
| Key Enzymes | BsaI, BpiI/BbsI | BsaI, BsmBI | EcoRI, XbaI, SpeI (traditional) |
| Modularity | High | High | Medium |
| Scalability | High (6+ genes) | Very High (iterative) | Limited |
| Primary Applications | Plant, yeast, bacterial engineering | Plant synthetic biology, CRISPR | Educational, basic genetic circuits |
| Part Reuse | Extensive libraries | Specialized collections | Large international repository |
Table 2: Available toolkit resources for each standard
| System | Toolkit Name | Organism | Key Components |
|---|---|---|---|
| MoClo | MoClo Plant Parts Kit | Plants | 95 standardized parts for plant synthetic biology [15] |
| MoClo-YTK | Yeast | 96 characterized parts for S. cerevisiae [15] | |
| EcoFlex MoClo Toolkit | Bacteria | 78 parts including promoters, RBS, tags for E. coli [15] | |
| GoldenBraid | GoldenBraid 2.0 Kit | Plants | 94 plasmids for plant synthetic biology, CRISPR/Cas9 [15] |
| iGEM | Registry of Standard Biological Parts | Multiple | Thousands of parts contributed by international teams |
Experimental Protocols
Golden Gate Assembly Protocol for Multigene Constructs
The following protocol adapts the NEBridge Golden Gate Assembly system for constructing multigene assemblies using standardized parts [21]:
Reagents and Materials:
- NEBridge Ligase Master Mix (NEB #M1100)
- Type IIS restriction enzyme (BsaI-HFv2 or similar)
- DNA fragments (approximately 0.05 pmol each)
- Molecular biology grade water
- PCR tubes and thermocycler
Procedure:
- Reaction Setup: In a PCR tube on ice, combine:
- 5 µL NEBridge Ligase Master Mix (3X)
- 0.05 pmol of each DNA fragment (including backbone)
- 1 µL BsaI-HFv2 restriction enzyme
- Molecular water to 15 µL total volume
For complex assemblies (7+ fragments), scale to 30 µL total volume with 10 µL Master Mix.
Thermocycling Conditions:
- For 3-6 fragment assembly: 30 cycles of (37°C for 1 min + 16°C for 1 min)
- For 7+ fragment assembly: 30 cycles of (37°C for 5 min + 16°C for 5 min)
- Final extension: 60°C for 5 minutes
- Hold at 4°C indefinitely
Post-Assembly Processing:
- Transform 2-5 µL directly into competent E. coli
- Alternatively, analyze assembly by agarose gel electrophoresis
- Store unused reaction at -20°C for future use
MoClo Level 0 Module Construction
This protocol describes the creation of standard Level 0 MoClo parts, which form the foundation of the hierarchical system [19]:
Part Identification and Design:
- Define the genetic element to be cloned (promoter, CDS, terminator)
- Verify absence of internal BsaI, BpiI, and BsmBI sites
- If sites are present, design strategies for domestication (silent mutation)
Primer Design:
- Design primers to amplify part with appropriate overhangs
- Include BsaI recognition sites flanking the part
- Ensure compatibility with destination vector
PCR Amplification:
- Amplify part with high-fidelity DNA polymerase
- Purify PCR product to remove enzymes and nucleotides
Golden Gate Cloning:
- Set up reaction with purified PCR product, Level 0 vector, BsaI enzyme, and ligase
- Incubate using standard thermocycling conditions (as above)
- Transform into competent cells and plate on selective media
Sequence Verification:
- Screen colonies by colony PCR or restriction digest
- Sequence validated parts to ensure fidelity
- For large parts, consider sub-cloning as Level -1 constructs first
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key research reagents for Golden Gate assembly workflows
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Type IIS Enzymes | BsaI-HFv2, BsmBI-v2, BbsI | Create 4-bp overhangs for fragment assembly; HF versions reduce star activity [17] |
| Ligase Master Mix | NEBridge Ligase Master Mix | Optimized blend of high-fidelity ligase and buffers for efficient Golden Gate assembly [21] |
| Modular Toolkits | MoClo Plant Parts, MoClo-YTK, EcoFlex | Pre-assembled collections of standardized genetic parts for specific applications [15] |
| Destination Vectors | Level 0, 1, 2 Vectors | Hierarchical acceptor plasmids with appropriate selection markers and replication origins [15] |
| Computational Tools | NEBridge Golden Gate Assembly Tool | Web-based tool for designing overhangs and identifying internal restriction sites [17] |
Workflow and System Architecture Visualization
Applications in Research and Drug Development
These standardized cloning systems have enabled significant advances across biological research and therapeutic development:
Pharmaceutical Protein Production: MoClo-YTK and related yeast toolkits allow rapid optimization of protein expression strains for producing therapeutic proteins, with systems available for S. cerevisiae, P. pastoris, and Y. lipolytica [15]. The modular nature enables systematic testing of promoter strength, secretion signals, and gene dosage effects.
CRISPR-Based Therapeutic Development: The ENABLE toolkit provides streamlined assembly of CRISPR-Cas9 vectors for plant and mammalian cell engineering, supporting both stable integration and transient expression systems for functional genomics and gene therapy development [15].
Metabolic Pathway Engineering: CIDAR MoClo and EcoFlex systems enable combinatorial assembly of biosynthetic pathways in E. coli, allowing rapid prototyping of enzyme variants and regulatory elements for producing small molecule therapeutics and natural products [15].
Microbial Consortia Development: Toolkits for non-model bacteria and fungi (Cultivarium POSSUM) facilitate engineering of microbial consortia for complex biotransformations, expanding the range of producible compounds for drug discovery and development [15].
These standardized systems collectively provide the synthetic biology foundation for next-generation biotherapeutics and sustainable biomedicine, accelerating the design-build-test cycle through reusable, well-characterized genetic parts and predictable assembly methods.
Golden Gate Assembly is a powerful molecular cloning technique that enables the seamless, one-pot assembly of multiple DNA fragments. Its efficiency stems from the use of Type IIS restriction enzymes, which cleave DNA outside of their recognition sites, creating unique, non-palindromic overhangs that facilitate the ordered ligation of fragments [5] [22]. This method is particularly vital for constructing complex multigene constructs in synthetic biology and metabolic engineering, where the precise assembly of genetic elements like promoters, coding sequences, and terminators is required [9] [12]. The process relies on a hierarchical workflow centered on three core components: entry vectors, which store standardized DNA parts; destination vectors, which accept assembled constructs; and the designed overhangs that dictate the order and orientation of assembly. This article details the function and application of these essential elements within the context of advanced research and drug development.
The Core Components of Golden Gate Systems
Entry Vectors: Libraries of Standardized Parts
Entry vectors serve as the foundational repository for basic, validated DNA modules. These modules can include promoters, 5' untranslated regions (UTRs), coding sequences (CDSs), and terminators [12]. A key principle in modular cloning (MoClo) systems is that each type of genetic part is flanked by specific, standardized fusion sites (overhangs). For instance, all promoter parts in a MoClo library might be flanked by 5'-GGAG and 3'-TACT overhangs, while CDS parts are flanked by 5'-AATG and 3'-GCTT, ensuring they are freely interchangeable and assemble in the correct order [23] [12].
Simplified Systems: Recent developments, such as the Golden EGG system, have simplified entry clone creation by using a single, universal entry vector for all DNA parts. This system employs a specialized primer design that allows any DNA fragment to be cloned into the same vector location, from which it can later be released with any desired overhang sequence using the same Type IIS enzyme, streamlining the process and enhancing compatibility with existing toolkits [6].
Table 1: Common Genetic Parts and Their Standardized Overhangs in MoClo Systems
| Genetic Part Type | 5' Overhang | 3' Overhang | Purpose and Notes |
|---|---|---|---|
| Promoter (P) | GGAG | TACT | Flanks regulatory elements; positioned upstream. |
| Coding Sequence (CDS) | AATG | GCTT | AATG contains the start codon; for cytosolic proteins [12]. |
| Coding Sequence (no stop) | AATG | ...* | Used for C-terminal fusions to tags or other proteins. |
| Terminator (T) | ...* | CGCT | Flanks the transcription termination sequence. |
| Signal Peptide (SP) | ...* | AGGT | AGGT encodes a glycine, common in signal peptides [12]. |
| 5' UTR (U) | TACT | AATG | Connects promoter to the coding sequence. |
Note: Specific overhangs may vary between different MoClo kits. The ellipsis (...) indicates that the overhang is defined by the specific standard being used and must be compatible with the adjacent part. Researchers must consult their specific kit's documentation.
Destination Vectors: Acceptors for Assembled Constructs
Destination vectors are engineered to accept the DNA fragments released from entry vectors. They typically contain two outward-facing Type IIS recognition sites that flank a "dropout" cassette, often a marker gene like lacZ or a toxic gene like ccdB [5] [6] [22]. During the Golden Gate reaction, digestion of the destination vector removes this cassette, leaving behind vector backbone ends with specific overhangs (OHL and OHR) that are complementary to the first and last fragments of the assembly, respectively [6].
The hierarchy of the cloning system is reflected in the destination vectors. In a MoClo system, Level 1 destination vectors are designed to accept basic parts from Level 0 modules to assemble single transcription units. Level 2 destination vectors are then used to accept multiple transcription units (from Level 1) to build multigene constructs [22] [12]. The use of different antibiotic resistance markers at each level allows for efficient selection of correctly assembled plasmids [23].
Overhangs: The Directors of Assembly
Overhangs, or fusion sites, are the 4-base single-stranded DNA ends generated by Type IIS enzyme cleavage. They are the critical feature that dictates the order and orientation of fragment assembly [5]. The principle is simple: two fragments will ligate only if their overhangs are complementary. By designing a series of unique, sequential overhangs, a researcher can direct multiple fragments to assemble in a predetermined linear order in a single reaction [22].
The design is scarless because the Type IIS recognition sites themselves are cleaved off and are not present in the final assembled construct. This leaves only the intended genomic or synthetic sequence, which is essential for creating functional proteins and genetic elements without unwanted amino acid additions or regulatory disruptions [5] [22]. The high fidelity of T4 DNA ligase for correctly matched overhangs ensures a low frequency of errors in the final product [24].
Diagram 1: This workflow illustrates the hierarchical nature of a standardized MoClo system. Level 0 involves the assembly of basic genetic parts into a single transcription unit (Level 1) using one Type IIS enzyme (e.g., BsaI). Multiple Level 1 units are then assembled into a complex multigene construct (Level 2) using a different Type IIS enzyme (e.g., BpiI/BbsI) [22] [12].
Essential Protocols for Golden Gate Assembly
Protocol 1: Designing and Domesticating DNA Parts
Principle: Successful assembly requires careful in silico planning to ensure overhangs are correctly designed and that internal Type IIS recognition sites within the fragments or vector backbone are eliminated—a process known as domestication [5] [23].
Methodology:
- Define Final Construct: Map the structure of the desired final multigene construct to identify all required basic parts (promoters, CDSs, etc.) [9].
- Select Overhangs: Assign standardized, sequential overhangs to each junction between parts, ensuring correct order and orientation. Use web tools like the NEBridge Golden Gate Assembly Tool to simplify this process [24].
- Check for Internal Sites: Use sequence analysis software to scan all DNA parts and the destination vector for the recognition site of the Type IIS enzyme being used (e.g., BsaI). Internal sites will be cleaved during the reaction, disrupting the assembly.
- Domesticate Internal Sites: Remove unwanted recognition sites by introducing silent mutations into coding sequences or by mutating non-coding regions if it does not affect function. This can be done via site-directed mutagenesis or, more efficiently, by employing gene synthesis services to supply pre-domesticated sequences [5] [23].
Protocol 2: Setting Up the Golden Gate Reaction
Principle: The one-pot restriction-ligation reaction cycles between the optimal temperatures for the Type IIS restriction enzyme (37°C) and T4 DNA ligase (16°C), allowing for iterative digestion and ligation until the correct, stable product is formed [5] [22].
Step-by-Step Methodology:
- Calculate Molar Ratios: To maximize ligation efficiency, calculate the volume of each plasmid (entry vectors and destination vector) based on its concentration and size to achieve an equimolar ratio of all fragments. A typical insert-to-vector molar ratio is 2:1 to 3:1 [23]. The equation for the volume of a given plasmid to use is: Volume (µL) = (Amount of Vector (fmol) × Molar Ratio × Insert Size (bp)) / (Vector Size (bp) × Plasmid Concentration (fmol/µL))
- Prepare Reaction Mix: Combine the following components in a single tube:
- DNA: ~100 fmol of destination vector and the calculated amounts of each entry vector/insert.
- Enzymes: 1 µL of Type IIS restriction enzyme (e.g., BsaI-HFv2 or BsmBI-v2) and 0.5-1 µL of T4 DNA ligase (or high-fidelity ligase master mixes) [24].
- Buffer: 2 µL of 10× T4 DNA ligase buffer (which contains ATP). The final reaction volume is typically 20 µL.
- Run Thermocycler Program: Place the tube in a thermocycler and run a program that cycles between digestion and ligation temperatures. A standard protocol is:
- Transform and Screen: Transform 1-5 µL of the reaction into competent E. coli. Screen resulting colonies by colony PCR, restriction digest, and/or sequencing. For complex multigene constructs, it is advisable to screen a minimum of 8-10 colonies [23].
Diagram 2: Architecture of Entry and Destination Vectors. Entry vectors hold DNA parts flanked by inward-facing Type IIS sites. Destination vectors contain an outward-facing Type IIS site at each end of a dropout cassette. Digestion releases the insert from the entry vector and the cassette from the destination vector, generating complementary overhangs (OHL/OHR) for ligation [5] [6] [22].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Golden Gate Assembly
| Reagent / Kit | Function / Application | Examples and Notes |
|---|---|---|
| Type IIS Restriction Enzymes | Cleaves DNA outside recognition site to generate defined overhangs. | BsaI-HFv2: Most common, 4-bp overhangs. BsmBI-v2: Common alternative. BbsI (BpiI): Used in hierarchical MoClo steps [22] [24]. |
| DNA Ligase | Joins DNA fragments via complementary overhangs. | T4 DNA Ligase: Standard enzyme. High-Fidelity T4 Ligase Master Mixes: Reduce mis-ligation of non-compatible ends, enabling >10 fragment assemblies [24]. |
| Modular Cloning (MoClo) Kits | Pre-assembled libraries of standardized genetic parts and vectors. | Yeast MoClo Kit, Plant MoClo Toolbox. Provide Level 0, 1, and 2 vectors with standardized overhangs for building multigene constructs [25] [23] [12]. |
| Golden Gate Entry Vectors | Plasmid for housing and storing individual standardized DNA parts. | pGGA, pEGG vectors. Often feature negative selection markers (e.g., ccdB) to counter-select against empty vectors [5] [6]. |
| Software & Web Tools | In silico design and simulation of assembly reactions. | SnapGene, NEBridge Golden Gate Assembly Tool. Crucial for planning overhangs, checking for internal restriction sites, and simulating final constructs [5] [23] [24]. |
| Competent E. coli Strains | Transformation of assembled plasmid after reaction. | High-Efficiency Strains: Recommended for complex assemblies with many fragments to increase colony yield [23]. |
Step-by-Step Protocol: Building Multigene Constructs with Golden Gate Assembly
Within the broader context of developing a robust Golden Gate assembly protocol for multigene constructs, the formulation of the master mix is a critical determinant of success. The Golden Gate method is a one-pot, one-step cloning procedure that uses Type IIS restriction enzymes and DNA ligase to seamlessly assemble multiple DNA fragments in a defined order [5]. This technique is particularly valuable for synthetic biology and advanced genetic engineering, enabling the construction of complex multigene pathways for applications in metabolic engineering and therapeutic development [9]. Precision in master mix preparation ensures efficient digestion and ligation, which is paramount for assembling multiple transcription units into functional genetic circuits. This application note provides detailed methodologies and calculations for formulating optimized Golden Gate assembly reactions, specifically tailored for complex multigene constructs.
Core Principles and Reaction Components
Golden Gate assembly relies on the coordinated activity of a Type IIS restriction enzyme and a DNA ligase within a single reaction tube. Type IIS enzymes, such as BsaI-HFv2 or BsmBI-v2, recognize non-palindromic sequences and cleave outside their recognition sites, generating unique, user-defined 4-base overhangs [26] [5]. The ligase then joins these complementary overhangs. Because the recognition sites are eliminated in the final assembled product, the reaction is driven to completion as correctly assembled plasmids cannot be re-digested and are thus stable [26]. This "scarless" assembly is ideal for hierarchical construction systems like MoClo (Modular Cloning) for building multigene constructs [9].
For complex assemblies involving many fragments, the reaction efficiency can be significantly increased by extending the number of thermal cycles, as the enzymes remain stable and active through extended cycling protocols [27].
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details the key reagents required for a standard Golden Gate assembly reaction.
