Optimizing Gene Expression Using Orthogonal Regulators: From Foundational Concepts to Therapeutic Applications
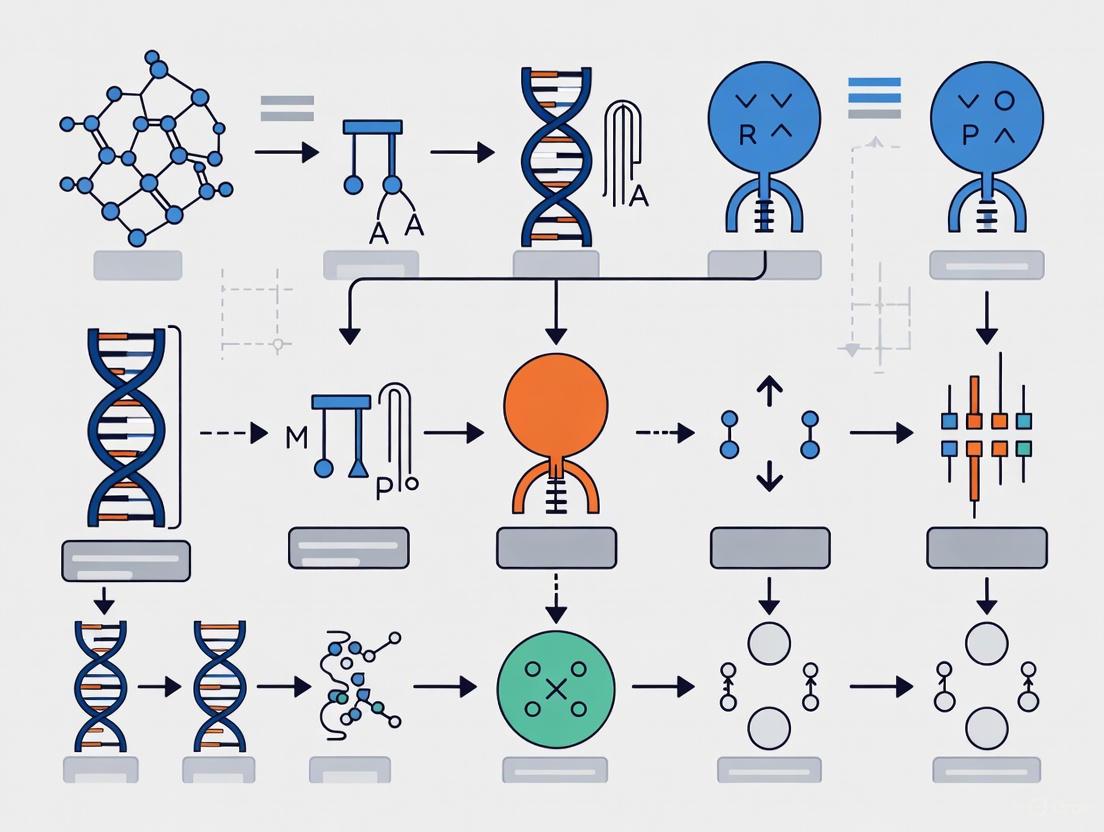
Orthogonal gene expression systems represent a transformative approach in synthetic biology, enabling precise, independent control of multiple genetic elements without cross-talk with host regulatory networks.
Optimizing Gene Expression Using Orthogonal Regulators: From Foundational Concepts to Therapeutic Applications
Abstract
Orthogonal gene expression systems represent a transformative approach in synthetic biology, enabling precise, independent control of multiple genetic elements without cross-talk with host regulatory networks. This article provides a comprehensive resource for researchers and drug development professionals, exploring the foundational principles of orthogonality from bacterial to mammalian systems and plants. It details cutting-edge methodological applications, including CRISPR-based transcription factors and synthetic promoter design, for programming complex cellular behaviors. The content further addresses critical troubleshooting and optimization strategies to enhance performance and reduce variability in experimental and therapeutic settings. Finally, it covers rigorous validation frameworks and comparative analyses of orthogonal tools, establishing a clear path for their use in advancing biomedical research, protein evolution, and next-generation gene therapies.
The Principles of Orthogonality: Decoupling Gene Expression from Host Networks
Core Concepts and Terminology: Frequently Asked Questions
What is orthogonal control in synthetic biology? Orthogonal control describes the design of biological systems that operate independently from the host's native cellular processes. The term "orthogonality" refers to the inability of two or more biomolecules, similar in composition or function, to interact with each other or affect one another's substrates. For example, two proteases are mutually orthogonal if they cannot cleave each other's target substrates, and two aminoacyl-tRNA synthetases are orthogonal if they do not inappropriately cross-aminoacylate non-cognate tRNAs. This independence prevents unintended interactions, or cross-talk, enabling more predictable and reliable circuit performance [1].
Why is achieving orthogonality crucial for engineered genetic circuits? Engineered gene circuits frequently repurpose components from natural biological systems and are often hampered by inadvertent interactions with host machinery. This cross-talk can:
- Deplete essential host resources, reducing host fitness and circuit performance [1].
- Create unpredictable circuit behavior due to interference from native signaling pathways [2].
- Limit the complexity of circuits that can be built, as the potential for harmful interactions grows with the number of components [1]. Orthogonal systems mitigate these issues by creating insulated functional modules.
At what levels can orthogonal control be implemented? Orthogonal control can be engineered to act at multiple stages of the central dogma and beyond, including:
- DNA Level: Using orthogonal DNA replication systems (e.g., OrthoRep in yeast) [1], site-specific recombinases [3], and synthetic epigenetic regulation with non-canonical nucleobases [1] [3].
- Transcriptional Level: Employing synthetic transcription factors (sTFs) and orthogonal RNA polymerases that recognize custom promoter sequences [3] [4] [5].
- Translational Level: Utilizing orthogonal ribosomes and genetic code expansion to incorporate non-canonical amino acids [1].
- Post-Translational Level: Implementing synthetic receptors that trigger user-defined, self-contained signaling pathways independent of native intracellular signaling [2].
What are the primary design principles for creating orthogonal systems? The design of orthogonal systems relies on several key principles:
- Modularity: Systems are built from discrete, interchangeable parts (e.g., DNA-binding domains, activation domains, and core promoters) that can be mixed and matched [5].
- Self-Contained Signaling: Ideal orthogonal systems function as closed loops. For instance, synthetic receptors like MESA and NatE MESA are designed so that ligand binding directly induces a synthetic intracellular signal (e.g., protease-mediated release of a transcription factor) without relying on native signaling cascades [2].
- Use of Heterologous Components: Sourcing parts from unrelated organisms (e.g., using bacterial LexA DNA-binding domains in yeast) reduces the likelihood of cross-talk with the host's machinery [5].
Troubleshooting Common Experimental Issues
Problem: High Background Expression (Leakiness) in an Off-State
- Potential Cause 1: Inadequate Insulation of Synthetic Promoters.
- Solution: Ensure synthetic promoters are designed with minimal sequence homology to host promoters. For systems using upstream activating sequences (UAS), verify that the core promoter has low basal activity on its own. Using a stronger or more specific UAS can improve the signal-to-noise ratio [4] [5].
- Potential Cause 2: Non-Specific Interaction of Synthetic Transcription Factors.
- Solution: Characterize the DNA-binding specificity of your sTF using methods like electrophoretic mobility shift assays (EMSAs). If using a system with multiple sTFs, confirm their mutual orthogonality. Directed evolution can be used to enhance the specificity of DNA-binding domains [5].
- Potential Cause 3: Overexpression of Circuit Components.
Problem: Low Induced Expression or Weak Signal Output
- Potential Cause 1: Suboptimal Component Matching.
- Solution: Systematically test different combinations of functional modules. The output of a synthetic transcription amplifier system, for example, is tunable by selecting different activation domains, varying the number of transcription factor binding sites in the promoter, and swapping the core promoter module [5]. The table below summarizes key tunable parameters from a study in Saccharomyces cerevisiae [5].
- Potential Cause 2: Poor Surface Expression of Synthetic Receptors.
- Solution: When engineering synthetic receptors (e.g., NatE MESA), the choice of transmembrane and juxtamembrane domains is critical. Test designs that retain native domains versus those with validated orthogonal domains (e.g., truncated CD28 TMD) to find the optimal configuration for proper trafficking and function [2].
- Potential Cause 3: Host-Specific Silencing or Instability.
- Solution: Check the stability of your genetic constructs. For eukaryotic hosts, consider using matrix attachment regions (MARs) or insulators to prevent positional effects and epigenetic silencing. Also, ensure that codon optimization is appropriate for your host chassis.
Problem: Unintended Effects on Host Cell Fitness or Growth
- Potential Cause 1: Resource Overload and Metabolic Burden.
- Solution: High-level expression of orthogonal circuits can deplete cellular resources like nucleotides, amino acids, and energy. To mitigate this, use lower-copy-number plasmids or genomic integration, and avoid excessively strong constitutive promoters for circuit components [1].
- Potential Cause 2: Toxicity of Orthogonal Components.
- Solution: Some synthetic parts, such as certain activation domains or proteases, may be toxic to the host. If a design fails to yield viable transformants (as was observed with a modified QF2 AD in Penicillium chrysogenum [4]), try alternative orthologous parts or use inducible systems to control the timing of expression.
- Potential Cause 3: Inadvertent Disruption of Native Pathways.
- Solution: Perform RNA-seq or proteomic analyses on your engineered strain compared to a wild-type control to identify global changes. This can help pinpoint unexpected interactions and guide the re-design of more specific orthogonal parts.
| Module | Function | Tuning Effect on Output |
|---|---|---|
| Activation Domain (AD) | Recruits the transcriptional machinery | Stronger ADs (e.g., VP64) produce higher maximal output. |
| Transcription Factor Binding Site (UAS) | Number and affinity of sTF binding sites | Increasing the number of high-affinity binding sites amplifies the output signal. |
| Core Promoter (CP) | Site for assembly of the general transcription machinery | Different core promoters provide a wide range of baseline and maximal expression levels. |
Experimental Protocols for Key Characterizations
Protocol: Characterizing Synthetic Transcription Factor Binding
- Objective: To validate the specificity and affinity of a synthetic transcription factor (sTF) for its target DNA sequence using an Electrophoretic Mobility Shift Assay (EMSA) [5].
- Procedure:
- Protein Purification: Express and purify the sTF, often with an affinity tag (e.g., 6xHis).
- DNA Probe Preparation: Synthesize and label double-stranded DNA oligonucleotides containing the target binding site with a fluorescent dye (e.g., Cy5).
- Binding Reaction: Incubate a fixed amount of the labeled DNA probe with increasing concentrations of the purified sTF in a binding buffer.
- Gel Electrophoresis: Load the reactions onto a non-denaturing polyacrylamide gel. The protein-DNA complex will migrate slower than the free DNA probe.
- Imaging and Analysis: Visualize the gel using a fluorescence scanner. A successful, specific interaction is indicated by a clear shift in the probe's migration, which increases with higher sTF concentration. Competition with unlabeled probe can confirm specificity.
- Troubleshooting Tip: If a shift is not observed, optimize buffer conditions (salt, pH, divalent cations) and confirm the activity of the purified sTF.
Protocol: Validating Orthogonality of a Synthetic Receptor
- Objective: To confirm that a synthetic receptor (e.g., a NatE MESA receptor) signals through its intended orthogonal pathway without activating native signaling.
- Procedure:
- Reporter Cell Line: Create a cell line that expresses the synthetic receptor and a reporter gene (e.g., GFP) under the control of the receptor's synthetic output promoter.
- Stimulus Application: Expose the cells to the target ligand. Include controls with no ligand and with a negative control ligand.
- Output Measurement: Quantify the reporter signal (e.g., via flow cytometry or fluorescence microscopy).
- Specificity Test: In a separate experiment, use pharmacological inhibitors or genetic knockouts of key components in the native signaling pathways that could be perceived as potential cross-talk targets. The output of the orthogonal receptor should be unaffected by these perturbations.
- Cross-Talk Test: Measure the activity of reporters for native pathways (e.g., NF-κB, MAPK) upon stimulation of your synthetic receptor. A truly orthogonal receptor will not activate these native reporters.
- Troubleshooting Tip: High background in the "no ligand" control may indicate leaky receptor dimerization; consider tuning the transmembrane domain or split protease affinity [2].
Essential Research Reagent Solutions
Table: Key Reagents for Orthogonal System Construction
| Reagent | Function | Example(s) |
|---|---|---|
| Orthogonal DNA-Binding Domains | Provides sequence-specific targeting to synthetic promoters. | LexA (bacterial) [5], QF (Neurospora crassa) [4], TALEs, dCas9. |
| Activation/Repression Domains | Recruits or blocks the cellular transcription machinery. | VP16, VP64 (strong activation) [4] [5], KRAB, SID (repression). |
| Synthetic Promoters | Drives expression of output genes in response to orthogonal regulators. | A core promoter combined with specific Upstream Activating Sequences (UAS) for a chosen sTF (e.g., QUAS for QF, LexAop for LexA) [4] [5]. |
| Orthogonal Replication System | Enables independent maintenance and evolution of genetic material. | OrthoRep system in yeast, based on cytoplasmic plasmids and an orthogonal DNA polymerase [1]. |
| Synthetic Receptor Platforms | Converts extracellular ligand sensing into a custom intracellular signal. | MESA (Modular Extracellular Sensor Architecture) [2], NatE MESA (using natural ectodomains) [2]. |
| Non-Canonical Amino Acids (ncAAs) | Enables genetic code expansion for novel chemical and biological properties. | Various ncAAs incorporated using orthogonal aminoacyl-tRNA synthetase/tRNA pairs [1]. |
System Architecture and Workflow Diagrams
Diagram Title: Orthogonal Synthetic Receptor Signaling Pathway
Diagram Title: Troubleshooting Guide for High Background Expression
The Critical Need for Orthogonality in Basic Research and Complex Phenotype Investigation
Troubleshooting Guides
Guide 1: Troubleshooting Low Transcriptional Output in Synthetic Orthogonal Systems
Problem: Your synthetic orthogonal transcriptional system is producing unexpectedly low levels of gene expression output.
Question Answer:
- Q: I've assembled an orthogonal CRISPR/dCas9 activation system, but my gene expression output is very low compared to expectations. What could be wrong?
- Solution: Low transcriptional output can stem from several issues in your orthogonal system. First, verify the number and positioning of gRNA binding sites upstream of your minimal promoter. Research shows that varying the number of gRNA binding sites (typically 3-4 repeats) significantly impacts activation strength [6]. Second, ensure your synthetic promoters maintain proper modular architecture with gRNA binding sites followed by a minimal 35S promoter [6]. Third, check the expression level of your dCas9 activator fusion (e.g., dCas9:VP64) and confirm nuclear localization. Finally, validate that your gRNA expression constructs using either Pol III (U6) or inducible Pol II promoters are functioning correctly [6].
Table: Key Variables to Test for Low Transcriptional Output
| Variable to Test | Suggested Test Range | Expected Impact |
|---|---|---|
| Number of gRNA binding sites | 2, 3, 4, 5 repeats | Increased sites typically boost activation [6] |
| dCas9-activator expression level | Low, Medium, High | Higher levels may increase target activation |
| gRNA expression strength | Different promoters (U6, inducible Pol II) | Stronger promoters enhance gRNA production [6] |
| Incubation time post-induction | 24h, 48h, 72h | Longer times may increase output accumulation |
Guide 2: Troubleshooting Cross-Talk in Orthogonal Genetic Circuits
Problem: Your designed orthogonal genetic circuits are exhibiting unexpected cross-talk between components that should function independently.
Question Answer:
- Q: My multi-gene circuit with supposed orthogonal regulators shows unexpected activation of off-target genes. How can I identify the source of cross-talk?
- Solution: Cross-talk violates the fundamental principle of orthogonality, where systems should function independently. To resolve this: First, conduct individual testing of each regulator-promoter pair to establish baseline specificity before combining them in full circuits [7] [6]. Second, verify the specificity of your DNA-binding elements (e.g., gRNA sequences for CRISPR systems) by examining potential off-target binding sites in your synthetic promoters through sequence alignment tools. Third, ensure you're using thoroughly characterized orthogonal parts with demonstrated minimal host interaction [6]. Implement proper controls including negative controls with non-targeting gRNAs and positive controls for each independent pathway. If cross-talk persists, consider increasing the sequence divergence between your orthogonal promoter elements or implementing additional insulation strategies with boundary elements.
Diagram: Cross-talk in Genetic Circuits
Guide 3: Troubleshooting Background Expression in Chemically-Induced Systems
Problem: Your chemically induced orthogonal regulation system shows significant background expression in the non-induced state.
Question Answer:
- Q: My chemically induced CRISPR/Cas9 activator shows high background expression even without the inducing ligand. How can I reduce this leakiness?
- Solution: Background expression in chemically induced systems compromises experimental control. To address this: First, optimize the chemical inducer concentration using a dose-response curve to identify the minimal concentration that provides robust induction with minimal background [8]. Second, consider implementing a dual-control system that combines transcriptional control with post-translational control, such as nuclear localization signal masking or conditional degrons. Third, verify the integrity of your inducible dimerization system components - ensure that your chemically induced dimerizing (CID) proteins are properly expressed and functional [8]. Fourth, examine your vector design for potential promoter interference that might cause basal expression of your activators. If using multiple inducible systems, ensure they are truly orthogonal and not cross-reacting with the same ligands.
Table: Troubleshooting Background Expression
| Possible Cause | Diagnostic Experiment | Potential Fix |
|---|---|---|
| Promoter leakiness | Measure uninduced expression with different promoters | Switch to tighter regulated promoters |
| Non-specific CID interaction | Test system with mutated CID domains | Optimize CID domains for orthogonality [8] |
| Non-optimal inducer concentration | Perform dose-response curve | Titrate inducer for optimal window |
| Protein misfunction | Express & purify components for in vitro testing | Use validated protein variants |
Frequently Asked Questions (FAQs)
Conceptual Questions
Q: What exactly does "orthogonality" mean in the context of genetic regulation? A: In genetic regulation, orthogonality refers to synthetic biological systems that operate independently from the host's native regulatory machinery and from each other [6]. This means orthogonal transcription factors should regulate only their intended synthetic promoters without affecting endogenous genes or other synthetic circuits. Similarly, orthogonal promoters should respond only to their cognate regulators. This concept ensures that complex genetic circuits can be built from modular parts that function predictably without unwanted interactions [3].
Q: Why is orthogonality critically important for investigating complex phenotypes? A: Orthogonality is essential for complex phenotype investigation because it enables precise dissection of individual pathway contributions without confounding cross-talk [7]. When studying multifactorial traits (like disease states or developmental processes), orthogonal systems allow researchers to selectively modulate specific genes or pathways while leaving others unaffected. This precise control is particularly valuable in drug development for validating therapeutic targets and understanding mechanism of action without the noise of pleiotropic effects [3].
Technical Implementation Questions
Q: What are the main advantages of CRISPR-based orthogonal systems compared to traditional transcription factor systems? A: CRISPR-based orthogonal systems offer several key advantages: (1) Programmability - specificity is determined by easily designed gRNA sequences rather than complex protein-DNA interactions; (2) Scalability - multiple orthogonal gRNAs can target different promoters using the same dCas9 activator; (3) Predictability - binding follows simple Watson-Crick base pairing rules; and (4) Versatility - the same dCas9 platform can be fused to various effector domains (activators, repressors, epigenomic modifiers) [6]. Traditional transcription factors require engineering of DNA-binding domains for each new target, which is more technically challenging.
Q: How many truly orthogonal regulatory systems can I realistically combine in a single cell? A: Current research demonstrates successful implementation of 3-4 mutually orthogonal systems in single plant cells [6] and human cells [8]. The theoretical limit is constrained by the number of available non-cross-reacting components (DNA-binding domains, small molecule inducers, etc.). The emerging toolkit includes orthogonal systems based on CRISPR/dCas9 with engineered gRNAs, synthetic zinc fingers, and recombinases with specific target sites [3]. When designing multi-system experiments, it's crucial to empirically validate orthogonality for your specific combination, as context-dependence can cause unexpected interactions in new cellular environments.
Q: Can I achieve temporal control with orthogonal systems? A: Yes, multiple strategies exist for temporal control. Chemically induced dimerization (CID) systems allow ligand-dependent temporal control of CRISPR/Cas9 activators with minimal background [8]. Light-controlled systems using photocleavable linkers or optogenetic dimerization domains provide even finer temporal resolution, enabling precise manipulation of gene expression patterns in the minute-to-hour range [9]. For example, recombinase activity has been made light-dependent by splitting the protein and reconstituting it through light-inducible dimerization systems [3].
Experimental Protocols
Protocol 1: Validating Orthogonality of Synthetic Promoter-Transcription Factor Pairs
Purpose: To empirically confirm that synthetic transcriptional components function without cross-talk.
Materials:
- Plasmids encoding orthogonal transcription factors (e.g., dCas9:VP64) [6]
- Reporter constructs with synthetic promoters containing corresponding gRNA binding sites [6]
- Appropriate host cells (Nicotiana benthamiana, Arabidopsis thaliana, or human cell lines) [7] [6]
- Transfection/transformation reagents
- Reporter assay reagents (microscopy, luciferase, or flow cytometry equipment)
Procedure:
- Clone transcriptional units using modular cloning systems like Golden Gate or MoClo to ensure standardized assembly [6].
- Design a test matrix where each transcription factor is paired with each synthetic promoter in separate experiments.
- Transferd/transform each combination into your host system alongside appropriate controls (empty vector, non-targeting gRNA).
- Measure reporter expression after 24-48 hours using appropriate detection methods.
- Analyze data for orthogonality: True orthogonality is demonstrated when each transcription factor strongly activates only its cognate promoter and shows minimal activation (<5-10% of cognate activation) of non-cognate promoters [6].
Diagram: Orthogonality Validation Workflow
Protocol 2: Implementing Chemically Induced Orthogonal CRISPR Activation
Purpose: To establish temporal control of gene expression using ligand-inducible CRISPR-based orthogonal regulators.
Materials:
- Plasmids encoding chemically inducible dCas9 fused to dimerization domains [8]
- gRNA expression constructs targeting synthetic promoters [6]
- Chemical inducer (specific to your dimerization system)
- Appropriate cell culture materials and reporter assays
Procedure:
- Design synthetic promoters with target sites for your gRNAs upstream of a minimal promoter.
- Clone gRNA expression cassettes under control of Pol III (U6) or inducible Pol II promoters.
- Co-transfect inducible dCas9 activator, gRNA, and reporter constructs into your target cells.
- Treat with chemical inducer at varying concentrations and timepoints to establish kinetic and dose-response profiles.
- Quantify gene expression using appropriate methods (RT-qPCR, fluorescence, luminescence) at multiple timepoints post-induction.
- Verify orthogonality by testing multiple inducible systems with their specific ligands to confirm no cross-activation [8].