Table 1: Essential Reagents for Golden Gate Assembly
| Reagent | Function | Key Considerations |
|---|---|---|
| Type IIS Restriction Enzyme (e.g., BsaI-HFv2) | Digests vector and insert DNA to generate specific overhangs. | Choose an enzyme with a longer recognition site (e.g., 7-base PaqCI) to minimize internal site issues [27]. |
| T4 DNA Ligase | Joins complementary overhangs from digested fragments. | Uses ATP; ensure buffer is supplemented accordingly. Highly stable during extended cycling [27]. |
| Reaction Buffer | Provides optimal ionic and pH conditions for both restriction and ligation. | T4 DNA Ligase Buffer is often optimal; other buffers (e.g., NEBuffer r1.1) can be used with 1 mM ATP and 5-10 mM DTT [27]. |
| Destination Vector | Backbone for the final assembled construct. | Must be free of internal Type IIS sites. Contains outward-facing sites flanking the cloning cassette [26] [5]. |
| Insert DNA(s) | DNA fragments to be assembled. | Can be PCR amplicons or pre-cloned "entry clones." Must be free of internal Type IIS sites for the enzyme used [5]. |
| pGGAselect Vector | A versatile destination vector included in some kits. | Compatible with BsaI, BsmBI, and BbsI assemblies and lacks internal sites for these enzymes [27] [26]. |
Master Mix Formulation and Calculations
A precisely calculated master mix ensures consistent enzyme activity and ligation efficiency across all reactions, which is especially critical for multigene assemblies with many fragments.
Standard Master Mix Protocol for a 20 µL Reaction
The following table outlines a standard setup for a Golden Gate assembly using commonly available enzymes. The volumes are calculated for a single reaction.
Table 2: Standard 20 µL Golden Gate Assembly Master Mix
| Component | Final Concentration/Amount | Volume per 20 µL Reaction (µL) |
|---|---|---|
| 10X T4 DNA Ligase Buffer | 1X | 2.0 |
| Destination Vector | 50-75 ng | x (variable, typically ~0.02-0.10 pmol) |
| Each Insert Fragment | 50-75 ng (for pre-cloned) | y (variable, typically ~0.02-0.10 pmol each) |
| Type IIS Enzyme (e.g., BsaI-HFv2, 20,000 U/mL) | 1.0 µL | 1.0 |
| T4 DNA Ligase (400,000 U/mL) | 0.5 µL | 0.5 |
| Nuclease-free Water | To 20 µL | To 20 µL |
Protocol Steps:
- Thaw and Prepare: Thaw all reagents on ice and briefly vortex and spin them down before use.
- Formulate Master Mix: In a sterile tube, combine the T4 DNA Ligase Buffer, Type IIS Enzyme, T4 DNA Ligase, and nuclease-free water. Mix thoroughly by pipetting. This "Master Mix" can be prepared for n+1 reactions to account for pipetting error.
- Aliquot DNA: Dispense the calculated volumes of destination vector and each insert DNA into the reaction tube(s).
- Assemble Reaction: Add the appropriate volume of the Master Mix to each DNA sample. Mix the complete reaction gently by pipetting.
- Thermal Cycling: Place the tube(s) in a thermocycler and run the following program [27]:
- Cycle Step 1: 25-30 cycles of:
- Digestion/Ligation: 37°C for 2-5 minutes (for BsaI)
- Digestion/Ligation: 16°C for 2-5 minutes
- Cycle Step 2: 5 minutes at 50°C (final digestion).
- Cycle Step 3: 5-10 minutes at 80°C (heat inactivation).
- Hold: 4°C hold.
- Cycle Step 1: 25-30 cycles of:
- Transformation: Transform 2-5 µL of the reaction into competent E. coli cells following standard protocols.
Workflow Diagram: Golden Gate Assembly for Multigene Constructs
The following diagram illustrates the hierarchical workflow for constructing multigene assemblies using systems like MoClo, from basic parts to the final construct.
Diagram 1: Hierarchical Multigene Assembly Workflow
Advanced Optimization for Complex Assemblies
Assembling more than 10 fragments presents unique challenges. The following adjustments to the standard protocol can enhance efficiency.
Table 3: Optimization Strategies for Complex Assemblies
| Parameter | Standard Condition | Optimized Condition for >10 Fragments | Rationale |
|---|---|---|---|
| Thermal Cycles | 25-30 cycles | 45-65 cycles [27] | Increases opportunity for correct fragment ligation. |
| Insert Amount | 75 ng each | 50 ng each [27] | Reduces mis-assembly from excess DNA. |
| Overhang Design | - | Use NEBridge Ligase Fidelity Tool [27] | Ensures high-fidelity ligation at all junctions. |
Reaction Kinetics and Temperature Profile
The success of Golden Gate assembly hinges on the precise temperature cycling between the optimal temperatures for digestion (37°C for BsaI) and ligation (16°C). The following diagram details this cyclic process.
Diagram 2: Golden Gate Reaction Kinetics Cycle
Troubleshooting and Quality Control
Even with optimized master mixes, issues can arise. Below are common problems and their solutions.
- Low Efficiency in Complex Assemblies: Ensure the plasmid preps for pre-cloned inserts are free of RNA, which can lead to overestimation of DNA concentration [27]. Verify the absence of primer dimers in PCR-amplified inserts, as these can compete in the assembly reaction [27].
- High Background of Empty Vectors: This often indicates incomplete digestion. Confirm that neither the vector nor the inserts contain internal recognition sites for the Type IIS enzyme used. Use site-directed mutagenesis to "domesticate" any such sites [5].
- Unexpected Assembly Products: Carefully redesign every overhang using tools like the NEBridge Ligase Fidelity Tool to ensure high specificity [27]. If using pre-cloned inserts that were previously functional, sequence them to check for mutations that may have occurred during propagation in E. coli [27].
Within the broader context of developing robust Golden Gate assembly protocols for multigene constructs, the precise control of thermocycler parameters is a critical determinant of success. Golden Gate cloning, which utilizes Type IIS restriction enzymes and DNA ligase in a simultaneous digestion-ligation reaction, enables the seamless assembly of multiple DNA fragments into a single construct. The efficiency of this one-pot reaction hinges on an optimized temperature cycling profile that balances the enzymatic activities for maximal product yield. This application note provides a detailed, optimized protocol for digestion-ligation temperature cycling, supported by quantitative data and tailored for the assembly of complex multigene constructs essential for advanced research and therapeutic development.
Principles of Temperature Cycling in Golden Gate Assembly
The fundamental principle of Golden Gate assembly involves using Type IIS restriction enzymes, which cleave DNA outside of their recognition sites, to create unique, user-defined overhangs on DNA fragments. These fragments are then ligated together in a defined order. In a one-pot reaction, temperature cycling is employed to promote the simultaneous digestion and ligation of DNA fragments.
The cycling drives the reaction forward by repeatedly denaturing incorrectly annealed fragments, cleaving any improperly ligated products, and creating opportunities for correct fragments to anneal via their complementary overhangs and be ligated into the final, stable assembly product. The recognition site for the Type IIS enzyme is eliminated in the correct final product, protecting it from further digestion and favoring its accumulation [6].
Optimized Thermocycler Protocol for Digestion-Ligation
The following protocol is optimized for a typical Golden Gate assembly using enzymes such as BsaI. The volumes and enzyme concentrations can be scaled as needed.
Reagent Setup
| Component | Final Concentration/Amount | Notes |
|---|---|---|
| DNA Parts (Entry Clones/PCR fragments) | 10-50 fmol each | Recommended total DNA: 100-200 ng |
| Type IIS Restriction Enzyme (e.g., BsaI-HFv2) | 1.0 µL (10 U/µL) | |
| T4 DNA Ligase (High-Concentration) | 1.0 µL (30 U/µL) | Ensure ligase buffer contains ATP |
| 10x T4 DNA Ligase Buffer | 2.0 µL | Provides ATP and DTT; critical for ligase activity |
| Nuclease-free Water | to 20 µL |
Temperature Cycling Parameters
The thermocycler program below is designed to maximize the efficiency of both digestion and ligation. The cyclical drop in temperature allows for the ligase to work efficiently, while the rise back to the digestion temperature cleaves any products that were incorrectly ligated, giving the correct assembly another chance to form.
| Step | Temperature | Time | Cycles | Purpose |
|---|---|---|---|---|
| 1. Initial Digestion | 37°C | 5-15 minutes | 1 | Initial digestion of DNA parts to release fragments with designed overhangs. |
| 2. Denaturation | 95°C | 2-5 minutes | 1 | Heat inactivation of the restriction enzyme (if applicable). Not always required. |
| 3. Annealing & Ligation | 55-60°C | 3-5 minutes | Critical step for correct annealing of overhangs. | |
| 4. Ligation | 16-25°C | 3-5 minutes | 20-50x | Optimal temperature for T4 DNA ligase activity. |
| 5. Final Digestion | 37°C | 5-15 minutes | 1 | Ensures complete digestion of any residual incorrect intermediates. |
| 6. Final Ligation | 16°C | 10-30 minutes | 1 | Stabilizes the final correct assembly product. |
| 7. Enzyme Inactivation | 80°C | 5-10 minutes | 1 | Heat inactivation of all enzymes before transformation. |
Workflow Visualization
Key Experimental Parameters and Optimization Data
Successful assembly depends on several key parameters. The data below summarize critical optimization points.
Table 1: Optimization of Critical Reaction Parameters
| Parameter | Recommended Range | Effect of Sub-Optimal Conditions | Optimization Evidence |
|---|---|---|---|
| Number of Cycles | 20 - 50 | <20 cycles: Incomplete assembly. >50 cycles: Potential degradation of products. | Increased colony counts observed with higher cycles (20-50) for complex assemblies [6]. |
| Annealing Temperature | 55°C - 60°C | Too low: Non-specific annealing. Too high: Reduced hybridization of overhangs. | System-specific; testing a gradient (55-65°C) is recommended for new designs [28]. |
| Ligation Temperature | 16°C - 25°C | Higher temperatures reduce stability of annealed fragments, decreasing efficiency. | T4 DNA ligase has optimal activity at 25°C, but 16°C stabilizes DNA interactions [29]. |
| Enzyme Ratio | Varies | High background if ligase is insufficient; incomplete digestion if RE is insufficient. | A typical ratio is 1:1 (RE:Ligase) by units, but should be empirically determined. |
Advanced Consideration: The "Cold Treatment" Method
Recent advancements suggest a modified approach to temperature cycling. Some Type IIS restriction enzymes, like BsaI, retain significant activity at lower temperatures (e.g., 16°C), which can lead to the destabilization of the desired ligated product in the entry clone during the assembly process. To circumvent this, a "cold treatment" can be applied.
Protocol for Cold Treatment [6]:
- Perform the standard digestion-ligation reaction at 37°C for 15 minutes.
- Instead of a standard thermal cycle, transfer the reaction to 4°C (on a cold block or in a refrigerator) for 15-30 minutes.
- This cold shift leverages the fact that T4 DNA ligase retains over 50% of its activity at 0-4°C, while the activity of the restriction enzyme is significantly reduced.
- This kinetic shift favors the circularization of the final ligation products, leading to higher transformation efficiency.
The Scientist's Toolkit: Essential Research Reagents
The following reagents are critical for setting up a successful Golden Gate digestion-ligation reaction.
Table 2: Key Research Reagent Solutions
| Reagent | Function | Critical Notes |
|---|---|---|
| Type IIS Restriction Enzyme (e.g., BsaI, BsmBI) | Cleaves DNA upstream/downstream of its recognition site to generate defined, complementary overhangs on DNA parts. | Use high-fidelity (HF) versions to reduce star activity. Must be compatible with ligase buffer or use a universal buffer [6]. |
| High-Concentration T4 DNA Ligase | Catalyzes the formation of a phosphodiester bond between the 3'-OH and 5'-Phosphate of adjacent DNA fragments. | Essential for one-pot reactions. The buffer must contain ATP [30] [29]. |
| 10x T4 DNA Ligase Buffer | Provides co-factors (Mg²⁺, ATP) and a reducing agent (DTT) essential for ligase activity. | Aliquot buffer to prevent degradation of ATP and DTT from freeze-thaw cycles [30] [29]. |
| DNA Parts (Entry Vectors or PCR Fragments) | The building blocks of the final assembly. Can be promoters, coding sequences, terminators, etc. | Must be devoid of internal recognition sites for the Type IIS enzyme used. Concentration and purity are critical [9] [6]. |
| Polyethylene Glycol (PEG) 4000 | Molecular crowding agent. Increases the effective concentration of DNA and enzymes, significantly boosting ligation efficiency. | Often included in proprietary ligase buffers. For blunt-ended ligations, its addition is highly recommended [30]. |
The implementation of this optimized thermocycler protocol for digestion-ligation temperature cycling provides a reliable foundation for the efficient assembly of multigene constructs via Golden Gate cloning. By carefully controlling the reaction parameters—specifically the alternating temperatures that facilitate digestion and ligation—researchers can achieve high yields of correct assemblies. The inclusion of advanced techniques, such as the "cold treatment," further enhances the robustness of the protocol. This detailed guide, complete with structured data and visual workflow aids, is designed to empower scientists in the fields of synthetic biology and drug development to accelerate their research on complex genetic circuits and pathways.
Golden Gate Assembly is a versatile, one-pot cloning method that enables the seamless, directional assembly of multiple DNA fragments into a single construct. This technique is particularly valuable for synthetic biology and the construction of complex multigene systems, as it allows for the modular, hierarchical assembly of DNA parts in a defined order [31]. The method was invented by Marillonnet and coworkers in 2008 and has since become a cornerstone technique for precision cloning, offering significant advantages over traditional restriction enzyme cloning and other homology-directed assembly methods like Gibson Assembly [31].
The core principle distinguishing Golden Gate Assembly is its use of Type IIS restriction enzymes [31]. Unlike traditional Type IIP restriction enzymes (e.g., EcoRI, BamHI) that cut within palindromic recognition sites and generate self-complementary overhangs, Type IIS enzymes recognize non-palindromic sequences and cut outside of their recognition sites. This produces unique, user-defined, 4-base overhangs that are independent of the enzyme's recognition sequence [31]. This key feature allows for the simultaneous digestion and ligation of multiple DNA fragments in a single reaction, generating a scarless final product devoid of restriction sites at the junctions [31].
The following diagram illustrates the complete experimental workflow, from initial part preparation through to the analysis of the final construct.
DNA Part and Vector Preparation
Designing and Sourcing DNA Parts
The first critical step is obtaining DNA fragments (inserts) compatible with Golden Gate Assembly. Each fragment must be flanked by the appropriate Type IIS restriction enzyme recognition sites, with the internal sequence lacking the recognition site for the enzyme used [31].
- PCR Amplification: The most common method for generating insert DNA is PCR amplification from a genomic or plasmid template. Primers are designed to add the required Type IIS restriction sites and the desired 4-base overhangs to the amplicon [31].
- DNA Synthesis: For complex projects or to avoid internal restriction sites, DNA fragments can be synthesized de novo (e.g., as gBlocks from IDT or gene fragments from Twist). This allows for in silico "domestication"—the removal of internal Type IIS recognition sites via silent mutations during the sequence design phase [31].
Selecting and Preparing the Vector
A suitable destination vector is essential for a successful assembly. The vector must contain the standard features for propagation and selection (e.g., origin of replication, antibiotic resistance marker) and must be engineered for Golden Gate compatibility [31].
- Key Vector Features: The vector must contain a Golden Gate cloning site with two Type IIS recognition sites arranged to point away from each other. When cut, this arrangement excises the fragment between the sites, allowing its replacement with the assembled insert [31]. Some vectors, like the pGGAselect vector included in NEBridge Kits, also incorporate counterselection markers (e.g., sfGFP) between the cloning sites to help identify successful clones by the loss of fluorescence [31].
- Source of Vectors: Compatible vectors are available from commercial suppliers (e.g., NEB) or repositories like Addgene. Researchers can also modify an existing vector by using site-directed mutagenesis to remove internal Type IIS sites and subsequently adding a Golden Gate cloning site [31].
Golden Gate Assembly Reaction Setup
The assembly reaction strategically combines the prepared vector and DNA fragments with the necessary enzymes in a single tube.
Reagent Setup and Calculations
The table below summarizes the key reagents and their roles in the assembly reaction.
Table 1: Key Reagents for Golden Gate Assembly
| Reagent | Function | Common Examples |
|---|---|---|
| Type IIS Restriction Enzyme | Digests DNA fragments and vector to generate complementary overhangs. | BsaI-HFv2, BsmBI-v2, SapI [31] [21] |
| DNA Ligase | Joins the compatible overhangs of the digested fragments. | T4 DNA Ligase [32] |
| DNA Fragments & Vector | The building blocks for the final assembly; must be free of internal restriction sites. | PCR products, synthesized genes, domesticated plasmids [31] |
| Reaction Buffer | Provides optimal ionic and pH conditions for both restriction and ligation activities. | T4 DNA Ligase Buffer, NEBridge Ligase Master Mix [21] [32] |
Accurate calculation of DNA quantities is crucial for high-efficiency assembly. The recommended amount is typically based on molar concentration (fmol) rather than mass (ng) to ensure all fragments are present in equimolar ratios [21] [32].