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Reagents for Orthogonal Genetic Regulation
| Reagent/Category | Function | Examples/Specific Instances |
|---|---|---|
| Programmable DNA-binding platforms | Provide targeting specificity to synthetic promoters | dCas9:VP64 [6], TALEs [7], Zinc finger proteins [3] |
| Synthetic promoters | Serve as orthogonal targets for artificial transcription factors | Engineered promoters with gRNA binding sites upstream of minimal promoters [6] |
| Chemical dimerization systems | Enable temporal control of regulator activity | Chemically induced dimerizing (CID) proteins [8] |
| Modular cloning systems | Facilitate standardized assembly of genetic circuits | Golden Gate assembly, MoClo framework [6] |
| Reporter genes | Enable quantification of orthogonal system performance | Fluorescent proteins (GFP, BFP, YFP, RFP), luciferase [6] |
| Cox-2-IN-26 | Cox-2-IN-26, MF:C23H21N7OS3, MW:507.7 g/mol | Chemical Reagent |
| Antibacterial agent 92 | Antibacterial Agent 92|Triple-site aaRS Inhibitor | Antibacterial agent 92 is a potent triple-site aminoacyl-tRNA synthetase (aaRS) inhibitor. For Research Use Only. Not for human use. |
Troubleshooting Guide: Common Experimental Issues
No or Low Expression Output
| Problem Cause | Underlying Principle | Solution |
|---|---|---|
| Non-Orthogonal Crosstalk | Circuit components (e.g., repressors, promoters) interact with host genome or other circuit parts, creating unintended regulation [3]. | Perform BLAST analysis to ensure component uniqueness; use highly orthogonal parts from different microbial species [3]. |
| Host Strain Mismatch | Expression of orthogonal components often requires specific host factors (e.g., T7 RNA polymerase in BL21(DE3) for pET systems) [10]. | Transfer plasmid to an appropriate expression host strain (e.g., BL21(DE3), BL21-AI) [10] [11]. |
| High Basal Expression (Leakiness) | Incomplete repression leads to resource drain and toxicity, compromising circuit function [3] [11]. | Use tighter regulation systems (e.g., BL21(DE3) pLysS/E); add glucose to repress basal T7 polymerase expression; consider arabinose-inducible pBAD system [11]. |
Unstable or Oscillating Expression Dynamics
| Problem Cause | Underlying Principle | Solution |
|---|---|---|
| Unintended Feedback Loops | Endogenous host regulators create unforeseen interactions with synthetic circuit [3] [12]. | Map host interactions using transcriptomics; insulate circuit with non-regulatory genetic elements [3]. |
| Insufficient Repressor Strength | Weak repressors cannot overcome strong promoter activity, failing to establish linearization [3] [13]. | Characterize repressor-promoter pairs; use chimeric repressors with stronger repression domains [13]. |
| Component Toxicity | Overexpressed regulatory proteins (e.g., repressors) burden the host, causing growth defects and instability [11] [14]. | Lower induction temperature (e.g., 18-30°C); reduce inducer concentration (e.g., 0.1-1 mM IPTG); use low-copy number plasmids [11]. |
High Cell-to-Cell Variability (Noise)
| Problem Cause | Underlying Principle | Solution |
|---|---|---|
| Chromatin Environment Effects | Genomic integration locus influences expression noise; repressed chromatin states associate with higher variability [15]. | Target genomic loci associated with low noise (e.g., open chromatin); use insulators to buffer positional effects [15]. |
| Low Burst Frequency | Expression noise is regulated by transcriptional burst frequency; low frequencies lead to higher variability [15]. | Engineer promoters for higher burst frequencies to reduce noise independent of mean expression levels [15]. |
| Resource Competition | Limited cellular resources (e.g., RNA polymerase, ribosomes) are shared between host and circuit, creating stochastic fluctuations [3]. | Use orthogonal resources (e.g., T7 RNA polymerase, orthogonal ribosomes) to decouple circuit from host [3]. |
Experimental Protocols for Key Characterizations
Protocol 1: Quantifying Orthogonality and Crosstalk
Objective: Measure interaction between orthogonal system components and host genome.
- Clone Reporter Arrays: Construct fluorescent reporter plasmids for each repressor-promoter pair in your system.
- Transform Single and Multiple Systems: Introduce individual reporters and all possible combinations into your expression host (e.g., BL21(DE3)).
- Flow Cytometry Measurement: Grow cultures to mid-log phase (OD600 ≈ 0.6), induce with appropriate concentration of inducer, and analyze using flow cytometry after 4-6 hours.
- Calculate Orthogonality Score: For each repressor-promoter pair (i, j), calculate the score as (Expression{ii} - Expression{ij}) / Expression_{ii}. A score of 1 indicates perfect orthogonality [3] [13].
Protocol 2: Characterizing Feedback Loop Dynamics
Objective: Analyze temporal dynamics and stability of repressor-based feedback loops.
- Circuit Assembly: Clone feedback architecture (e.g., autorepression loop) into appropriate vector with fluorescent reporter.
- Time-Course Monitoring: Transform into host, induce, and monitor both OD600 and fluorescence over 12-24 hours using plate readers.
- Single-Cell Analysis: Sample at key time points for flow cytometry to assess population heterogeneity.
- Model Fitting: Fit data to mathematical models (e.g., delayed differential equations: dx/dt = α/(1+x(t-τ)^h) - βx) to parameters like repression strength (h) and critical delay (τ*) [12].
Pathway and Workflow Visualizations
Diagram: Orthogonal Repressor System Architecture
Diagram: Troubleshooting Expression Problems
Research Reagent Solutions
| Research Need | Recommended Reagents | Function & Rationale |
|---|---|---|
| Tightly Regulated Expression | BL21(DE3) pLysS/pLysE strains; BL21-AI for T7 and pBAD systems [11]. | pLysS/E expresses T7 lysozyme to inhibit basal T7 RNA polymerase; BL21-AI tightly controls T7 pol with arabinose [11]. |
| Orthogonal Transcriptional Parts | Synthetic transcription factors based on TALEs or dCas9; orthogonal RNA polymerases and sigma factors from bacteriophages [3] [13]. | Programmable DNA-binding domains target synthetic promoters without affecting host genes; phage polymerases only transcribe specific promoters [3]. |
| Noise Control & Characterization | Dual-reporter lentiviral systems for noise quantification; chromatin environment modifiers [15]. | Dual reporters distinguish intrinsic vs. extrinsic noise; epigenetic tools control burst frequency and size to tune variability [15]. |
| Circuit Memory & Logic | Serine integrases (Bxb1, PhiC31); tyrosine recombinases (Cre, Flp); CRISPR base/prime editors [3]. | Enable stable, heritable genetic memory through DNA inversion/excision; record stimulus history via sequential DNA edits [3]. |
FAQs: Core Concepts and Troubleshooting
Q1: What is orthogonality in genetic engineering, and why is it critical for synthetic biology? Orthogonality refers to the design of synthetic biological systems that function independently from the host organism's native cellular processes. In practice, this means creating genetic regulators, promoters, and circuits that do not recognize the host's natural sequences and are not recognized or interfered with by the host's machinery. This is critical because it prevents unwanted cross-talk that can lead to:
- Circuit Failure: Host interference can disrupt the intended logic of synthetic gene circuits.
- Host Fitness Costs: Unprogrammed interactions can burden the host, causing severe growth and developmental defects, as observed in plants [6].
- Unpredictable Behavior: Without orthogonality, synthetic circuits behave inconsistently across different organisms or even different cell types, hindering reliable engineering [6] [16].
Q2: I am experiencing high basal expression (leakiness) in my orthogonal system. What are the primary causes and solutions? High basal expression is a common challenge. The causes and mitigation strategies are often system-dependent:
- Insufficient Promoter Insulation: Synthetic promoters may not be fully shielded from the host's transcriptional machinery.
- Solution: Ensure the use of well-characterized minimal promoters and consider increasing the number of engineered binding sites for your orthogonal transcription factor to enhance specificity [6].
- Leaky Inducer Systems: The inducible systems themselves may have background activity.
- Solution: Employ a dual-input cascade system. Research in human cell lines using a Tunable Noise Rheostat (TuNR) showed that a serial arrangement of two inducible transcriptional activators can significantly attenuate basal leakiness while achieving over 1000-fold induction [16].
- Context Sensitivity (for Riboswitches): The sequence surrounding a riboswitch, especially the coding region immediately downstream, can drastically affect its folding and function, leading to an "always ON" state [17].
- Solution: Systematically engineer the N-terminal sequence of the gene. Using a Design of Experiments (DoE) approach to optimize the ribosome-binding site (RBS), anti-RBS, and downstream codons can yield riboswitches with a 550-fold dynamic range over basal expression [17].
Q3: My orthogonal system works in one organism but fails in another. How can I improve portability? This typically stems from host-specific factors.
- Cause: Differences in transcriptional/translational machinery, chromatin environments, or innate immune responses against foreign DNA.
- Solutions:
- Use Highly Orthogonal Parts: Leverage regulators derived from distantly related species (e.g., yeast regulators in plants) to minimize recognition by the new host [7].
- Universal Programmable Systems: Adopt CRISPR/dCas9-based transcription factors. These can be programmed to target completely synthetic promoters, making the system inherently portable across eukaryotes. The core dCas9 activator is universal, and only the gRNA needs to be redesigned for new synthetic promoters [6].
- Validate Parts in the Target Host: Always characterize genetic parts (promoters, terminators) in the specific host organism of interest, as performance can vary significantly [6].
Troubleshooting Guide: Common Experimental Issues
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Low Signal/Induction | Weak synthetic promoter design | Increase the number of transcription factor binding sites (e.g., from 3 to 4 repeats) upstream of the minimal promoter [6]. |
| Sub-optimal expression of orthogonal regulators | Use stronger constitutive promoters to express the orthogonal transcription factor (e.g., dCas9 fusion) and ensure efficient nuclear localization [6]. | |
| High Cell-to-Cell Variability | Stochastic expression of circuit components | Use the TuNR circuit or similar to titrate the expression of the orthogonal components with small molecules, allowing you to find an induction regime that minimizes noise [16]. |
| Host Growth Defects | Toxicity or burden from circuit components | Implement inducible systems to only activate the circuit during experimentation. Use orthogonal systems to minimize pervasive interference with host genes [6]. |
| Poor Dynamic Range | Context-dependent part performance (esp. riboswitches) | Re-engineer the genetic context. For riboswitches, combine codon optimization of the downstream sequence with RBS and anti-RBS library screening to isolate high-performance variants [17]. |
Experimental Protocols
Protocol 1: Implementing a CRISPR/dCas9-Based Orthogonal Control System (OCS) in Plants
This protocol is adapted from the work published in Plant Methods for constructing a fully orthogonal gene expression system in plants using synthetic promoters and dCas9 [6].
1. Principle An artificial transcription factor (dCas9:VP64) is programmed with guide RNAs (gRNAs) to activate synthetic promoters (pATFs). These promoters are designed from scratch with binding sites for the gRNA, making them unresponsive to the plant's native transcription factors, thus achieving orthogonality.
2. Materials
- Molecular Cloning: Modular Cloning (MoClo) framework with Type IIS restriction enzymes (e.g., BsaI).
- Vectors: Agrobacterium shuttle vectors with plant selection markers (e.g., BASTA, hygromycin).
- Genetic Parts:
- Transcription Factor: dCas9:VP64 gene under a strong constitutive promoter.
- Synthetic Promoters (pATFs): Minimal 35S CaMV promoter upstream of 3-4 repeats of a gRNA-binding site.
- gRNA Expression Cassettes: Driven by Pol III (e.g., U6) or inducible Pol II promoters.
- Host: Nicotiana benthamiana for transient expression or Arabidopsis thaliana for stable transformation.
3. Step-by-Step Workflow
- Step 1: Design and Assemble Synthetic Promoters.
- Design oligonucleotides containing the desired gRNA target sequences in tandem repeats (3-4x).
- Clone these repeats directly upstream of a minimal 35S promoter in a MoClo-compatible vector.
- Step 2: Assemble Transcriptional Units.
- Using Golden Gate assembly, construct the following units:
- TU1: Constitutive Promoter -> dCas9:VP64 -> Terminator
- TU2: gRNA Promoter (U6 or inducible) -> gRNA scaffold -> Terminator
- TU3: Synthetic pATF -> Your Gene of Interest -> Terminator
- Using Golden Gate assembly, construct the following units:
- Step 3: Assemble Final Construct.
- Combine the transcriptional units (TUs) into a single final expression vector using the MoClo framework's connector sequences.
- Step 4: Transformation and Validation.
- Transform the final construct into Agrobacterium.
- Infiltrate Agrobacterium into N. benthamiana leaves for transient expression.
- Quantify gene expression (e.g., via fluorescence or luminescence) 2-4 days post-infiltration to validate orthogonal activation.
4. Data Analysis and Interpretation
- Orthogonality Test: Co-express multiple orthogonal pATF-gRNA pairs. Confirm that each gRNA only activates its cognate pATF and shows minimal cross-activation of others.
- Leakiness: Measure the output signal (e.g., fluorescence) in the absence of the gRNA. Compare this to the signal when the gRNA is present. A high signal-to-background ratio indicates a well-insulated promoter.
Diagram 1: Orthogonal system architecture preventing host cross-talk.
Protocol 2: Decoupling Mean and Variability of Gene Expression in Mammalian Cells
This protocol is based on the TuNR (Tunable Noise Rheostat) system described in Nature Communications for the orthogonal control of mean expression and cell-to-cell heterogeneity in a human cell line [16].
1. Principle A synthetic circuit with two orthogonal, small molecule-inducible transcriptional activators connected in a cascade allows for independent titration of a gene's mean expression level and its population variability.
2. Materials
- Cell Line: Mammalian cells (e.g., PC9).
- Inducers: Abscisic Acid (ABA) and Gibberellic Acid (GA).
- Genetic Components:
- Node 1: ABA-inducible system (Gal4 DNA-binding domain fused to a split ABA-binding domain).
- Node 2: GA-inducible system (dCas9 fused to a split GA-binding domain and a VPR activation domain).
- gRNA targeting a promoter of choice (e.g., pTRE).
- Reporter Genes: mRuby (Node 1 reporter) and mAzamiGreen (Node 2/target gene reporter).
- Equipment: Flow cytometer for measuring fluorescence and population variance.
3. Step-by-Step Workflow
- Step 1: Circuit Integration.
- Stably integrate the complete TuNR circuit, along with a gRNA cassette and a reporter gene, into the genome of your mammalian cell line. Isolate single-cell clones to ensure homogeneity.
- Step 2: Characterization of the First Node.
- Treat cells with a dose range of ABA (e.g., 0-400 µM). Replenish media every 24 hours.
- Use flow cytometry to measure mRuby fluorescence daily until steady-state is reached (typically ~3 days). This characterizes the input-output relationship for Node 1.
- Step 3: Characterization of the Second Node.
- Prime cells with a saturating dose of ABA (400 µM) for 3 days.
- While maintaining ABA, treat with a dose range of GA. Measure mAzamiGreen fluorescence daily until steady-state (~6 days).
- Step 4: Isomean and Iso-variance Analysis.
- Treat cells with a full matrix of ABA and GA concentrations.
- For each (ABA, GA) combination, measure the mean fluorescence and the coefficient of variation (CV) or Fano factor of the population.
- Identify pairs of inducer concentrations that yield the same mean expression (isomeans) but different variabilities, and vice-versa.
4. Data Analysis and Interpretation
- Plot the mean expression and population variance (e.g., CV) against the two inducer concentrations.
- Successful implementation is demonstrated by finding at least two different input combinations (e.g., High ABA/Low GA and Low ABA/High GA) that produce statistically identical mean expression but significantly different levels of cell-to-cell variability.
Diagram 2: Dual-input TuNR circuit for orthogonal control of mean and noise.
The Scientist's Toolkit: Research Reagent Solutions
| Research Goal | Essential Reagents / Tools | Function in Orthogonal Control |
|---|---|---|
| Orthogonal Transcription | dCas9 Activator (e.g., dCas9:VP64, dCas9:VPR) [6] [16] | Programmable DNA-binding scaffold that recruits transcriptional activation machinery to synthetic targets without cleaving DNA. |
| Synthetic Promoters (pATFs) [6] | Engineered DNA sequences containing binding sites for gRNA/dCas9 complexes, minimizing recognition by host transcription factors. | |
| Multiplexed & Tunable Control | Small Molecule Inducers (e.g., ABA, GA) [16] | Orthogonal inputs that control the dimerization of synthetic transcription factors, allowing for precise, external titration of circuit activity. |
| Design of Experiments (DoE) [17] | A statistical framework for efficiently optimizing multiple factors (e.g., RBS strength, temperature, inducer concentration) simultaneously to maximize system performance. | |
| Context Insulation | Engineered Riboswitches [17] | Structured RNA elements placed in the 5' UTR that undergo ligand-induced conformational changes to control translation initiation orthogonally from host regulation. |
| Standardized Assembly | Modular Cloning (MoClo) Toolkits [6] | A standardized cloning framework using Type IIS restriction enzymes that enables rapid, modular, and reproducible assembly of complex genetic circuits. |
| LpxC-IN-9 | LpxC-IN-9|LpxC Inhibitor | LpxC-IN-9 is a potent LpxC inhibitor with antibacterial activity. This product is for research use only and not for human use. |
| Ferroportin-IN-1 | Ferroportin-IN-1|Ferroportin Inhibitor|For Research Use | Ferroportin-IN-1 is a potent and selective ferroportin inhibitor for iron homeostasis research. This product is for research use only (RUO). Not for human or veterinary use. |
Table 1: Performance metrics of orthogonal regulatory systems across different hosts.
| System / Host | Key Regulator | Induction / Dynamic Range | Key Performance Metrics | Citation |
|---|---|---|---|---|
| OCS / Plants | dCas9:VP64 + Synthetic Promoters | N/A (Validated Orthogonality) | Successful concerted expression of multiple genes; tissue-specific and environmentally responsive. | [6] |
| TuNR / Human Cells | ABA & GA Inducible Cascade | >1000-fold (Transgene) | Decouples mean expression from population variability (noise); 7.2-fold induction of endogenous NGFR. | [16] |
| ORS / Bacteria | PPDA Riboswitch | 72-fold (Riboswitch-dependent); 550-fold (over basal) | Achieved via systematic engineering of RBS and N-terminal codon context to reduce sensitivity. | [17] |
Troubleshooting Common Experimental Issues
FAQ: My orthogonal transcription factor system shows high background expression (leakiness) in the absence of an inducer. What could be the cause and how can I resolve this?
High background expression often stems from non-specific interactions or insufficient promoter specificity. To address this:
- Optimize Promoter Design: Ensure your synthetic promoter has minimal sequence identity to the host's endogenous promoters. Using completely synthetic, de novo designed promoters can significantly reduce cross-talk with the host's regulatory machinery [18] [19].
- Verify Transcription Factor Orthogonality: Test your transcription factors for off-target binding to native genomic sequences. For CRISPR/dCas9-based systems, confirm the specificity of the gRNA and check for unintended PAM (protospacer adjacent motif) sites near the target [19].
- Adjust Effector Domains: The choice of activator or repressor domain (e.g., VP64, KRAB) fused to your DNA-binding protein can influence leakage. Screening different effector domains may help identify one with a more favorable ON/OFF ratio [19] [3].
FAQ: I am not achieving a strong enough induction (low dynamic range) with my orthogonal system. How can I improve the output?
A low dynamic range can be caused by weak promoter activation or inefficient transcription factor recruitment.
- Increase Binding Site Multiplicity: Incorporating multiple copies of the transcription factor binding site upstream of a minimal promoter can amplify the transcriptional output. For instance, synthetic promoters for dCas9:VP64 were designed with varying repeats of gRNA binding sites to enhance activation [18].
- Enhance Transcription Factor Activity: Consider using engineered, high-performance variants of your transcription factor. For example, an optimized λ cI mutant (cIopt) was shown to have significantly greater activity than the wild-type protein [20].
- Optimize Inducer Concentration: Systematically titrate the chemical inducer to find the concentration that yields maximum expression without causing cellular toxicity. The performance of some inducible systems is highly dependent on inducer concentration [21] [22].
FAQ: My system works in a model organism like E. coli, but fails in a non-model host. What steps can I take?
Failure in non-model organisms is often due to inefficient parts or host-specific incompatibilities.
- Use Broad-Host-Range Parts: Employ genetic parts and polymerases known to function across diverse species. For example, broad-host expression systems based on MmP1, K1F, and VP4 phage RNA polymerases have been successfully used in non-model organisms like Halomonas bluephagenesis and Pseudomonas entomophila where T7 RNAP fails [21].
- Decouple Regulation from Host Metabolism: Use inducers that the host cannot metabolize. For the rhaBAD promoter, switching the inducer from L-rhamnose (which is metabolized, causing transient expression) to L-mannose provided sustained, strong induction because E. coli could not use it as a carbon source [22]. This principle can be applied to other hosts.
- Check for Orthogonal Replication and Selection: Ensure your plasmid vectors contain origins of replication and selection markers that are functional in the new host [18].
Performance Data for Selected Orthogonal Systems
The following tables summarize key performance metrics for various orthogonal systems as reported in recent literature, providing a benchmark for experimental expectations.
Table 1: Performance of CRISPR/dCas9-Based Orthogonal Systems in Different Hosts
| Host Organism | System Description | Key Performance Metric | Reported Value | Reference |
|---|---|---|---|---|
| Plants (e.g., Nicotiana benthamiana) | dCas9:VP64 + synthetic promoters (pATFs) | Demonstrated complete orthogonality; ratiometric expression output | Ethylene-driven, multiplexed control achieved | [18] |
| Saccharomyces cerevisiae (Yeast) | dCas9/synTALE + synthetic promoters (synPs) | Induction Factor (ON/OFF ratio) | Up to 400-fold | [19] |
| Escherichia coli | dCas9-Mxi1 repressor | Circuit response | High orthogonality and digital response | [19] |
Table 2: Performance of Orthogonal Mutagenesis and Protein Evolution Systems
| System Name | Host Organism | Core Components | Key Performance Metric | Reported Value | |
|---|---|---|---|---|---|
| Orthogonal Transcription Mutator (OTM) | H. bluephagenesis & E. coli | PmCDA1-UGI fused to MmP1 RNAP | On-target mutation frequency increase vs. control | >80,000-fold | [21] |
| Mutation Rate | 2.9 x 10â»âµ substitutions per base (s.p.b.) | [21] | |||
| Off-target rate (genomic) increase vs. control | 5-fold | [21] | |||
| Phagemid-assisted Evolution | E. coli | M13 phagemid + λ cI variants | Enrichment efficiency (strong vs. weak activator) | Enriched from 10³-fold excess in 6 rounds | [20] |
Table 3: Characteristics of Engineered Transcription Factor / Promoter Pairs
| TF / Promoter Class | Number of Orthogonal Pairs | Functionality | Notable Features |
|---|---|---|---|
| Bacteriophage λ cI variants | 12 TFs on up to 270 promoters | Activator, Repressor, Dual Activator-Repressor | First set of dual activator-repressor switches for orthogonal logic gates [23] [20]. |
| synTALE / synP (Yeast) | A characterized set of functional pairs | Activator | Wide range of expression outputs, low background [19]. |
Essential Experimental Protocols
Protocol: Assembling Multi-Transcriptional Unit Constructs Using a Modular Cloning (MoClo) Framework
This protocol is adapted from a plant synthetic biology study for constructing complex genetic circuits with orthogonal parts [18].
Principle: The MoClo framework uses Type IIS restriction enzymes (e.g., BsaI) to assemble standardized genetic parts (Promoters, Coding Sequences, Terminators) into transcriptional units (TUs), which are then assembled into a final vector in a single pot reaction [18].
Workflow Diagram: Modular Cloning (MoClo) Workflow
Step-by-Step Procedure:
- Part Preparation: Clone each genetic part (e.g., orthogonal promoter, gene of interest, terminator) into specific "entry" vectors with flanking BsaI sites and unique 4-bp overhangs.
- Level 1 Assembly (Single TU): Mix a promoter, gene, and terminator plasmid in a single tube with BsaI-HFv2 restriction enzyme and T4 DNA ligase. Perform a Golden Gate reaction (e.g., 37°C for 5 mins, then 16°C for 5 mins, cycled 50 times, then 60°C for 10 mins, 80°C for 10 mins). This assembles a single, correct TU in an intermediate vector.
- Final Assembly (Multiple TUs): Mix the Level 1 TU plasmids (now flanked by connector sequences for multi-TU assembly) with the final destination vector in another Golden Gate reaction. This stitches all TUs into the final vector in the correct order.
- Screening: Transform the final assembly reaction into E. coli. Screen colonies for the loss of a GFP dropout cassette present in the backbone to easily identify correct assemblies (white colonies instead of green) [18].
Protocol: Measuring Orthogonality and Cross-Talk in a Multi-Component System
Principle: Systematically test each transcription factor against all non-cognate promoters to ensure that activation or repression only occurs with the intended partner [20].
Workflow Diagram: Orthogonality Testing Matrix
Step-by-Step Procedure:
- Construct Reporter Library: Create reporter constructs where each orthogonal promoter drives the expression of a easily quantifiable output protein (e.g., GFP, YFP, RFP, luciferase).
- Express Individual Transcription Factors: Construct plasmids for the inducible expression of each orthogonal transcription factor (e.g., dCas9:VP64 with specific gRNAs, synTALEs, or λ cI variants).
- Co-transformation and Assay: Co-transform a single transcription factor plasmid with each member of the promoter-reporter library into your host organism. Include controls with no transcription factor.
- Quantification: For each combination, measure the reporter output under induced and uninduced conditions. Calculate the induction ratio (ON/OFF) for the intended pair.