Table 2: DNA Quantities for Golden Gate Assembly
| Component | Recommended Quantity | Notes |
|---|---|---|
| Each DNA Insert | 0.05 pmol per reaction [21] or 50 fmol/μL final concentration [32] | Use online calculators (e.g., NEBioCalculator) to convert molar amount to mass (ng). |
| Vector Backbone | 0.05 pmol per reaction [21] or 25 fmol/μL final concentration [32] | Using half the molar amount of inserts is a common strategy to favor correct assembly. |
Master Mix Assembly and Thermocycling
The following diagram details the step-by-step procedure for setting up the Golden Gate reaction and the subsequent thermocycling program that drives the assembly.
Protocol Note:
- For a standard reaction with 3-6 fragments, a typical protocol uses 30 cycles of (37°C for 1-2 minutes + 16°C for 1-5 minutes), followed by a final digestion at 55°C for 10 minutes and heat inactivation at 80°C for 10 minutes [21] [32]. The duration of the digestion and ligation steps can be increased for assemblies involving a higher number of fragments (e.g., 5 minutes each for 7+ fragments) [21] [32].
Bacterial Transformation and Clone Validation
Transformation of Assembled Constructs
The product of the Golden Gate reaction is then introduced into competent E. coli cells for propagation. The choice between heat shock and electroporation depends on the desired efficiency and available equipment.
Table 3: Comparison of Transformation Methods
| Aspect | Heat Shock Transformation | Electroporation |
|---|---|---|
| Principle | Cells are made permeable using cations (e.g., CaCl₂) and a brief 42°C heat pulse [33] [34]. | A high-voltage electric pulse creates transient pores in the cell membrane [33] [34]. |
| Efficiency | 1.0 x 10⁵ – 2.0 x 10⁹ CFU/μg [34]. Suitable for obtaining a few positive clones. | 5.0 x 10⁹ – 2.0 x 10¹⁰ CFU/μg [34]. Preferred for complex libraries or low-yield reactions. |
| Key Considerations | Simple, requires only a water bath. Use chemically competent cells [33]. | Higher efficiency but requires an electroporator. Use electrocompetent cells washed in glycerol to prevent arcing [33] [34]. |
Transformation Protocol:
- Thaw Competent Cells: Thaw 50-100 µL of competent cells on ice [33].
- Add DNA: Add 1-10 ng of the Golden Gate reaction product (or 1-5 µL of the ligation mixture) to the cells. Mix gently by flicking the tube. Do not vortex [33].
- Heat Shock or Electroporation:
- Recovery: Add 250 µL to 1 mL of pre-warmed SOC medium to the cells and incubate at 37°C with shaking for 1 hour. This allows the bacteria to express the antibiotic resistance gene on the plasmid [33].
- Plating: Spread 50-100 µL of the cell culture onto an LB agar plate containing the appropriate antibiotic. Incubate the plate overnight at 37°C [33].
Screening and Validation of Transformants
After overnight incubation, colonies should be visible on the plate. These transformed colonies require screening to identify those containing the correct construct.
- Primary Screening (Colony PCR or Visual Screening): For vectors with a counterselection marker like sfGFP, successful clones can be identified under blue light by the loss of fluorescence [31] [32]. Alternatively, colony PCR using primers flanking the insert site can quickly verify the presence and approximate size of the insert.
- Plasmid Isolation (Miniprep): Inoculate a single colony into a small culture (2-5 mL) of antibiotic-containing LB medium and grow overnight [35]. Isolate the plasmid DNA using a commercial miniprep kit (e.g., QIAprep Spin Miniprep Kit). The protocol typically involves cell resuspension, lysis, neutralization, and column-based purification of plasmid DNA [35].
- Final Validation (Restriction Digest and Sequencing): Perform a diagnostic restriction digest on the isolated plasmid using enzymes that will release the insert, confirming the assembly by the resulting fragment pattern on a gel [32]. For ultimate confirmation, especially with multigene constructs, Sanger sequencing of the assembly junctions and the entire insert is recommended.
For large-scale plasmid production required for downstream applications, a maxiprep can be performed from a larger (50-200 mL) bacterial culture using kits such as the ZymoPURE Plasmid Maxiprep Kit [35].
The assembly of multi-gene constructs is a cornerstone of modern synthetic biology, enabling the engineering of complex biological systems. Among the various techniques available, the Golden Gate assembly protocol has emerged as a powerful and efficient method for constructing sophisticated genetic circuits and metabolic pathways. This cloning strategy utilizes Type IIS restriction enzymes, which cleave DNA outside of their recognition sites, allowing for the seamless assembly of multiple DNA fragments in a single reaction [9] [6]. The protocol's simplicity, efficiency, and modularity facilitate the creation of complex multigene constructs through a series of one-pot assembly steps starting from libraries of standardized, sequenced parts [9].
This application note details advanced implementations of the Golden Gate protocol, focusing on its application in large-scale metabolic pathway assembly. We provide a detailed comparative analysis of modern Golden Gate systems, a step-by-step protocol for a simplified assembly workflow, and an innovative strategy for enhancing metabolic flux through enzyme scaffolding. The information is structured to provide researchers, scientists, and drug development professionals with the practical tools necessary to implement these techniques in their own work.
Comparative Analysis of Golden Gate Cloning Systems
Several Golden Gate cloning systems have been developed, each with unique advantages for specific applications. The table below summarizes the key features of prominent systems.
Table 1: Comparison of Golden Gate Cloning Systems
| System Name | Key Features | Primary Type IIS Enzyme(s) | Advantages | Ideal Applications |
|---|---|---|---|---|
| Modular Cloning (MoClo) [9] | Standardized assembly strategy, facilitates variant generation | Not explicitly stated | Well-established, efficient for multigene constructs | Plant synthetic biology, metabolic engineering |
| Golden EGG [6] | Single entry vector, simplified primer design, single-enzyme workflow | Eco31I (BsaI) | User-friendly, cost-effective, compatible with existing toolkits | Streamlined construction of entry clones and assemblies |
| Multi-Kingdom (MK) System [36] | Unified platform, standardized parts and overhangs | BpiI (for entry), BsaI (for Level 1 assembly) | Cross-kingdom compatibility, streamlined workflow for diverse organisms | Research involving multiple model organisms (Fungi, Plantae, Animalia, etc.) |
Protocol: Simplified Multigene Assembly with Golden EGG
Golden EGG offers a streamlined workflow that reduces the complexity and cost of traditional Golden Gate assembly by employing a single universal entry vector and a single Type IIS enzyme for both entry clone creation and final assembly [6].
Research Reagent Solutions
Table 2: Essential Reagents for the Golden EGG Protocol
| Reagent / Material | Function / Description | Example / Note |
|---|---|---|
| pEGG Entry Vector | Universal entry vector with a negative selection cassette (e.g., ccdB and cat genes) flanked by outward-facing BsaI sites. | Provides a consistent backbone for all DNA parts, simplifying design and storage [6]. |
| Type IIS Restriction Enzyme | Enzyme for digestion-ligation. Cleaves outside its recognition site to generate custom overhangs. | Eco31I (BsaI) is used in the original study. BsaI-HFv2 is a common commercial alternative [6] [37]. |
| T4 DNA Ligase | Enzyme for ligating the compatible overhangs of digested DNA fragments. | Crucial for seamless assembly. Often used in the same buffer as the restriction enzyme [6] [37]. |
| PCR Primers with Extensions | Primers designed to amplify DNA parts with 5' extensions containing the BsaI site and desired overhang sequence. | Format: 5'-NGGTCTCNn1n2n3n4-[gene-specific sequence]-3', where n1-n4 define the 4-nt overhang [6]. |
| Thermocycler | Instrument to perform the digestion-ligation reaction with precise temperature cycling. | Enforces the cyclical digestion and ligation necessary for high-efficiency assembly. |
Experimental Workflow
The following diagram illustrates the streamlined Golden EGG cloning workflow, from part preparation to final multigene assembly.
Diagram 1: Golden EGG Assembly Workflow
Step 1: Primer Design and PCR Amplification
- Design primers to amplify each DNA part (e.g., promoters, coding sequences). Each primer must have a 5' extension:
NGGTCTCNn1n2n3n4, wheren1-n4is the desired 4-nucleotide overhang sequence. - Amplify all parts using a high-fidelity DNA polymerase to avoid PCR-induced errors [37].
Step 2: Construction of Entry Clones
- Digest the universal pEGG entry vector and the PCR-amplified parts with BsaI.
- Perform a ligation reaction. A critical optimization is a cold treatment (4°C for 15 minutes) after an initial incubation at 37°C. This shifts reaction kinetics toward ligation, maximizing the yield of circularized entry clones even though the product contains functional BsaI sites [6].
- Transform the ligation product into E. coli and sequence-validate the entry clones.
Step 3: One-Pot Multigene Assembly
- Combine the validated entry clones (now containing the parts flanked by inward BsaI sites) with a destination vector containing outward-facing BsaI sites.
- The destination vector's overhangs (OHL and OHR) must be complementary to the left overhang of the first fragment and the right overhang of the last fragment, respectively [6].
- Set up a Golden Gate digestion-ligation reaction containing BsaI and T4 DNA Ligase.
- Use an extended thermocycling protocol (e.g., 45-65 cycles of 37°C and 16°C) to enhance assembly efficiency for complex mixtures [37].
- Transform the final reaction product to obtain the multigene construct.
Advanced Application: Metabolic Pathway Assembly with Enzyme Scaffolding
While Golden Gate excels at DNA-level assembly, optimizing metabolic flux in engineered pathways requires control at the protein level. The mimic PKS Enzyme Assembly Line (mPKSeal) strategy addresses this by recruiting cascade enzymes into complexes using docking domains (DDs) from type I cis-AT polyketide synthases (PKSs) [38].
The mPKSeal Strategy and Workflow
This approach assembles key metabolic enzymes in close proximity to enhance intermediate channeling and overall biocatalytic efficiency.
Diagram 2: mPKSeal Metabolic Pathway Assembly
Step 1: Selection of Docking Domains
- DDs are short, independently folding regions from PKSs that mediate specific protein-protein interactions.
- DDs from different PKSs (e.g., DEBS, RAPS) or from different classes within the same PKS can serve as orthogonal interaction pairs, allowing for the specific recruitment of different enzymes [38].
Step 2: Construction of Enzyme-DD Fusions
- Genetically fuse a C-terminal DD (CDD) to the upstream enzyme in a metabolic pathway.
- Fuse a compatible N-terminal DD (NDD) to the downstream enzyme.
- These fusions can be constructed using Golden Gate assembly to ensure precise, seamless integration.
Step 3: In Vivo Assembly and Pathway Validation
- Co-express the engineered Enzyme-CDD and Enzyme-NDD fusions in the production host (e.g., E. coli).
- The DDs interact spontaneously, forming a multi-enzyme complex that mimics natural assembly lines.
- In a case study applying mPKSeal to the astaxanthin biosynthetic pathway in E. coli, this strategy increased astaxanthin production by 2.4-fold, demonstrating its efficacy in enhancing metabolic flux [38].
Technical Optimization and Troubleshooting
Successful implementation of complex Golden Gate assemblies requires attention to several technical details.
- Eliminating Internal Restriction Sites: Always check DNA sequences for internal recognition sites of the Type IIS enzyme used. For multi-fragment assemblies, these sites should be removed ("domesticated") via silent mutation or by selecting an enzyme with a longer, rarer recognition site like PaqCI (7-base pair site) [37].
- Ensuring High Fidelity of Overhangs: The design of the 4-base pair overhangs is critical. Using sets of vetted, high-fidelity junction sequences can prevent mis-assembly. Tools like the NEBridge Ligase Fidelity Tool can predict optimal junctions for accurate assembly [39].
- Handling Complex Assemblies: For assemblies involving more than 10 fragments, consider decreasing the amount of each pre-cloned insert from 75 ng to 50 ng to maintain high efficiency. Furthermore, increasing the total thermocycling cycles to 45-65 can significantly improve the yield of correct assemblies without sacrificing fidelity [37].
- Troubleshooting Pre-cloned Parts: If a previously functional entry clone suddenly fails to assemble correctly, verify its sequence. Mutational events, such as frameshifts in homopolymer runs, can occur during propagation in E. coli [37].
Troubleshooting Golden Gate Assembly: Maximizing Efficiency and Accuracy
Golden Gate Assembly is a one-pot, one-step cloning procedure that utilizes Type IIS restriction enzymes to enable the seamless assembly of multiple DNA fragments. This method is particularly valuable for constructing complex multigene constructs, a common requirement in synthetic biology and pharmaceutical development. The technique's efficiency stems from its use of Type IIS restriction enzymes, which cleave DNA outside of their recognition sequences, generating unique, user-defined overhangs that facilitate the ordered assembly of DNA fragments. Unlike traditional restriction cloning, Golden Gate Assembly is considered "scarless" or "seamless" because it does not leave unwanted nucleotides between assembled fragments. The method's robustness and flexibility have led to its adoption in various standardized cloning systems, such as the Modular Cloning (MoClo) system, which enables researchers to assemble multigene constructs through a series of hierarchical, one-pot assembly steps [9] [5].
For research on multigene constructs, Golden Gate Assembly provides a systematic approach for combining basic genetic modules—such as promoters, coding sequences, and terminators—into transcription units, which can then be further assembled into complete multigene constructs. This capability is crucial for metabolic engineering, gene stacking, and the development of complex biological systems in both basic and applied research contexts [9] [5].
Fundamental Principles of Overhang Design
The Role of Type IIS Restriction Enzymes
Type IIS restriction enzymes are the cornerstone of Golden Gate Assembly, possessing unique properties that differentiate them from conventional Type IIP restriction enzymes. Unlike Type IIP enzymes that recognize palindromic sequences and cleave within them, Type IIS enzymes recognize asymmetric sequences and cleave outside of these recognition sites at specific offsets. This external cleavage generates predictable, sticky ends (overhangs) whose sequences are determined by the adjacent DNA sequence rather than the recognition site itself. For example, the commonly used enzyme BsaI recognizes the sequence GGTCTC and cleaves one nucleotide downstream on the top strand and five nucleotides downstream on the bottom strand, generating 4-base overhangs. The length of these overhangs can vary depending on the specific Type IIS enzyme used, with 4-base overhangs being particularly popular as they provide 256 (4^4) possible sequence combinations, enabling the simultaneous assembly of numerous DNA fragments [5].
Core Rules for Overhang Design
Table 1: Fundamental Rules for Overhang Design in Golden Gate Assembly
| Rule Number | Rule Name | Description | Consequence of Violation |
|---|---|---|---|
| 1 | Unique Overhangs | Each fusion point must use a distinct overhang sequence | Assembly of incorrect fragment orders and formation of chimeric constructs |
| 2 | Reverse Complement Avoidance | Avoid overhangs that are reverse-complements of others in the same assembly | Undesired fragment interactions and reduced assembly efficiency |
| 3 | Self-Complementarity Prevention | Avoid overhangs that are self-complementary | Fragment homodimerization and depletion of available parts |
| 4 | Sequence Imposition | Accommodate biologically required sequences when necessary | Incompatibility with functional genetic elements (start codons, etc.) |
| 5 | Ligase Compatibility | Consider enzymatic preferences of DNA ligase | Reduced ligation efficiency despite perfect complementarity |
Designing effective overhangs requires adherence to several fundamental rules that prevent misassembly and ensure high efficiency. Rule 1 (Unique Overhangs) mandates that each fusion point between fragments must have a distinct overhang sequence. If the same overhang is used for different fusion points, the assembly products will lack the correct fragment order, producing incomplete or "monster" DNA molecules. Rule 2 (Reverse Complement Avoidance) requires that no overhang in the set should be the reverse-complement of another overhang in the same assembly. Violating this rule leads to non-productive interactions between fragments not intended to be adjacent, reducing the efficiency of correct assembly. Rule 3 (Self-Complementarity Prevention) prohibits the use of overhangs that are self-complementary (palindromic), as these enable individual fragments to dimerize, effectively removing them from the assembly reaction [40].
In practice, these rules require careful planning and verification. For instance, if Part A connects to Part B using overhang sequence ATGC, and Part C connects to Part D using GGCA, these appear different at first glance. However, if the reverse complement of Part C's overhang is TGCC, which complements Part A's overhang (ATGC), unintended annealing can occur between Part A and Part C, compromising assembly efficiency. Advanced projects often face additional constraints, such as sequence imposition, where specific overhang sequences must be incorporated to maintain biological function. For example, in protein-coding sequences, an overhang might need to include an ATG start codon or specific amino acid codons at fusion points, limiting the available sequence options [40].
Figure 1: Logical workflow for designing compatible overhangs in Golden Gate Assembly, incorporating the fundamental rules to prevent misassembly.
Advanced Considerations in Overhang Design
Biochemical Constraints and Ligase Specificity
The enzymatic specificity of DNA ligase introduces additional constraints in overhang design. Not all perfectly complementary overhangs ligate with equal efficiency due to the biochemical preferences of DNA ligase. For example, ligase demonstrates poor efficiency when joining overhangs consisting of only A-T base pairs (e.g., AAAA and TTTT), despite their perfect complementarity. Furthermore, certain non-identical overhangs with minimal sequence differences may be mistakenly fused by ligase, a phenomenon known as misligation. Research by Potapov et al. (2018) systematically characterized these misligation frequencies, revealing that overhangs with only single-nucleotide differences can sometimes be fused, particularly when the differences occur at specific positions within the overhang. To minimize these issues, experts recommend selecting overhang sequences that contain at least one G/C and one A/T nucleotide, and ensuring that any pair of overhangs in the set (including their reverse complements) differ by at least two nucleotides [40].
Computational Approaches for Complex Assemblies
As the number of fragments in an assembly increases, manually designing compatible overhangs that satisfy all constraints becomes computationally challenging. For assemblies involving 10, 20, or more fragments, computational design tools are essential for generating optimal overhang sets. The most straightforward algorithm follows a constructive branch-and-bound approach: starting with a single overhang, additional compatible overhangs are incrementally added until the required set is complete or no further compatible overhangs can be added. When this approach fails to yield sufficient overhangs, backtracking can be implemented by removing some overhangs and trying alternative candidates [40].
A more sophisticated approach utilizes graph theory algorithms. In this method, all permissible overhangs (following basic sequence constraints) are represented as nodes in a graph. Edges are drawn between nodes (overhangs) that are compatible according to specified criteria (e.g., differing by at least two nucleotides). The largest set of mutually compatible overhangs then corresponds to the maximum clique in the graph—a subset of nodes where every pair is connected by an edge. Advanced clique-finding algorithms, such as Bron-Kerbosch or Tarjan-Trojanowski, can efficiently identify these maximal compatible sets. Tools like NEB's GetSet app implement these computational approaches to help researchers design overhang sets for complex assemblies [40].
Primer Design for Golden Gate Assembly
Incorporating Type IIS Recognition Sites
In Golden Gate Assembly, primers must be designed to add Type IIS recognition sites and the desired overhang sequences to DNA fragments. The standard approach involves designing forward and reverse primers that include, from 5' to 3': (1) the desired 4-nucleotide overhang sequence, (2) the Type IIS enzyme recognition site, and (3) the target-specific sequence that anneals to the DNA fragment of interest. The recognition sites must be oriented to face "inward" toward the insert so that digestion removes the recognition sites and releases the fragment with the desired overhangs. For example, a forward primer might follow the pattern: 5'- [4-nt overhang] + [Type IIS site] + [18-25 nt target-specific sequence] -3' [5].
The Golden EGG system, a simplified Golden Gate approach, utilizes a specialized primer design that includes an NGGTCTCNn1n2n3n4 extension 5' to the target-specific sequence, where n1-n4 represents one of the possible non-palindromic four-nucleotide overhang sequences. This design, combined with a universal entry vector, allows for the use of a single Type IIS enzyme for both entry clone construction and final assembly, streamlining the cloning process [6].
Table 2: Primer Design Specifications for Golden Gate Assembly
| Parameter | Optimal Range | Rationale | Special Considerations for Golden Gate |
|---|---|---|---|
| Total Length | 18-24 nucleotides (target-specific portion) | Balances specificity and annealing efficiency | Additional 5' extensions for overhangs and Type IIS sites |
| Melting Temperature (Tm) | 54°C - 65°C | Ensures specific binding during PCR | Tm calculation should exclude non-target 5' extensions |
| GC Content | 40% - 60% | Provides balanced binding strength | Avoid >3 G/C at 3' end to prevent non-specific binding |
| 3' End Design | No self-complementarity | Precludes primer-dimer formation | Critical for assembly efficiency |
| Overhang Addition | 4 nt overhang + Type IIS site | Creates compatibility for Golden Gate | Orientation must be "inward" for proper excision |
Ensuring Primer Specificity and Efficiency
Well-designed primers are essential for successful Golden Gate Assembly, as they must specifically amplify the target fragments while introducing the necessary sequences for subsequent assembly. The target-specific portion of primers (typically 18-25 nucleotides) should be designed following established principles for PCR primer design. This includes maintaining a melting temperature (Tm) between 54°C and 65°C, with forward and reverse primers having similar Tm values (within 2°C of each other). The GC content should ideally range between 40% and 60%, with a balanced distribution of nucleotides. Particularly important is avoiding self-complementarity at the 3' end, which can lead to primer-dimer formation and reduce amplification efficiency. A "GC clamp" (the presence of G or C bases in the last five nucleotides at the 3' end) can promote specific binding, but more than three G/C residues at the 3' end should be avoided as they may promote non-specific amplification [41].
When designing primers for Golden Gate Assembly, researchers must remember that the Tm calculations should primarily consider the target-specific portion, as the 5' extensions (overhangs and restriction sites) do not participate in the initial annealing during PCR amplification. Additionally, to maintain the reading frame in protein-coding sequences, additional nucleotides may need to be inserted between the Type IIS site and the target-specific portion of the primer. Verifying that the final primers do not introduce internal Type IIS recognition sites within the amplified fragment is also crucial, as these could interfere with the assembly process [5].
Experimental Protocols
Basic Golden Gate Assembly Workflow
The standard Golden Gate Assembly protocol involves setting up a single reaction mixture containing all components necessary for both digestion and ligation. The following protocol is adapted from established methods for assembling multigene constructs using the MoClo system [9] [5]:
Reaction Setup: In a single tube, combine:
- 50-100 ng of destination vector
- Equimolar amounts of each DNA fragment (inserts)
- 1× T4 DNA ligase buffer
- 1 μL (10 U) of Type IIS restriction enzyme (e.g., BsaI-HFv2)
- 1 μL (400 U) of T4 DNA ligase
- Nuclease-free water to 20 μL total volume
Thermocycling Conditions:
- Cycle between digestion and ligation temperatures 25-30 times:
- 37°C for 2-5 minutes (digestion)
- 16°C for 2-5 minutes (ligation)
- Final incubation:
- 50°C for 5 minutes (to complete ligation)
- 80°C for 5 minutes (to inactivate enzymes)
- Cycle between digestion and ligation temperatures 25-30 times:
Transformation:
- Transform 2-5 μL of the assembly reaction into competent E. coli cells
- Plate on selective media and incubate overnight
- Screen resulting colonies for correct constructs by colony PCR or sequencing
For assemblies involving more than 3-4 fragments, increasing the number of cycles to 50-60 can improve efficiency. The Golden EGG system modifies this basic protocol by including a cold treatment step (4°C for 15 minutes) after the initial digestion-ligation cycles to shift reaction kinetics toward ligation, particularly important when the final construct contains recognition sites for the Type IIS enzyme used [6].
Figure 2: Golden Gate Assembly experimental workflow showing the key steps from reaction setup through transformation.
Simplified Golden Gate Using the Golden EGG System
The Golden EGG system provides a streamlined approach that uses a single Type IIS enzyme for both entry clone construction and final assembly. The following protocol details this simplified method [6]:
Entry Clone Construction:
- Design primers with the structure: 5'-NGGTCTCNn1n2n3n4 + target-specific sequence-3'
- Amplify DNA parts by PCR using these primers
- Set up Golden Gate reaction with:
- 50 ng of pEGG entry vector
- 20-50 ng of PCR product
- 1× T4 DNA ligase buffer
- 0.5 μL (5 U) of Eco31I (or BsaI)
- 0.5 μL (200 U) of T4 DNA ligase
- Water to 10 μL total
- Thermocycling conditions:
- 37°C for 5 minutes
- 4°C for 15 minutes (cold treatment)
- 80°C for 5 minutes
- Transform into competent cells and verify sequences
Multifragment Assembly:
- Set up Golden Gate reaction with:
- 50 ng of destination vector
- Equimolar amounts (20-50 ng each) of entry clones
- 1× T4 DNA ligase buffer
- 0.5 μL (5 U) of Eco31I (or BsaI)
- 0.5 μL (200 U) of T4 DNA ligase
- Water to 10 μL total
- Thermocycling conditions:
- 25 cycles of: 37°C for 3 minutes + 16°C for 4 minutes
- 50°C for 5 minutes
- 80°C for 5 minutes
- Transform and screen as above
- Set up Golden Gate reaction with:
The key innovation in this system is the universal entry vector with a specific cloning site (EGG site) that allows any DNA part to be cloned using the same vector and subsequently released with any desired overhangs for final assembly.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Golden Gate Assembly
| Reagent Category | Specific Examples | Function | Notes for Selection |
|---|---|---|---|
| Type IIS Restriction Enzymes | BsaI, BsmBI, Eco31I, BbsI | Digest DNA fragments to generate specific overhangs | Select based on recognition site length and overhang size; BsaI most common |
| DNA Ligase | T4 DNA Ligase | Joins DNA fragments via complementary overhangs | High-concentration formulations recommended for one-pot reactions |
| Vectors | MoClo Kit vectors, pEGG vectors, Custom destination vectors | Provide backbone for assembled constructs | Ensure compatibility with selection markers and replication origins |
| Polymerases | Q5 High-Fidelity DNA Polymerase, Kapa Polymerase | Amplify DNA fragments with minimal errors | High-fidelity enzymes critical for error-free parts |
| Competent Cells | E. coli DH5α, NEB Stable, other cloning strains | Propagate assembled constructs | High-efficiency strains (>10^8 cfu/μg) recommended for complex assemblies |
Successful implementation of Golden Gate Assembly requires access to specific research reagents and tools. Type IIS restriction enzymes form the core of the technique, with BsaI being the most commonly used enzyme due to its specific recognition sequence and generation of 4-base overhangs. However, alternative enzymes such as BsmBI or BbsI may be preferable when internal recognition sites are present in the fragments to be assembled. High-quality DNA ligase is equally important, with T4 DNA ligase being the standard choice for its efficiency in joining cohesive ends [6] [5].
Specialized vector systems have been developed for Golden Gate Assembly, including the MoClo toolkit vectors that facilitate hierarchical assembly of multigene constructs, and simplified systems like the Golden EGG vectors that use a universal entry vector for all DNA parts. When selecting a vector system, researchers should consider factors such as antibiotic resistance markers, replication origins, and the presence of negative selection markers (e.g., ccdB) that reduce background from empty vectors. For PCR amplification of DNA parts, high-fidelity DNA polymerases such as Q5 or Kapa are essential to minimize introduction of mutations during part generation. Finally, high-efficiency competent cells are crucial for obtaining sufficient clones after transformation, particularly when assembling complex constructs with multiple fragments [9] [6].
Troubleshooting and Optimization
Even with careful design, Golden Gate Assembly may require troubleshooting to achieve optimal results. Common issues include incomplete assemblies, high background of empty vectors, and incorrectly assembled constructs. Several strategies can address these problems:
Increasing Cycle Numbers: For assemblies involving more than 3-4 fragments, increasing the thermocycling steps to 50-60 cycles can significantly improve efficiency by providing more opportunities for correct assembly.
Optimizing Fragment Ratios: While equimolar ratios of fragments are standard, adjusting these ratios (e.g., using 2-3× molar excess of inserts relative to vector) can improve assembly efficiency for complex constructs.
Addressing Internal Restriction Sites: If vector backbones or insert fragments contain unwanted Type IIS restriction sites, these can be removed through site-directed mutagenesis before assembly. Alternatively, selecting a different Type IIS enzyme with a longer recognition site (e.g., BaeI with 7-base recognition) may circumvent the problem.
Reducing Empty Vector Background: The use of destination vectors with negative selection markers (e.g., ccdB toxin gene) between the Type IIS sites can dramatically reduce background from undigested or re-ligated vector.
Verifying Part Quality: Sequencing all entry clones before final assembly is crucial to ensure that mutations have not been introduced during part generation. Even high-fidelity polymerases introduce errors at low frequencies, and these can compromise final construct function [5].
When troubleshooting failed assemblies, systematic analysis of intermediate products can identify the specific point of failure. For example, testing individual fragment combinations can identify problematic fusions, while sequencing incorrectly assembled constructs can reveal specific misligation events that may inform redesign of overhang sequences.
In the context of Golden Gate assembly, a powerful molecular cloning technique for constructing complex multigene constructs, 'domestication' refers to the crucial process of removing internal Type IIS restriction enzyme recognition sites from the DNA sequences of interest [42]. This process is fundamental to the success of the assembly because Golden Gate cloning utilizes Type IIS restriction enzymes, which cut outside of their recognition sequences to generate user-defined overhangs for seamless fragment assembly [11] [12]. If a target DNA fragment (insert or vector) contains an internal recognition site for the enzyme being used (e.g., BsaI), it will be cleaved at that location during the assembly reaction. This internal cleavage generates unwanted DNA ends with incompatible overhangs, which compete with the intended assembly, leading to incorrect assemblies, rearrangements, or significantly reduced efficiency [6] [43]. Therefore, domesticating genetic parts by eliminating these internal sites is an essential preparatory step to ensure the high fidelity and efficiency required for assembling multigene constructs, a common task in synthetic biology and drug development research [9] [25].
The principle behind Golden Gate assembly is the use of a Type IIS restriction enzyme and a DNA ligase in a single-reaction "restriction-ligation" [12]. The correctly assembled final product is stable because the designed fusion sites between parts are seamless and no longer contain the recognition sequence for the restriction enzyme. In contrast, any DNA fragment with an internal recognition site remains a substrate for repeated cleavage, disrupting the ligation equilibrium and compromising the assembly [6]. For research scientists engineering pathways for therapeutic compound production, the reliability of assembling multiple transcription units is paramount, and domestication provides this reliability.
Strategies for Sequence Domestication
Several strategies can be employed to domesticate DNA sequences, each with its own advantages and procedural considerations. The choice of strategy depends on factors such as the number of internal sites, the length of the DNA fragment, and the available resources.
Silent Mutagenesis by Site-Directed Mutagenesis
The most precise and recommended method for domestication is the introduction of silent mutations that abolish the restriction site without altering the amino acid sequence of the encoded protein.
- Principle: The recognition sequence of the Type IIS enzyme is mutated at the nucleotide level without changing the protein sequence. This is possible due to the degeneracy of the genetic code.
- Workflow: This is typically performed via site-directed mutagenesis on the basic part cloned into an entry vector. The process involves designing primers that incorporate the desired nucleotide changes, followed by a PCR-based mutagenesis protocol. The success of mutagenesis must be confirmed by DNA sequencing of the domesticated module before it is incorporated into a Golden Gate assembly pipeline [11].
- Advantages: This method is scarless and preserves the exact protein function, which is critical for downstream applications in functional genomics and protein expression studies.
PCR-Based Domestication with Custom Primers
For de novo generation of domesticated parts, a common strategy is to use PCR amplification with primers designed to omit or alter internal restriction sites.
- Principle: When amplifying a coding sequence from genomic DNA or a cDNA template, primers are designed so that the amplified product simply lacks the internal restriction site. This often involves the same principle of silent mutagenesis but is integrated into the initial part-creation step.
- Workflow: The gene of interest is amplified using primers that contain the necessary 5' extensions for Golden Gate assembly (e.g., the appropriate fusion sites like "AATG" for the start codon) and that incorporate nucleotide changes to disrupt any internal BsaI sites within the gene body [6] [12]. The PCR product is then cloned into a universal entry vector for validation and storage.
- Advantages: This method combines part acquisition and domestication into a single step, streamlining the workflow for creating new basic modules for a toolkit.
Alternative Assembly Strategies to Bypass Domestication
In some cases, extensive domestication requirements can be a practical bottleneck. Recent methodological developments offer strategies to mitigate this challenge.
- Enzyme Switching: If a DNA part contains an internal site for one Type IIS enzyme (e.g., BsaI), one can switch to a different Golden Gate assembly toolkit that uses an enzyme with a different recognition sequence (e.g., AarI, BsmBI) [11] [6].
- The Golden EGG System: The simplified "Golden EGG" system uses a specific primer and vector design that allows for a more flexible approach [6]. In this system, the internal recognition site is not entirely avoided. However, the authors note that the reaction can still proceed with high efficiency, especially if the internal site's cleavage produces an overhang that does not match any of the predefined fusion sites used in the assembly, thus preventing incorrect ligations [6]. For difficult assemblies with internal sites, an extra ligation step after the restriction-ligation reaction can be performed to improve the yield of correct constructs [6] [43].
The following workflow diagram illustrates the decision-making process for selecting and implementing a domestication strategy.