- Data Analysis: A system is considered orthogonal if the induction ratio for the intended TF-promoter pair is high, while the output for all non-cognate pairs remains at baseline (uninduced) levels [18] [20].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Orthogonal System Development
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| dCas9 Effector Fusions | Programmable DNA-binding chassis (e.g., dCas9:VP64 for activation, dCas9:KRAB for repression). | Core component for building CRISPR-based orthogonal transcription factors [18] [19]. |
| Synthetic Promoters (synPs / pATFs) | Minimal, de novo designed promoters containing specific binding sites for orthogonal TFs, with minimal host cross-talk. | Providing the regulatory DNA element that is selectively controlled by a synthetic TF and not by host factors [18] [19]. |
| Modular Cloning Toolkit (e.g., MoClo) | A standardized assembly system using Type IIS restriction enzymes (BsaI) for rapid, modular construction of multi-gene circuits. | Accelerating the design-build-test cycle for assembling complex genetic circuits with orthogonal parts [18]. |
| Orthogonal RNA Polymerases | Phage-derived RNAPs (e.g., T7, MmP1, K1F) that specifically transcribe genes under their cognate promoters. | Enabling orthogonal gene expression streams and in vivo mutagenesis systems, especially in non-model hosts [21]. |
| Non-Metabolizable Inducers | Chemical inducers that trigger expression but are not consumed by the host's metabolism (e.g., L-mannose for the engineered rhaBAD system). | Providing sustained induction and avoiding transient expression profiles caused by inducer depletion [22]. |
| Phagemid-Assisted Continuous Evolution (PACE) Systems | A platform for the directed evolution of biomolecules, linking desired activity to phage propagation. | Engineering non cross-reacting, orthogonal variants of transcription factors like λ cI [20]. |
| Pbrm1-BD2-IN-1 | Pbrm1-BD2-IN-1, MF:C17H19ClN2O, MW:302.8 g/mol | Chemical Reagent |
| Sos1-IN-12 | Sos1-IN-12|Potent SOS1 Inhibitor|For Research | Sos1-IN-12 is a potent SOS1 inhibitor that disrupts KRAS activation. This product is for research use only (RUO) and not for human or veterinary diagnosis or therapy. |
Implementing Orthogonal Systems: A Toolkit for Multi-Gene Control Across Chassis
CRISPR/dCas9-Based Orthogonal Transcription Factors for Programmable Gene Activation
Orthogonal control in gene regulation refers to the design of synthetic genetic systems that operate independently from the host's native regulatory machinery. For CRISPR/dCas9-based transcription factors, this means creating activator-promoter pairs that function with minimal cross-talk with the host genome or with each other. This orthogonality is crucial for constructing complex genetic circuits and for precise manipulation of gene expression without unintended side effects [6] [24].
The foundational technology uses a catalytically dead Cas9 (dCas9) protein, which retains its DNA-binding capability but lacks nuclease activity. When fused with transcriptional activation domains and guided by specific sgRNAs, dCas9 can be programmed to activate target genes. Orthogonal systems expand this concept by creating multiple, non-interacting dCas9/sgRNA/promoter combinations that can function simultaneously within the same cell [25] [26].
Frequently Asked Questions (FAQs)
Q1: What constitutes an orthogonal transcription factor system in CRISPR/dCas9 applications? An orthogonal transcription factor system consists of multiple synthetic transcription factors and their corresponding synthetic promoters that function independently without cross-activation or cross-repression. True orthogonality requires that each dCas9/sgRNA complex activates only its intended synthetic promoter and does not interact with other synthetic promoters in the system or endogenous genomic regions. This is typically achieved through careful computational design of sgRNA sequences and their binding sites to minimize off-target interactions [6] [24].
Q2: Why is my dCas9 activator showing low activation efficiency despite optimal sgRNA design? Low activation efficiency can result from several factors. The binding position within the promoter region is critical - sites between -50 to -100 bp upstream of the transcription start site typically work best. The chromatin accessibility of the target region also significantly impacts efficiency. Additionally, using a single activation domain like VP64 may provide insufficient activation strength; consider upgraded systems like VPR (VP64-p65-Rta) or SAM (Synergistic Activation Mediator) that incorporate multiple activation domains for stronger effects [27] [28].
Q3: How can I design multiple orthogonal CRISPR/dCas9 systems for simultaneous use? Designing orthogonal systems requires a multi-step computational approach. First, generate candidate sgRNA sequences and screen them against the host genome and circuit components using weighted Hamming distance metrics that account for mismatch sensitivity in the seed region. Select sequences with minimum off-target potential, then test mutual orthogonality between candidate pairs. Experimental validation should include testing all possible combinations to confirm absence of cross-talk [24].
Q4: What strategies can reduce off-target effects in CRISPR/dCas9 activation systems? Multiple strategies can mitigate off-target effects. Bioinformatic selection of sgRNAs with high specificity scores and appropriate GC content (40-60%) is essential. Using Cas9 variants with enhanced specificity, such as eSpCas9 or SpCas9-HF1, can reduce off-target binding. Fine-tuning the expression levels of dCas9 and sgRNA components also helps, as high concentrations increase off-target potential. Additionally, consider incorporating inducible systems that limit the duration of dCas9 expression [25] [29].
Q5: How can I make my orthogonal CRISPR system responsive to specific inducters? Inducible orthogonal systems can be created using chemically-induced dimerization domains. For example, coupling dCas9 activation to small molecules like rapamycin, abscisic acid, or gibberellin allows temporal control. These systems typically split the transcription factor components and fuse them to domains that dimerize only in the presence of the specific inducer, enabling precise control over the timing and duration of gene activation [26].
Troubleshooting Guides
Problem: Low Gene Activation Efficiency
Potential Causes and Solutions:
- Cause: Suboptimal sgRNA binding position
- Cause: Weak activation domain
- Solution: Upgrade from basic VP64 to stronger activation systems like VPR (VP64-p65-Rta) or SAM (dCas9-VP64 with MS2-p65-HSF1 recruitment). The SAM system has demonstrated 10-100 fold improvements over dCas9-VP64 alone [28].
- Cause: Epigenetic silencing at target locus
Problem: Lack of Orthogonality (Cross-Talk Between Systems)
Potential Causes and Solutions:
- Cause: Insufficient specificity in sgRNA design
- Solution: Implement stricter computational screening using position-weighted mismatch scoring, with particular emphasis on seed region specificity (nucleotides 10-20 of the sgRNA). Ensure a minimum weighted Hamming distance of at least 5 against the host genome and 9 against circuit components [24].
- Cause: Overexpression of dCas9 components
- Cause: Similar promoter architecture across synthetic promoters
- Solution: Design synthetic promoters with varying architectures in terms of gRNA binding site numbers (3-4 sites shown effective) and spatial arrangements. Incorporate different core promoter elements to enhance orthogonality [6].
Problem: High Cytotoxicity or Cellular Stress
Potential Causes and Solutions:
- Cause: Excessive transcriptional burden
- Cause: Off-target activation of oncogenes or stress response genes
- Solution: Conduct comprehensive off-target prediction using multiple algorithms and validate potential off-target sites using RNA-seq or specific PCR assays. Consider using cell-specific promoters to restrict expression to target cells [29].
- Cause: Viral vector toxicity
Key Experimental Protocols
Protocol: Design and Assembly of Orthogonal CRISPR/dCas9 Systems
Objective: Create multiple non-interacting dCas9 activator-promoter pairs for orthogonal gene regulation.
Materials:
- dCas9-VPR or dCas9-SAM expression vector
- Modular cloning system (e.g., Golden Gate MoClo)
- sgRNA expression scaffolds with appropriate RNA aptamers (for SAM system)
- Synthetic promoter parts with minimal 35S core and transcription factor binding sites
Procedure:
- Computational Design: Generate candidate sgRNA sequences using orthogonal design algorithms that screen against host genome and circuit components with weighted Hamming distance metrics [24].
- Vector Assembly: Use modular cloning framework to assemble transcriptional units:
- Assemble synthetic promoters with 3-4 binding sites for corresponding sgRNAs upstream of minimal core promoter [6].
- Clone sgRNA expression cassettes with appropriate aptamer modifications (e.g., MS2, PP7) for recruiter systems.
- Assemble dCas9-activator fusions (dCas9-VPR or dCas9-VP64 for recruiter systems).
- Orthogonality Testing: Co-transfect each dCas9/sgRNA pair with all synthetic promoter reporters and measure activation specificity. Select pairs showing high on-target activation with minimal cross-talk.
- Validation: Test selected orthogonal pairs in the context of the intended genetic circuit or pathway manipulation.
Protocol: Quantitative Assessment of Orthogonal System Performance
Objective: Measure activation strength and specificity of orthogonal transcription factor systems.
Materials:
- Reporter constructs with orthogonal synthetic promoters driving fluorescent proteins (GFP, RFP, etc.)
- dCas9 activator constructs
- sgRNA expression vectors
- Flow cytometer or fluorescence plate reader
Procedure:
- Transfection Setup: Plate cells in multi-well plates and co-transfect with:
- dCas9 activator construct
- sgRNA expression vector(s)
- Reporter construct(s)
- Control Samples: Include controls with individual components missing to establish baseline.
- Incubation: Incubate cells for 48-72 hours to allow gene expression.
- Quantification: Harvest cells and measure fluorescence intensity using flow cytometry or plate reader.
- Data Analysis:
- Calculate fold-activation for each pair compared to controls.
- Compute orthogonality score as the ratio of on-target to off-target activation.
- Assess crosstalk by comparing unintended activation to intended activation.
Performance Data and System Comparisons
Table 1: Comparison of CRISPR/dCas9 Activation Systems
| System | Components | Activation Fold | Key Advantages | Limitations |
|---|---|---|---|---|
| dCas9-VP64 | dCas9-VP64 + sgRNA | 2-5x [27] | Simple design; Low size | Weak activation; Limited efficiency |
| dCas9-VPR | dCas9-VP64-p65-Rta + sgRNA | 50-300x [29] | Strong activation; Single sgRNA sufficient | Larger size; Potential cytotoxicity |
| SAM | dCas9-VP64 + MS2-p65-HSF1 + engineered sgRNA | 100-1000x [28] | Very high activation; Synergistic effect | Complex 3-component system |
| CRISPR-Assisted Trans Enhancer | dCas9-VP64 + csgRNA + sCMV enhancer | High (specific numbers not provided) | Uses strong CMV enhancer; Works on endogenous genes | Requires special csgRNA and sCMV components [30] |
| SunTag | dCas9-GCN4 + scFv-VP64 + sgRNA | Similar to VPR/SAM [30] | Modular recruitment; Amplified signal | Multi-component; Large genetic payload |
| Anticancer agent 51 | Anticancer agent 51, MF:C22H20F3N3O2S, MW:447.5 g/mol | Chemical Reagent | Bench Chemicals | |
| Neuroinflammatory-IN-3 | Neuroinflammatory-IN-3|NLRP3 Inflammasome Inhibitor | Neuroinflammatory-IN-3 is a potent NLRP3 inflammasome inhibitor for neuroscience research. This product is For Research Use Only. Not for human or veterinary diagnostic or therapeutic use. | Bench Chemicals |
Table 2: Design Parameters for Orthogonal Systems
| Parameter | Optimal Range | Considerations |
|---|---|---|
| sgRNA binding position | -50 to -200 bp from TSS [28] | Avoid positions downstream of TSS; Consider chromatin accessibility |
| Number of sgRNA binding sites | 3-4 sites per promoter [6] | More sites increase activation but may reduce orthogonality |
| sgRNA specificity score | Weighted Hamming distance ≥9 against circuit components [24] | Seed region (positions 10-20) requires perfect matching |
| sgRNA GC content | 40-60% [25] | Higher GC near PAM improves efficiency |
| Activation domain selection | VPR or SAM for strong activation [28] [29] | Balance strength with size constraints |
Signaling Pathways and System Architectures
Orthogonal CRISPR/dCas9 System Architecture
Research Reagent Solutions
Table 3: Essential Research Reagents for Orthogonal CRISPR/dCas9 Systems
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| dCas9 Activation Domains | VP64, VP160, VPR (VP64-p65-Rta), p300 core | Transcriptional activation with varying strengths; VPR provides strongest activation [27] [31] |
| Synthetic Promoter Parts | Minimal 35S core, synthetic TF binding arrays, orthogonal core promoters | Create target promoters for orthogonal systems; minimal cores reduce background [6] |
| sgRNA Scaffold Modifications | MS2, PP7, com aptamers; tetraloop and stem-loop 2 engineering | Enable recruitment of additional activation domains (e.g., in SAM system) [28] |
| Inducible Systems | Light-inducible, chemically-inducible (rapamycin, gibberellin), tetracycline-controlled | Provide temporal control over dCas9 activity; reduce off-target effects [26] [29] |
| Delivery Tools | AAV vectors (size-optimized), lentiviral vectors, lipid nanoparticles | Enable efficient delivery of multi-component systems; AAV requires compact systems [30] [29] |
| Orthogonality Design Tools | Weighted Hamming distance algorithms, off-target prediction software | Computational design of specific sgRNAs with minimal cross-talk [24] |
Design and Engineering of Fully Synthetic Orthogonal Promoters
Fundamental Concepts and FAQs
What are fully synthetic orthogonal promoters?
Fully synthetic orthogonal promoters are artificially designed DNA sequences that control gene expression without interacting with the host's native regulatory networks. Unlike natural promoters, they are built from scratch using modular components: a core promoter region (containing TATA box and transcription start site) and synthetically arranged cis-regulatory elements (CREs) upstream. Their "orthogonal" nature means they are exclusively controlled by engineered transcription factors, such as CRISPR-based systems, ensuring minimal cross-talk with host cellular machinery [6] [32].
Why is orthogonality critical for synthetic biology applications?
Orthogonality prevents unwanted interactions between synthetic genetic circuits and the host organism's native genes. This isolation is crucial for predictable circuit behavior, as cross-talk can lead to:
- Severe growth and developmental defects in plants and other organisms [6]
- Loss of circuit function and unreliable performance [6]
- Unintended pleiotropic effects or epigenetic silencing of transgenes [32]
What are the key advantages of synthetic over native promoters?
- Precise Control: Enable spatiotemporal and inducible expression patterns unattainable with constitutive native promoters [32] [33]
- Reduced Metabolic Burden: Avoid unnecessary negative feedback and energy loss [32]
- Genetic Stability: Minimal sequence homology to the native genome reduces recombination and silencing events [34]
- Design Flexibility: Cis-element arrangement can be optimized for specific strength and inducibility [35] [32]
Troubleshooting Common Experimental Challenges
Low or No Transcriptional Activation
Problem: Your synthetic promoter shows weak or no activity despite the presence of your orthogonal transcription factor.
| Potential Cause | Diagnostic Experiments | Recommended Solutions |
|---|---|---|
| Suboptimal cis-element spacing | Test constructs with 10-50 bp variations in spacing between CREs [32]. | Maintain minimum 50 bp between CREs and core promoter to avoid steric hindrance [32]. |
| Insufficient copy number of CREs | Build a series with 1-6 copies of the target CRE and measure reporter output [35]. | Use 3-4 copies of CREs for strong inducibility; higher copies may increase background [35]. |
| Inefficient gRNA binding (CRISPR systems) | Verify gRNA expression with Northern blot and test different gRNA scaffolds [6]. | Use validated Pol III promoters (e.g., U6) for gRNA expression and optimize gRNA binding sites [6]. |
High Background Expression (Leakiness)
Problem: Significant expression occurs even in the uninduced state or without the orthogonal transcription factor.
| Potential Cause | Diagnostic Experiments | Recommended Solutions |
|---|---|---|
| Too many CRE copies | Compare leakiness in constructs with 2 vs. 6 copies of the same CRE [35]. | Reduce CRE copies to 2-3; find the "sweet spot" balancing inducibility and background [35]. |
| Cryptic host TF binding sites | Scan synthetic sequence for host transcription factor motifs using databases like JASPAR. | Redesign synthetic promoter sequence to eliminate cryptic binding sites [34]. |
| Core promoter too strong | Test your CRE array with different minimal promoters (e.g., 35S mini vs. mas) [6]. | Use a weaker core promoter or incorporate synthetic insulators [32]. |
Lack of Orthogonality (Cross-talk)
Problem: Your synthetic promoter responds to endogenous cellular signals or affects native gene expression.
| Potential Cause | Diagnostic Experiments | Recommended Solutions |
|---|---|---|
| CRE similarity to native elements | Test promoter activity in knockout strains of suspected endogenous TFs. | Use heterologous CREs from distant species and validate orthogonality systematically [36] [34]. |
| dCas9-VP64 off-target effects | Perform RNA-seq in cells expressing dCas9-VP64 without gRNAs. | Include control with catalytically dead dCas9 only; use higher-fidelity Cas9 variants [6]. |
Step-by-Step Experimental Protocols
Protocol: Designing and Testing a Novel Synthetic Orthogonal Promoter
Principle: Create a synthetic promoter by assembling specific cis-regulatory elements upstream of a minimal core promoter, then validate its orthogonality and inducibility.
Materials:
- Modular Cloning (MoClo) system with Type IIS restriction enzymes (BsaI) [6]
- Agrobacterium shuttle vectors with selection markers (e.g., BASTA, hygromycin) [6]
- Reporter genes: GFP, YFP, RFP, or firefly luciferase (F-luc) [6]
- Nicotiana benthamiana plants for transient expression [6]
Procedure:
- Design Phase:
Assembly Phase:
- Use Golden Gate assembly with BsaI sites for modular construction [6]
- Assemble transcriptional unit in order: Synthetic promoter - Reporter gene - Terminator
- Clone into Agrobacterium binary vector with plant selection marker
Validation Phase:
- Transform Agrobacterium with final construct [6]
- Infiltrate Nicotiana benthamiana leaves for transient expression [6]
- Quantify reporter expression 2-4 days post-infiltration:
- Fluorescence imaging for fluorescent proteins
- Luciferase assays with substrate injection
- Test orthogonality by measuring expression without induction and against different orthogonal transcription factors
Troubleshooting Tips:
- If assembly fails: Verify BsaI site elimination in final construct and use fresh competent cells
- If expression is low: Test different copy numbers (3-6) of CREs and optimize spacing [35]
- If background is high: Reduce CRE copy number or try a weaker minimal promoter
Protocol: Establishing CRISPR/dCas9-Based Orthogonal Control
Principle: Implement orthogonal control by programming dCas9-VP64 artificial transcription factors to target synthetic promoters containing specific gRNA binding sites.
Materials:
- dCas9-VP64 fusion construct [6]
- gRNA expression vectors with U6 or other Pol III promoters [6]
- Synthetic promoters with programmed gRNA binding sites upstream of minimal 35S promoter [6]
Procedure:
- System Design:
- Engineer synthetic promoters with 3-4 repeats of specific gRNA binding sites [6]
- Place binding sites 50-100 bp upstream of minimal core promoter
- Design multiple gRNAs with different target sequences for orthogonal control
Vector Construction:
- Clone synthetic promoters upstream of reporter genes (GFP, YFP, RFP)
- Express dCas9-VP64 from a strong constitutive promoter (e.g., 35S)
- Express gRNAs from U6 or other Pol III promoters
Validation:
- Co-express dCas9-VP64, gRNAs, and reporter constructs in Nicotiana benthamiana
- Measure reporter expression 2-4 days post-infiltration
- Test specificity by measuring cross-activation between different gRNA-promoter pairs
- For inducible systems, place gRNA under chemical or hormone-inducible promoters (e.g., ethylene-responsive promoters) [6]
Research Reagent Solutions
Table: Essential Research Tools for Synthetic Orthogonal Promoter Development
| Reagent/Tool | Function | Examples & Specifications |
|---|---|---|
| Modular Cloning Systems | Enables rapid assembly of genetic constructs | MoClo system with BsaI sites; YTK (Yeast Toolkit) adapted for plants [6] |
| Orthogonal Transcription Factors | Programmable regulators for synthetic promoters | dCas9-VP64 fusions; synthetic activators from Saccharomyces spp. [6] [36] |
| Minimal Core Promoters | Basal transcription machinery recruitment | 35S CaMV minimal promoter (-46 to +1 or -90 to +1); TATA-box containing minimal promoters [32] [33] |
| Reporter Genes | Quantitative measurement of promoter activity | GFP, YFP, RFP, BFP for fluorescence; Firefly luciferase (F-luc) for sensitive detection [6] |
| gRNA Expression Systems | Delivery of targeting molecules for CRISPR systems | Pol III promoters (U6, U3); ethylene-inducible Pol II promoters for conditional control [6] |
| Bioinformatics Tools | Prediction and design of synthetic elements | AI models for expression prediction; CRE databases; motif analysis tools [37] [34] |
Dual Orthogonal Linearizer Circuits for Independent Control of Two Genes in Mammalian Cells
A fundamental challenge in synthetic biology and drug development is the precise, independent control of multiple gene expression levels within the same mammalian cell. Traditional methods, such as simple overexpression or knockdown, often lack the precision and uniformity needed to dissect complex phenotypic outcomes. Dual orthogonal linearizer circuits represent a significant advancement, enabling researchers to simultaneously and independently tune the expression of two genes with linear dose responses and low cell-to-cell variability. These systems utilize chemically inducible synthetic gene circuits built with orthogonal repressor proteins, allowing for the exploration of synergistic or epistatic gene interactions critical for therapeutic development and basic research. This technical support center provides troubleshooting and methodological guidance for implementing these powerful tools in your experiments.
Frequently Asked Questions (FAQs)
Q1: What is the primary advantage of using a linearizer circuit over a standard inducible system like Tet-On? Standard inducible systems (e.g., Tet-On) often have a steep, non-linear dose-response curve, making it difficult to titrate a specific, intermediate level of gene expression. Linearizer circuits incorporate negative feedback to create a linear relationship between the inducer concentration and the output protein level. This results in precise, uniform expression across a cell population, which is essential for mapping gene expression levels to phenotypic outcomes [38] [39].
Q2: What does "orthogonal" mean in the context of these gene circuits? Orthogonality means that the two control systems operate independently without cross-talk. In a dual orthogonal linearizer system, the regulator protein (e.g., TetR) of one circuit does not interact with the promoter or inducer (e.g., Doxycycline) of the second circuit, and vice-versa. This allows you to modulate the expression of two different genes in the same cell without the systems interfering with each other [38] [1].
Q3: My circuit has high basal expression in the uninduced state. What could be the cause? High basal expression, or "leakiness," can occur due to several factors:
- Insufficient Repression: The repressor protein may not be expressed at a high enough level or may not bind strongly enough to its operator sites in your promoter construct. Using a promoter with two operator sites (e.g., PhlFd-mLin) instead of one can improve repression and lower basal levels [38].
- Promoter Strength: The chosen promoter may be inherently too strong. Consider testing a promoter with a different basal strength.
- Integration Site Effects: The genomic location where the circuit is integrated can influence its expression. Using a system like Flp-In to integrate the circuit into a consistent, well-characterized genomic locus can help minimize this variability [38].
Q4: I am not observing a linear dose-response in my experiment. How can I troubleshoot this? A non-linear response can be caused by:
- Incorrect Inducer Concentration Range: The linear range of the circuit might be outside the inducer concentrations you are testing. Consult the literature for your specific repressor protein and perform a broad dose-response experiment to identify the linear regime [38].
- Circuit Saturation: At very high inducer concentrations, the circuit will saturate, and the response will plateau. Ensure you are measuring within the dynamic, non-saturated range.
- Insufficient Negative Feedback: The linear response depends on effective negative autoregulation. Verify the integrity of your construct, ensuring the repressor gene and the target gene are under identical promoter control as required by the linearizer design [38] [39].
Troubleshooting Guide
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| No Expression Upon Induction | - Non-functional repressor/producer construct.- Wrong inducer or degraded inducer.- Cell line not compatible.- Toxic gene product causing cell death. | - Sequence verify the plasmid construct.- Prepare a fresh inducer stock.- Use validated cell lines (e.g., HEK Flp-In-293).- Check cell viability; use inducible system for toxic genes. |
| High Cell-to-Cell Variability (High CV) | - Inefficient genomic integration leading to copy number variation.- Weak or inconsistent negative feedback. | - Use site-specific integration (e.g., Flp-In system) for a single copy.- Ensure strong, identical promoters for the repressor and reporter [38]. |
| Cross-Talk Between Circuits | - Lack of orthogonality between repressor/inducer pairs.- Shared cellular resources causing burden. | - Validate orthogonality of repressors (e.g., TetR/PhlF) in your cell type.- Use lower expression strength promoters to reduce metabolic load [38] [40]. |
| Low Maximum Fold Induction | - High basal expression.- Weak promoter strength in the induced state.- Saturation at a low expression level. | - Use a double-operator promoter design to reduce leakage [38].- Consider a stronger core promoter element.- Extend the inducer concentration range tested. |
Performance Data for Common Orthogonal Systems
The table below summarizes key performance metrics for two orthogonal linearizer circuits, TetR-mLin and PhlFd-mLin, when integrated into HEK 293 cells. This data can serve as a benchmark for your experiments [38].
| Gene Circuit | Inducer | Range of Linearity | Maximum Fold Induction | Slope of Linear Regime | Average Coefficient of Variation (CV) | Basal Expression (A.U.) |
|---|---|---|---|---|---|---|
| TetR-mLinearizer | Doxycycline | 0 to 10 ng/mL | 43.8 | 683 A.U. per ng/mL | 0.47 | 212 ± 17.3 |
| PhlFd-mLinearizer | DAPG | 0 to 5 µM | 12.0 | 6870 A.U. per µM | 0.38 | 3805 ± 364 |
Experimental Protocols
Protocol 1: Implementing a Dual Orthogonal Linearizer System
This protocol outlines the key steps for establishing a dual orthogonal linearizer system in mammalian cells, based on the methodology from [38].