Experimental Protocols
Protocol A: Domestication via Silent Mutagenesis
This protocol details the domestication of a basic part (e.g., a Coding Sequence - CDS) already present in an entry vector, using a standard site-directed mutagenesis kit.
Materials:
- Plasmid DNA containing the part with the internal Type IIS site.
- High-fidelity DNA polymerase (e.g., Q5 Hot Start High-Fidelity DNA Polymerase).
- DpnI restriction enzyme.
- Oligonucleotide primers designed for mutagenesis.
Primer Design:
- Design forward and reverse primers that are complementary to the region of the internal site.
- Introduce 1-3 nucleotide substitutions within the enzyme's recognition sequence (e.g., for BsaI, 5'-GGTCTC-3'). Ensure the mutations are silent and do not change the amino acid sequence.
- Primers should typically be 25-45 bases long, with the desired mutation located in the middle.
Procedure:
- Set up the PCR Reaction:
- Combine template DNA (10-50 ng), primers (0.5 µM each), dNTPs, and high-fidelity polymerase in its recommended buffer.
- Run the PCR with an appropriate annealing temperature based on the primer Tm.
Digest Template:
- Following PCR, add 1 µL of DpnI enzyme directly to the reaction mix.
- Incubate at 37°C for 1 hour to digest the methylated parental template DNA.
Transform:
- Transform 2-5 µL of the DpnI-treated DNA into competent E. coli cells.
- Plate cells on selective media and incubate overnight.
Validate:
- Pick several colonies for culture and plasmid isolation.
- Verify the mutation by Sanger sequencing across the entire domesticated region.
Protocol B: Golden Gate Assembly with Domesticated Parts
Once all parts are domesticated, they can be assembled into a multigene construct. The following protocol is adapted from established MoClo workflows [9] [25] and lab protocols [43].
Materials:
- Domesticated Level 0 modules (e.g., Promoter, CDS, Terminator) as plasmid DNA.
- Appropriate Level 1 destination vector.
- Type IIS Restriction Enzyme (e.g., BsaI-HF v2).
- T4 DNA Ligase (400 U/µL, includes 10x T4 Ligase Buffer).
- Thermocycler.
Procedure:
- Set Up Restriction-Ligation Reaction:
- In a 0.2 mL PCR tube, assemble the following reaction on ice:
- 75 ng of each plasmid-based Level 0 module
- 75 ng of the destination vector
- 1 µL 10x T4 DNA Ligase Buffer
- 1 µL (400-500 U) T4 DNA Ligase
- 0.5-1 µL BsaI-HF v2
- Nuclease-free water to a final volume of 10 µL
- In a 0.2 mL PCR tube, assemble the following reaction on ice:
Run Restriction-Ligation Cycle:
- Place the tube in a thermocycler and run one of the following programs:
Transform:
- Transform 2 µL of the reaction mixture into chemically competent E. coli cells.
- Plate onto LB agar plates with the appropriate antibiotic and incubate overnight at 37°C.
Screen and Verify:
- Screen colonies by colony PCR or restriction digest.
- For critical constructs, validate the final assembly by sequencing.
The Scientist's Toolkit: Research Reagent Solutions
The table below summarizes key reagents and tools essential for performing sequence domestication and Golden Gate assembly.
Table 1: Essential Reagents for Golden Gate Domestication and Assembly
| Reagent / Tool | Function / Description | Example Products / Kits |
|---|---|---|
| Type IIS Restriction Enzymes | Core enzyme for Golden Gate; cuts DNA outside recognition site to create custom overhangs. | BsaI-HF, BsmBI-v2, AarI (NEB) [11] [43] |
| High-Fidelity DNA Polymerase | For accurate PCR amplification during domestication and part generation. | Q5 Hot Start High-Fidelity DNA Polymerase (NEB) |
| Site-Directed Mutagenesis Kit | Facilitates introduction of specific base changes to remove internal restriction sites. | Q5 Site-Directed Mutagenesis Kit (NEB) |
| T4 DNA Ligase | Joins DNA fragments with compatible overhangs in the one-pot restriction-ligation. | T4 DNA Ligase (400 U/µL or 2,000 U/µL) [43] |
| Golden Gate Toolkits | Pre-made libraries of standardized, domesticated parts and vectors for specific organisms. | MoClo Toolkit [12], GoldenBraid Kit [11], CIDAR MoClo [11] |
| Universal Entry Vectors | Plasmid backbones for cloning and storing domesticated basic parts (Level 0). | pL0 series vectors [12], pEGG vectors [6] |
Data Presentation and Analysis
The efficiency of domestication directly impacts the success rate of Golden Gate assembly. The following table quantifies key aspects and expected outcomes based on published methodologies.
Table 2: Quantitative Data and Expected Outcomes for Domestication and Assembly
| Parameter | Typical Value / Observation | Context / Notes |
|---|---|---|
| Golden Gate Assembly Efficiency | 95-100% correct assemblies for 10-fragment assembly [12] | Achievable with fully domesticated parts and optimized protocols. |
| Reaction Volume | 5-20 µL (10 µL standard) [43] | Smaller volumes can be used to conserve reagents. |
| Cycle Number (Thermocycler) | 30 cycles (simple) to 99 cycles (complex/difficult) [43] | More cycles can help with assemblies containing undomesticated internal sites. |
| Amount of Vector per Reaction | 75 ng (for plasmid fragments) [43] | A 2:1 molar ratio of insert:vector is recommended for PCR fragments. |
| Ligase Amount | 400-500 Units per 10 µL reaction [43] | Critical for high efficiency; ensure sufficient enzyme is added. |
The domestication of DNA sequences is a critical, non-negotiable preparatory step in the Golden Gate assembly workflow for constructing multigene plasmids. By systematically removing internal Type IIS restriction sites through silent mutagenesis or PCR-based strategies, researchers can ensure the high efficiency and fidelity required for complex genetic engineering projects. The adoption of standardized, domesticated toolkits like MoClo and GoldenBraid, along with robust experimental protocols, provides a reliable framework for scientists in drug development and synthetic biology to accelerate their research, from metabolic engineering of production strains to the development of novel gene therapies. As the field progresses, methods like Golden EGG that offer simplified workflows with reduced domestication burdens will further enhance the accessibility and throughput of this powerful cloning technology.
Within the broader context of developing robust Golden Gate assembly protocols for constructing multigene constructs, the assembly of ten or more DNA fragments presents a distinct set of challenges. As the number of fragments increases, the reaction efficiency can decline due to factors such as imperfect ligation fidelity, suboptimal stoichiometry, and the increased statistical probability of misassembly [44] [45]. This application note provides detailed methodologies and optimized reaction conditions to overcome these hurdles, enabling researchers to reliably execute complex assemblies essential for advanced applications in synthetic biology and drug development, such as building entire metabolic pathways or comprehensive gene libraries [13].
The following workflow outlines the core experimental process for a complex Golden Gate assembly, from initial setup to the final product.
Key Reagents and Research Tools
Successful complex assemblies depend on the quality and compatibility of core reagents. The table below details the essential components for a robust Golden Gate reaction.
Table 1: Essential Reagents for Complex Golden Gate Assembly
| Reagent | Function & Key Features | Specific Recommendations for 10+ Fragments |
|---|---|---|
| Type IIS Restriction Enzyme | Digests vector and inserts to generate specific overhangs; its recognition site is absent from the final construct, making the reaction irreversible [5] [46]. | BsaI-HFv2 or BsmBI-v2 are optimized for assembly [45] [46]. For fewer internal cut sites, use enzymes with longer recognition sites like PaqCI (7 bp) [44] [45]. |
| DNA Ligase | Joins complementary overhangs of digested fragments. High fidelity is critical for accurate assembly of multiple fragments [45]. | T4 DNA Ligase is standard. For highest efficiency and fidelity, use a master mix like NEBridge Ligase Master Mix, which includes ligase in an optimized buffer [45] [21]. |
| Vector Backbone | Accepts the assembled inserts. Must be free of internal Type IIS sites used in the assembly [5] [44]. | Use domesticated vectors like pGGAselect, which lack internal BsaI, BsmBI, or BbsI sites [44]. |
| DNA Fragments (Inserts) | The genetic parts to be assembled. Can be PCR amplicons or pre-cloned in entry vectors [5]. | Ensure they are free of primer dimers (for amplicons) or RNA contamination (for plasmids) to avoid concentration errors and misassemblies [44]. Pre-cloned, sequenced-validated fragments are preferred for complex assemblies. |
Optimized Reaction Setup and Cycling Conditions
For assemblies involving a high number of fragments, the reaction volume and component ratios must be adjusted to ensure sufficient interaction and correct assembly.
Reaction Setup
The following table provides a detailed protocol for setting up a Golden Gate assembly reaction for 10 or more fragments, based on the use of NEBridge Ligase Master Mix [21]. This master mix is highly recommended as it contains T4 DNA Ligase in an optimized buffer with a proprietary ligation enhancer, simplifying the reaction setup and improving outcomes [45].
Table 2: Reaction Setup for Assemblies with 10+ Fragments
| Component | Volume per Reaction | Final Amount / Notes |
|---|---|---|
| NEBridge Ligase Master Mix (3X) | 10 µL | Provides ligase, buffer, and ATP. |
| DNA Fragments (each) | Variable | 0.05 pmol of each fragment [21]. Calculate volumes based on concentration and length. |
| Type IIS Restriction Enzyme (e.g., BsaI-HFv2) | 1 µL | - |
| Molecular Biology-Grade Water | To a final volume of 30 µL | - |
| Total Volume | 30 µL | - |
Key Considerations:
- Fragment Molarity: Using an equimolar amount (e.g., 0.05 pmol each) of all fragments, including the vector backbone, is crucial for balanced assembly and to prevent biased representation of shorter fragments [21].
- Concentration Calculations: Precisely calculate the mass of each DNA fragment required using its molecular weight. The equation below can be used to determine the volume of each fragment to add: ( V = \frac{(0.05 \times 10^{-12} \, \text{mol}) \times (\text{Length in bp} \times 660 \, \text{g/mol/bp})}{\text{Concentration in ng/µL}} \times 10^3 ) [23] [21].
- Negative Control: Always include a control reaction without inserts to monitor vector re-ligation background.
Thermocycling Parameters
The thermocycling protocol is designed to efficiently drive the digestion and ligation reactions to completion. For complex assemblies, an increased number of cycles is a key optimization strategy [44].
Table 3: Optimized Thermocycling Protocol for Complex Assemblies
| Step | Temperature | Time | Cycles | Purpose |
|---|---|---|---|---|
| Digestion/Ligation Cycling | 37°C | 5 minutes | 30-65 [44] | Optimal temperature for Type IIS restriction enzyme activity. |
| 16°C | 5 minutes | Optimal temperature for T4 DNA ligase activity. | ||
| Final Digestion | 37°C | 5 minutes | 1 | Optional step to digest any residual misassembled products. |
| Enzyme Inactivation | 60°C | 5 minutes | 1 | Heat-inactivates the enzymes, stabilizing the final product. |
| Hold | 4°C | ∞ | - | For short-term storage of reactions. |
Critical Optimization: For assemblies with >10 fragments, simply increasing the total number of digestion-ligation cycles from a standard 30 to 45-65 cycles can significantly enhance assembly efficiency without sacrificing fidelity, as the enzymes remain stable and active during extended cycling [44].
Critical Design and Optimization Strategies
Beyond the core protocol, several strategic considerations are vital for the success of high-complexity assemblies.
In Silico Design and Domestication
- Comprehensive Sequence Checking: Prior to the experiment, scrutinize all vector and insert sequences for internal recognition sites of the Type IIS enzyme being used. These sites must be removed through a process called domestication—introducing silent mutations without changing the amino acid sequence—to prevent undesired cleavage [5] [44].
- Leverage Design Tools: Utilize free web-based tools like the NEBridge Golden Gate Assembly Tool and NEBridge Ligase Fidelity Tool to design overhangs, predict assembly accuracy, and identify optimal junctions. These tools use data on ligase fidelity to minimize mis-ligation [44] [45].
- In Silico Simulation: Always simulate the entire assembly using sequence analysis software before starting wet-lab work. This verifies the correct order and orientation of fragments and confirms that all overhangs are compatible [23].
Fragment Preparation and Quality Control
- Source High-Quality DNA: For PCR-amplified inserts, use a high-fidelity DNA polymerase (e.g., Q5 DNA Polymerase) and avoid over-cycling to prevent PCR-induced errors [44]. Purify amplicons to remove primer dimers, which can compete in the assembly reaction and lead to misassemblies [44].
- Accurate Quantification: For pre-cloned inserts, ensure plasmid preparations are free of RNA, as its presence leads to overestimation of DNA concentration via spectrophotometry [44]. Use fluorometric methods for the most accurate DNA quantification.
Post-Assembly Validation
- Screen Multiple Colonies: The efficiency of complex assemblies is lower than that of simple ones. Therefore, screen a minimum of 8-10 colonies to increase the likelihood of identifying a correct clone [23].
- Sequence the Entire Insert Region: While colony PCR can confirm the presence of an insert, it cannot verify the correct order, direction, or sequence of multiple fragments. Sequencing the entire assembled region is strongly recommended to confirm the integrity of the final construct [23].
Golden Gate Assembly is a powerful, one-pot cloning method that uses Type IIS restriction enzymes and DNA ligase to seamlessly assemble multiple DNA fragments in a defined order [47]. Its efficiency stems from the ability of Type IIS enzymes to cut outside their recognition sequences, creating unique, user-defined overhangs, and the simultaneous ligation of these fragments into a destination vector [48]. Despite its robustness, researchers often encounter three major pitfalls: low assembly yield, incorrect constructs, and a high background of empty vectors. These issues become increasingly prevalent with complex multigene assemblies and often originate from suboptimal reaction design, fragment preparation, or cycling conditions. This application note details the root causes of these common problems and provides validated protocols to overcome them, enabling reliable construction of complex DNA libraries and multigene constructs for therapeutic development.
Troubleshooting Common Pitfalls
The following tables summarize the major pitfalls, their underlying causes, and specific solutions to optimize Golden Gate Assembly outcomes.
Table 1: Addressing Low Assembly Yield
| Root Cause | Specific Issue | Recommended Solution |
|---|---|---|
| Suboptimal Enzyme/Robot Conditions | Inefficient restriction or ligation in one-pot reaction. | Use master mixes optimized for Golden Gate (e.g., NEBridge Ligase Master Mix) [49] [21]. |
| Inefficient Cycling | Insufficient time for complex assemblies with many fragments. | Increase cycles to 45-65 for assemblies involving >10 fragments [49]. |
| Incorrect Fragment Stoichiometry | Molar imbalance of fragments reduces correct assembly. | Use 0.05 pmol of each fragment, including the backbone [21]. For >10 fragments, reduce pre-cloned insert amount to 50 ng each [49]. |
| Fragment Quality and Purity | PCR inhibitors or enzymes degraded by repeated freeze-thaw. | Use high-fidelity polymerase (e.g., Q5), avoid over-cycling PCR, and use fresh, high-quality DNA [49]. |
Table 2: Preventing Incorrect Constructs
| Root Cause | Specific Issue | Recommended Solution |
|---|---|---|
| Mispaired Overhangs | Ligation of non-complementary overhangs creates sequence errors. | Design overhangs using fidelity tools (e.g., NEBridge Ligase Fidelity Tools) to predict and avoid error-prone junctions [47] [49]. |
| Internal Restriction Sites | Unintended cutting within insert or vector sequences. | Domesticate sequences by removing internal Type IIS sites via mutagenesis or codon optimization [47] [6]. |
| PCR-Induced Errors | Mutations introduced during fragment amplification. | Use a high-fidelity, proofreading DNA polymerase and avoid excessive PCR cycles [49]. |
| Primer Dimers | Short, unintended PCR products with overhangs cause mis-assembly. | Optimize PCR to ensure a specific product and eliminate primer dimers before assembly [49]. |
Table 3: Reducing Empty Vector Background
| Root Cause | Specific Issue | Recommended Solution |
|---|---|---|
| Inefficient Vector Digestion | Undigested or self-ligated vector transforms successfully. | Use a vector with a negative selection marker (e.g., ccdB, sfGFP) that is removed upon successful insertion [47] [6]. |
| Fragment Issues | Missing, degraded, or low-concentration inserts. | Accurately quantify fragments; ensure RNA-free plasmid preps for pre-cloned inserts to avoid concentration overestimation [49]. |
| Final Product Re-Cleavage | Assembled product retains internal restriction sites. | Verify final construct is free of the Type IIS enzyme's recognition site to prevent re-digestion after assembly [6]. |
Workflow for Troubleshooting Golden Gate Assembly
The diagram below outlines a logical workflow for diagnosing and resolving the most common Golden Gate Assembly failures.