1. Circuit Design and Cloning:
- Select Orthogonal Repressors: Choose two non-interacting repressor proteins, such as TetR and PhlF.
- Design Linearizer Constructs: For each repressor, design a construct where the repressor gene is transcriptionally fused via a P2A self-cleaving peptide to a fluorescent reporter (e.g., eGFP or mCherry) and your gene of interest (GOI). This entire operon must be under the control of a promoter containing the repressor's specific operator sequence(s) (e.g., 2xtetO2 for TetR, phlFO for PhlF).
- Promoter Configuration: For lower basal expression and better linearity, use promoters with two flanking operator sites (e.g., PhlFd-mLin) rather than a single site.
2. Cell Line Engineering and Validation:
- Stable Cell Line Generation: Use a cell line with a defined genomic landing pad, such as Flp-In-293, to allow for single-copy, site-specific integration of your constructs. This minimizes variability due to copy number and position effects.
- Clone Selection: After transfection and selection, isolate single-cell clones. Polyclonal populations can be highly heterogeneous.
- Dose-Response Characterization: For each clone, seed cells and treat with a broad range of inducer concentrations (e.g., 0-100 ng/mL Doxycycline for TetR; 0-25 µM DAPG for PhlF). Allow expression to reach steady-state (typically ~48 hours).
- Flow Cytometry Analysis: Analyze the cells using flow cytometry to measure the mean fluorescence intensity and the coefficient of variation (CV) for each inducer dose. Plot the mean fluorescence against the inducer concentration to identify the linear range and calculate the fold induction.
Protocol 2: Troubleshooting a Poor Linear Dose-Response
If your initial characterization does not yield a linear response, follow this systematic troubleshooting workflow.
The Scientist's Toolkit: Key Research Reagents
| Research Reagent | Function in the Experiment |
|---|---|
| Flp-In-293 Cell Line | Host cell line with a single defined FRT genomic landing pad for reproducible, single-copy integration of genetic circuits [38]. |
| TetR-mLin & PhlF-mLin Constructs | The core genetic circuits that provide linear, low-noise gene expression control in response to Doxycycline and DAPG, respectively [38]. |
| Doxycycline (Dox) | Inducer molecule for TetR-based circuits; binds TetR and relieves repression of the target promoter [38] [39]. |
| 2,4-diacetylphloroglucinol (DAPG) | Inducer molecule for PhlF-based circuits; binds PhlF and prevents its binding to the phlFO operator site [38]. |
| P2A Self-Cleaving Peptide | A short peptide sequence that allows for the co-translation of multiple proteins (e.g., repressor and reporter) from a single mRNA transcript [38]. |
| Operator Sites (tetO, phlFO) | Specific DNA sequences inserted into promoters that are bound by their cognate repressor proteins (TetR, PhlF) to confer regulation [38]. |
| Aldh3A1-IN-2 | Aldh3A1-IN-2, MF:C11H14N2O3, MW:222.24 g/mol |
| Anti-inflammatory agent 21 | Anti-inflammatory agent 21, MF:C24H21FO6, MW:424.4 g/mol |
Orthogonal Circuit Design and Mechanism
The following diagram illustrates the core architecture and mechanism of a single linearizer gene circuit, which is replicated and modified with orthogonal parts to create the dual-gene control system.
Directed evolution is a powerful protein engineering technique that mimics natural selection in a laboratory setting to develop proteins with enhanced or entirely new functions [41]. This process involves creating genetic diversity within a target gene and then screening or selecting for variants that exhibit improved properties [42]. Orthogonal Transcription Mutation Systems represent a cutting-edge advancement in this field, enabling targeted, in vivo hypermutation with unprecedented specificity and efficiency [43].
These systems function by leveraging phage RNA polymerases (such as T7 RNAP) fused to mutagenic enzymes like deaminases. This combination creates an orthogonal transcription-mutation machinery that specifically introduces mutations into target genes without significantly affecting the host genome [43] [44]. Recent developments have demonstrated the capability to achieve over 1,500,000-fold increased mutation rates while maintaining high specificity and minimal off-target effects [43]. This technical support document provides comprehensive guidance for researchers implementing these systems in their protein evolution workflows.
Troubleshooting Guide: Common Experimental Issues and Solutions
Low Mutation Rate or Diversity
Problem: Inadequate mutation frequency or limited diversity in the generated mutant library.
| Possible Cause | Verification Method | Solution |
|---|---|---|
| Inefficient polymerase-deaminase fusion activity | Measure mutation frequency in a control target gene using sequencing analysis. | Optimize fusion linker length and composition; test different deaminase orthologs [43]. |
| Suboptimal expression of orthogonal system | Check protein expression via Western blot or activity assays. | Modify promoter strength or ribosome binding site; tune expression level [45]. |
| Insufficient mutagenesis time | Monitor mutation accumulation over time with sequencing. | Extend mutagenesis period; implement continuous evolution approaches [43]. |
| Unfavorable host factors | Compare mutation rates in different host strains. | Use mutator strains as hosts; engineer chaperone co-expression [42]. |
High Off-Target Mutagenesis
Problem: Significant mutations occurring outside the intended target gene.
| Possible Cause | Verification Method | Solution |
|---|---|---|
| Non-specific activity of mutagenic enzyme | Whole-genome sequencing of evolved clones. | Use engineered deaminases with enhanced specificity; adjust expression levels [43]. |
| Leaky expression of orthogonal system | Assess off-target transcription with RNA-Seq. | Implement tighter regulatory systems; improve promoter orthogonality [45]. |
| Cross-talk with host systems | Evaluate host stress responses and mutation profiles. | Use more orthogonal phage polymerases; engineer systems for reduced host interaction [44]. |
Host Toxicity or Reduced Viability
Problem: Decreased host cell fitness during mutagenesis experiments.
| Possible Cause | Verification Method | Solution |
|---|---|---|
| Burden of protein overexpression | Measure growth rates with and without system induction. | Titrate expression using inducible systems; use lower-copy plasmids [46]. |
| Critical gene disruption | Identify mutation locations through whole-genome sequencing. | Implement more specific targeting; use conditionally essential genes as counterscreens [43]. |
| DNA damage response activation | Monitor SOS response and other stress pathways. | Incorporate rest periods between mutagenesis cycles; use error-prone DNA repair mutants [42]. |
Frequently Asked Questions (FAQs)
Q1: What types of mutations do orthogonal transcription mutation systems primarily generate?
These systems are engineered to generate all transition mutations (C:G to T:A and A:T to G:C) uniformly across target genes [43]. This differs from earlier systems that were limited to specific mutation types and provides more comprehensive coverage of mutational space for protein evolution.
Q2: How does this system achieve orthogonality and minimize off-target effects?
Orthogonality is achieved through the use of phage-derived RNA polymerases (such as T7, T3, or SP6) that recognize specific promoter sequences not utilized by the host's transcription machinery [43] [44]. The fusion deaminases then act specifically on transcripts generated by these polymerases, creating a highly targeted mutagenesis system.
Q3: What host organisms are compatible with these orthogonal mutation systems?
While initially developed for E. coli, these systems have been successfully implemented in both model and non-model organisms, including the industrially relevant bacterium Halomonas bluephagenesis [43]. Recent work has also adapted T7 RNAP-based systems for eukaryotic hosts like yeast and mammalian cells through engineering of capping enzyme fusions [44].
Q4: What is the typical mutation rate achievable with these systems?
Published reports demonstrate >1,500,000-fold increased mutation rates compared to baseline, enabling rapid protein evolution within a single day of mutagenesis process [43]. Actual rates can be tuned by adjusting expression levels and mutagenesis duration.
Q5: How do I control mutation rate and spectrum in my experiments?
Mutation rates can be controlled by:
- Modulating expression levels of the polymerase-deaminase fusion
- Varying the duration of mutagenesis periods
- Using different deaminase orthologs with varying activities
- Employing engineered versions with temperature-sensitive or otherwise regulable activities [43] [42]
Q6: What protein classes have been successfully evolved using these systems?
This technology has been successfully applied to evolve diverse protein classes including:
- Fluorescent proteins and chromoproteins
- Cytoskeletal proteins
- Cell division-related proteins
- Global sigma factors
- Transporters (e.g., LysE exporter) [43]
Research Reagent Solutions
Table: Essential Components for Orthogonal Transcription Mutation Systems
| Component | Function | Examples & Notes |
|---|---|---|
| Phage RNA Polymerase | Orthogonal transcription | T7, T3, or SP6 RNAP; determines promoter specificity [43] [44] |
| Deaminase Enzyme | Introduces point mutations | Cytidine/adenine deaminases; defines mutation spectrum [43] |
| Expression Vector | Host expression of system | Tunable promoters (e.g., arabinose, T7lac) recommended [45] |
| Target Plasmid | Contains gene to be evolved | Must include specific phage promoter (e.g., T7 promoter) [43] |
| Host Strain | System propagation | E. coli BL21; Halomonas; engineered for specific applications [43] |
Experimental Workflow and Protocol
System Setup and Validation
The following diagram illustrates the core mechanism of an orthogonal transcription mutation system:
Step-by-Step Implementation Protocol:
System Assembly:
- Clone the polymerase-deaminase fusion construct into an appropriate expression vector with inducible promoter [43]
- Clone your target gene into a separate plasmid containing the corresponding phage-specific promoter
- Co-transform both plasmids into your selected host strain
Validation of Orthogonality:
- Sequence the host genome before and after limited mutagenesis to assess off-target effects
- Use transcriptomics (RNA-Seq) to verify specific targeting of desired genes [43]
- Measure mutation frequency in target vs. non-target genes using sequencing-based methods
Mutation Rate Calibration:
- Induce system expression for varying durations (2-24 hours)
- Sequence target regions to quantify mutation frequency
- Adjust expression levels or induction time to achieve desired mutation rate [42]
Directed Evolution Workflow
The complete directed evolution cycle using orthogonal transcription mutation systems involves the following steps, visualized in the workflow below:
Key Considerations for Successful Evolution:
Library Size Planning:
- Calculate theoretical diversity based on your mutation rate
- Ensure screening capacity exceeds library diversity by 3-10x
- For larger diversity than screenable space, implement sequential screening strategies [42]
Screening Methodology Selection:
Balancing Activity and Stability:
- Monitor stability during evolution campaigns
- Incorporate stability screens if activity-stability trade-offs emerge
- Consider consensus mutations or backbone stabilization if destabilization occurs [46]
Advanced Applications and Optimization
Eukaryotic System Adaptation
For implementation in eukaryotic hosts, additional engineering is required:
- Capping enzyme fusion: Addresses 5' methyl guanosine capping requirement in eukaryotes [44]
- Nuclear localization signals: Ensures proper cellular compartment targeting
- Codon optimization: Enhances expression in non-native hosts
- Episomal maintenance: Maintains target genes on separate replicons
Recent studies have demonstrated two orders of magnitude higher protein expression in yeast and mammalian cells using engineered T7 RNAP-capping enzyme fusions compared to wild-type systems [44].
Managing Activity-Stability Trade-offs
A common challenge in directed evolution is the emergence of variants with enhanced activity but compromised stability [46]. Strategies to address this include:
- Incorporating stability screens alongside activity selection
- Using consensus design to introduce stabilizing mutations
- Implementing backbone stabilization between evolution rounds
- Employing computational prediction of stabilizing mutations
The orthogonal transcription mutation system platform provides a powerful and versatile tool for accelerating protein evolution across diverse host organisms and target protein classes. By following these troubleshooting guidelines and optimized protocols, researchers can effectively harness this technology to overcome traditional bottlenecks in directed evolution and achieve rapid protein optimization.
Troubleshooting Guide: Common Issues with Orthogonal Systems
1. Problem: Low or No Expression from an Orthogonal Transcription Factor
- Potential Cause: The synthetic promoter may not be optimally designed, or the Transcription Factor (TF) may not be functioning in the host.
- Solution:
- Verify the design of your synthetic promoter. Ensure it contains the correct number and sequence of TF binding sites upstream of a minimal core promoter [6].
- For plant systems using CRISPR/dCas9-based TFs, confirm the gRNA is correctly expressed and targets the designed synthetic promoter sequence [6].
- In yeast, check the fusion between the DNA-binding domain (e.g., from plant NAC TFs) and the activation domain (e.g., EDLL or VP64). The EDLL domain has been shown to be significantly stronger than the GAL4 AD in some contexts [47].
2. Problem: High Background Expression (Leakiness)
- Potential Cause: The orthogonal system has crosstalk with the host's endogenous regulatory machinery, or the synthetic parts are not sufficiently insulated.
- Solution:
3. Problem: Unstable Expression or Gene Silencing
- Potential Cause: Repeat sequences in genetic constructs can trigger host defense mechanisms like silencing or lead to homologous recombination [34].
- Solution:
- Engineer synthetic promoters to minimize repetitive sequences. Use high sequence diversity by incorporating functionally equivalent cis-regulatory elements (CREs) from diverse organisms [34].
4. Problem: Inconsistent Results Between Technical Replicates
- Potential Cause: The genetic circuit is sensitive to small fluctuations in cellular conditions or is not genetically stable.
- Solution:
- Ensure the orthogonal system is stably integrated into the chromosome rather than on a plasmid, where possible. In yeast, chromosomally integrated plant TF systems have demonstrated a wide range of transcriptional output [47].
- Follow a structured "Design-Build-Test-Learn" (DBTL) cycle, utilizing high-throughput cloning techniques like Golden Gate (MoClo) to rapidly prototype and test different constructs [48] [6].
5. Problem: The Observed Phenotype Does Not Match the Genetic Perturbation
- Potential Cause: Off-target effects of the gene-modulation tool (e.g., RNAi) can lead to misinterpretation of the gene's function [49].
- Solution:
- Employ orthogonal validation: use a different, complementary technique to confirm your results. For example, if a phenotype is observed with RNAi knockdown, validate it with a CRISPR-based knockout (CRISPRko) or interference (CRISPRi) system, and vice-versa [49].
Frequently Asked Questions (FAQs)
Q1: What does "orthogonality" mean in synthetic biology? In synthetic biology, an orthogonal system is one that functions independently from the host's native systems. It is engineered to have minimal crosstalk with the host's regulatory networks, allowing for precise and predictable control of gene expression without unintended interference [6].
Q2: Why should I use orthogonal regulators instead of native ones? Orthogonal regulators are crucial for building complex genetic circuits because they provide isolated control channels. This prevents your synthetic circuit from disrupting essential host functions and protects it from host interference, leading to more reliable and predictable performance [6].
Q3: Can I use the same orthogonal system across different organisms, like in both yeast and plants? While some components may be portable, orthogonal systems often need to be tailored to the specific host. For example, plant-derived TFs can function orthogonally in yeast [47], and synthetic promoters for plants are designed to work within the plant's transcriptional machinery [6] [34]. Porting systems between distantly related organisms typically requires validation and optimization.
Q4: What are the key advantages of synthetic promoters over natural ones?
- Reduced Crosstalk: They can be designed from scratch to avoid recognition by native transcription factors.
- Genetic Stability: Their minimal and non-repetitive sequences reduce the risk of recombination and gene silencing [34].
- Customizability: They offer a high degree of tuning for expression strength, inducibility, and tissue-specificity [34].
- Scalability: They allow for the creation of multiple, mutually orthogonal parts for complex circuits [6].
Q5: When should I use CRISPRi/CRISPRa instead of RNAi or CRISPR knockout?
- Use RNAi for a simple, reversible gene knockdown.
- Use CRISPR knockout (CRISPRko) for complete, permanent gene disruption.
- Use CRISPR interference (CRISPRi) for reversible gene repression without altering the DNA, useful for studying essential genes.
- Use CRISPR activation (CRISPRa) to overexpress a gene. Employing multiple methods (e.g., RNAi followed by CRISPRi) provides orthogonal validation and strengthens the confidence in your findings [49].
Experimental Protocol: Establishing an Orthogonal Control System in Plants
This protocol outlines the key steps for implementing a CRISPR/dCas9-based Orthogonal Control System (OCS) in plants, as described in the research [6].
1. Design and Assembly of Genetic Constructs
- Design Synthetic Promoters (pATFs): Design synthetic promoters by assembling unique gRNA binding sites upstream of a minimal promoter (e.g., the 35S CaMV minimal promoter). Vary the number of binding sites (e.g., 3x or 4x repeats) to modulate expression strength [6].
- Clone Transcriptional Units (TUs): Use a standardized modular cloning (MoClo) framework [6].
- Assemble the final TUs in the format: Promoter (pATF) -> Gene of Interest -> Terminator.
- The key TUs are:
- TU1: A constitutive promoter (e.g., 35S) driving the expression of the artificial transcription factor dCas9:VP64.
- TU2: A promoter (Pol II or Pol III) driving the expression of the gRNA that targets your synthetic pATF.
- TU3: The synthetic promoter (pATF) itself, driving your output gene (e.g., a fluorescent reporter).
- Assemble Final Vector: Stitch the multiple TUs together into a final Agrobacterium shuttle vector using the modular cloning system [6].
2. Plant Transformation and Transient Expression
- Introduce the assembled vector into Agrobacterium tumefaciens.
- For initial testing, use transient expression in Nicotiana benthamiana leaves via agroinfiltration [6].
- For stable expression, generate transgenic plants (e.g., in Arabidopsis thaliana).
3. System Validation and Testing
- Measure Output: Quantify the expression of the output gene (e.g., fluorescence intensity or luciferase activity).
- Test Orthogonality: Co-transform different gRNA/pATF pairs to ensure they act independently without cross-activation [6].
- Inducibility Test: If the gRNA is under an inducible promoter (e.g., ethylene-responsive), apply the stimulus and measure the dynamic change in output [6].
The workflow for this protocol is summarized in the following diagram:
Table 1: Orthogonal System Output Strength in Yeast using Plant TFs
This table summarizes quantitative data from a study testing plant-derived transcription factors in Saccharomyces cerevisiae [47].
| Transcription Factor (TF) Type | Activation Domain | Promoter Type | Relative Expression Output (yEGFP) | Notes |
|---|---|---|---|---|
| Arabidopsis NAC TF | EDLL | Cognate Synthetic | 6 to 10-fold stronger than TDH3 | Strongest performers in the library [47]. |
| Arabidopsis NAC TF | GAL4 AD | Cognate Synthetic | Wide spectrum of output | Output varied based on specific TF [47]. |
| Arabidopsis NAC TF | VP64 | Cognate Synthetic | Wide spectrum of output | Output varied based on specific TF [47]. |
| Various Plant TFs | Various | Cognate Synthetic | Wide range | 106 different combinations tested [47]. |
Table 2: Comparison of Gene Modulation Techniques for Orthogonal Validation
This table compares different techniques used to manipulate gene expression, highlighting their utility for orthogonal validation [49].
| Technique | Mechanism of Action | Key Features | Best Used For |
|---|---|---|---|
| RNA Interference (RNAi) | Knocks down gene expression at the mRNA level. | Reversible silencing; potential for off-target effects. | Initial, reversible gene silencing studies [49]. |
| CRISPR Knockout (CRISPRko) | Permanently disrupts the gene at the DNA level. | Complete, permanent gene silencing. | Confirming a phenotype is due to complete loss of a gene [49]. |
| CRISPR Interference (CRISPRi) | Represses gene expression without altering DNA. | Reversible "dimmer switch" knockdown; targets essential genes. | Reversible knockdown without genomic damage [49]. |
| CRISPR Activation (CRISPRa) | Overexpresses gene expression without altering DNA. | Targeted gene activation. | Studying gene function through overexpression [49]. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Orthogonal Biology
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Modular Cloning (MoClo) Toolkit | A standardized assembly method using Type IIS restriction enzymes (e.g., BsaI) for high-throughput, modular construction of genetic circuits [6]. | Rapid assembly of multi-gene constructs for orthogonal control systems in plants and yeast [6]. |
| Artificial Transcription Factors (ATFs) | Programmable DNA-binding proteins fused to activation/repression domains. Examples include dCas9:VP64 and plant NAC-EDLL fusions [47] [6]. | dCas9:VP64 serves as a universal ATF in plants; NAC-EDLL fusions provide strong orthogonal activation in yeast [47] [6]. |
| Synthetic Promoter Libraries | A collection of engineered promoters with varying strengths and specificities, designed to be orthogonal to native regulation [6] [34]. | Providing multiple, non-interfering control channels for complex genetic circuits in plants [6]. |
| Orthogonal Array Testing (OAT) | A statistical method to test multiple variables and their interactions with a minimal number of experimental runs [50]. | Systematically testing the performance of different TF-promoter pairs, gRNA designs, and circuit components in a high-throughput manner [50]. |
| CRISPR/dCas9 Reagents | Tools for CRISPR interference (CRISPRi) and activation (CRISPRa), allowing for targeted gene repression or overexpression without cutting DNA [49]. | Validating RNAi hits orthogonally or tuning gene expression in a reversible manner for functional studies [49]. |
| Stat3-IN-9 | Stat3-IN-9, MF:C22H21N3O4, MW:391.4 g/mol | Chemical Reagent |
Enhancing Performance: Strategies to Boost Efficiency and Reduce Noise
Optimizing Dynamic Range and Linearity in Dose-Response Relationships
FAQs on Dynamic Range and Linearity
1. What defines the 'dynamic range' and 'linear range' in a dose-response experiment?
The dynamic range refers to the entire span of doses over which a biological system produces a measurable response, from the minimum detectable effect up to the maximum effect ((E_{max})) [51]. For example, in VIPAR polymer gel dosimetry, a linear response was observed up to approximately 40 Gy, while the dynamic range extended to at least 250 Gy [52]. The linear range is a subset of the dynamic range, typically between 20% and 80% of the maximal response, where the relationship between the logarithm of the dose and the effect is approximately linear. This is the most useful and interpretable region of the curve for accurate calculation of parameters like EC50 or IC50 [53] [51].
2. Why are my dose-response curves shallow or incomplete, and how can I optimize them?
Shallow slopes or incomplete curves often result from suboptimal experimental design or biological complexity. To optimize them:
- Number and Spacing of Doses: Use 5-10 concentrations that are log-spaced to adequately define the bottom plateau, top plateau ((E_{max})), and the central linear portion of the curve [53].
- Non-linear Regression Model: Ensure you are using an appropriate model, such as the four-parameter logistic (4PL) model, which fits the Bottom, Top, Hill Slope, and EC50/IC50 parameters [53].
- Check for Underlying Biology: In gene expression studies, complex regulatory networks with feedback loops can lead to non-monotonic (e.g., U-shaped) dose-response curves. Re-examine your system for such biological nuances [54].
3. How can I ensure my dose-response data in gene expression studies is robust?
- Leverage Orthogonal Systems: Using orthogonal transcriptional regulators (e.g., sigma factor toolboxes) allows you to independently control different pathway modules without interference from the host's native regulatory machinery. This reduces noise and provides clearer, more interpretable dose-response relationships [45] [55].
- Employ Biosensors: Integrate metabolite-responsive biosensors to enable high-throughput screening of pathway output. This allows you to rapidly characterize a diverse subset of pathway variants (e.g., with different promoters or enzyme variants) and collect high-quality data for model training [55].
- Normalization: Normalize response values to a percentage scale (0% to 100%) based on control values to correct for inter-experiment variability and facilitate comparison across different studies [53].
Troubleshooting Guide: Common Issues and Solutions
| Issue | Potential Cause | Recommended Solution |
|---|---|---|
| Shallow Hill Slope | The drug has multiple, non-specific targets. The assay lacks sensitivity. | Use a more specific agonist/antagonist. Validate assay sensitivity and check for confounding factors in the experimental system [53]. |
| Incomplete Curve (No Upper or Lower Plateau) | The tested dose range is too narrow. | Extend the range of concentrations tested to capture the full span of the response, from minimal to maximal effect [53]. |
| High Variability in Replicate Data Points | Technical errors in dilution or dispensing. Cell culture contamination or inconsistency. | Use precise liquid handling systems. Maintain consistent and sterile cell culture practices. Perform outlier analysis, but do not exclude points without cause [53]. |
| EC50/IC50 Outside Tested Range | The initial dose range estimate was incorrect. | Perform a pilot experiment with a very broad dose range (e.g., spanning several orders of magnitude) to bracket the expected potency before running the definitive assay [53]. |
| Non-Sigmoidal or Biphasic Curve | Presence of multiple receptor subtypes with different affinities. Activation of opposing pathways at different concentrations. | Consider alternative non-linear regression models. Investigate the biology of the system for evidence of multiple targets or complex network interactions [53] [54]. |
Quantitative Parameters for Dose-Response Analysis
The table below summarizes the key parameters used to quantify dose-response relationships, derived from non-linear regression analysis of sigmoidal curves [53] [54].
| Parameter | Symbol | Definition | Interpretation in Gene Expression Context |
|---|---|---|---|
| Potency | EC({50}) / IC({50}) | The concentration that produces 50% of the maximal effect. | Indicates the efficiency of a transcriptional activator/repressor. A lower EC50 means higher potency [53] [54]. |
| Efficacy | (E_{max}) | The maximum possible response achievable by the drug. | Reflects the maximum level of gene expression activation or repression achievable by the orthogonal regulator [51]. |
| Hill Slope | (n_H) | A measure of the steepness of the curve at its midpoint. | A steeper slope suggests higher cooperativity in the system, such as multi-step transcriptional activation [53]. |
| Dynamic Range | - | The dose range from the lowest measurable effect to (E_{max}). | The operational range of your orthogonal system—the span of inducer concentrations over which you can reliably tune expression [52] [51]. |
| Linear Range | - | The range (typically 20%-80% of (E_{max})) where the log-dose vs. response is linear. | The most reliable range for predicting gene expression levels based on inducer concentration [53] [51]. |
Experimental Protocol: Establishing a Dose-Response Curve for an Orthogonal Gene Expression System
This protocol outlines the steps to characterize the dose-response relationship of an orthogonal transcriptional activator controlling a gene of interest, using a reporter like GFP.
1. Experimental Design and Setup
- Construct Design: Clone your gene of interest (or a fluorescent reporter) downstream of a promoter controlled by your chosen orthogonal regulator (e.g., a sigma factor-specific promoter) [55].
- Cell Culture: Transform the construct into your host organism (e.g., E. coli or A. thaliana). Prepare a master culture and then aliquot into a multi-well plate to ensure consistent starting conditions.
- Dosing Scheme: Prepare a logarithmic series of inducer concentrations (e.g., 0, 0.1, 1, 10, 100, 1000 µM). Ensure you have sufficient replicates (n=3 or more) for each concentration [53].
2. Data Collection
- Treatment and Incubation: Add the inducer to the cultures and incubate under optimal growth conditions for a predetermined period.
- Endpoint Measurement: Measure the response (e.g., fluorescence intensity for GFP) and a normalization metric (e.g., optical density at 600nm for bacterial cultures) for each well [53].
3. Data Analysis and Curve Fitting
- Normalization: Normalize the fluorescence readings to the cell density. Then, transform the data to a percentage scale, where the minimum response (e.g., no inducer) is 0% and the maximum observed response is 100% [53].
- Non-linear Regression: Input the log(concentration) and normalized response values into a software tool (e.g., GraphPad Prism) and fit the data to a four-parameter logistic (4PL) model: ( Y = Bottom + \frac{Top - Bottom}{1 + 10^{(\log{EC_{50}} - X) * HillSlope}} ) where (X) is the log(concentration) and (Y) is the response [53].
- Parameter Interpretation: Extract and record the fitted values for EC({50}), (E{max}) (Top), Hill Slope, and assess the goodness-of-fit.
The workflow for this experimental and computational process is summarized in the following diagram:
Research Reagent Solutions for Pathway Optimization
The table below lists key tools and reagents essential for optimizing dose-response in gene expression studies, particularly those involving synthetic orthogonal systems.
| Research Reagent | Function & Application |
|---|---|
| Orthogonal Sigma Factors & Promoters | Enables independent, tunable control of multiple gene modules without host crosstalk. Crucial for deconvoluting complex dose-response relationships in metabolic pathways [45] [55]. |
| Small Molecule Biosensors | Provides a high-throughput link between metabolite concentration (output) and a measurable signal (e.g., fluorescence). Essential for screening large libraries of pathway variants to generate robust dose-response data [55]. |
| Gene Expression Databases (e.g., GDSC, CMap) | Reference databases containing gene expression profiles linked to drug response data (e.g., IC50). Allows for computational prediction of drug efficacy or repurposing based on input gene signatures [56] [57]. |
| Dose-Response Analysis Software | Tools like GraphPad Prism implement non-linear regression models (e.g., 4PL) to accurately calculate potency, efficacy, and other critical parameters from experimental data [53]. |
| Computational Prediction Models (ML/Statistical) | Machine learning models trained on characterized pathway variants can predict optimal genetic configurations (promoter strength, enzyme variants) to maximize product titer, guiding a more efficient design-build-test-learn cycle [55]. |
Advanced Strategy: Integrating Dose-Exposure-Response (DER) Modeling
For a more precise understanding, especially in drug development, moving from a simple Dose-Response (DR) model to a Dose-Exposure-Response (DER) model can be beneficial [58]. This approach sequentially models the relationship between the administered dose and the internal drug/exposure concentration (PK), and then between the internal exposure and the biological effect (PD). This is particularly useful when the internal exposure is variable between individuals. The DER framework can provide more accurate parameter estimates and predictions for dose optimization, especially when paired with methods that use randomization as an instrumental variable to control for unobserved confounding factors [58].
The logical flow of this advanced modeling approach is shown below:
Minimizing Gene Expression Noise and Cell-to-Cell Variability
Gene expression noise, the random fluctuation in protein and RNA levels between genetically identical cells under the same environmental conditions, is a fundamental challenge in genetic engineering and synthetic biology. For researchers and drug development professionals working with orthogonal regulators, controlling this variability is paramount to achieving predictable and reliable system behavior. This technical support guide provides troubleshooting advice and methodologies to help you minimize cell-to-cell variability in your experiments, enhancing the robustness of your genetic constructs.
FAQs and Troubleshooting Guides
Fundamental Concepts and Definitions
What is gene expression noise and why is it important in synthetic biology? Gene expression noise refers to random fluctuations in gene expression levels within a population of genetically identical cells. This variability impacts cellular fitness and can compromise the function of synthetic genetic circuits. It consists of two primary components:
- Intrinsic Noise: Arises from stochastic biochemical reactions involved in the transcription and translation of an individual gene.
- Extrinsic Noise: Stems from cell-to-cell differences in global factors such as cell size, cell cycle stage, and concentrations of transcription factors or ribosomes, which affect the expression of all genes in a cell [59].
For applications requiring precise control over protein dosage, such as metabolic engineering or gene therapy, high noise levels can lead to inconsistent outcomes and reduced product yields [60].
Our circuit performance is inconsistent between cell cultures. Could gene expression noise be the cause? Yes. Inconsistent performance often stems from unacceptably high levels of gene expression noise. Before troubleshooting your specific circuit, we recommend you first quantify the noise in your system. A common and powerful method is to use a dual-reporter system, where two identical reporter genes (e.g., GFP and Venus) are placed in the same cell. The correlation between their expressions reveals the nature of the noise:
- Low correlation suggests high intrinsic noise (local, gene-specific fluctuations).
- High correlation suggests high extrinsic noise (global, cell-wide fluctuations) [59].
Experimental Design and Genetic Architecture
How can we independently control the mean and noise of gene expression? A powerful strategy is to express a gene from two separate, orthogonally inducible promoters within the same cell. The total protein output is the sum of the expressions from both promoters. By using one low-noise and one high-noise promoter, you can tune the inducer concentrations to adjust the mean expression (by changing total output) and the noise (by changing the proportion of expression from each promoter) independently [61].
Table 1: Strategies for Independent Control of Mean and Noise
| Strategy | Mechanism | Key Consideration |
|---|---|---|
| Dual Promoter Convolution [61] | Express gene from two orthogonally regulated promoters (one low-noise, one high-noise). | Requires careful characterization of individual promoter properties. |
| Promoter Engineering [60] [62] | Modify promoter sequence to alter its activation kinetics (e.g., from slow ON-OFF switching to fast). | Can be labor-intensive to develop and test. |
| Epigenetic & Locus Control [15] [63] | Integrate transgenes into genomic locations with specific chromatin environments (e.g., open chromatin for low noise). | Genomic position effects can be strong and unpredictable. |
Does the genomic location of our transgene integration affect expression noise? Absolutely. Epigenetic features, such as chromatin environment, at different genomic loci orthogonally control the mean and variance of gene expression. Genomic locations associated with more repressed chromatin (heterochromatin) are linked to higher expression noise. This is because repressed chromatin leads to infrequent, large transcriptional bursts, which increases cell-to-cell variability. When possible, target genomic "safe-harbor" loci known for stable and predictable expression [15] [63].
We are designing a multi-gene circuit. How does gene arrangement impact noise? Gene order and orientation significantly influence expression covariance. For genes whose products need to be maintained in a strict stoichiometry (e.g., subunits of a protein complex), a divergent gene pair (DGP) configuration can be beneficial. Co-regulated DGPs show more synchronized transcription firing, leading to higher expression covariance and lower uncorrelated noise between the two genes. This synchronization is specific to the divergent configuration and is not observed in tandem or convergent arrangements [64].
Conversely, for differentially regulated genes, a DGP configuration can be detrimental, as regulatory signals can stochastically "leak" to the adjacent promoter, thereby increasing noise. In such cases, separating the genes is the preferred strategy [64].
Measurement and Quantification
What is the best method to quantify gene expression noise in my scRNA-seq data? A comparative analysis of 14 variability metrics recommends scran as a robust metric for quantifying cell-to-cell variability from single-cell RNA-sequencing data. It performs well across different sequencing platforms (e.g., Smartseq2 vs. 10X Genomics) and is less sensitive to sample size variations compared to other metrics like CV, DESeq2, or edgeR [65].
Our flow cytometry data shows a wide distribution. How do we interpret the noise level? The standard metric for noise from flow cytometry or fluorescence microscopy data is the coefficient of variation (η), which is the standard deviation of the fluorescence distribution divided by its mean (η = σ/μ). This metric normalizes the spread of the distribution to the average expression level, allowing for comparison across different conditions and promoters [61].
Advanced Control and Circuit-Level Solutions
How can we reduce noise in a multi-module circuit competing for cellular resources? Resource competition (e.g., for RNA polymerases, ribosomes) is a major source of extrinsic noise in complex circuits. To mitigate this, consider implementing a multi-module antithetic control strategy. Research shows that a Negatively Competitive Regulation (NCR) controller outperforms other controllers in reducing intrinsic noise under resource competition. This controller uses antisense RNAs that are promoted by the module proteins and co-degrade each other, effectively stabilizing the system [66].
Table 2: Summary of Antithetic Controllers for Noise Reduction
| Controller Type | Mechanism | Noise Reduction Efficiency |
|---|---|---|
| Single-Module Controller (SMC) | Antithetic mechanism applied to only one gene module. | Moderate |
| Local Controller (LC) | Two distinct antisense RNAs, each controlling one module. | Good |
| Global Controller (GC) | A common antisense RNA controls both module mRNAs. | Good |
| NCR Controller | Two antisense RNAs control their respective modules and co-degrade each other. | Best |
Can we build orthogonal systems to completely isolate our circuit from host regulation? Yes. In various organisms, you can create fully orthogonal control systems (OCS). For example, in plants, synthetic promoters have been designed to be activated only by custom CRISPR-based transcription factors (dCas9:VP64) programmed with specific guide RNAs. This system creates a regulatory layer that is completely insulated from the host's native transcription factors, minimizing unwanted cross-talk and enhancing predictability [6]. Similar principles using orthogonal RNA polymerases or ribosomes can be applied in bacterial and mammalian systems.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Investigating Gene Expression Noise
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Dual Fluorescent Reporters (e.g., GFP, Venus, mCherry) [64] [59] | Quantify intrinsic vs. extrinsic noise by measuring expression correlation. | Fundamental noise characterization in a new cell line or chassis. |
| Orthogonal Inducible Promoters [61] | Allow independent control of two gene copies from different regulatory systems. | Convolution method for independent mean and noise control. |
| CRISPR-dCas9 Transcriptional Regulators [6] | Provide programmable activation/repression orthogonal to host machinery. | Creating synthetic gene circuits with minimal host cross-talk. |
| smFISH (single-molecule RNA FISH) Probes [64] [15] | Visualize and quantify individual mRNA molecules in single cells. | Investigating transcriptional bursting and nascent transcript dynamics. |
| Antithetic Control RNAs [66] | Engineered RNAs that promote degradation of target mRNAs and each other. | Implementing NCR control in multi-gene circuits to reduce resource competition noise. |
Essential Experimental Protocols
- Construct Design: Clone two identical, codon-optimized fluorescent reporter genes (e.g., GFP and Venus) under the control of identical promoters. Integrate this construct into a defined genomic locus.
- Cell Culture & Imaging: Grow a clonal population of cells and image them using time-lapse fluorescence microscopy over multiple cell generations.
- Single-Cell Analysis: Use image analysis software to extract the fluorescence intensities of both reporters for each individual cell over time.
- Noise Decomposition: Calculate the total noise (ηtot) as the standard deviation divided by the mean of the total fluorescence. The correlation coefficient (R) between the two reporters' intensities indicates the extrinsic noise component. A low R value signifies high intrinsic noise.
- Promoter Selection: Characterize a low-noise and a high-noise inducible promoter (e.g., one with positive feedback leading to bistability) driving the same output gene.
- Strain Construction: Create a strain where your gene of interest is expressed from both promoters simultaneously.
- Calibration: For each promoter individually, measure the mean (μ) and noise (η) of the expression output across a range of inducer concentrations.
- Fine-Tuning: To achieve a desired mean expression level with a specific noise, use combinations of the two inducers. The total mean is the sum of the means from each promoter. The total noise is a weighted average of the individual noises, allowing for independent control.
Addressing Metabolic Burden and Genetic Instability in Engineered Pathways
Frequently Asked Questions
What are the first signs of metabolic burden in my culture? The most common symptoms include a decreased growth rate, an aberrant or enlarged cell size, and genetic instability such as plasmid loss or mutations over many generations [67] [68]. These symptoms indicate that the host cell is under significant stress from the engineered pathway.
My protein expression is high initially but drops off over time. What could be causing this? This is a classic sign of genetic instability often triggered by metabolic burden. Cells that inactivate or lose the engineered pathway can outgrow the productive cells. Using genetically stable parts from a combinatorial assembly platform can help maintain production over the long term [67].
How can orthogonal regulators help reduce metabolic burden? Orthogonal regulatory systems, derived from organisms like Saccharomyces spp., are designed to function independently of the host's native regulation. This allows for precise modulation of gene expression without interfering with essential cellular processes, thereby minimizing the stress on the host [45].
Why is my transformation efficiency low or why are no clones growing? If your competent cells are functional (verified with a positive control), the issue often lies with the plasmid DNA. Possible causes include low plasmid concentration or degradation. Always verify DNA quality and quantity via gel electrophoresis and spectrophotometry [69].
Troubleshooting Guide
The following table outlines common problems, their underlying causes, and potential solutions.
| Symptom | Possible Root Cause | Recommended Solution |
|---|---|---|
| Decreased growth rate & low productivity | Resource competition: depletion of amino acids, ATP, and charged tRNAs for native processes [68] | Use weaker, tunable promoters; implement inducible systems; employ orthogonal parts to decouple expression from host regulation [45] |
| Genetic instability (plasmid loss, mutation) | Stress from metabolic burden favors non-productive mutants; burden from (over)expression of heterologous proteins [67] [68] | Use combinatorial libraries to screen for genetically stable constructs; optimize codon usage without disrupting rare codon regions for folding [67] [68] |
| High levels of misfolded proteins | Translation errors due to rare codons depleting charged tRNAs; improper codon optimization disrupting translation pacing [68] | Verify codon usage includes necessary rare codons for correct folding; fine-tune expression levels to match cellular capacity [68] |
| No PCR product in diagnostic assay | Incorrect primer design, degraded template, or suboptimal PCR conditions [70] | Redesign primers (18-22 bp, 45-60% GC content); use additives like DMSO or GC enhancer for difficult templates; check template quality [70] |
| Low CRISPR editing efficiency | Poor crRNA design; low transfection efficiency; inaccessible target genomic locus [70] | Ensure crRNA avoids off-targets; optimize transfection; use selection markers (antibiotics/FACS) to enrich edited cells [70] |
Experimental Protocols
Protocol 1: Assembling a Combinatorial Library for Stable Pathway Engineering
This methodology is based on a platform that enabled high-level, stable lycopene production in cyanobacteria [67].
- Part Library Generation: Create large libraries of characterized biological parts, including synthetic promoters and Ribosome Binding Sites (RBSs) with varying strengths.
- Combinatorial Assembly: Assemble your pathway genes (e.g., for a terpenoid like lycopene) using these parts in a sparse, combinatorial fashion to generate millions of construct variants.
- Library Transformation: Transform the library into your host organism (e.g., Synechocystis sp. PCC 6803).
- Long-Term Selection: Culture transformations photoautotrophically over many generations under selective pressure.
- Screening: Screen randomly chosen variants for stable accumulation of the target product (e.g., lycopene) from COâ‚‚. A high success rate (e.g., 80%) indicates effective library design.
Protocol 2: Applying a Systematic Troubleshooting Framework
This general-purpose protocol can be adapted to diagnose various experimental failures [69].
- Identify the Problem: Clearly define the issue without assuming causes (e.g., "No clones on agar plate").
- List Possible Causes: Brainstorm all potential explanations (e.g., faulty competent cells, incorrect antibiotic, low plasmid DNA concentration, flawed heat-shock procedure).
- Gather Data: Review controls, reagent storage conditions, and your laboratory notebook against the standard protocol.
- Eliminate Causes: Use data to rule out incorrect explanations (e.g., if positive control worked, competent cells are not the cause).
- Test Hypotheses: Design and run simple experiments to test remaining causes (e.g., run a gel to check plasmid DNA integrity and concentration).
- Identify Root Cause: Analyze experimental results to pinpoint the definitive issue and implement a fix.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function |
|---|---|
| Synthetic Promoter/RBS Library | Provides a set of characterized parts with varying strengths to fine-tune the expression of each gene in a pathway, balancing flux and minimizing burden [67]. |
| Orthogonal Transcriptional Regulators | Synthetic activators and repressors from other species (e.g., Saccharomyces spp.) that function independently of the host machinery for precise, isolated control of gene circuits [45]. |
| GeneArt Genomic Cleavage Detection Kit | A tool to verify the efficiency of CRISPR-Cas9 cleavage at the target genomic locus, which is critical for assessing the success of genome editing experiments [70]. |
| PureLink HQ Mini Plasmid Purification Kit | Used to obtain high-quality, purified plasmid DNA, which is essential for reliable sequencing results and efficient transformations [70]. |
Pathway and Workflow Visualizations
Balancing On-Target Mutation Efficiency with Off-Target Effects in Evolutionary Systems
Troubleshooting Guides
Guide 1: Addressing High Off-Target Effects in CRISPR Experiments
Problem: My CRISPR experiment shows unexpected phenotypic outcomes, potentially due to high off-target activity.
Question: Why are off-target effects a critical concern in my research? Answer: Off-target effects refer to nonspecific and unintended genetic modifications that can confound experimental results and reduce reproducibility [71]. In therapeutic development, they pose significant safety risks, including potential disruption of vital coding regions that could lead to genotoxic effects such as cancer [71] [72].
Question: How can I predict where off-target effects might occur? Answer: Utilize in silico prediction tools that identify potential off-target sites based on sequence similarity to your guide RNA:
- Cas-OFFinder: Widely applied with high tolerance for various sgRNA lengths, PAM types, and numbers of mismatches or bulges [73]
- CRISPOR: Provides off-target scores and rankings based on predicted on-target to off-target activity [72]
- FlashFry: High-throughput tool that characterizes thousands of CRISPR target sequences quickly and provides GC content information [73]
Question: What experimental strategies can minimize off-target effects? Answer: Implement these validated approaches:
- Use High-Fidelity Cas Variants: Engineered Cas9 variants like HypaCas9, eSpCas9(1.1), SpCas9HF1, and evoCas9 have reduced mismatch tolerance [74]
- Employ Cas9 Nickases: Use paired nickases that require two single-strand breaks in close proximity to create a double-strand break, dramatically reducing off-target indels [71]
- Optimize gRNA Design: Select guides with low similarity to other genomic sites and consider truncating gRNAs to 17-18 nucleotides for increased specificity [71] [72]
- Utilize Chemical Modifications: Incorporate 2'-O-methyl analogs (2'-O-Me) and 3' phosphorothioate bond (PS) modifications in synthetic gRNAs to reduce off-target editing [72]
Question: How do I properly detect and quantify off-target events? Answer: Based on your experimental needs:
- For comprehensive analysis: Use whole genome sequencing (WGS) for complete assessment of off-target editing and chromosomal aberrations [72]
- For targeted approaches: Implement GUIDE-seq, CIRCLE-seq, or DISCOVER-seq to sequence specific sites where Cas binding or NHEJ repair has occurred [73] [72]
- For candidate validation: Perform sequencing of predicted off-target sites identified during gRNA design [74]
Guide 2: Optimizing Orthogonal Regulatory Systems
Problem: My orthogonal gene expression system shows leakage or insufficient regulation.
Question: What are the key components of orthogonal regulatory systems? Answer: Orthogonal systems use synthetic transcriptional regulators that function independently of host cellular machinery. These typically include:
- Synthetic Transcription Factors: Programmable DNA-binding domains (e.g., dCas9, TALEs) fused to effector domains [3] [13]
- Synthetic Promoters: Engineered DNA sequences responsive to specific synthetic transcription factors [13]
- Regulatory Devices: Sensing and actuation components that operate at transcriptional, translational, or post-translational levels [3]
Question: How can I improve the precision of my orthogonal regulators? Answer:
- Implement Aptazyme Strategies: Use ligand-dependent ribozymes in your sgRNA design to dramatically decrease off-target mutation frequency [75] [76]
- Employ Dimerization Systems: Utilize Fok1-dCas9 fusions that require dimerization to activate, reducing off-target effects by up to 10,000-fold [71]
- Leverage Epigenetic Regulators: Implement CRISPRoff/CRISPRon systems that combine dCas9 with DNA methyltransferases or demethylases for stable, programmable epigenetic control [3]
Question: What design principles ensure orthogonal circuit functionality? Answer:
- Ensure Orthogonality: Select DNA-binding domains with minimal cross-reactivity to host genomes [3]
- Address Context-Dependence: Characterize parts in your specific experimental system [3]
- Manage Complexity: Start with simple circuits and progressively increase complexity [3]
- Consider Noise: Implement signal amplification devices where necessary [3]
Frequently Asked Questions (FAQs)
FAQ 1: What is the fundamental difference between on-target and off-target effects? On-target effects are the intended, specific modifications at the desired genomic location, while off-target effects are unintended modifications at sites with sequence similarity to the target [77]. In toxicology, on-target refers to exaggerated pharmacologic effects at the target of interest, while off-target refers to adverse effects from modulation of other targets [77].
FAQ 2: How many base pair mismatches can CRISPR-Cas9 tolerate? The commonly used Streptococcus pyogenes Cas9 (SpCas9) can tolerate between three and five base pair mismatches between the gRNA and target DNA, particularly in the PAM-distal region [75] [71] [72].
FAQ 3: Are off-target effects more concerning for basic research or therapeutic applications? The concern level depends on application. For basic research, off-targets can confound results but might be manageable. For human therapies, they pose critical safety risks and are subject to rigorous FDA scrutiny, requiring comprehensive characterization [72].
FAQ 4: What is the most effective strategy to reduce off-target effects? Studies indicate that using ligand-dependent ribozymes (aptazymes) is particularly effective for avoiding unwanted mutations [75] [76]. Additionally, high-fidelity Cas variants and paired nickase systems provide substantial improvements [71].
FAQ 5: How does the choice of delivery method affect off-target rates? The duration of CRISPR component activity significantly impacts off-target effects. Short-term expression via transient delivery methods (such as mRNA or ribonucleoprotein complexes) reduces the window for off-target activity compared to stable plasmid transfection [72].