Optimized Protocols for Complex Assemblies
Standard Golden Gate Assembly Protocol
This protocol is designed for assembling 3-6 DNA fragments into a destination vector using NEBridge Ligase Master Mix, which is optimized for high-efficiency Golden Gate Assembly [21].
Research Reagent Solutions
| Item | Function/Description |
|---|---|
| NEBridge Ligase Master Mix (NEB #M1100) | Optimized buffer containing T4 DNA Ligase and ATP for high-fidelity ligation. |
| Type IIS Restriction Enzyme (e.g., BsaI-HFv2) | Cleaves DNA outside recognition site to generate specific, sticky ends for assembly. |
| pGGAselect Destination Plasmid | Versatile vector with no internal BsaI, BsmBI, or BbsI sites; allows blue-white or counterselection [47] [49]. |
| Q5 High-Fidelity DNA Polymerase | Generates high-quality, mutation-free PCR fragments for assembly [49]. |
| NEBridge Golden Gate Assembly Tool | Free web tool for designing primers and optimizing overhangs [47] [48]. |
Procedure
- Reaction Setup: On ice, combine the following reagents in a PCR tube in the order listed:
Note: For assemblies with 7 or more fragments, scale the reaction to 30 µL, using 10 µL of Master Mix and proportionally more enzyme and fragments [21].Component Volume (µL) NEBridge Ligase Master Mix (3X) 5.0 DNA Fragments (each at 0.05 pmol) X BsaI-HFv2 1.0 Molecular Biology-Grade Water to 15.0 µL - Thermocycling: Place the tube in a thermocycler and run the following program:
- 30 cycles of:
- 37°C for 1 minute (digestion)
- 16°C for 1 minute (ligation)
- 60°C for 5 minutes (enzyme inactivation)
- 4°C hold (indefinite)
- 30 cycles of:
- Transformation: Transform 2-5 µL of the reaction into competent E. coli cells using standard methods.
Advanced Protocol for High-Complexity Assemblies (>10 Fragments)
Assembling a large number of fragments requires adjustments to the standard protocol to maintain efficiency and accuracy [49] [21].
Procedure
- Reaction Setup: Use a scaled-up reaction mixture.
Component Volume (µL) NEBridge Ligase Master Mix (3X) 10.0 DNA Fragments (each at 0.05 pmol) X BsaI-HFv2 1.0 Molecular Biology-Grade Water to 30.0 µL - Extended Thermocycling: Use longer cycle times and more cycles.
- 30-65 cycles of:
- 37°C for 5 minutes (digestion)
- 16°C for 5 minutes (ligation)
- 60°C for 5 minutes (enzyme inactivation)
- 4°C hold (indefinite) Note: Increasing the cycle number to 45-65 can significantly boost the efficiency of complex assemblies without sacrificing fidelity [49].
- 30-65 cycles of:
- Fragment Stoichiometry Adjustment: For pre-cloned inserts, reduce the amount of each fragment to 50 ng without significantly decreasing assembly efficiency [49].
Successful Golden Gate Assembly for complex multigene constructs hinges on meticulous experimental design and optimization. Key strategies include the use of high-fidelity design tools to ensure proper overhang pairing, careful domestication of internal restriction sites, selection of appropriate vectors with counterselection markers, and optimization of cycling conditions for assemblies with high fragment numbers. By systematically addressing the pitfalls of low yield, incorrect constructs, and empty vectors through the detailed protocols provided herein, researchers can robustly leverage Golden Gate technology to accelerate synthetic biology and therapeutic development pipelines.
Beyond the Protocol: Validating Constructs and Comparing DNA Assembly Methods
The successful integration and expression of multigene constructs, assembled via techniques such as Golden Gate assembly, is a cornerstone of advanced biological research and therapeutic development. This application note provides a detailed framework for the validation of these complex constructs, focusing on a two-pronged approach that combines next-generation sequencing (NGS) for analytical validation with functional cell-based assays to confirm biological activity. The protocols outlined herein are designed to ensure the integrity of the genetic assembly and provide confirmation of its intended function within a cellular context, which is critical for applications in synthetic biology, pathway engineering, and drug development. The workflow summarized in Figure 1 provides a high-level overview of this integrated validation strategy.
Figure 1. Integrated validation workflow for multigene constructs, combining NGS and functional assays.
Analytical Validation: Next-Generation Sequencing
Next-generation sequencing provides a comprehensive method to verify the sequence integrity, copy number, and correct assembly of multigene constructs. Targeted sequencing panels are particularly well-suited for this application, as they offer a cost-effective and efficient means to achieve high-depth coverage of specific regions of interest [50].
NGS Method Selection and Design
Two major library preparation methods are available for targeted NGS, each with distinct advantages as detailed in Table 1.
Table 1. Comparison of Targeted NGS Library Preparation Methods
| Feature | Hybrid Capture-Based | Amplification-Based (Amplicon) |
|---|---|---|
| Principle | Solution-based biotinylated probes hybridize to target regions [51] | PCR primers amplify specific target regions [51] |
| Variant Detection | SNVs, Indels, CNAs, SVs/Gene Fusions [51] | Primarily SNVs and small Indels [51] |
| Advantages | Tolerates probe mismatches, reduces allele dropout, comprehensive variant detection [51] | Fast, cost-effective for small target regions [51] [50] |
| Ideal Use Case | Large, complex multigene panels; detection of diverse variant types [51] | Small, focused panels for rapid screening of known hotspots [50] |
Detailed Protocol: Targeted NGS Sequencing
This protocol is adapted for validating multigene constructs and assumes the availability of high-quality plasmid DNA.
- Sample Preparation (DNA Extraction): Isolate plasmid DNA from bacterial cultures using a standard maxiprep kit. Assess DNA concentration and purity via spectrophotometry (e.g., Nanodrop) and fluorometry (e.g., Qubit). Verify DNA integrity by agarose gel electrophoresis.
- Library Preparation:
- For Hybrid Capture: Fragment 100-200 ng of plasmid DNA by acoustic shearing or enzymatic fragmentation. Perform end-repair, A-tailing, and adapter ligation. Hybridize the library with biotinylated probes designed against the entire multigene construct. Capture probe-bound targets using streptavidin-coated magnetic beads, and wash away non-specific fragments [51].
- For Amplicon-Based: Dilute plasmid DNA to a working concentration. Amplify target regions using a multiplexed PCR primer pool designed to tile across the entire assembled construct. Clean up the PCR products using magnetic beads [50].
- Sequencing: Pool the uniquely indexed libraries in equimolar ratios. Sequence on an appropriate NGS platform (e.g., Illumina MiSeq or NextSeq) to a depth sufficient for the detection of low-frequency variants. For plasmid validation, a minimum mean coverage of 500x-1000x is recommended to confidently identify potential minor contaminants or sequence errors.
- Data Analysis:
- Primary Analysis: Demultiplex sequenced reads and generate FASTQ files.
- Secondary Analysis: Align reads to the reference multigene construct sequence using a suitable aligner (e.g., BWA). Call sequence variants (SNVs, indels) and analyze copy number variations (CNVs) using established bioinformatics pipelines and software (e.g., CLC Genomics Workbench, GATK) [50].
- Tertiary Analysis: Annotate variants and filter against expected sequence. Visually inspect alignment files (e.g., using IGV) to verify critical regions such as assembly junctions and promoter/terminator sequences.
Functional Validation: Cell-Based Assays
Sequencing confirms the structure of the construct, but functional assays are required to verify its biological activity in a relevant cellular context. The workflow for functional validation is illustrated in Figure 2.
Figure 2. Workflow for functional validation beginning with single-cell clone isolation.
Detailed Protocol: Single-Cell Clone Isolation and Validation
This protocol ensures the derivation of clonal cell populations expressing the multigene construct for downstream functional analysis [52].
- Cell Transfection: Transfect the target cell line (e.g., HEK-293, CHO) with the validated multigene construct plasmid using a standard method (e.g., PEI, Lipofectamine). Include a negative control (mock transfection).
- Single-Cell Clone Isolation (Two Methods):
- Limiting Dilution Cloning (LDC):
- 48-72 hours post-transfection, wash cells with PBS and dissociate with TrypLE or trypsin/EDTA.
- Neutralize the dissociation reagent, centrifuge to pellet cells (300 x g for 5 min), and resuspend in pre-warmed growth medium.
- Perform a cell count and dilute the cell suspension to a density of 8 cells/mL. Seed 100 µL per well into 96-well plates (targeting 0.8 cells/well) [52].
- Incubate plates at 37°C, 5% CO₂ for 1-3 weeks, monitoring regularly for single-cell-derived colony formation.
- Fluorescence-Activated Cell Sorting (FACS):
- Prepare a single-cell suspension as described for LDC.
- Resuspend 1 x 10⁶ cells in 1 mL of ice-cold FACS buffer (PBS + 1% FBS). Add propidium iodide (PI) to a final concentration of 1 µg/mL to exclude dead cells.
- Filter the cell suspension through a suitable strainer and sort single, PI-negative cells directly into the wells of a 96-well plate containing 100 µL of complete growth medium [52].
- Limiting Dilution Cloning (LDC):
- Clone Expansion and Genotypic Validation:
- Once colonies are visible and sufficiently large, expand the clonal populations for 2-3 weeks [52].
- Harvest a portion of the cells from each clone for genotypic validation. This can include:
- Genotyping PCR: Use primers specific to the transgene to confirm its presence.
- Next-Generation Sequencing: As described in Section 2.2, to confirm the sequence integrity of the integrated construct.
- Western Blotting: To detect the expression of proteins encoded by the multigene construct [52].
- Functional/Phenotypic Assay:
- The specific assay is dictated by the function of the multigene construct.
- Example 1 (Reporter/Signaling Pathway): If the construct contains a fluorescent reporter gene (e.g., GFP) under a specific promoter, analyze cells using flow cytometry or fluorescence microscopy to quantify the reporter signal.
- Example 2 (Therapeutic Protein Production): For constructs designed to produce a secreted protein, collect conditioned medium from validated clones and quantify protein production using an ELISA.
- Example 3 (Cell Survival/Proliferation): If the construct confers resistance to a selective agent, challenge the clones with the agent and measure cell viability using assays like MTT or CellTiter-Glo.
Performance Metrics and Quality Control
Rigorous assessment of assay performance is critical for generating reliable validation data. Key metrics for the NGS assay are summarized in Table 2.
Table 2. Key Analytical Performance Metrics for NGS Validation [51]
| Performance Metric | Target | Calculation / Notes |
|---|---|---|
| Positive Percentage Agreement (PPA) | ≥99% for SNVs/Indels | (True Positives / (True Positives + False Negatives)) x 100 |
| Positive Predictive Value (PPV) | ≥99% for SNVs/Indels | (True Positives / (True Positives + False Positives)) x 100 |
| Coverage Depth | Minimum 500x mean coverage | Ensures high confidence in variant calling for plasmid/clone validation. |
| Variant Type | SNVs, small Indels, CNAs | Core set of variants a NGS test should reliably detect [53]. |
| Limit of Detection (LOD) | Varies by variant type | Heavily dependent on sample purity and sequencing depth [51]. |
For functional assays, key quality control measures include:
- Clonality Assurance: Document the origin of each clone from a single cell via microscopy imaging immediately after isolation [52].
- Assay Controls: Always include appropriate positive, negative, and vehicle controls in every functional experiment.
- Replication: Perform functional assays with a sufficient number of biological and technical replicates (e.g., n ≥ 3) to ensure statistical significance.
Research Reagent Solutions
A curated list of essential reagents and tools for the validation of multigene constructs is provided in Table 3.
Table 3. Essential Research Reagents and Materials for Validation
| Reagent / Material | Function / Application | Example / Note |
|---|---|---|
| NGS Library Prep Kit | Prepares DNA fragments for sequencing on NGS platforms. | Nextera XT DNA Library Prep Kit (for amplicon) or Nextera Rapid Capture Custom Enrichment Kit (for hybrid capture) [50]. |
| NGS Sequencing System | Generates sequencing data. | Illumina MiSeq or similar benchtop sequencer [50]. |
| Cell Dissociation Reagent | Creates single-cell suspensions for cloning. | TrypLE or trypsin-EDTA [52]. |
| FACS Buffer | Protects cells during flow cytometry and sorting. | Phosphate-buffered saline (PBS) supplemented with 1-5% fetal bovine serum (FBS). |
| Propidium Iodide (PI) | Fluorescent dye for identifying and excluding dead cells during FACS. | Added to cell suspension at 1 µg/mL final concentration before sorting [52]. |
| Genotyping Primers | Amplify specific transgene sequences for PCR validation. | Must be designed to be unique to the integrated construct. |
| Bioinformatics Software | Analyzes NGS data for alignment, variant calling, and visualization. | CLC Genomics Workbench, GATK, IGV [50]. |
| Cell Viability Assay | Quantifies functional outcomes like proliferation or cytotoxicity. | MTT, CellTiter-Glo, or similar assays. |
Within synthetic biology and advanced genetic engineering, the construction of multigene constructs is a foundational step for pathway engineering, therapeutic development, and functional genomics. The selection of an appropriate DNA assembly method is critical for efficiency and success. Golden Gate Assembly and Gibson Assembly represent two of the most powerful and widely adopted techniques for this purpose [54] [55]. This application note provides a scenarios-based comparison of these methods, with a specific focus on the context of multigene construct research, detailing their principles, protocols, and optimal applications to guide researchers in selecting and implementing the most suitable technique.
Golden Gate Assembly utilizes Type IIS restriction enzymes, which cleave DNA outside of their recognition sites, creating unique, non-palindromic overhangs that facilitate the seamless, one-pot, ordered assembly of multiple DNA fragments [54] [56]. In contrast, Gibson Assembly is an isothermal, single-tube reaction that employs a cocktail of three enzymes—an exonuclease, a DNA polymerase, and a DNA ligase—to join multiple overlapping DNA fragments via homologous recombination [57] [58].
The table below summarizes the key characteristics of each method to provide a direct comparison.
Table 1: Key Characteristics of Golden Gate and Gibson Assembly
| Feature | Golden Gate Assembly | Gibson Assembly |
|---|---|---|
| Core Mechanism | Restriction-ligation using Type IIS enzymes [54] | Homologous recombination with an enzyme cocktail [59] [58] |
| Key Enzymes | Type IIS Restriction Enzyme (e.g., BsaI), DNA Ligase [56] | 5' Exonuclease, DNA Polymerase, DNA Ligase [54] [57] |
| Seamless/Scarless | Yes [54] [59] | Yes [59] [58] |
| Typical Fragment Capacity | High (up to 30+ fragments in a single reaction) [59] [56] | Moderate (typically up to 6-15 fragments) [59] [57] |
| Optimal Overlap/Homology | Short, defined 4-bp overhangs [56] | Longer homologous overlaps (20-40 bp) [57] [58] |
| Primary Advantage | Superior for high-throughput, modular assembly of many fragments [59] | Flexibility in vector choice and assembly of large fragments [54] [59] |
Scenario-Based Selection Guide
Use Golden Gate Assembly when: Your priority is assembling a large number of DNA fragments (more than 6) in a single, highly efficient reaction [59]. It is the preferred method for modular cloning, building complex multigene pathways, and creating combinatorial libraries where standardized parts are mixed and matched [9] [60]. It is also ideal for assembling DNA fragments with internal homologous or repetitive sequences, which can be problematic for other methods [56].
Use Gibson Assembly when: You are assembling a moderate number of fragments (2-6) and require flexibility in your vector choice, as it does not require specific recognition sites in the final construct [59] [58]. It is also well-suited for assembling very large DNA fragments and is a robust choice for applications like seamless plasmid construction, CRISPR vector creation, and site-directed mutagenesis [58].
Experimental Protocols for Multigene Constructs
Golden Gate Assembly Protocol for Multigene Constructs
The Modular Cloning (MoClo) system is a standardized framework for using Golden Gate Assembly to build complex multigene constructs [9] [61]. The process involves a hierarchical series of one-pot reactions, starting from basic parts and assembling them into transcriptional units, which are then further assembled into multigene constructs.
Detailed Methodology:
- Define Construct and Identify Parts: The structure of the final multigene construct must be defined first. This involves identifying all required basic biological parts (promoters, coding sequences, terminers) from a pre-validated library [9] [61].
- Prepare DNA Fragments: Each basic part must be cloned into a specific MoClo vector, flanked by the appropriate Type IIS recognition sites (e.g., BsaI). The overhangs generated upon digestion are designed to be unique and dictate the order of assembly with adjacent parts [9] [56].
- Set Up Level 1 (Transcriptional Unit) Assembly:
- Reaction Setup: In a single tube, combine:
- Incubation: The reaction is performed in a thermal cycler. For assemblies of 5-10 fragments, a typical program is 30 cycles of (37°C for 1 minute + 16°C for 1 minute), followed by a final 5-minute incubation at 60°C to inactivate the enzymes [56].