Experimental Protocols & Data Presentation
Table 1: Comparison of Off-Target Detection Methods
| Method | Principle | Sensitivity | Advantages | Limitations |
|---|---|---|---|---|
| Whole Genome Sequencing | Sequences entire genome before and after editing | Comprehensive | Detects all mutation types including chromosomal rearrangements | Expensive; requires high sequencing coverage [73] |
| GUIDE-seq | Integrates dsODNs into DSB sites during repair | Highly sensitive, low false positive rate [73] | Cost-effective; sensitive | Limited by transfection efficiency [73] |
| CIRCLE-seq | Circularizes sheared DNA; incubates with Cas9 RNP; linearizes for sequencing | High validation rate | Biochemical method; works with purified DNA | Does not account for cellular context [73] |
| DISCOVER-seq | Uses DNA repair protein MRE11 for ChIP-seq | High precision in cells | Utilizes native repair machinery; works in various cell types | May have some false positives [73] |
| Digenome-seq | Digests purified genomic DNA with Cas9 RNP then performs WGS | Highly sensitive | Cell-free method; comprehensive | Requires high sequencing coverage; expensive [73] |
Table 2: Research Reagent Solutions for Optimizing Gene Editing
| Reagent Type | Specific Examples | Function | Application Context |
|---|---|---|---|
| High-Fidelity Cas Variants | HypaCas9, eSpCas9(1.1), SpCas9HF1, evoCas9 [74] | Reduce off-target cleavage while maintaining on-target activity | Therapeutic development where specificity is critical |
| Cas9 Nickases | Paired Cas9n systems [71] | Create single-strand breaks requiring dual recognition for DSB formation | Reducing off-target indels while maintaining editing efficiency |
| Base Editors | Cas9 nickase-cytidine deaminase fusions [76] | Enable precise single nucleotide changes without DSBs | Applications requiring precise point mutations |
| Orthogonal RNA Polymerases | T7, T3 polymerases with specific promoters [3] | Enable independent regulation of multiple genetic circuits | Multiplexed gene expression systems |
| Aptazyme-Embedded sgRNAs | Ligand-dependent ribozyme designs [75] | Provide small molecule control over sgRNA activity | Inducible genome editing systems |
Protocol 1: GUIDE-seq for Comprehensive Off-Target Detection
- Transfection: Co-transfect cells with Cas9-sgRNA RNP complex and dsODN donors using appropriate method for your cell type.
- Genomic DNA Extraction: Harvest cells 72 hours post-transfection and extract genomic DNA using standard methods.
- Library Preparation:
- Fragment DNA to ~400 bp size
- End-repair and A-tail fragments
- Ligate Illumina sequencing adapters
- Enrich for dsODN-integrated fragments via PCR
- Sequencing & Analysis:
- Perform high-throughput sequencing (Illumina)
- Align sequences to reference genome
- Identify dsODN integration sites as potential off-target loci
- Validation: Verify top candidate off-target sites using targeted amplicon sequencing [73].
Protocol 2: Implementing Paired Nickases for Specific Editing
- gRNA Design: Design two gRNAs targeting adjacent sites (10-30 bp apart) on opposite DNA strands.
- Vector Preparation: Clone both gRNAs into appropriate expression vector with Cas9 nickase (Cas9n).
- Delivery: Transfect target cells with paired nickase system.
- Efficiency Assessment:
- Extract genomic DNA 48-72 hours post-transfection
- Amplify target region by PCR
- Analyze editing efficiency via T7E1 assay or sequencing
- Off-Target Screening: Use targeted sequencing of predicted off-target sites for both gRNAs to verify specificity improvement [71].
Visualization Diagrams
Diagram 1: Orthogonal Regulatory System with CRISPR-Based Control
Diagram 2: Off-Target Effect Management Workflow
In the field of orthogonal regulator research, achieving precise control over gene expression is paramount for applications ranging from metabolic engineering to therapeutic development. Orthogonal control systems (OCS) are specifically designed to function independently of the host's native regulatory machinery, thereby minimizing cross-talk and enabling predictable circuit behavior [18]. The performance of these systems is heavily dependent on two fundamental parameters: inducer concentration and induction timing.
Proper optimization of these parameters directly influences key experimental outcomes, including the strength of transcriptional output, the minimization of metabolic burden, and the successful implementation of complex genetic programs such as bistable switches and logic gates [3]. This guide provides detailed troubleshooting protocols and technical specifications to help researchers systematically optimize these critical parameters for robust and reproducible results in their orthogonal control systems.
Quantitative Parameter Optimization
Case Study: Fine-Tuning the DDI2 Promoter in Budding Yeast
Research with the DDI2 promoter in Saccharomyces cerevisiae provides a clear framework for parameter optimization. This promoter is induced by cyanamide and demonstrates how systematic testing of time and concentration variables can lead to optimal expression conditions.
Table 1: Optimization of DDI2 Promoter Induction with Cyanamide [78]
| Cyanamide Concentration (mM) | Induction Time (hours) | Relative Expression Level | Impact on Cell Growth |
|---|---|---|---|
| 0 | 3-5 | Baseline | No adverse effects |
| <5 | 3-5 | Increasing | Minimal impact |
| 5 | 5 | High (Optimal) | Minimal impact |
| 8 | 5 | Highest | Moderate growth inhibition |
Comparative Analysis of Promoter Systems
Different promoter systems exhibit distinct induction profiles and performance characteristics. Understanding these differences is crucial for selecting the appropriate system for specific experimental needs.
Table 2: Comparison of Common Inducible Promoter Systems [78]
| Promoter | Inducer | Optimal Induction Conditions | Fold Induction | Key Characteristics |
|---|---|---|---|---|
| PDDI2 | Cyanamide | 5 mM, 4 hours | 8.72x over PADH1 | High inducibility, low leakiness |
| PGAL1 | Galactose | Medium change, 4 hours | High (lower than PDDI2) | Requires medium change, expensive |
| PCUP1 | CuSO4 | 0.5 mM, 4 hours | 4.3x over PADH1 | High basal expression, metal toxicity concerns |
| PADH1 | None | Constitutive | Baseline (1x) | Stable, no inducer needed |
Figure 1: Experimental workflow for optimizing inducer concentration and timing parameters. The process involves systematic screening of variables, measurement of outputs, and balancing expression with cellular health.
Experimental Protocols for Parameter Optimization
Protocol: Determining Optimal Inducer Concentration and Timing
This protocol provides a systematic approach for optimizing induction parameters for any inducible promoter system, based on established methodologies in orthogonal control research [78].
Materials Required:
- Reporter strain (e.g., with fluorescent protein under promoter control)
- Appropriate induction medium
- Inducer stock solution (concentration known)
- Flow cytometer or fluorescence plate reader
- Spectrophotometer for OD600 measurements
Procedure:
- Culture Preparation:
- Inoculate reporter strain in appropriate medium and grow overnight.
- Dilute culture to low OD600 (typically 0.1) in fresh medium and grow to mid-log phase (OD600 ≈ 0.5-0.6).
Induction Matrix Setup:
- Prepare a series of inducer concentrations (e.g., 0, 1, 2, 5, 8 mM) in culture tubes.
- Add equal volumes of mid-log culture to each tube.
- Incubate at appropriate temperature with shaking.
Time-Course Sampling:
- Collect samples at multiple time points (e.g., 2, 3, 4, 5 hours post-induction).
- For each sample:
- Measure OD600 to assess cell growth.
- Analyze reporter expression (fluorescence via flow cytometry or plate reader).
- Record data in structured format for analysis.
Data Analysis:
- Normalize expression levels to cell density.
- Plot expression versus time for each concentration.
- Identify concentration and time that provide desired expression level with minimal growth impact.
Troubleshooting Notes:
- If expression is low even at high inducer concentrations, verify promoter functionality and inducer stability.
- If growth inhibition is severe, consider lower inducer concentrations or alternative induction windows.
- For autoinducible systems, monitor growth phase-dependent induction carefully [79].
Protocol: Implementing Two-Stage Fermentation with Optimized Induction
For metabolic engineering applications, a two-stage fermentation process separates growth and production phases, requiring precise timing of induction [79].
Procedure:
- Growth Phase:
- Grow culture under non-inducing conditions until target cell density is reached (typically late log phase).
- Monitor growth carefully to identify the optimal induction point.
Induction Trigger:
- Add predetermined optimal inducer concentration based on prior optimization.
- Alternatively, for autoinducible systems, allow natural transition to production phase.
Production Phase:
- Maintain culture conditions to support production rather than growth.
- Monitor product formation over time to confirm successful induction.
Troubleshooting Guides
FAQ 1: Why is my gene expression level low even with high inducer concentrations?
Potential Causes and Solutions:
Inducer Uptake Issues: Some inducers may not efficiently enter cells. Consider:
- Using chemical analogs with better membrane permeability (e.g., IPTG instead of lactose) [79]
- Checking for transporter mutations in your strain
- Testing induction at different cell densities
Metabolic Burden: High expression demands may overwhelm cellular machinery.
- Reduce expression level by lowering inducer concentration
- Use weaker promoters or ribosome binding sites in your construct
- Implement a two-stage fermentation to separate growth and production [79]
Promoter Incompatibility: The promoter may not function optimally in your host.
- Verify promoter specificity for your host organism
- Consider orthogonal promoters designed for minimal cross-talk [13]
- Test multiple promoter options to identify the best performer
Catabolite Repression: Sugar-based inducers may be subject to carbon catabolite repression.
- Use non-repressing carbon sources in your medium
- Consider CRP* mutant strains that reduce catabolite repression [79]
- Switch to non-metabolizable inducer analogs (e.g., IPTG)
FAQ 2: How do I reduce basal expression (leakiness) before induction?
Potential Causes and Solutions:
Promoter Selection:
Genetic Circuit Design:
- Implement repression systems that actively silence pre-induction expression
- Incorporate additional regulatory layers (e.g., riboswitches, toehold switches) [3]
- Use degradation tags to reduce stability of leaky expression products
Inducer Optimization:
- Verify inducer purity and stability in your medium
- Consider adding repressing agents during growth phase where applicable
- Optimize medium composition to minimize unintended induction
FAQ 3: My induction is causing severe growth inhibition. How can I address this?
Potential Causes and Solutions:
Toxic Inducer Effects:
- Test inducer toxicity independently by measuring growth with inducer but without expression construct
- Switch to less toxic inducer analogs (e.g., IPTG instead of lactose) [79]
- Reduce inducer concentration and accept lower expression levels
Product Toxicity:
- Implement inducible systems that delay production until sufficient biomass is achieved [79]
- Use weaker promoters to reduce metabolic burden
- Consider product secretion or compartmentalization strategies
Expression Timing:
- Shift induction to later growth phase when higher cell density is achieved
- Use autoinducible systems that naturally activate at appropriate cell densities [79]
- Implement gradual induction protocols rather than abrupt induction
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Orthogonal Control System Optimization
| Reagent/Category | Function | Examples & Notes |
|---|---|---|
| Chemical Inducers | Activate expression from inducible promoters | Cyanamide (DDI2 promoter), IPTG (Lac-based), Galactose (GAL promoters), Tetracycline (Tet systems) [78] [79] |
| Reporter Systems | Quantify expression levels and kinetics | Fluorescent proteins (sfGFP, RFP), Luciferase enzymes, Chromogenic enzymes [18] [78] |
| Orthogonal Promoters | Minimize host cross-talk | Synthetic promoters for dCas9:VP64, Hybrid eukaryotic/prokaryotic promoters [18] [13] |
| Programmable TFs | Provide specific DNA binding | dCas9 fusions, Synthetic zinc fingers, TAL effectors [18] [3] |
| Assembly Systems | Enable modular construct building | Golden Gate (MoClo) systems, Type IIS restriction enzymes [18] |
Figure 2: Orthogonal control system architecture showing key modules and optimization parameters. Induction timing and concentration primarily affect the sensor and processing modules respectively.
Advanced Applications and Future Directions
As orthogonal control systems evolve, several emerging approaches show promise for more precise temporal and quantitative control of gene expression:
Quorum Sensing Integration: Autoinducible systems that activate at critical cell densities eliminate the need for external inducer addition, facilitating more scalable bioprocesses [79]. These systems naturally link induction timing to culture density, creating self-regulating expression control.
CRISPR-Based Orthogonal Systems: Synthetic promoters designed for CRISPR-based transcription factors enable multiplexed orthogonal control with minimal host interference [18]. These systems offer unprecedented programmability and scalability for complex genetic circuits.
Hybrid Inducer Systems: Combining chemical inducers with environmental triggers (temperature, light) provides multi-dimensional control over expression timing and amplitude [3]. These approaches enable more sophisticated dynamic control strategies for metabolic engineering and therapeutic applications.
The continuing characterization of genetic elements that dictate transcriptional output provides a foundation for the rational design of increasingly refined synthetic regulators, moving the field toward more predictable and robust control of biological systems [13].
Benchmarking Orthogonal Tools: Validation Frameworks and Cross-System Analysis
Troubleshooting Guides
FAQ: How do I quantify and improve the dynamic range of an orthogonal gene expression system?
Problem: Your synthetic biological circuit shows only a small difference between its fully "ON" and fully "OFF" states, limiting its utility.
Explanation: Dynamic range is the ratio between the maximum induced output and the minimum basal output (leakiness) of a system. A wide dynamic range is crucial for creating robust, sensitive genetic circuits that produce a clear signal.
Solution:
- Quantification: Measure the system's output (e.g., reporter protein level) under both fully induced and uninduced conditions. The dynamic range is calculated as: Dynamic Range = [Output]{ON} / [Output]{OFF} In qRT-PCR data, this can be equivalent to a 4- to 6-fold dynamic range in cycle threshold (Ct) values for a well-defined system. [80]
- Improvement Strategies:
- Enhancer Engineering: To reduce leakiness, engineer multi-domain steric hindrance. For example, a transcription factor flanked by two pleckstrin homology (PH) domains on each side (
PHx2-TF-PHx2) can be docked to the cell membrane, dramatically reducing basal activity. Upon protease cleavage, the TF is liberated, achieving a high induction fold change (e.g., 33.6-fold). [81] - Promoter/Operator Optimization: Use promoters with low basal activity and high inducibility. Fine-tune the affinity of repressor or activator binding sites.
- Enhancer Engineering: To reduce leakiness, engineer multi-domain steric hindrance. For example, a transcription factor flanked by two pleckstrin homology (PH) domains on each side (
FAQ: My engineered receptor is non-functional inside the secretory pathway. What is the cause and how can I fix it?
Problem: A receptor that functions in the cytoplasm loses activity when targeted to the endoplasmic reticulum (ER) or plasma membrane.
Explanation: The secretory pathway introduces post-translational modifications (PTMs) like disulfide bond formation and N-linked glycosylation. These modifications can misfold synthetic proteins or block their ligand-binding domains. [81]
Solution:
- Identify Problematic Residues: Inspect the protein sequence for motifs susceptible to PTMs, particularly asparagine (N) in N-X-S/T motifs (for glycosylation) and cysteine (C) residues (for disulfide bonds).
- Systematic Mutagenesis: Perform site-directed mutagenesis to eliminate these sites. For example, mutating cysteine and asparagine residues in chemically induced dimerization (CID) domains has successfully restored the function of orthogonal chemically activated cell-surface receptors (OCARs) in the ER and on the plasma membrane. [81]
FAQ: How do I validate that my gene expression measurements are specific and comparable across experiments?
Problem: Gene expression data from qRT-PCR appears inconsistent, making it difficult to compare results from different labs or experimental batches.
Explanation: Traditional relative quantification in qRT-PCR uses separate standards for the marker and reference genes. Inefficient reverse transcription or inequalities in the molar concentration of these standards can lead to non-comparable data. [82]
Solution:
- Adopt Absolute Quantification: Use a Single Standard for Marker and Reference genes (SSMR). This is a single DNA molecule containing the amplicon sequences for all target and reference genes ligated together. [82]
- Implementation: The SSMR is serially diluted to create a standard curve. This method directly quantifies the absolute number of mRNA molecules for both target and reference genes from the same standard, ensuring data are independent of variations in cDNA sample concentration and are directly comparable across different labs. [82]
The table below summarizes target values and experimental outcomes for key metrics in orthogonal system optimization.
Table 1: Target Benchmarks for Key Performance Metrics
| Metric | Definition | Calculation Method | Target / Example Value |
|---|---|---|---|
| Dynamic Range | Ratio of induced (ON) to basal (OFF) output. | ( \text{Dynamic Range} = \frac{[\text{Output}]{\text{ON}}}{[\text{Output}]{\text{OFF}}} ) | 33.6-fold (for a membrane-docked TF system after optimization) [81] |
| Leakiness | Unwanted basal output of a system in the "OFF" state. | Measured as reporter output (e.g., SEAP, fluorescence) without inducer. | Output level comparable to background/reporter-only control (for PHx2-TF-PHx2 system) [81] |
| Orthogonality | Specificity of interaction within a system, without crosstalk to other systems. | Functional independence tested by activating one system and measuring output in others. | 10 orthogonal systems (e.g., collection of split inducible Cas13 orthologs operating without crosstalk) [83] |
Experimental Protocols
Protocol: Measuring Leakiness and Dynamic Range of an Inducible Expression System
Objective: To quantitatively assess the performance of an inducible gene switch (e.g., a synthetic receptor) in mammalian cells.
Materials:
- Plasmids encoding the inducible expression system.
- Reporter plasmid (e.g., SEAP, GFP).
- Appropriate cell line (e.g., HEK-293).
- Transfection reagent.
- Inducer molecule (e.g., rapamycin, abscisic acid).
- Equipment for reporter quantification (e.g., plate reader, qRT-PCR machine).
Method:
- Cell Seeding and Transfection: Seed cells in a multi-well plate. Co-transfect cells with a fixed amount of the receptor plasmid and the reporter plasmid. Include controls: cells with reporter plasmid only (background control) and cells with a constitutively active promoter driving the reporter (maximum expression control).
- Induction: Divide the transfected cells into two groups. Treat one group with the inducer (+Inducer) and the other with the vehicle alone (-Inducer).
- Harvest and Quantification:
- For secreted reporters (e.g., SEAP): Collect cell culture supernatant at a defined time point and measure reporter activity. [81]
- For intracellular reporters (e.g., GFP) or mRNA: Lyse cells and measure fluorescence or extract RNA for qRT-PCR analysis.
- Data Analysis:
- Leakiness = Reporter output (-Inducer) - Background control.
- Induced Output = Reporter output (+Inducer) - Background control.
- Dynamic Range = Induced Output / Leakiness.
Protocol: Orthogonal Validation via Genetic Knockout/Knockdown
Objective: To confirm antibody specificity in immunodetection (WB, IHC) by eliminating the target protein.
Materials:
- Wild-type cell line.
- Isogenic cell line with target gene knocked out (e.g., via CRISPR/Cas9) or knocked down (e.g., via siRNA).
- Antibody against the target protein.
- Standard reagents for Western Blot or Immunohistochemistry.
Method:
- Sample Preparation: Prepare protein lysates or fix cell samples from both wild-type and knockout cell lines.
- Parallel Processing: Process both samples simultaneously using the same batch of antibodies and reagents.
- Detection and Analysis:
- Expected Result for Validated Antibody: A strong signal in the wild-type sample and a absent (for KO) or significantly reduced (for KD) signal in the modified sample. [84]
- A persistent signal in the knockout sample indicates non-specific antibody binding.
Visualization of Core Concepts
Signaling Pathway of an Orthogonal Chemically Activated Receptor
Experimental Workflow for System Validation
Logic of an Orthogonal Cas13 Circuit
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Orthogonal System Development
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Chemically-Induced Dimerization (CID) Domains (e.g., FKBP/FRB, PYL1/ABI) | Protein parts that dimerize upon addition of a small molecule (e.g., rapamycin, abscisic acid). | Serve as the ligand-sensing domain in synthetic receptors to trigger downstream signaling. [81] |
| Split TEV Protease (scTEVp) | A site-specifically split protease that reconstitutes upon dimerization of its fused parts. | Acts as a signal transducer in cytoplasmic and ER-localized synthetic receptors. [81] |
| Notch1 Transmembrane Domain | A core component from a natural signaling receptor. | Used as the transmembrane and signaling core for engineering cell-surface orthogonal receptors (OCARs). [81] |
| Single Standard for Marker and Reference (SSMR) | A single DNA standard containing amplicons for multiple genes. | Enables absolute, comparable quantification of gene expression in qRT-PCR, critical for validation. [82] |
| CRISPR/Cas9 Knockout Cell Lines | Isogenic cell lines with a specific gene permanently deleted. | Used in genetic strategies to validate antibody specificity by confirming loss of signal. [84] |
Orthogonal genetic systems are engineered to function independently from the host's native cellular machinery, enabling precise control over biological functions such as gene expression without cross-reactivity or interference. This technical support center provides a comprehensive resource for researchers employing three powerful orthogonal regulators: the T7 RNA Polymerase (T7RNAP) system, Matrix Metallopeptidase-1 (MMP-1) signaling, and CRISPR-based technologies. The content is framed within the broader thesis of optimizing gene expression, focusing on practical experimental protocols, troubleshooting common issues, and providing comparative data to inform system selection. Below you will find detailed FAQs, troubleshooting guides, summarized quantitative data, and essential research reagents to support your work in synthetic biology and drug development.
The table below summarizes the core characteristics, applications, and key performance metrics of the three orthogonal regulator systems.
Table 1: Comparative Analysis of Orthogonal Regulator Systems
| Feature | T7 RNA Polymerase (T7RNAP) System | MMP-1 Signaling | CRISPR-Based Systems |
|---|---|---|---|
| Primary Function | High-level, specific transcription of target genes [85] [86] | Extracellular signaling via cleavage of PAR1 [87] | Targeted genome editing, transcriptional regulation [88] |
| Key Components | T7RNAP, T7 promoter, host machinery [85] | proMMP-1, collagen, PAR1 receptor [87] | Cas nuclease, guide RNA (gRNA), repair template (for HDR) [88] |
| Typical Hosts | E. coli (e.g., BL21(DE3), W3110), attempted in yeast [85] [86] | Human platelets, cancer cells [87] | Mammalian cells, plants, in vivo models [88] [89] |
| Orthogonality Mechanism | Specific recognition of T7 promoter sequence [86] | Cleavage of PAR1 at a novel, non-thrombin site (P...Y...) [87] | gRNA-guided DNA targeting via complementary base pairing [88] |
| Key Quantitative Metrics | Cadaverine production: Up to 36.9 g/L in engineered E. coli W3110 [85] | Cleaves PAR1 at P...Y... site; Blockade inhibits thrombosis in vivo [87] | Typical editing efficiency (NHEJ): 30-60%; HDR: typically single-digit % [88] |
| Main Advantages | High processivity and specificity; strong expression [85] [86] | Direct signaling capability; novel therapeutic target [87] | High programmability and versatility across applications [88] |
Troubleshooting FAQs and Experimental Protocols
T7 RNA Polymerase (T7RNAP) System
Q1: I am attempting to implement a T7RNAP system in a eukaryotic yeast host, but while I detect mRNA transcripts, I see no protein expression. What is the likely cause and how can I resolve this?
A: This is a well-documented challenge. The primary issue is that T7RNAP, being prokaryotic in origin, produces transcripts that lack the 5' cap structure essential for recognition by the eukaryotic translation machinery in yeast [86].
- Recommended Solutions:
- Implement Cap-Dependent Translation: Fuse the T7RNAP to capping enzymes (RNA triphosphatase, guanylyltransferase, and methyltransferase) to ensure the mRNA is properly capped co-transcriptionally [86].
- Utilize Cap-Independent Translation: Incorporate an Internal Ribosome Entry Site (IRES) from viruses like EMCV or HCV upstream of your target gene in the transcript. This allows the ribosome to bind internally, bypassing the need for a 5' cap [86].
- Ensure Proper Transcript Processing: Verify that your transcript includes necessary eukaryotic signals for nuclear export and polyadenylation to enhance stability and transport [86].
Q2: My heterologous protein is not expressing in E. coli, or is expressed only in an insoluble form. What steps should I take?
A: This is a common problem in heterologous expression. Follow this systematic troubleshooting guide [90]:
- Verify Your Construct: Sequence the entire expression cassette to confirm there are no unintended mutations or stop codons.
- Use a Sensitive Detection Method: Do not rely solely on SDS-PAGE/Coomassie staining. Use a Western blot or an activity assay to detect low expression levels.
- Check Protein Solubility: Centrifuge your cell lysate and analyze both the supernatant (soluble fraction) and the resuspended pellet (insoluble fraction).
- Modulate Expression Speed: If the protein is insoluble, slow down expression by lowering the induction temperature (e.g., to 25-30°C) or reducing the concentration of your inducer (e.g., IPTG).
- Co-Express Chaperones: Co-express plasmid sets encoding chaperone proteins (e.g., GroEL/GroES) to assist with proper protein folding.
- Use a Soluble Fusion Tag: Fuse your protein to a highly soluble tag like MBP (Maltose Binding Protein) or Trx (Thioredoxin) to improve solubility.
- Optimize Codon Usage: Check the codon adaptation index (CAI) of your gene for E. coli. Use strains like Rosetta 2 that supply tRNAs for codons rarely used in E. coli.
- Try a Different Expression System: If all else fails, consider switching to a different host system (e.g., yeast, insect cells) that may be better suited for your protein.
CRISPR-Based Systems
Q3: The editing efficiency in my cell population is low and heterogeneous. How can I improve the rate of knockout or knock-in?
A: Low and heterogeneous editing is often due to variable delivery, inefficient cleavage, or the competing DNA repair pathways.
For Knocking Out Genes (via NHEJ):
- Validate Cas9 Activity: Use a positive control, such as a GFP-reporter cell line transfected with a GFP-targeting sgRNA. A functional system should produce a high percentage of GFP-negative cells [88].
- Optimize sgRNA Design: Use validated bioinformatics tools to select sgRNAs with high predicted on-target activity and minimal off-target potential.
- Ensure Efficient Delivery: Use viral transduction (lentivirus, AAV) for hard-to-transfect cells. Verify Cas9 and sgRNA expression levels in your target cells [88].
- Allow Sufficient Time: Wait at least one week post-transfection/transduction to allow for the accumulation of edits in the cell population [88].
For Knocking In Genes (via HDR):
- Synchronize Cell Cycle: Favor HDR by using cell cycle inhibitors to arrest cells in S/G2 phase, when HDR is most active [88].
- Inhibit NHEJ: Use small molecule inhibitors (e.g., Scr7) of key NHEJ pathway components to skew repair toward HDR [88].
- Optimize Template Design: Provide a single-stranded or double-stranded DNA repair template with sufficient homology arms (typically >500 bp).
- Isolate Clones: HDR efficiency is typically low (single-digit percentages). You must isolate and expand single-cell clones, then genotype them to identify those with the desired edit [88].
Q4: How do I design a high-quality sgRNA for a CRISPR experiment?
A: Careful sgRNA design is critical for success. Key considerations include [88]:
- Target Sequence: The ~20 base sgRNA sequence must be complementary to your genomic target and located immediately 5' to a Protospacer Adjacent Motif (PAM). For the common S. pyogenes Cas9 (SpCas9), the PAM is 5'-NGG-3'.
- On-Target Efficiency: Use established algorithms (e.g., from the Broad Institute or MIT) to predict and select sgRNAs with high on-target activity scores.
- Off-Target Specificity: The tool should scan the genome for sequences with high similarity, especially those with mismatches in the "seed" region near the PAM. Choose sgRNAs with minimal predicted off-target sites.
- Genomic Context: Ensure the target site is within an exon for gene knockouts and is accessible (not in tightly packed heterochromatin).
MMP-1 Signaling
Q5: How can I experimentally demonstrate that MMP-1 is cleaving and activating PAR1 in my cellular model?
A: To conclusively demonstrate this specific protease-receptor interaction, employ the following methodological approach [87]:
- Cleavage Assay: Treat cells or purified PAR1 with active MMP-1. Use Western blotting with an antibody against the N-terminal extracellular domain of PAR1 to detect a cleavage-induced shift in molecular weight.
- Site-Specific Validation: Utilize mass spectrometry to analyze the cleaved PAR1 N-terminal fragment. This will confirm the exact cleavage site, which for MMP-1 is between P...Y..., distinct from thrombin's LDPR41↓S42 site [87].
- Functional Signaling Assay: Measure downstream signaling events after MMP-1 treatment. Specifically, look for activation of Rho-GTPase pathways and MAPK signaling, which are characteristic of MMP-1-PAR1 signaling, rather than the canonical Gq-coupled pathways strongly activated by thrombin [87].
- Inhibition Controls: Use specific MMP-1 inhibitors (e.g., FN-439) or blocking antibodies. These should abrogate PAR1 cleavage and the subsequent signaling events, confirming the role of MMP-1 [87].
Essential Research Reagents and Materials
The table below lists key reagents and resources essential for working with these orthogonal systems.
Table 2: Research Reagent Solutions for Orthogonal Regulator Experiments
| Reagent / Resource | System | Function and Application Notes |
|---|---|---|
| CRIM Plasmid System [85] | T7RNAP | Enables stable, site-specific integration of genetic elements (e.g., T7RNAP gene) into the host chromosome using phage attachment (attP/attB) sites and integrase helper plasmids. |
| lentiCRISPRv2 Vector [88] | CRISPR | An all-in-one lentiviral vector for delivery of both SpCas9 and your sgRNA of interest into a wide range of cell types, including primary and hard-to-transfect cells. |
| FN-439 Inhibitor [87] | MMP-1 | A broad-spectrum, peptide metalloprotease inhibitor used to specifically block MMP-1 collagenase activity in experimental settings. |
| Chaperone Plasmid Sets (e.g., Takara) [90] | T7RNAP | A set of plasmids for co-expressing various chaperone proteins (e.g., GroEL/GroES, DnaK/DnaJ/GrpE) to improve the solubility and proper folding of recombinant proteins in E. coli. |
| Rosetta / Origami E. coli Strains [90] | T7RNAP | Engineered host strains: Rosetta supplies tRNAs for rare codons, enhancing translation of genes with non-optimal codon usage. Origami promotes disulfide bond formation in the cytoplasm, aiding soluble expression of proteins requiring correct disulfide bridging. |
| PAR1 N-terminal Antibody [87] | MMP-1 | A monoclonal antibody targeting the thrombin-cleavage peptide region (residues 32-46) of PAR1. Used to detect receptor cleavage and the release of the N-terminal peptide upon MMP-1 activation. |
Signaling Pathways and Experimental Workflows
MMP-1 Mediated Activation of PAR1 in Platelets
This diagram illustrates the signaling pathway by which collagen exposure leads to platelet activation through MMP-1 and PAR1.
CRISPR-Cas9 Gene Editing Workflow
This diagram outlines the key experimental steps for performing a CRISPR-Cas9 gene editing experiment, from design to validation.
T7RNAP System Implementation in Eukaryotic Yeast
This diagram visualizes the core challenge and potential solutions for implementing a functional T7RNAP system in yeast.
Troubleshooting Guides
Why am I detecting unexpected phenotypic changes or genetic mutations in my edited cells?
Problem: Following a CRISPR-based intervention using orthogonal regulators, your cell lines or model organisms exhibit unexpected growth defects, metabolic changes, or transcriptional profiles that don't align with your intended genetic modification.
Solution:
- Perform comprehensive off-target prediction using multiple computational tools before experimentation. The limited specificity of RNA-guided nucleases means Cas9 can tolerate multiple mismatches between the gRNA and target DNA, particularly in the PAM-distal region [24] [73]. Use both alignment-based (Cas-OFFinder, FlashFry) and scoring-based (CFD, DeepCRISPR) algorithms to identify potential off-target sites [73] [91].
Validate with empirical detection methods. Computational prediction alone has limitations as it primarily identifies sgRNA-dependent off-target effects and may miss sites influenced by chromatin context or epigenetic factors [73]. Employ methods like:
Implement high-fidelity Cas variants such as HypaCas9, eSpCas9, SpCas9-HF1, or evoCas9, which have been engineered for enhanced specificity through reduced tolerance for gRNA-DNA mismatches [74] [91].
Table 1: Comparison of Off-Target Detection Methods
| Method | Type | Key Features | Limitations | Best Use Cases |
|---|---|---|---|---|
| Cas-OFFinder | Computational/Alignment | Adjustable sgRNA length, PAM types, mismatch/bulge tolerance [73] | Does not consider chromatin accessibility or epigenetic factors [73] | Initial sgRNA screening and design phase |
| FlashFry | Computational/Alignment | High-throughput analysis of thousands of targets; provides GC content data [73] [91] | Alignment-based limitations apply | Large-scale sgRNA library design |
| CFD Scoring | Computational/Scoring | Uses experimentally validated dataset for predictions [73] [91] | Limited to sgRNA-dependent effects | Refining sgRNA selection from initial candidates |
| GUIDE-seq | Empirical/Cell-based | Highly sensitive, low false positive rate, cost-effective [73] | Limited by transfection efficiency [73] | Comprehensive off-target profiling in cell cultures |
| CIRCLE-seq | Empirical/Cell-free | Circularized DNA library with Cas9 cleavage; no reference genome needed [73] | Lower validation rate; does not account for cellular context [73] | Unbiased off-target identification without cellular constraints |
| WGS | Empirical/Cell-based | Comprehensive analysis of entire genome [74] [73] | Expensive; requires high sequencing coverage [74] [73] | Critical applications like clinical trial preparation |
My orthogonal regulator system shows inconsistent repression/activation across different cell types – what could be wrong?
Problem: Your engineered orthogonal transcription factors (e.g., dCas9-based regulators, zinc finger proteins, or TALEs) demonstrate variable efficiency in different cellular environments or when targeting different genomic loci.
Solution:
- Evaluate chromatin accessibility and epigenetic context. The same DNA target sequence can have different accessibility depending on cell type, nuclear organization, and epigenetic modifications [73] [26]. Perform:
- ATAC-seq to assess regional chromatin openness
- ChIP-seq for histone modification markers
- Cas9 ChIP-seq using catalytically inactive dCas9 to map binding sites genome-wide [73]
Optimize delivery mechanisms to reduce variable expression of regulator components. Consider:
Employ position-independent strategies by incorporating chromatin insulators or utilizing landing pad approaches with validated genomic safe harbors to minimize position effect variegation [89].
How can I distinguish true off-target effects from background genetic variation or experimental noise?
Problem: When analyzing next-generation sequencing data from your gene editing experiments, you're uncertain whether detected variants represent genuine off-target effects or artifacts.
Solution:
- Implement appropriate controls:
- Include non-treated controls from the same cell population
- Use non-targeting gRNAs as negative controls, though note these have limitations as different gRNAs have distinct off-target profiles [74]
- Employ multiple clonal isolates when working with cell lines to distinguish true off-target events from clonal variations [74]
Apply rigorous statistical thresholds and validation:
- Use independent validation methods like Sanger sequencing for putative off-target sites
- Implement biological replicates to distinguish consistent off-target effects from random noise
- Consider molecular validation through in vitro cleavage assays for high-priority candidates [73]
Utilize orthogonal detection methods to confirm findings:
Table 2: Research Reagent Solutions for Specificity Assessment
| Reagent/Category | Specific Examples | Function & Application | Key Considerations |
|---|---|---|---|
| High-Fidelity Cas Variants | eSpCas9, SpCas9-HF1, HypaCas9, evoCas9 [74] | Engineered for reduced mismatch tolerance; decreases off-target editing while maintaining on-target activity | Different variants may have varying efficiency trade-offs; test multiple options |
| Computational Prediction Tools | Cas-OFFinder, CCTop, CRISPOR, DeepCRISPR [74] [73] [91] | Algorithmic nomination of potential off-target sites during guide RNA design phase | Use multiple tools with different algorithms for comprehensive coverage |
| Empirical Detection Kits | GUIDE-seq, CIRCLE-seq, SITE-seq kits [73] | Experimental validation of off-target sites through next-generation sequencing | Varying sensitivity/specificity trade-offs; choose based on experimental needs |
| Delivery Modalities | RNP complexes, IDLV, AAV vectors [73] [91] | Influences duration and concentration of editor exposure; affects off-target rates | RNP delivery generally shows faster clearance and reduced off-target effects |
| Control Materials | Non-targeting gRNAs, Wild-type controls [74] | Essential for distinguishing true off-target effects from background variation | Non-targeting gRNAs control for transfection but not off-target specificity |
Frequently Asked Questions (FAQs)
What are the most critical factors in gRNA design to minimize off-target effects?
Optimal gRNA design incorporates multiple factors:
- Low sequence similarity to other genomic regions, particularly in the seed region (PAM-proximal 10-12 nucleotides) [24] [73]
- Moderate GC content (40-60%) as extremely high or low GC can affect specificity [91]
- Minimizing potential self-complementarity that could affect gRNA structure and function
- Using truncated gRNAs (17-18 nt instead of 20 nt) which can reduce off-target effects while sometimes maintaining on-target efficiency [91]
- Leveraging dual gRNA approaches with Cas9 nickases, which require two adjacent off-target events for double-strand breaks, dramatically reducing off-target mutation rates [74]
How do I choose between the various off-target detection methods for my specific experiment?
Selection depends on your experimental goals and resources:
- For preliminary sgRNA screening: Start with computational tools like Cas-OFFinder or FlashFry for cost-effective initial assessment [73] [91]
- For therapeutic development: Employ multiple orthogonal methods including GUIDE-seq or CIRCLE-seq followed by targeted validation of identified sites [73]
- For clinical trial applications: Implement WGS on multiple clonal isolates when possible, despite higher costs, for the most comprehensive assessment [74] [73]
- For high-throughput screens: Utilize computational prediction with selective empirical validation of highest-risk candidates to balance comprehensiveness with practical constraints [91]
What are the key trade-offs between specificity and efficiency in orthogonal regulator systems?
The balance between specificity and efficiency involves several considerations:
- High-fidelity Cas variants typically show reduced off-target effects but may have slightly decreased on-target efficiency in some contexts [74] [91]
- RNP delivery offers reduced off-target rates compared to plasmid-based delivery but can be less efficient for some cell types [91]
- Truncated gRNAs improve specificity but may require testing multiple designs to maintain sufficient on-target activity [91]
- Dual nickase systems dramatically improve specificity but require two functional gRNAs and careful spacing optimization [74]
- Promoter strength for gRNA expression affects both on-target efficiency and off-target rates, with moderate expression often providing the best balance [24]
How do I assess off-target effects for CRISPR-based transcriptional regulators (dCas9) that don't cut DNA?
For catalytically impaired dCas9 systems used in transcriptional regulation:
- Focus on binding-level assessment rather than cleavage detection:
- Consider that high-fidelity mutations developed for nuclease-active Cas9 may not reduce binding-level off-target effects, as they primarily prevent cleavage at mismatched sites [74]
- Evaluate functional consequences through comprehensive transcriptome analysis rather than relying solely on binding data [26]
Phylogenetic Expression Profiling (PEP) is an advanced computational method that identifies functionally related genes by analyzing the coordinated evolution of their expression levels across a wide phylogenetic range of species. Unlike traditional phylogenetic profiling, which relies on gene presence/absence patterns, PEP utilizes quantitative gene expression data to uncover functional linkages, making it particularly valuable for studying essential genes that are rarely lost during evolution [93].
Within research focused on optimizing gene expression using orthogonal regulators, PEP serves as a powerful in silico validation tool. It can predict whether genes potentially regulated by your orthogonal system are part of the same functional pathway, complex, or biological process based on their shared evolutionary history. This guide provides troubleshooting and methodological support for implementing PEP in your research workflow.
Key Research Reagent Solutions
The following table outlines essential computational "reagents" and resources required for a successful PEP analysis.
Table 1: Essential Research Reagents and Resources for PEP
| Item | Function in PEP Analysis | Examples & Notes |
|---|---|---|
| Multi-Species Expression Data | Provides the foundational matrix of gene expression values across different organisms. | Marine Microbial Eukaryotic Transcriptome Project (MMETSP) database [93]; Ancestral transcriptome reconstructions [94]. |
| Ortholog Identification Tool | Maps genes across different species to their corresponding evolutionary counterparts. | BLAST-based approaches; Custom pipelines for transcriptome assemblies [93]. |
| Normalization Pipeline | Standardizes expression data from different species/experiments to enable valid cross-species comparison. | R-value normalization using self-alignment scores [95]; Transformation of TPM to binary states [94]. |
| Profile Similarity Metric | Quantifies the co-evolutionary relationship between the expression profiles of two or more genes. | Spearman correlation; Inner product of expression vectors [93] [95]. |
| Statistical Testing Framework | Distinguishes significant coordinated evolution from background correlations caused by phylogenetic relatedness. | Permutation-based tests that account for phylogenetic structure [93]. |
Standard Experimental Protocol and Workflow
Implementing a robust PEP analysis involves a defined sequence of steps. The diagram below outlines the core workflow.
Detailed Methodologies for Key Steps:
Step 1: Dataset Selection and Ortholog Identification
- Reference Collection: Compile a gene expression dataset from a wide range of species. The Marine Microbial Eukaryotic Transcriptome Project (MMETSP), for example, provides 657 RNA-seq profiles from 309 diverse eukaryotic microbes [93]. For studies on specific traits, you may select species that represent key evolutionary transitions [94].
- Ortholog Mapping: This is a critical step. For genomes or transcriptomes without pre-computed ortholog groups, a two-step approach is effective:
- Match each transcript to a cluster of related proteins (e.g., UniProt100 IDs) using BLASTP.
- Iteratively merge redundant hits. If a transcript's top match is uninformative, proceed to the next best match. Sum read counts for multiple matches to the same UniProt ID within a sample to represent total expression of that gene family [93].
Step 2: Constructing the Unified Expression Matrix
- Generate a single matrix where rows represent orthologous genes and columns represent samples from different species.
- Include only genes with detectable expression in a sufficient number of samples (e.g., >100 samples in a large dataset) to ensure robust analysis [93].
Step 3: Data Normalization and Encoding
- Normalization: Normalize raw expression values (e.g., TPM - Transcripts Per Million) to account for technical variations. One method transforms the best BLAST bit score (S-value) into a normalized R-value:
R_ab = S_ab / S_aa, whereS_abis the bit score of geneain organismb, andS_aais the score of geneaaligned to itself. Values below 50 are typically trimmed to zero [95]. - Binary Encoding (Optional): To reduce noise, continuous expression values (TPM) can be converted to binary states:
1for TPM ≥ 2.0 (expressed) and0for TPM < 2.0 (not expressed). This can significantly enhance phylogenetic signal [94].
Step 4: Measuring Profile Similarity and Statistical Testing
- Similarity Calculation: Compute all pairwise similarities between gene profiles. The Spearman correlation is commonly used for this purpose [93].
- Statistical Validation: To correct for phylogenetic inertia (where closely related species share similar expression patterns by default), use a permutation-based null model. Select random sets of genes and calculate their background correlation distribution. A gene set is deemed significant if its observed correlation exceeds this expected distribution (e.g., FDR < 5%) [93].
Frequently Asked Questions (FAQs)
Q1: My PEP analysis identified a gene of unknown function as being co-evolved with a known pathway. How confident can I be in this functional prediction? The prediction is a strong hypothesis, not a confirmation. Confidence increases if: a) the correlation is statistically significant after phylogenetic correction, b) the gene set is a known functional module (e.g., the proteasome or ribosome), and c) the result is consistent with other evidence, such as protein-protein interaction data. You should use PEP as a prioritization tool for downstream experimental validation, such as testing the gene's role using your orthogonal regulator system [93] [96].
Q2: Why does PEP sometimes perform poorly for eukaryotic genes compared to prokaryotic genes? Traditional phylogenetic profiling (based on presence/absence) struggles with eukaryotes due to the historically smaller number of sequenced genomes and greater genomic complexity. PEP, which uses expression levels, is better suited. However, challenges remain, such as accurately identifying orthologs across large evolutionary distances and the quality of gene annotations, especially for plant-specific secondary metabolism pathways [95] [96]. Ensuring a high-quality, phylogenetically broad reference collection is key to success.
Q3: What is the minimum number of species required for a reliable PEP analysis? There is no universally agreed-upon minimum, but the power of PEP increases with the number and phylogenetic diversity of the species in your dataset. Studies have successfully used datasets ranging from dozens to hundreds of species [93] [94]. The critical factor is to have sufficient independent data points (species) to detect a significant correlation signal over phylogenetic noise. It is more important to avoid taxonomic redundancy than to maximize species count with very closely related strains.
Q4: How does "Partial Phylogenetic Profiling" (PPP) improve the method? Partial Phylogenetic Profiling (PPP) is a heuristic that optimates the analysis without relying on pre-defined, fixed protein family boundaries. It works by examining an ordered list of BLAST hits from various genomes against a query profile, choosing an optimal cut-off that maximizes the statistical significance of the match. This is especially useful for analyzing protein families whose members have diverged in sequence but may retain functional coherence [97] [98].
Troubleshooting Guide
Table 2: Common PEP Issues and Solutions
| Problem | Potential Cause | Solution |
|---|---|---|
| Weak or non-significant co-evolution signals | 1. Reference dataset has low phylogenetic diversity.2. High noise in expression data.3. Incorrect ortholog assignment. | 1. Expand species selection to cover broader evolutionary range.2. Apply binary encoding to expression values to reduce noise [94].3. Manually curate ortholog groups or use more stringent clustering thresholds. |
| Correlations are inflated by phylogenetic relatedness | Failure to account for the shared evolutionary history of species, which causes non-functional correlation. | Implement a permutation-based test that randomizes gene assignments while preserving the species relationship structure to create a valid null model [93]. |
| Too few orthologs detected for my gene of interest | Gene is rapidly evolving, or the reference dataset lacks relevant species. | 1. Use a less stringent orthology detection method (e.g., profile HMMs).2. Incorporate single-gene transcriptomes or expand the reference dataset to include more closely related species. |
| PEP results conflict with within-species co-expression data | PEP and within-species co-expression capture different biological phenomena. | This is expected. PEP reveals deep evolutionary constraints, while within-species co-expression reflects condition-specific regulation. The two methods are largely orthogonal and can reveal different aspects of gene function [93]. |
Advanced Data Analysis and Visualization
For advanced users, the following diagram illustrates the core logic of how PEP infers functional linkages, which is key to interpreting results.
Interpreting PEP Results in the Context of Orthogonal Regulators:
When you identify a set of genes that show coordinated evolution, you have uncovered a group of genes that have been under shared evolutionary pressure. In the context of your thesis, if you are designing an orthogonal regulator to control one gene in this set, it is plausible that the regulator might impact or be impacted by the other members of the functionally linked module. This insight can guide the design of more sophisticated multi-gene regulatory systems or help anticipate potential off-pathway effects in your engineered system.
Comparative Performance of Expression Systems
Table 1: Key Characteristics of Recombinant Protein Expression Systems
| Parameter | Bacterial (E. coli) | Mammalian (CHO/HEK293) | Plant-Based Systems |
|---|---|---|---|
| Typical Yields | High (grams/L) [99] | Lower than bacteria; HEK293: up to 140 mg/L for scFv [100]; CHO: can be high with engineering [100] | Varies; up to 10x higher for mutant proteins (e.g., hGAD65mut) [101] |
| Process Speed | Very Fast (hours to days) [99] [102] | Slow (weeks); Transient: 3-7 days; Stable: weeks/months [99] | Fast transient expression in plants (e.g., days) [101] |
| Cost & Scalability | Low cost, highly scalable [99] [103] | High cost, scalable but challenging/expensive [99] [103] | Cost-effective, simple media, good scalability [101] [104] |
| Key Advantage | Simplicity, speed, high yield for simple proteins [99] [102] | Complex Post-Translational Modifications (PTMs), proper folding of human proteins [99] [102] | Cost-effective, functional PTMs, low risk of human pathogens [101] [104] |
| Major Limitation | Lack of complex PTMs, inclusion body formation [99] [102] | High cost, complex media, longer timelines [99] [103] | Different glycosylation patterns (plant glycans) [104] |
Table 2: Protein Folding, Modifications, and Typical Applications
| Parameter | Bacterial (E. coli) | Mammalian (CHO/HEK293) | Plant-Based Systems |
|---|---|---|---|
| Glycosylation | Cannot perform complex glycosylation [102] | Can add complex, human-like glycans [99] [102] | Adds glycans, but with plant-specific patterns (e.g., α(1,3)-fucose, β(1,2)-xylose) [104] |
| Disulfide Bond Formation | Possible, but may be incorrect; strains available to help [99] | Excellent, occurs in endoplasmic reticulum [102] | Can perform disulfide bond formation [104] |
| Protein Folding & Solubility | Prone to misfolding and inclusion bodies, especially for complex proteins [99] [102] | Chaperones and organelles assist proper folding [102] | Can produce properly folded, active complex human proteins [104] |
| Typical Applications | Research-grade proteins, industrial enzymes, simpler therapeutics (e.g., insulin) [99] [102] | Therapeutic antibodies, complex glycoproteins, multi-subunit proteins [99] [103] | Enzymes (e.g., Taliglucerase alfa), vaccines, non-glycosylated antibody fragments [101] [104] |
Troubleshooting Guides & FAQs
Frequently Asked Questions
Q1: My target protein is accumulating in inclusion bodies in E. coli. What strategies can I use to improve soluble expression?
- Lower Induction Temperature: Shifting the culture temperature to 18-25°C after induction can slow down protein synthesis, allowing more time for proper folding [105] [99].