- Set Up Level 2 (Multigene Construct) Assembly: The process is repeated using the assembled Level 1 plasmids as parts for the final multigene assembly into a Level 2 vector, following the same reaction setup and cycling conditions [9].
- Transformation and Screening: The final assembly reaction is transformed into competent E. coli. Positive clones are screened by colony PCR or restriction digest, and the sequence of the final construct is verified [61].
Gibson Assembly Protocol for Multigene Constructs
Gibson Assembly joins multiple overlapping DNA fragments in a single, isothermal reaction, making it suitable for assembling multigene constructs without the need for hierarchical assembly.
Detailed Methodology:
- Design and Generate DNA Fragments:
- Overlap Design: Each DNA fragment must be designed with 20-40 base pair homologous overlaps to its neighboring fragments. These overlaps are added via PCR primers [57] [58]. Using software tools like NEBuilder or SnapGene is highly recommended for accurate primer design [57] [58].
- Fragment Preparation: Amplify all DNA fragments and the linearized vector backbone using a high-fidelity DNA polymerase. Purify the PCR products to remove enzymes, buffers, and primers, which can significantly increase assembly efficiency, especially for complex assemblies with three or more fragments [57].
- Perform Gibson Assembly Reaction:
- Reaction Setup: Combine the prepared DNA fragments and linearized vector with the Gibson Assembly Master Mix. For assemblies involving 1-2 fragments, use 0.02-0.5 pmols of total DNA. For more complex assemblies of 4-6 fragments, use 0.2-1.0 pmols [57]. The concentration of insert fragments should be 2-3 times higher than that of the vector backbone [57].
- Incubation: Incubate the reaction at 50°C. The incubation time can range from 15 minutes for simple assemblies to 60 minutes for more complex ones involving multiple fragments [57] [58].
- Transformation and Screening: Transform 2 µL of the assembly reaction into high-efficiency competent E. coli cells, such as NEB 5-alpha. Plate the cells on selective media. Screen resulting colonies by colony PCR, restriction digest analysis, or sequencing to confirm the correct assembly of the multigene construct [57] [58].
Essential Research Reagent Solutions
The following table lists key reagents and their critical functions for successfully executing Golden Gate and Gibson Assembly protocols.
Table 2: Essential Reagents for DNA Assembly Methods
| Reagent / Kit | Function in Assembly | Example Products |
|---|---|---|
| Type IIS Restriction Enzymes | Cleaves DNA outside recognition site to generate specific overhangs for Golden Gate. | BsaI-HFv2, BsmBI-v2, BbsI-HF [56] |
| DNA Ligase | Catalyzes the formation of phosphodiester bonds to seal nicks in DNA backbone. | T4 DNA Ligase [56] |
| Gibson Assembly Master Mix | Pre-mixed cocktail of exonuclease, polymerase, and ligase for seamless fragment assembly. | Gibson Assembly HiFi Master Mix [58], NEB Gibson Assembly Kit (E5510) [57] |
| High-Fidelity DNA Polymerase | Amplifies DNA fragments with minimal errors for high-quality inserts and vectors. | Phusion DNA Polymerase, Platinum SuperFi II PCR Master Mix [58] |
| High-Efficiency Competent Cells | Essential for robust transformation of assembled DNA constructs into E. coli. | NEB 5-alpha Competent E. coli, One Shot TOP10 Competent E. coli [57] [58] |
Golden Gate and Gibson Assembly are both powerful methods for constructing multigene plasmids, yet they serve complementary roles in the research workflow. Golden Gate, particularly through the MoClo system, is unparalleled for standardized, high-throughput assembly of numerous fragments, making it a cornerstone for large-scale synthetic biology projects. Gibson Assembly offers exceptional flexibility and ease of use for projects involving a moderate number of fragments, especially when vector compatibility or the presence of internal restriction sites is a concern. By understanding the strengths and protocols of each method, researchers can strategically select and optimize the right cloning technique to accelerate their multigene construct research and drug development pipelines.
In molecular biology and synthetic biology, the construction of recombinant DNA constructs is a foundational technique. For complex endeavors, such as assembling multigene constructs for metabolic engineering or cellular pathway analysis, the choice of cloning method is critical. Two powerful, yet philosophically distinct, methods have become predominant: Golden Gate and Gateway cloning. This application note provides a structured comparison of these two technologies, framing them within the context of a broader research thesis on advanced assembly protocols. It details their mechanisms, provides actionable protocols, and evaluates their suitability for specific research applications, helping scientists and drug development professionals make an informed choice.
Golden Gate cloning is a restriction-ligation-based method that uses Type IIS restriction enzymes to create and assemble DNA fragments with custom overhangs in a single reaction [62]. Its key advantage is seamless assembly, leaving no residual "scar" sequences at the junctions between fragments [6] [62]. This makes it ideal for applications where the precision of the final DNA sequence is paramount.
In contrast, Gateway cloning is a site-specific recombination-based system. It uses proprietary enzyme mixes (Clonases) to catalyze the exchange of DNA fragments between specialized vectors by recognizing specific attachment (att) sites [63] [64]. Its primary strength is high-throughput versatility, allowing a pre-validated DNA fragment ("entry clone") to be rapidly transferred into a wide variety of "destination vectors" designed for different expression contexts (e.g., different cell lines, tags, or promoters) [64].
Table 1: Core Characteristics of Golden Gate and Gateway Cloning
| Feature | Golden Gate Assembly | Gateway Cloning |
|---|---|---|
| Core Mechanism | Type IIS restriction enzyme digestion and ligation [62] | Site-specific recombination (BP and LR reactions) [63] [64] |
| Reaction Output | Seamless, scarless constructs [6] | Leaves short "scar" sequences (attB sites) in the final construct [63] |
| Multigene Assembly | Highly efficient for one-pot assembly of many fragments (e.g., 1-20) [9] [65] | Possible via Multisite Gateway (typically up to 4 fragments) [64] |
| Typical Speed | Fast, single-tube reaction [62] | Fast, but often involves two sequential reactions (BP and LR) [64] |
| Flexibility/Reusability | High flexibility in fragment order; parts are reusable in other Golden Gate assemblies [9] | High throughput; once an entry clone is made, it can be used in many destination vectors [64] |
| Cost Considerations | Lower cost per reaction; uses standard enzymes [6] | Higher cost; requires proprietary Clonase enzyme mixes and specific vectors [63] [65] |
| Key Limitation | Requires domestication (removal of internal Type IIS sites) from parts [62] | Presence of att site "scars" can interfere with gene function or regulation [63] |
Experimental Protocols
Golden Gate Assembly Protocol for Multigene Constructs
The following protocol, adapted from the Modular Cloning (MoClo) system, is designed for the assembly of multiple genetic elements into a single destination vector [9].
Step 1: Define Construct and Identify Parts
- Determine the final structure of the multigene construct.
- Identify all basic biological parts (promoters, coding sequences, terminers) from a sequenced library.
- Define the assembly strategy, ensuring each part is in a MoClo-compatible entry vector with appropriate flanking Type IIS sites (e.g., for BsaI enzyme).
Step 2: Perform Hierarchical Golden Gate Reaction
- Reaction Setup: In a single tube, combine:
- DNA: ~50-100 fmol of each entry vector and the destination vector.
- Enzyme: BsaI-HFv2 restriction enzyme (or similar Type IIS enzyme).
- Ligase: High-connectivity T4 DNA Ligase.
- Buffer: A compatible buffer supplied with the enzymes (e.g., T4 DNA Ligase Buffer).
- Thermocycling:
- Cycle between 37°C (digestion) and 16°C (ligation) 25-50 times. This cycling drives the reaction toward complete assembly.
- Final enzyme inactivation: 60°C for 5-10 minutes.
- Reaction Setup: In a single tube, combine:
Step 3: Transform and Screen
- Transform the final reaction product into competent E. coli cells.
- Screen colonies for correct constructs. The high efficiency of Golden Gate assembly typically results in a high percentage of correct clones, which can be verified by colony PCR, diagnostic digest, or sequencing.
Gateway Cloning Protocol for Expression Clone Generation
This protocol outlines the standard two-step process to create an expression clone using Gateway technology [63] [64].
Step 1: Create an Entry Clone (BP Reaction)
- Objective: Recombine a DNA fragment flanked by attB sites with a Donor Vector containing attP sites.
- Reaction Setup: Mix ~50 femtomoles of your attB-flanked PCR product or fragment with an equal molar amount of the Donor Vector (e.g., pDONR). Add BP Clonase enzyme mix.
- Incubation: Incubate at 25°C for 1 hour. (Note: Overnight incubation can improve efficiency for some fragments).
- Transformation: Transform the reaction into competent E. coli and select on kanamycin plates. The negative selection marker ccdB in the Donor Vector is replaced by your insert, allowing only correct recombinants to grow.
Step 2: Create an Expression Clone (LR Reaction)
- Objective: Recombine the Entry Clone (with attL sites) with a Destination Vector (with attR sites) to create the final expression construct.
- Reaction Setup: Mix ~20 femtomoles of the verified Entry Clone with ~20 femtomoles of the Destination Vector. Add LR Clonase enzyme mix.
- Incubation: Incubate at 25°C for 1 hour to overnight.
- Transformation: Transform the reaction into competent E. coli and select on ampicillin plates. The ccdB gene in the Destination Vector is replaced by your insert, enabling positive selection of the expression clone.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Golden Gate and Gateway Cloning
| Reagent/Kit | Function | Example Products |
|---|---|---|
| Type IIS Restriction Enzymes | Cuts DNA outside recognition site to generate custom overhangs for seamless assembly. | BsaI-HFv2, BsmBI-v2, PaqCI [62] |
| DNA Ligase | Joins DNA fragments with complementary overhangs. | T4 DNA Ligase [62] |
| Golden Gate Assembly Kits | Pre-optimized master mixes for simplified workflow. | NEBridge Golden Gate Assembly Kit (BsaI-HFv2) [62] |
| Clonase Enzyme Mixes | Proprietary enzyme mixes that catalyze the BP and LR recombination reactions. | BP Clonase II, LR Clonase II [63] [64] |
| Gateway Vectors | Specialized plasmids for the different stages of cloning (Donor, Entry, Destination). | pDONR vectors, pDEST vectors [63] [64] |
| Synthetic DNA Fragments | Custom double-stranded DNA for use as inserts, allowing in silico domestication. | IDT gBlocks Gene Fragments [63] |
| Competent E. coli Cells | For transformation and propagation of recombinant DNA constructs. | Commercially available high-efficiency strains |
Workflow and Mechanism Visualization
Golden Gate Assembly Flow
Gateway Cloning Flow
The choice between Golden Gate and Gateway cloning is not a matter of one being universally superior, but rather which is optimal for a specific research goal.
Choose Golden Gate Assembly when your priority is the precision and quality of the final DNA construct. It is the method of choice for building complex, multigene constructs seamlessly, for modular synthetic biology projects where parts are reused in different configurations, and when minimizing cost per reaction is important [9] [6] [62]. Its one-pot, hierarchical nature is exceptionally powerful for ambitious assembly projects.
Choose Gateway Cloning when your priority is high-throughput and versatility in functional screening. It is ideal for projects where a single gene needs to be expressed in many different contexts (e.g., with different tags, in different cell lines, or under different promoters) [63] [64]. The ability to leverage pre-made libraries of entry clones can dramatically accelerate research workflows focused on functional analysis of many genes.
For a research thesis focused on developing and optimizing protocols for multigene constructs, Golden Gate assembly, particularly the MoClo framework, offers a powerful, scalable, and precise foundation. However, integrating Gateway technology for downstream functional screening of individual genes from these constructs can create a comprehensive and highly efficient research pipeline.
Golden Gate Assembly is a one-pot, one-step cloning method that uses Type IIS restriction enzymes and DNA ligase to seamlessly assemble multiple DNA fragments in a defined order [5]. Its advantages over traditional cloning include a simplified protocol, low hands-on time, no requirement for purification between restriction and ligation, scarless ligation, low background, and easy scalability to multiple fragments and combinatorial libraries [66]. The key differentiator from traditional restriction cloning is the use of Type IIS restriction enzymes, which recognize non-palindromic sequences and cut outside their recognition sites, enabling the creation of custom overhangs for directional assembly [66]. This technical note provides a decision framework for selecting the optimal Golden Gate system for constructing multigene constructs, comparing established systems like MoClo with newer simplified approaches, and detailing essential protocols and reagents.
Comparative Analysis of Golden Gate Systems
The selection of an appropriate Golden Gate system depends on multiple project parameters, including the number of fragments to assemble, desired throughput, and available laboratory resources. The table below provides a quantitative comparison of the primary systems.
Table 1: Decision Framework for Selecting a Golden Gate Cloning System
| System Feature | Modular Cloning (MoClo) | Golden EGG | Expanded Golden Gate (ExGG) | Commercial Kits (e.g., NEBridge) |
|---|---|---|---|---|
| Primary Application | Large, hierarchical multigene constructs [9] | Simplified, cost-effective single-pot assembly [6] | Compatibility with existing, non-Golden Gate vectors [67] | Routine, single-vector cloning [66] |
| Number of Fragments per Reaction | High (hierarchical levels allow virtually unlimited assembly) [9] [5] | Multiple fragments in a single reaction [6] | Multiple fragments [67] | 3+ fragments in a single reaction [66] |
| Entry Clone Requirement | Yes (Level 0, Level 1, Level 2 vectors) [9] [5] | Single universal entry vector for all parts [6] | Not required for destination vector [67] | Can use PCR fragments or entry clones [66] [5] |
| Key Restriction Enzyme | BsaI commonly used [9] | Single Type IIS enzyme (e.g., BsaI) for all steps [6] | BsaI [67] | BsaI-HFv2, BsmBI-v2, PaqCI [66] |
| Typical Assembly Efficiency | High, facilitates construct variant libraries [9] | High, retains key Golden Gate attributes [6] | High, retains strengths of Golden Gate [67] | High efficiency with low empty vector background [66] |
| Ideal Use Case | Metabolic engineering, gene stacking [5] | Projects with limited budget or requiring quick start [6] | Projects needing to clone into specific, pre-existing vectors [67] | Standardized cloning for typical molecular biology labs [66] |
The workflow diagram below illustrates the logical decision-making process for selecting the most appropriate Golden Gate system based on project goals.
Detailed Experimental Protocols
Hierarchical Assembly for Multigene Constructs (MoClo)
The Modular Cloning (MoClo) system is a hierarchical, scalable approach for assembling complex multigene constructs. The following protocol outlines the key steps [9].
Step 1: Construct Planning and Part Identification
- Define the final structure of the multigene construct to identify all basic parts (promoters, coding sequences, terminators) and vectors required.
- Adhere to the standard MoClo assembly rules, which typically involve three hierarchical levels:
- Level 0: Assembly of basic parts into entry vectors.
- Level 1: Assembly of Level 0 modules into single transcription units.
- Level 2: Assembly of multiple Level 1 transcription units into the final multigene construct.
Step 2: Domesticate Parts
- Ensure that all basic parts and vectors lack internal recognition sites for the Type IIS enzyme being used (e.g., BsaI). This process, called domestication, is critical for reaction efficiency.
- If internal sites are present, remove them via site-directed mutagenesis, introducing silent mutations if the site is within a coding sequence [66] [5].
Step 3: Perform One-Pot Golden Gate Reactions
- For each hierarchical level, set up a single-tube digestion-ligation reaction.
- Reaction Mix: Combine equimolar amounts of DNA parts (e.g., 50-100 fmol each), 1 µL of Type IIS restriction enzyme (e.g., BsaI-HFv2), 1 µL of T4 DNA ligase, 2 µL of 10x T4 DNA ligase buffer, and nuclease-free water to a total volume of 20 µL [66] [5].
- Thermocycling: Incubate the reaction in a thermocycler. A typical protocol is 30-50 cycles of (37°C for 2-5 minutes + 16°C for 5-10 minutes), followed by a final digestion step at 60°C for 10 minutes and a hold at 4°C [66] [5]. This cycling promotes iterative digestion and ligation, driving the assembly toward the desired product.
Step 4: Transformation and Screening
- Transform 2-5 µL of the final assembly reaction into competent E. coli cells.
- Screen resulting colonies by colony PCR or restriction digest. The high efficiency of the system facilitates the generation of libraries of construct variants [9].
The following workflow provides a visual guide to the hierarchical MoClo assembly process.
Simplified Assembly with Golden EGG
The Golden EGG system simplifies the initial cloning steps, making it more accessible. The protocol deviates from MoClo in its approach to entry clone creation [6].
Step 1: Universal Entry Vector Preparation
- Use the dedicated pEGG universal entry vector, which contains a cassette with the ccdB and cat genes for negative selection, flanked by outward-directed BsaI recognition sites [6].
Step 2: PCR Amplification of DNA Parts with Special Primers
- Design PCR primers with a specific 5' extension: NGGTCTCNn1n2n3n4, where
n1-n4is the desired 4-nucleotide overhang sequence. - This extension contains an inward-facing BsaI recognition site. After digestion, this design releases the insert with the desired sticky ends [6].