- Use of Fusion Tags & Chaperones: Co-express molecular chaperones (e.g., GroEL/GroES) or fuse the target protein to solubility-enhancing tags (e.g., MBP, Trx) [99].
- Promoter and Strain Engineering: Employ cold-shock promoters (e.g., cspA) to control expression at lower temperatures or use engineered E. coli strains designed to enhance disulfide bond formation in the cytoplasm [105] [99].
Q2: I am getting low transient transfection yields in HEK293 cells. What factors should I optimize?
- Cell Line Modification: Use engineered variants like Expi293F or HEK293-6E, which are adapted for high-density suspension culture and high-level protein expression [100] [99].
- Expression Vector Engineering: Implement a dual-promoter system for multi-subunit proteins (e.g., antibodies) and incorporate cis-acting regulatory elements like UCOE (ubiquitous chromatin opening elements) to enhance transcriptional activity and stability [100].
- Culture Process Optimization: Use high-quality, serum-free media and optimize the cell density at the time of transfection (e.g., 2-3 x 10^6 cells/mL for Expi293F) [100] [99].
Q3: How can I humanize glycosylation patterns on a therapeutic protein produced in plant cells?
- Glyco-Engineering: Use CRISPR/Cas9 or other gene-editing tools to knock out genes responsible for adding plant-specific sugar residues, such as β(1,2)-xylosyltransferase and α(1,3)-fucosyltransferase [104].
- Subcellular Targeting: Target the protein to a specific cellular compartment where desired glycosylation patterns can be achieved.
- Clinical Experience: Note that for some approved products (e.g., Elelyso), the plant-derived glycans have not raised significant clinical concerns, potentially reducing the necessity for extensive engineering [104].
Q4: When should I choose a stable mammalian cell line over transient expression?
- Choose Transient Transfection when you need milligram to gram quantities of protein rapidly (within 3-7 days) for research or early-stage clinical trials [99].
- Choose Stable Transfection when you require sustained, high-volume, and predictable protein expression for commercial manufacturing (e.g., monoclonal antibody production). Be prepared for a time-intensive process (weeks to months) to develop and characterize the clonal cell line [99].
Advanced Troubleshooting: Orthogonal Regulator Systems
Q5: The basal (leaky) expression from my inducible mammalian promoter is too high. How can I achieve tighter regulation?
- Repressor Configuration: Use a system like the cumate gene-switch in its repressor configuration. The CymR repressor binds to the operator (CuO) site downstream of the promoter and strongly represses transcription. Adding cumate relieves this repression, providing low background and high induction [106].
- Combined Systems: For multi-gene applications, combine orthogonal systems (e.g., Tet-ON/OFF, cumate, streptogramin) to avoid crosstalk and achieve independent, tight control of different genes [106].
Q6: How can I co-express a protein of interest alongside a folding helper gene in a regulated manner?
- Dual-Vector Transactivation System: Use a two-vector system where both genes are under the control of the same inducible promoter (e.g., a cumate-dependent transactivator). Transfect both vectors simultaneously to ensure coordinated induction of both the target gene and the helper gene (e.g., chaperone, foldase) upon adding the inducer [100] [106].
Detailed Experimental Protocols
Protocol 1: Transient Transfection in HEK293 Cells for Recombinant Antibody Production
This protocol is adapted for high-yield, research-scale production using suspension-adapted HEK293 cells [100] [99].
Workflow: Transient Transfection in HEK293 Cells
Key Research Reagent Solutions:
- Cell Line: HEK293F or Expi293F, adapted to serum-free suspension culture [100] [99].
- Expression Vector: Vector with strong promoter (e.g., hCMV), dual-promoter for antibody heavy and light chains, and elements like a polyadenylation signal [100].
- Transfection Reagent: Linear polyethylenimine (PEI) is commonly used due to its high efficiency and cost-effectiveness [100].
- Culture Medium: Chemically defined, serum-free medium optimized for high-density growth [100].
Step-by-Step Methodology:
- Cell Culture: Maintain HEK293F cells in an appropriate serum-free medium. On the day before transfection (Day 0), seed cells at a density of 0.5 - 1.0 x 10^6 cells/mL in a shake flask [99].
- Transfection Complex (Day 1): When cell density reaches 2-3 x 10^6 cells/mL and viability is >95%, prepare the transfection mix. For 1 L of culture, combine your purified expression vector(s) (e.g., 1 mg total DNA) with PEI (e.g., 3 mg) in a separate tube of sterile, pre-warmed medium. Incubate at room temperature for 10-20 minutes to allow complex formation.
- Transfection: Add the DNA-PEI complex dropwise to the cell culture with gentle agitation.
- Process Enhancement: 24 hours post-transfection, add enhancers such as valproic acid or sodium butyrate to boost protein production [100].
- Harvest: Culture the cells for typically 3-7 days. Centrifuge the culture (e.g., 4,000 x g for 20 min) to separate the cells from the supernatant, which contains your secreted recombinant antibody [101].
Protocol 2: Expression and Purification of Plant Proteins in E. coli
This protocol focuses on expressing mature plant enzymes, which often requires removing organelle-targeting sequences [107] [108].
Workflow: Express Plant Protein in E. coli
Key Research Reagent Solutions:
- Bioinformatics Tool: Use TargetP or similar software for transit peptide prediction [107].
- Expression Vector: A standard prokaryotic vector like pET, which often includes an in-frame His-tag for purification (e.g., pET-28a) [107].
- Expression Strain: E. coli BL21(DE3) is the gold standard; use derivatives like Rosetta for proteins with rare codons [107] [99].
- Purification Resin: Immobilized metal affinity chromatography (IMAC) resin, such as Ni-NTA Agarose, for purifying His-tagged proteins [107].
Step-by-Step Methodology:
- Construct Design:
- Identify the coding sequence for your plant protein (e.g., Arabidopsis NAXD).
- Use prediction tools (e.g., TargetP) to identify the N-terminal chloroplast or mitochondrial transit peptide [107].
- Design a construct that encodes only the mature protein sequence (without the transit peptide) to facilitate functional expression in the bacterial cytoplasm [107].
- Cloning: Clone the optimized mature sequence into an E. coli expression vector that provides an N- or C-terminal affinity tag (e.g., 6xHis-tag) for simplified purification [107].
- Expression Testing: Transform the plasmid into a suitable E. coli strain (e.g., BL21(DE3)). Test small-scale expression cultures (5-10 mL) by inducing with IPTG (e.g., 0.1 - 1.0 mM) at different temperatures (e.g., 16°C, 25°C, 37°C) and for different durations (e.g., 4-16 hours). Analyze the solubility of the expressed protein by SDS-PAGE [107].
- Large-Scale Expression and Purification:
- Inoculate a large culture (e.g., 1 L) and grow to mid-log phase (OD600 ~0.6-0.8). Induce with IPTG under the optimal conditions determined in the test scale.
- Harvest cells by centrifugation (e.g., 4,000 x g for 20 min) [101].
- Resuspend the cell pellet in lysis buffer and lyse using sonication or a homogenizer.
- Clarify the lysate by centrifugation and apply the supernatant to an appropriate affinity chromatography column (e.g., Ni-NTA for His-tagged proteins) to purify the recombinant protein [107].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Expression System Optimization
| Reagent / Solution | Function | Example(s) |
|---|---|---|
| Inducible Promoter Systems | Provides temporal control over gene expression, crucial for toxic proteins and optimizing yield. | Tetracycline (Tet)-ON/OFF, Cumate gene-switch [106], Cold-shock (cspA) promoters [105] |
| Cis-Regulatory Elements | Enhances transcriptional activity and stabilizes expression by preventing epigenetic silencing. | Ubiquitous Chromatin Opening Elements (UCOE), Matrix Attachment Regions (MAR) [100] |
| Engineered Cell Lines | Host cells modified for improved protein titer, specific PTMs, or growth characteristics. | CHO cells with Bax/Bak knockout (reduced apoptosis) [100], Glyco-engineered P. pastoris (humanized glycosylation) [99] |
| Affinity Tags | Facilitates purification and detection of the recombinant protein. | Polyhistidine (His-tag), Glutathione-S-transferase (GST) tag [107] |
| Transfection Reagents | Enables delivery of foreign nucleic acids into mammalian cells. | Linear polyethylenimine (PEI), Liposomes [100] |
Conclusion
The development and refinement of orthogonal gene expression systems provide an unprecedented capacity to probe biological complexity and engineer novel cellular functions. By enabling precise, independent, and predictable control over multiple genetic elements, these tools are revolutionizing basic research into multigene contributions to phenotype and accelerating applied biotechnology. Key takeaways include the universal applicability of core design principles—such as negative feedback for linear control and CRISPR programmability for flexibility—across diverse organisms. Future directions will focus on increasing the multiplexing capacity of these systems, enhancing their orthogonality and integration in vivo for therapeutic applications, and leveraging machine learning for de novo design of regulatory parts. The continued evolution of these toolkits promises to unlock new frontiers in drug development, gene and cell therapies, and sustainable bioproduction, ultimately bridging the gap between sophisticated genetic programming and clinical impact.
References
- 1. Circuits Within an Synthetic Central Dogma... Biological Orthogonal [https://pmc.ncbi.nlm.nih.gov/articles/PMC7746572/]
- 2. Conversion of natural cytokine receptors into orthogonal ... synthetic [https://www.nature.com/articles/s41589-025-01986-1?error=cookies_not_supported&code=2ad76e5a-a8ee-4bda-9891-6685b01a774e]
- 3. Frontiers | Genetic circuits in synthetic biology: broadening the toolbox... [https://www.frontiersin.org/journals/synthetic-biology/articles/10.3389/fsybi.2025.1548572/full]
- 4. devices for gene regulation in Penicillium... Synthetic control [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-019-1253-3]
- 5. Transcription Amplifier System for Synthetic of... Orthogonal Control [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0148320]
- 6. control of Orthogonal in plants using synthetic... gene expression [https://plantmethods.biomedcentral.com/articles/10.1186/s13007-022-00867-1]
- 7. Design of orthogonal systems for modulating regulatory ... gene [https://www.nature.com/articles/s41589-020-0547-4?error=cookies_not_supported&code=ce4ca9b0-7c32-485b-b4f0-937ead4e60a6]
- 8. in Human Cells Using Chemically... Orthogonal Genetic Regulation [https://pubmed.ncbi.nlm.nih.gov/28054767/]
- 9. Photo-controllable multiple and orthogonal of regulation ... gene [https://link.springer.com/article/10.1007/s11426-024-2285-7]
- 10. Troubleshooting Recombinant Protein Expression | VectorBuilder [https://en.vectorbuilder.com/resources/faq/failed-bacterial-vector.html]
- 11. Bacteria Expression Support—Troubleshooting | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/protein-expression-support-center/bacterial-expression-support/bacterial-expression-support-troubleshooting.html]
- 12. Limit cycles in models of circular gene networks regulated by negative... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-020-03598-z]
- 13. Design of orthogonal systems for modulating regulatory ... gene [https://www.nature.com/articles/s41589-020-0547-4?error=cookies_not_supported&code=21ac2fdf-a268-485b-b5e6-cf7a96f2b4df]
- 14. Protein Expression Basics Support—Troubleshooting | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/protein-expression-support-center/expression-basics-support/expression-basics-support-troubleshooting.html]
- 15. control of Orthogonal mean and variance by epigenetic... expression [https://pubmed.ncbi.nlm.nih.gov/25943345/]
- 16. Orthogonal control of mean and variability of endogenous genes in a human cell line | Nature Communications [https://www.nature.com/articles/s41467-020-20467-8]
- 17. An Interdisciplinary, Data-Driven Approach to Re-Engineering... [https://www.benchling.com/blog/an-interdisciplinary-data-driven-approach-to-re-engineering-orthogonal-riboswitches-for-enhanced-function]
- 18. control of Orthogonal in plants using synthetic... gene expression [https://pmc.ncbi.nlm.nih.gov/articles/PMC8966344/]
- 19. Frontiers | Synthetic Promoters and Transcription for... Factors [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2017.00063/full]
- 20. Engineering orthogonal dual transcription factors for multi-input synthetic promoters | Nature Communications [https://www.nature.com/articles/ncomms13858]
- 21. An orthogonal mutation... | Nature Communications transcription [https://www.nature.com/articles/s41467-025-61354-4?error=cookies_not_supported&code=cbb5aacb-3037-4f27-a0fa-ecd1e39c4134]
- 22. Synthetic chemical inducers and genetic decoupling enable... [https://pure.au.dk/portal/en/publications/synthetic-chemical-inducers-and-genetic-decoupling-enable-orthogo]
- 23. Engineering orthogonal dual transcription factors for multi-input synthetic promoters - PubMed [https://pubmed.ncbi.nlm.nih.gov/27982027/]
- 24. modular Orthogonal repression in E. coli using engineered... gene [https://pmc.ncbi.nlm.nih.gov/articles/PMC5462464/]
- 25. CRISPR/dCas9-Based Systems: Mechanisms and Applications in Plant Sciences - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8540305/]
- 26. Complex transcriptional modulation with orthogonal and inducible... [https://www.nature.com/articles/nmeth.4042?error=cookies_not_supported]
- 27. Multiplexed activation of endogenous genes by CRISPR -on, an... [https://www.nature.com/articles/cr2013122?error=cookies_not_supported&code=57640fd0-2aff-44fe-bd67-6913fd958b08]
- 28. -scale transcriptional Genome by an engineered... activation [https://pmc.ncbi.nlm.nih.gov/articles/PMC4420636/]
- 29. Spatiotemporal control of CRISPR/Cas9 gene editing | Signal Transduction and Targeted Therapy [https://www.nature.com/articles/s41392-021-00645-w]
- 30. by a Gene -assisted trans enhancer | eLife activation CRISPR [https://elifesciences.org/articles/45973]
- 31. CRISPR/dCas9 Tools: Epigenetic Mechanism and Application in Gene Transcriptional Regulation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10573330/]
- 32. Designing artificial synthetic promoters for accurate, smart, and versatile gene expression in plants - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10363483/]
- 33. Frontiers | A Fruitful Decade Using Synthetic Promoters in the ... [https://www.frontiersin.org/articles/10.3389/fpls.2019.01433/full]
- 34. Frontiers | Recent advances in designing plant regulatory... synthetic [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1567659/full]
- 35. What Have We Learned About Synthetic Promoter Construction? - PubMed [https://pubmed.ncbi.nlm.nih.gov/27557757/]
- 36. Design of orthogonal systems for modulating regulatory ... gene [https://escholarship.org/uc/item/95x093wc]
- 37. Gene Expression Market Size and YoY Growth Rate, 2025-2032 [https://www.coherentmarketinsights.com/industry-reports/gene-expression-market]
- 38. Multiplexed Gene Tuning with Expression Synthetic... Orthogonal [https://pmc.ncbi.nlm.nih.gov/articles/PMC7197936/]
- 39. with Negative Feedback... | SpringerLink Linearizer Gene Circuits [https://link.springer.com/protocol/10.1007/978-1-61779-086-7_5]
- 40. Application of combinatorial optimization ... | Nature Communications [https://www.nature.com/articles/s41467-020-16175-y?error=cookies_not_supported]
- 41. Directed Evolution [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/protein-biology/protein-expression/directed-evolution]
- 42. A primer to directed evolution: current methodologies and future directions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10074555/]
- 43. An orthogonal generating all... transcription mutation system [https://pubmed.ncbi.nlm.nih.gov/40593783/]
- 44. of an Directed engine for... evolution orthogonal transcription [https://scholars.houstonmethodist.org/en/publications/directed-evolution-of-an-orthogonal-transcription-engine-for-prog]
- 45. Design of orthogonal systems for modulating regulatory ... gene [https://pubmed.ncbi.nlm.nih.gov/32424304/]
- 46. Directed evolution methods for overcoming trade-offs between protein activity and stability - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7384606/]
- 47. -Derived Transcription Factors for Orthologous Plant of... Regulation [https://pubmed.ncbi.nlm.nih.gov/28531348/]
- 48. åˆæˆ 生 å¦æ—¶ä»£åŸºäºŽ 物 细èŒçš„工业底盘细胞 éž çŽ°çŠ¶ä¸Žå±•æœ› 模 å¼ ç ” 究 [https://cjb.ijournals.cn/html/cjbcn/2021/3/gc21030874.htm]
- 49. Parallel lines: why orthogonal validation strengthens gene-modulation research [https://www.nature.com/articles/d42473-021-00353-7]
- 50. What is Orthogonal Array Testing? | Why & How to Perform? [https://testsigma.com/blog/orthogonal-array-testing/]
- 51. Graded dose - response curves | Deranged Physiology [https://derangedphysiology.com/main/cicm-primary-exam/pharmacodynamics/Chapter-412/graded-dose-response-curves]
- 52. Wide dynamic of VIPAR polymer gel dosimetry dose range [https://pubmed.ncbi.nlm.nih.gov/11512616/]
- 53. How Do I Perform a Dose-Response Experiment? - FAQ 2188 - GraphPad [https://www.graphpad.com/support/faq/how-do-i-perform-a-dose-response-experiment/]
- 54. Dose–response relationship - Wikipedia [https://en.wikipedia.org/wiki/Dose%E2%80%93response_relationship]
- 55. Biosensor-driven, model-based optimization of the orthogonally ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-022-01775-8]
- 56. DeSigN: connecting gene expression with therapeutics for drug repurposing and development - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5310278/]
- 57. The role of gene expression profiling in drug discovery - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1471489211000828]
- 58. The benefit of dose -exposure- response modeling in the estimation of... [https://arxiv.org/html/2508.04186v1]
- 59. Cell-to-cell variability in the propensity to transcribe explains correlated fluctuations in gene expression - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4662655/]
- 60. Mastering Gene in Expression Engineering Noise Genetic [https://www.numberanalytics.com/blog/mastering-gene-expression-noise]
- 61. Independent control of mean and noise by convolution of gene ... [https://www.nature.com/articles/s41467-021-27070-5?error=cookies_not_supported&code=6a067cc8-6fe0-4e50-8c41-b0d5a255c4cb]
- 62. in Eukaryotic Noise | PLOS Biology Minimization Gene Expression [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.0020137]
- 63. [PDF] Orthogonal control of expression mean and... | Semantic Scholar [https://www.semanticscholar.org/paper/Orthogonal-control-of-expression-mean-and-variance-Dey-Foley/d141ff0f31ca86a5c016914b17477e154e0432f5]
- 64. of Regulation - cell - to in divergent cell variability gene expression [https://www.nature.com/articles/ncomms11099?error=cookies_not_supported&code=ac72dc3e-f114-4f09-ad11-647e9865fa78]
- 65. Measuring cell-to-cell expression variability in single-cell RNA-sequencing data: a comparative analysis and applications to B cell aging | Genome Biology | Full Text [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-03036-2]
- 66. Reduction in Resource-Coupled Multi-Module Noise Circuits... Gene [https://pmc.ncbi.nlm.nih.gov/articles/PMC11987709/]
- 67. Combinatorial assembly platform enabling engineering of genetically ... [https://pubmed.ncbi.nlm.nih.gov/34554258/]
- 68. “ Metabolic †explained: stress symptoms and its related... burden [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02370-9]
- 69. How to be a Better Troubleshooter in Your Laboratory | GoldBio [https://goldbio.com/articles/article/how-to-be-a-better-troubleshooter]
- 70. CRISPR-Based Genome Editing Support—Troubleshooting | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/genome-editing-support-center/crispr-based-genome-editing-support/crispr-based-genome-editing-support-troubleshooting.html]
- 71. Off-target genome editing - Wikipedia [https://en.wikipedia.org/wiki/Off-target_genome_editing]
- 72. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 73. Off-target effects in CRISPR/Cas9 gene editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10034092/]
- 74. CRISPR 101: Off-Target Effects [https://blog.addgene.org/crispr-101-off-target-effects]
- 75. Strategies to Increase On - Target and Reduce Off - Target of... Effects [https://pubmed.ncbi.nlm.nih.gov/31366028/]
- 76. Strategies to Increase On-Target and Reduce Off-Target Effects of the CRISPR/Cas9 System in Plants - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6696359/]
- 77. On-target and off-target-based toxicologic effects - PubMed [https://pubmed.ncbi.nlm.nih.gov/23085982/]
- 78. - Fine the expression of target genes using a DDI2 promoter... tuning [https://www.nature.com/articles/s41598-019-49000-8?error=cookies_not_supported&code=71b7551c-9e92-4b26-b8a9-5621c75fbf75]
- 79. Delaying production with prokaryotic inducible expression systems [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02523-w]
- 80. A longitudinal study of gene in healthy individuals expression [https://bmcmedgenomics.biomedcentral.com/articles/10.1186/1755-8794-2-33]
- 81. Engineering receptors in the secretory pathway for orthogonal ... [https://www.nature.com/articles/s41467-022-35161-0?error=cookies_not_supported&code=ff17c213-943e-4c47-b13d-bbce2543f102]
- 82. Standardization of Gene Expression Quantification by Absolute Real-Time qRT-PCR System Using a Single Standard for Marker and Reference Genes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2935814/]
- 83. inducible control of Cas13 circuits enables programmable... Orthogonal [https://pmc.ncbi.nlm.nih.gov/articles/PMC10055290/]
- 84. Antibody Enhanced- Validation Strategies [https://www.sigmaaldrich.com/CA/en/technical-documents/technical-article/protein-biology/immunohistochemistry/antibody-enhanced-validation]
- 85. Development of chromosome-based T RNA polymerase and... 7 [https://bioresourcesbioprocessing.springeropen.com/articles/10.1186/s40643-020-00342-6]
- 86. Towards T RNA polymerase ( 7 )-based expression system in... T 7 RNAP [https://pmc.ncbi.nlm.nih.gov/articles/PMC10234217/]
- 87. Platelet Matrix Metalloprotease-1 Mediates Thrombogenesis By... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2807741/]
- 88. Resources for the design of CRISPR gene editing experiments [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0823-x]
- 89. Design of orthogonal regulatory systems for modulating gene expression in plants | Nature Chemical Biology [https://www.nature.com/articles/s41589-020-0547-4]
- 90. Solved: Heterologous Gene Expression Problems [https://bitesizebio.com/10285/heterologous-gene-expression-problems/]
- 91. Latest Developed Strategies to Minimize the Off-Target Effects in CRISPR-Cas-Mediated Genome Editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7407193/]
- 92. Frontiers | Troubleshooting to Guide Intrinsically... Expressing [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2018.00118/full]
- 93. Comparative expression profiling reveals widespread coordinated evolution of gene expression across eukaryotes | Nature Communications [https://www.nature.com/articles/s41467-018-07436-y]
- 94. Gene expression and ancestral transcriptome... phylogenies [https://elifesciences.org/articles/74297]
- 95. Combining Phylogenetic -Based and Machine... | PLOS One Profiling [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0075940]
- 96. Frontiers | Metabolic Pathway Assignment of Plant Genes based on... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2017.01831/full]
- 97. Exopolysaccharide-associated protein sorting in environmental organisms: the PEP-CTERM/EpsH system. Application of a novel phylogenetic profiling heuristic - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1569441/]
- 98. ProPhylo: partial phylogenetic profiling to guide protein family construction and assignment of biological process | BMC Bioinformatics | Full Text [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-12-434]
- 99. Overview of Recombinant Protein Expression Systems – Future Fields [https://futurefields.io/blogs/flylab/overview-of-recombinant-protein-expression-systems]
- 100. Improvement strategies for transient gene in expression ... mammalian [https://link.springer.com/article/10.1007/s00253-024-13315-y]
- 101. A Comparative Analysis of Recombinant Protein Expression in Different Biofactories: Bacteria, Insect Cells and Plant Systems - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4401374/]
- 102. Bacterial Protein Expression: Key Differences... Mammalian vs [https://synbio-tech.com/mammalian-vs-bacterial-protein-expression]
- 103. Differences between microbial fermentation & mammalian culture cell [https://www.susupport.com/blogs/knowledge/differences-between-microbial-fermentation-mammalian-cell-culture]
- 104. Culture for Recombinant Proteins - BioProcess International Plant Cell [https://www.bioprocessintl.com/cell-line-development/plant-cell-cultures-and-cell-lines-for-recombinant-protein-expression]
- 105. The potential of cold-shock promoters for the expression of... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9800685/]
- 106. The cumate gene -switch: a system for regulated expression in... [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/1472-6750-6-43]
- 107. Systems and strategies for plant protein expression - ScienceDirect [https://www.sciencedirect.com/science/chapter/bookseries/abs/pii/S0076687922003275]
- 108. Systems and strategies for plant protein expression - PubMed [https://pubmed.ncbi.nlm.nih.gov/36710015/]