- Design PCR primers with a specific 5' extension: NGGTCTCNn1n2n3n4, where
Step 3: Cloning into pEGG Entry Vector
- Set up a Golden Gate reaction with the pEGG vector and the purified PCR product.
- Critical Modification: Use a specialized temperature profile that includes a cold treatment (e.g., 4°C for 15 minutes) after the initial digestion-ligation phase. This step shifts the reaction kinetics toward ligation, maximizing the yield of circularized entry clones even though the product contains the restriction sites [6].
Step 4: Assembly into Destination Vector
- Release the insert from the entry clone and assemble it with other fragments in a standard Golden Gate reaction. The same Type IIS enzyme is used for both creating entry clones and the final assembly [6].
Research Reagent Solutions
Successful implementation of Golden Gate assembly requires specific, high-quality reagents. The table below details the essential materials and their functions.
Table 2: Essential Reagents for Golden Gate Assembly
| Reagent Category | Specific Examples | Function & Critical Features |
|---|---|---|
| Type IIS Restriction Enzymes | BsaI-HFv2, BsmBI-v2, PaqCI [66] | Creates unique, non-palindromic overhangs for directional assembly. High-fidelity (HF) versions reduce star activity. |
| DNA Ligase | T4 DNA Ligase [66] [5] | Joins DNA fragments via complementary overhangs. Must be active in the restriction enzyme buffer for a one-pot reaction. |
| Vectors & Cloning Kits | MoClo Toolkit (Level 0, 1, 2), pEGG vector, pGGAselect vector [9] [6] [66] | Provide standardized, validated backbones with optimized Type IIS sites for receiving inserts. May include selection/counterselection markers. |
| Competent Cells | MegaX DH10B T1 R Electrocomp Cells [68] | High transformation efficiency is critical for maintaining library diversity and obtaining sufficient colony yields. |
| Selection/Counter-selection Markers | Antibiotic Resistance (Kanamycin, Ampicillin), ccdB toxin gene, sfGFP [6] [66] | Select for successful transformants. Negative selection markers (e.g., ccdB) in empty vectors dramatically reduce background [6] [66]. |
Critical Technical Considerations
- Managing Internal Restriction Sites: The presence of internal Type IIS recognition sites in the vector backbone or insert sequences is a major cause of assembly failure. These sites must be removed through site-directed mutagenesis in a process called domestication [66] [5]. If domestication is not feasible, consider switching to a Type IIS enzyme with a longer, less frequent recognition site [5].
- Optimizing Fragment Order and Orientation: The order and orientation of DNA fragments in the final construct are determined by the designed overhangs. Carefully plan the assembly strategy and annotate all fusion sequences before primer design [5]. Software tools like Geneious Prime can assist in automating this design process and checking for compatibility [69].
- Troubleshooting Low Efficiency: If assembly efficiency is low, verify the absence of internal restriction sites, ensure the use of high-quality DNA, optimize the molar ratio of fragments (a 1:1 to 3:1 insert:vector ratio is a common starting point), and confirm the thermocycling conditions. For complex assemblies, increasing the number of cycles may improve the yield [5].
Conclusion
Golden Gate Assembly stands as a powerful, versatile, and efficient method for constructing complex multigene systems, fundamental to advancing synthetic biology and therapeutic development. By mastering its principles, methodological nuances, and optimization strategies, researchers can reliably engineer sophisticated genetic constructs. Future directions will likely focus on enhancing automation, increasing the fidelity and scale of assemblies, and further standardizing parts to accelerate the design of novel biosynthetic pathways and genetically engineered therapies for clinical applications.
References
- 1. Scarless Cloning with Golden Gate Assembly [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/scarless-cloning-larger-dna-sequences-type-iis.html]
- 2. Type IIS Restriction Enzymes | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/thermo-scientific-restriction-modifying-enzymes/restriction-enzymes-thermo-scientific/fastdigest-thermo-scientific/type-iis-restriction-enzymes.html]
- 3. Type II Restriction Enzymes: What You Need to Know [https://www.neb.com/en-us/tools-and-resources/feature-articles/everything-you-ever-wanted-to-know-about-type-ii-restriction-enzymes?srsltid=AfmBOorXT77kfwN-mRWk9TuvIlS7DVTqWEa2RtQ6rPqIXafw7VuDTlIW]
- 4. Structures, activity and mechanism of the Type IIS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10201449/]
- 5. Golden Gate Assembly [https://www.snapgene.com/guides/golden-gate-assembly]
- 6. Golden EGG, a simplified Golden Gate cloning system to ... [https://www.nature.com/articles/s41598-024-77327-4]
- 7. FAQ: Which restriction enzymes are used in Golden Gate ... [https://www.neb.com/en-us/faqs/2017/07/17/which-restriction-enzymes-are-used-in-golden-gate-assembly?srsltid=AfmBOoqNe7tNc3jj5_pmnF2sMS91L0UoBTJ_ESByckrLGwXq-5-GAzDi]
- 8. Type IIS Restriction Enzymes [https://www.neb.com/en-us/tools-and-resources/selection-charts/type-iis-restriction-enzymes?srsltid=AfmBOoobG-EvMHE8Gfvc__KrFWLJnq0_9fD0qcqkakiqAfB7RUG697Pt]
- 9. Golden Gate Cloning of Multigene Constructs Using the ... [https://pubmed.ncbi.nlm.nih.gov/39363064/]
- 10. NEBridge® Golden Gate Assembly [https://www.neb.com/en-us/applications/cloning-and-synthetic-biology/dna-assembly-and-cloning/golden-gate-assembly?srsltid=AfmBOoor2B7a4KPINTOS8GQaQPT6atbo8EfNLP1MFtRl1X3Q-UE_vV9Y]
- 11. A User's Guide to Golden Gate Cloning Methods and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9680027/]
- 12. A Modular Cloning System for Standardized Assembly of ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0016765]
- 13. Cloning strategies: Golden Gate cloning protocol and tips [https://www.idtdna.com/page/support-and-education/decoded-plus/cloning-strategies-golden-gate-cloning/]
- 14. Applications of Golden Gate cloning to protein production ... [https://www.sciencedirect.com/science/article/abs/pii/S0076687921002251]
- 15. Addgene: Modular Cloning Guide [https://www.addgene.org/cloning/moclo/]
- 16. Advancements in Golden Gate Cloning: A Comprehensive ... [https://pubmed.ncbi.nlm.nih.gov/39363089/]
- 17. NEBridge® Golden Gate Assembly [https://www.neb.com/en-us/applications/cloning-and-synthetic-biology/dna-assembly-and-cloning/golden-gate-assembly?srsltid=AfmBOorGEc1Waw2p_NIxUyyS0dm4aGC52tswaPQb98ByGZJVUTS7ESbi]
- 18. Modular Cloning (MoClo) Guide [https://www.addgene.org/mol-bio-reference/cloning/moclo/]
- 19. Golden Gate Cloning of MoClo Standard Parts - PubMed [https://pubmed.ncbi.nlm.nih.gov/39363063/]
- 20. Short Review: Classical Cloning vs. Gibson Assembly [https://2015.igem.org/Team:Freiburg/Project/Classic_vs_Gibson]
- 21. Golden Gate Assembly with NEBridge Ligase Master Mix [https://2025.igem.wiki/ubc-vancouver/protocols/golden-gate-assembly-with-nebridge-ligase-master-mix]
- 22. Golden Gate Cloning [https://en.wikipedia.org/wiki/Golden_Gate_Cloning]
- 23. Tips and Tricks for Using Golden Gate Modular Cloning ... [https://blog.addgene.org/tips-and-tricks-for-using-golden-gate-modular-cloning-moclo]
- 24. NEBridge® Golden Gate Assembly [https://www.neb.com/en-us/applications/cloning-and-synthetic-biology/dna-assembly-and-cloning/golden-gate-assembly?srsltid=AfmBOooTIV2Py_3u2Pnih6Ru8R9HSI8R97cbcnCW7zg3iRsiUge_7QlA]
- 25. Rapid Assembly of Multi-Gene Constructs using Modular ... [https://pubmed.ncbi.nlm.nih.gov/33616121/]
- 26. Getting Started with Golden Gate Assembly [https://www.neb.com/en-us/nebinspired-blog/getting-started-with-golden-gate?srsltid=AfmBOoortdT6Wp7AwTbRbw088Vi5T2AT0ubccFpU6SAcFP3paU093S1l]
- 27. Technical Tips For Optimizing Golden Gate Assembly ... [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/technical-tips-for-optimizing-golden-gate-assembly-reactions?srsltid=AfmBOooIml2eBL5BuIm_nKdi1B6bjSBjrQw3u8wZgMFwlnAKa37pfr3b]
- 28. PCR Cycling Parameters—Six Key Considerations for ... [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html]
- 29. DNA Ligation: 6 easy tips to improve your reactions [https://bitesizebio.com/10279/how-dna-ligation-works/]
- 30. Optimize Your DNA Ligation with 7 Must-Have Tips [https://www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/optimize-dna-ligase.html]
- 31. Getting Started with Golden Gate Assembly [https://www.neb.com/en-us/nebinspired-blog/getting-started-with-golden-gate?srsltid=AfmBOoq-AIw71tukgjhnR02yEIeMKZkVf0q2rgmwOvyi5X0iecgSQyKp]
- 32. Level 1 and Level 2 Golden Gate Assembly Protocol [https://bio-protocol.org/exchange/minidetail?id=16730010&type=30]
- 33. Bacterial Transformation Workflow [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/molecular-cloning/transformation/bacterial-transformation-workflow.html]
- 34. Understanding Competent Cells for Bacterial Transformation [https://www.goldbio.com/blogs/articles/understanding-competent-cells-for-bacterial-transformation?srsltid=AfmBOoo43sLTdooynx92WNsl30aJeyon4lfrXiIzo61sAxHcriXVrp9V]
- 35. Bacterial Transformation Maxi Prep & Mini Prep [https://www.protocols.io/view/bacterial-transformation-maxi-prep-amp-mini-prep-dxcc7isw.html]
- 36. A unified multi-kingdom Golden Gate cloning platform [https://www.nature.com/articles/s41598-019-46171-2]
- 37. Technical Tips For Optimizing Golden Gate Assembly ... [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/technical-tips-for-optimizing-golden-gate-assembly-reactions?srsltid=AfmBOorxQ2XAaVZD4MxYbzFIUg_uIFdxYHX59oVEwpOyLiRy3ceSpDsl]
- 38. Metabolic pathway assembly using docking domains from ... [https://www.nature.com/articles/s41467-022-33272-2]
- 39. Optimization of Golden Gate assembly through application ... [https://www.synoptics.co.uk/technical-paper/optimization-of-golden-gate-assembly-through-application-of-ligation-sequence-dependent-fidelity-and-bias-profiling/]
- 40. Strange dominos: the fun and perils of DNA overhang design [https://zulko.github.io/bricks_and_scissors/posts/overhangs/]
- 41. Primer design guide - 5 tips for best PCR results [https://the-dna-universe.com/2022/09/05/primer-design-guide-the-top-5-factors-to-consider-for-optimum-performance/]
- 42. Golden Gate Assembly Domestication Tutorial [https://www.neb.com/en-us/tools-and-resources/video-library/golden-gate-assembly-domestication-tutorial?srsltid=AfmBOop4ufe42pOu8lpf0rW_nLx5Ac-_d4Peeh05eL3djIKBSo6rRX1w]
- 43. Golden Gate assembly — Galloway Lab Protocols ... [https://gallowaylabmit.github.io/protocols/en/latest/protocols/cloning/golden_gate.html]
- 44. Technical Tips For Optimizing Golden Gate Assembly ... [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/technical-tips-for-optimizing-golden-gate-assembly-reactions?srsltid=AfmBOopvj2CTAhYZ7RMHOe46OCD5Bg2MnJzjsW_gBBcaClqR_h8rlNz6]
- 45. Golden Gate Assembly [https://www.neb.com/en-us/golden-gate/golden-gate?srsltid=AfmBOoqRCdSjIXcAq4CIuhGjvVxc5AQI3C6kFEqKEfXWVCfkIefg6e1H]
- 46. NEBridge® Golden Gate Assembly [https://www.neb.com/en-us/applications/cloning-and-synthetic-biology/dna-assembly-and-cloning/golden-gate-assembly?srsltid=AfmBOopRcGLqY3kbKW4x1qaRp02jut7K5XIvGLSJzY2dJRK6sLT_hBSU]
- 47. Getting Started with Golden Gate Assembly [https://www.neb.com/en-ca/nebinspired-blog/getting-started-with-golden-gate?srsltid=AfmBOopA6CSlevqcIDpBFwFv5PrJc69vF_asBrqUO_4ZgeqeM7PaBIVj]
- 48. NEBridge® Golden Gate Assembly [https://www.neb.com/en-us/applications/cloning-and-synthetic-biology/dna-assembly-and-cloning/golden-gate-assembly?srsltid=AfmBOorT8s7VU0KcQTlpN8H1aAVVpm0XVCKqus2cOVbNGi9LygGHlWhY]
- 49. Technical Tips For Optimizing Golden Gate Assembly ... [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/technical-tips-for-optimizing-golden-gate-assembly-reactions?srsltid=AfmBOopuWiCOkHEAs3lk8RvhaMqbf2FIDAoxCoykwe6llyqi_sN4hZ86]
- 50. Targeted Multigene Sequencing Panels Enable Fast, Cost ... [https://www.illumina.com/science/customer-stories/icommunity-customer-interviews-case-studies/targeted-multigene-sequencing-panels-enable-fast--cost-effective.html]
- 51. Guidelines for Validation of Next-Generation Sequencing– ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6941185/]
- 52. Guidelines For Clone Isolation and Validation [https://www.thermofisher.com/us/en/home/life-science/genome-editing/genome-editing-learning-center/genome-editing-resource-library/crispr-validated-protocols/guidelines-clone-isolation-validation.html]
- 53. Best practices for the analytical validation of clinical whole ... [https://www.nature.com/articles/s41525-020-00154-9]
- 54. Golden Gate Assembly vs. Gibson Assembly: DNA Cloning ... [https://synapse.patsnap.com/article/golden-gate-assembly-vs-gibson-assembly-dna-cloning-methods-compared]
- 55. A comparative review of DNA assembly strategies [https://www.sciencedirect.com/science/article/pii/S2452014425002493]
- 56. Comparative Analysis of Golden Gate Assembly and ... [https://www.biosynsis.com/blog/comparative-analysis-of-golden-gate-assembly-and-gibson-assembly-techniques/]
- 57. Gibson Assembly® Protocol (E5510) Milena 07/ 2025 [https://www.protocols.io/view/gibson-assembly-protocol-e5510-milena-07-2025-g385byry7.html]
- 58. Gibson Assembly 101: Expert Cloning Tips You Need to ... [https://www.thermofisher.com/blog/life-in-the-lab/gibson-assembly-101-expert-cloning-tips-you-need-to-know/]
- 59. Gibson Assembly vs Golden Gate: A Complete Guide to ... [https://www.clyte.tech/post/gibson-assembly-vs-golden-gate-a-complete-guide-to-choosing-the-right-cloning-method?srsltid=AfmBOorHfK0VSBcmbwUS4QsqfNnLp-3rD3E1m6-7JrM4ufHkgYEGYYPZ]
- 60. Recent advances in DNA assembly technologies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4257898/]
- 61. of Assembly Using Multigene Cloning Constructs Golden Gate [https://link.springer.com/protocol/10.1007/978-1-4939-2760-9_19]
- 62. Getting Started with Golden Gate Assembly [https://www.neb.com/en/nebinspired-blog/getting-started-with-golden-gate?srsltid=AfmBOoqYAZriO_jlXSnG9bXHAJwd8YpaUeX_w8YXmYOCBtByGXB5J-f_]
- 63. Gateway™ cloning: Definitions, tips, and tricks [https://eu.idtdna.com/page/support-and-education/decoded-plus/gateway-cloning-definitions-tips-and-tricks/]
- 64. Gateway Cloning Technique [https://www.snapgene.com/guides/gateway-cloning]
- 65. Introduction to Molecular Cloning Methods [https://www.geneious.com/guides/molecular-cloning-methods]
- 66. Getting Started with Golden Gate Assembly [https://www.neb.com/en-us/nebinspired-blog/getting-started-with-golden-gate?srsltid=AfmBOooobOkvV8zReo6_-aaHOv0plkKmvCmUvMgpR3tPrf4Tp7Rlkw-l]
- 67. An efficient cloning method to expand vector and restriction ... [https://www.sciencedirect.com/science/article/pii/S2667237523002084]
- 68. Protocol for multiplexed transcription factor activity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12491162/]
- 69. Practice Golden Gate Cloning [https://www.geneious.com/tutorials/golden-gate-cloning]